
Introduction
While patients diagnosed with germinal center B (GCB) cell of origin (COO) diffuse large B cell lymphoma (DLBCL)/high grade B cell lymphoma (HGBL) may experience more favorable survival outcomes following receipt of first-line rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) as compared to those diagnosed with activated B cell (ABC) COO DLBCL/HGBL [1], comprehensive genomic analyses have revealed that tumors from approximately 50% of newly-diagnosed GCB DLBCL/HGBL patients can be assigned to a poor-risk subgroup (Cluster 3, EZB) which is associated with inferior survival following receipt of first line R-CHOP [2, 3]. Additionally, 10–25% of tumors from newly-diagnosed GCB DLBCL/HGBL patients demonstrate poor-risk gene expression profiles which assign them to a subgroup (double hit signature, molecular high grade) which is also associated with inferior survival following receipt of first line R-CHOP [4, 5].
These poor-risk GCB DLBCL/HGBL subgroups are characterized by recurring genetic mutations, which can be detected by clinical laboratory mutation analysis (CLMA). Thus, we sought to analyze the results of CLMA performed at our institution on tumors from patients with newly-diagnosed GCB DLBCL/HGBL previously-treated with first line immunochemotherapy to determine the frequency and predictive value of the presence of individual genetic mutations associated with these experimentally-defined poor-risk subgroups.
Results
CLMA was performed on 59 tumor biopsies (48 formalin-fixed paraffin-embedded tissue, 11 bone marrow aspirate or biopsy, 2 body fluid) obtained from 2015–21, with 58 successful assays (98% success rate) and a median result turnaround time (TAT) of 16 days. Five biopsies were excluded due to documented request by treating clinician (3) or diagnosing pathologist (2) for purposes of medical decision making and 2 additional biopsies were excluded due to lack of clinical follow-up, resulting in analysis of 51 biopsies from 51 patients, for which Lymphoma Sequencing Panel (LSP) was performed on 32 and PennSeq™ Lymphoma Panel PSLP on 19. Baseline clinicopathologic characteristics are listed in Table 1 and a histogram of detected mutation number/frequency in Figure 1. Of note, 46 specimens expressed CD10 by IHC or flow cytometry and 35 harbored at least one mutation in a gene of interest. All patients treated with intensive immunochemotherapy (n = 15) received rituximab, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin (R-EPOCH).
Table 1: Baseline characteristics
| Characteristic | n (%) |
|---|---|
| Median age | 65 years |
| International Prognostic Index Score | |
| <3 | 28 (55) |
| ≥3 | 23 (45) |
| Histology | |
| DLBCL | 43 (84) |
| HGBL | 8 (16) |
| Transformed indolent lymphoma | |
| No | 43 (84) |
| Yes | 8 (16) |
| MYC IHC | |
| <40% | 23 (45) |
| ≥40% | 21 (41) |
| Unknown | 7 (14) |
| Double expressor lymphoma | |
| No | 35 (69) |
| Yes | 10 (20) |
| Unknown | 6 (11) |
| MYC rearrangement by FISH | |
| No | 40 (78) |
| Yes | 10 (20) |
| Unknown | 1 (2) |
| Double hit lymphoma | |
| No | 45 (88) |
| Yes | 5 (10) |
| Unknown | 1 (2) |
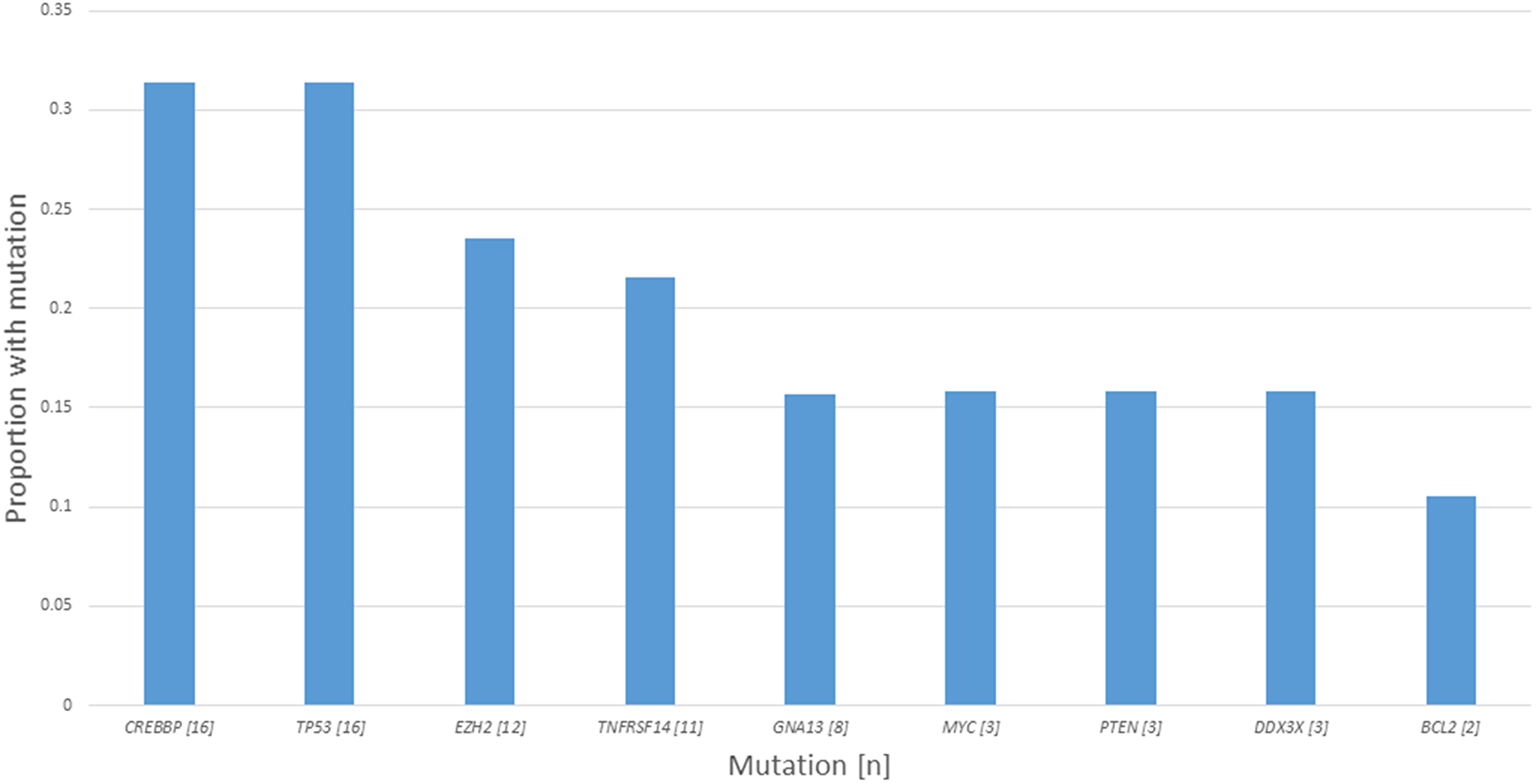
Figure 1: Number and frequency of detected mutations of interest.
For all 51 biopsies analyzed, there were a total of 87 mutations characterized as 56 missense, 17 frameshift, 11 nonsense and 3 splice site, and 35 biopsies harbored a mutation of a gene of interest with 32 biopsies a mutation of a gene of interest with gain or loss of function predicted. The median number of mutations of genes of interest was 1 (range 0–6) and mutations of genes of interest with gain or loss of function predicted was 1 (range 0–4). In total, there were 74 occurrences of mutations of genes of interest (counting duplicate mutations in the same gene in the same biopsy only once), with 60 predicted to result in gain or loss of gene function. For CREBBP, 21 mutations were characterized as 10 missense, 6 frameshift, 4 nonsense and 1 splice site with loss of function predicted to result in 15/16 biopsies. For TP53, 18 mutations were characterized as 16 missense, 1 frameshift and 1 nonsense with loss of function predicted to result in 15/16 biopsies. For EZH2, 12 mutations were characterized as 11 missense and 1 frameshift with gain of function predicted to result in 11/12 biopsies. For TNFRSF14, 11 mutations were characterized as 2 missense, 5 frameshift, 3 nonsense, 1 splice site with loss of function predicted to result in 8/11 biopsies. For other genes of interest (GNA13, BCL2, DDX3X, MYC and PTEN), 24 mutations were characterized as 17 missense, 4 frameshift, 2 nonsense and 1 splice site with gain or loss of function predicted in 11/19 biopsies. A summary of mutation characteristics is depicted in Figure 2 and a summary of tumor characteristics by patient is depicted in Figure 3.
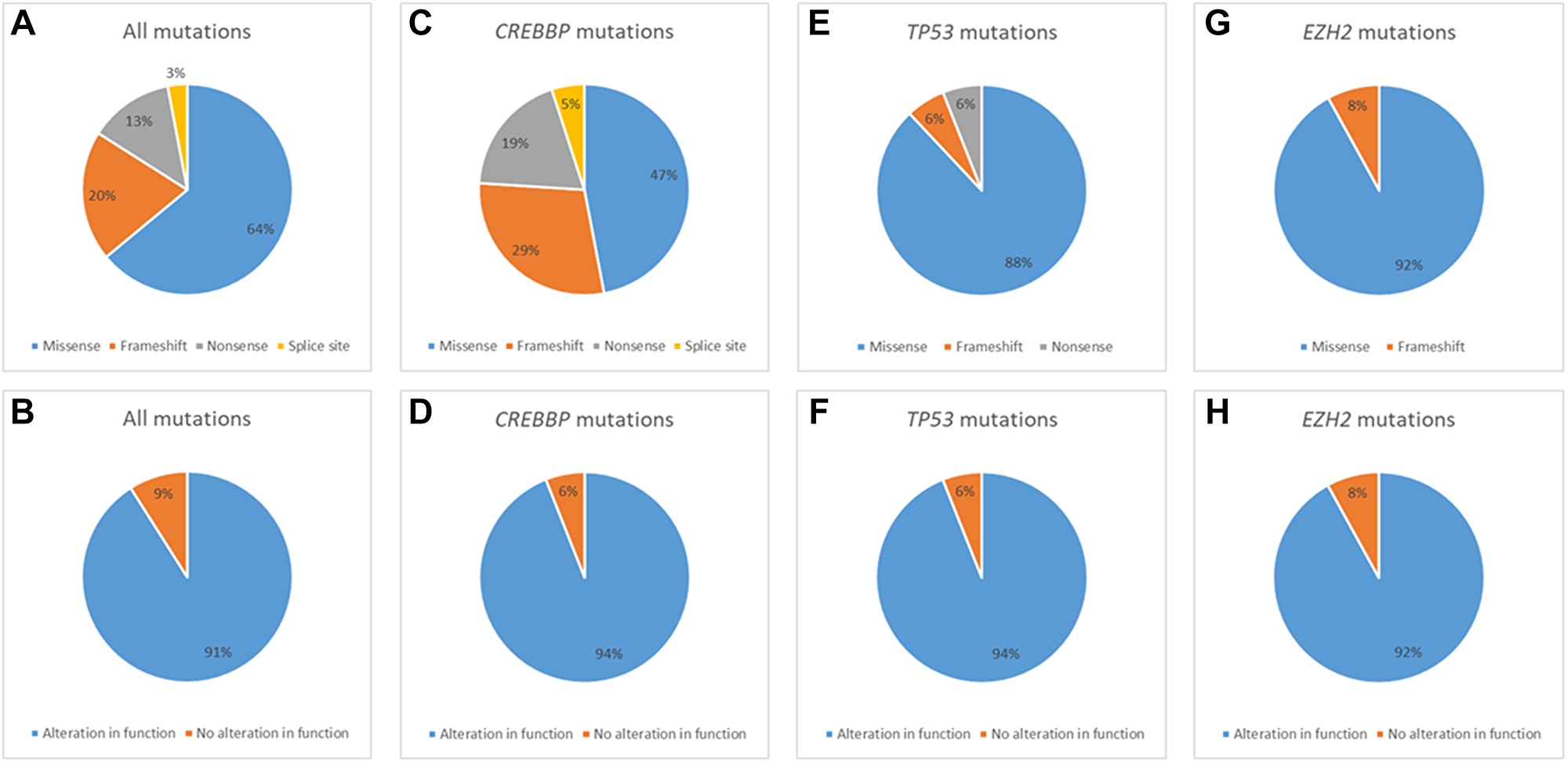
Figure 2: Mutation type and predicted impact of mutation on alteration of gene function for all mutations (A, B), CREBBP mutations (C, D), TP53 mutations (E, F) and EHZ2 mutations (G, H).
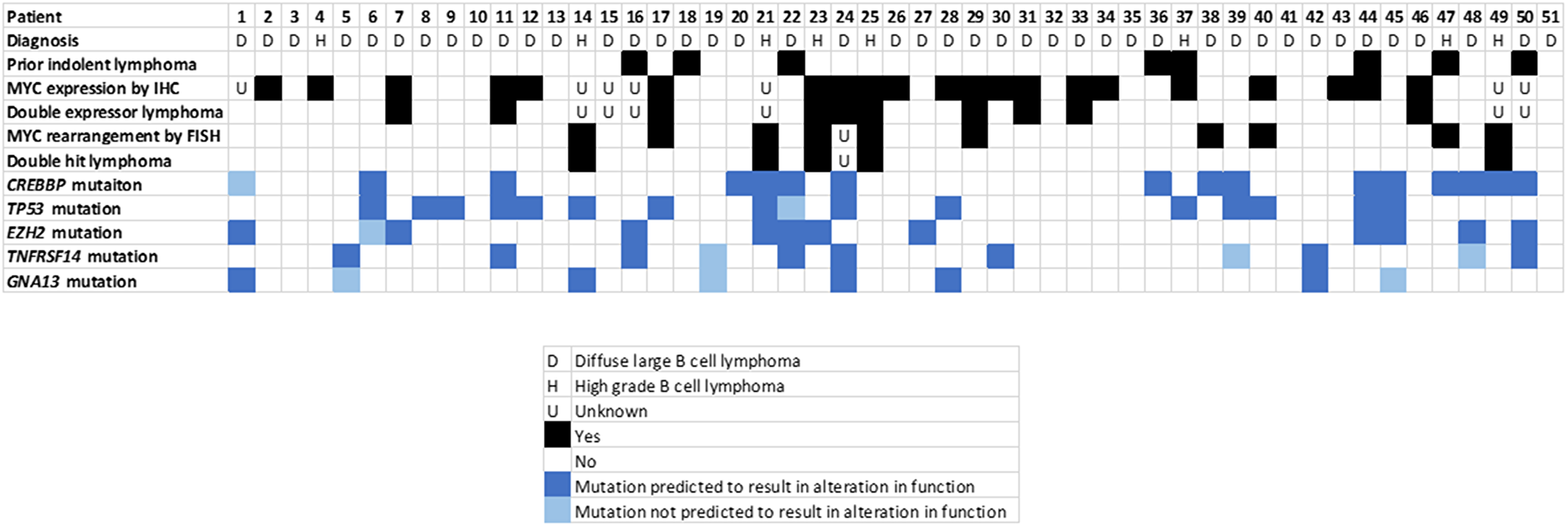
Figure 3: Tumor characteristics analyzed, by patient.
With a median follow-up of 25.2 months, the Kaplan Meier estimate of 2 year (y) DFS was 60% (95% confidence interval [CI] 41–74%) and 2y OS 80% (95% CI 64-89%) for all patients as depicted in Figure 4A and 4B, respectively. Characteristics listed in Table 1 as well as mutations detected in both LSP and PSLP predictive of disease relapse at 2y with P < 0.10 by univariate analysis were International Prognostic Index (IPI) score ≥3 vs. <3 (hazard ratio [HR] 2.3, 95% CI 0.86–6.5, P = 0.098), HGBL vs. DLBCL histology (HR 5.4, 95% CI 1.8–15.7, P = 0.002), double hit lymphoma vs. not (HR 9.0, 95% CI 2.6–30.8, P < 0.001) and CREBBP mutation vs. not (HR 2.6, 95% CI 0.98–7.0, P = 0.054). Univariate Cox regression analysis for death at 2 years performed for these factors revealed IPI score ≥3 vs. <3 (hazard ratio [HR] 11.3, 95% CI 1.4–90.5, P = 0.002), HGBL vs. DLBCL histology (HR 5.0, 95% CI 1.3–18.6, P = 0.02), double hit lymphoma vs. not (HR 8.0, 95% CI 1.8–36.0, P = 0.007) and CREBBP mutation vs. not (HR 2.8, 95% CI 0.76–10.5, P = 0.12). A meaningful multivariate analysis for factors predictive of disease relapse or death could not be carried out due to small sample size.
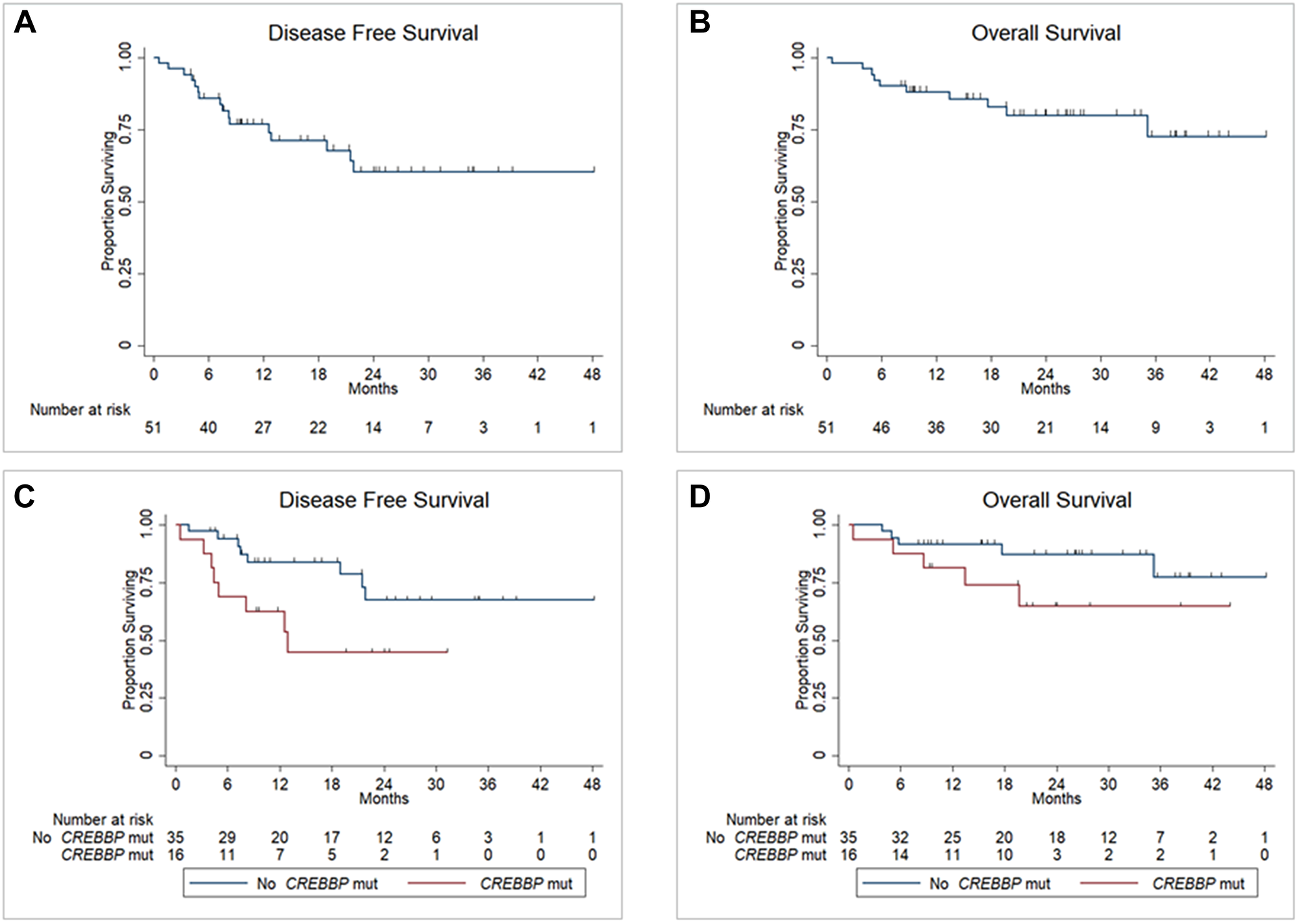
Figure 4: Disease free survival (A) and overall survival (B) for all patients; disease free survival (C) and overall survival (D) for all patients by CREBBP mutation status. Abbreviation: Mut: mutation.
Estimated 2y DFS was significantly lower for patients whose tumors demonstrated CREBBP mutation vs. not (45% [95% CI 18–68%] vs. 67% [95% CI 44–83%], P = 0.045) as depicted in Figure 4C, but not based upon the presence or absence of other mutations analyzed. Estimated 2y OS for patients whose tumors demonstrated CREBBP mutation vs. not did not differ significantly (65% [95% CI 34–84%] vs. 87% [95% CI 68–95%], P = 0.11) as depicted in Figure 4D. All patients with CREBBP mutation who relapsed did so ≤12 months from diagnosis. Characteristics analyzed from Table 1 and co-mutation frequency did not differ significantly when comparing patients whose tumors demonstrated CREBBP mutation vs. not, with the exception of EZH2 co-mutation (50% with CREBBP mutation vs. 11% without CREBBP mutation, P = 0.005). Estimated 2y DFS did not differ significantly for patients treated with R-CHOP (n = 36) as compared to R-EPOCH (n = 15) in the entire cohort (61% [95% CI 37–78%] vs. 58% [95% CI 29–79%], P = 0.72), nor for those whose tumors demonstrated CREBBP mutations (n = 12 vs. n = 4, 53% [95% CI 20–78%] vs. 25% [95% CI 9–67%], P = 0.42).
DISCUSSION
Our analysis demonstrates that the presence of CREBBP mutations in tumor biopsies from patients with newly-diagnosed GCB DLBCL/HGBL is associated with poorer DFS following treatment with front-line immunochemotherapy. CREBBP encodes an acetyltransferase protein which is a transcription factor responsible for several cellular functions including activation of p53 and repression of BCL6 [6], and therefore CREBBP mutations resulting in loss of function can promote lymphomagenesis. A high frequency of CREBBP mutations has also been demonstrated in biopsies from newly-diagnosed DLBCL/HGBL patients undergoing large-scale mutation analysis from the SAAK 38/07 prospective clinical trial cohort [7] as well as those treated in the GOYA study [8], although the predictive value of CREBBP mutations for DFS in GCB DLBCL/HGBL patients was not clearly stated in either analysis.
In terms of other mutations analyzed, the presence of TP53 mutations has been associated with a poor prognosis for patients diagnosed with lymphoid malignancies, including those with newly diagnosed GCB DLBCL treated with R-CHOP [9]. While our analysis does not support this finding, 2y DFS was 42% for patients with TP53 mutation with CREBBP co-mutation vs 63% for patients with TP53 mutation without CREBBP co-mutation analyzed in our series. This raises the possibility that TP53 mutations may predict for poorer DFS only in the presence of CREBBP co-mutation, perhaps due to a dual impact on TP53 transcription by inhibition of acetylation-driven TP53 activation mediated by loss-of-function CREBBP mutations as well as direct damage to TP53 mediated by loss-of-function TP53 mutations. Additionally, the other commonly detected mutated genes in our series, EZH2 and TNFRS14, were also not associated with DFS in patients with GCB DLBCL treated in the GOYA study [8].
Strengths of our analysis include a moderate sample size of biopsies on which CLMA was performed routinely without known selection bias at a single center on tumors from patients with comparable baseline characteristics to those of larger unselected DLBCL patient cohorts, including 45% with IPIs score ≥3 [10], 20% with double expressor lymphoma [11] and 10% with double hit lymphoma [12], which suggests that our finding may be applicable to the general population of newly-diagnosed GCB DLBCL/HGBL patients. Additionally, we demonstrate the high success rate and rapid result turnaround time of CLMA, which implies that results from this assay could be feasibly be incorporated into initial management decisions for these patients.
Weaknesses of our analysis include the use of two sequencing panels over time with one panel not detecting mutations in BCL2, DDX3X, MYC and PTEN and neither in KTM2D, both which limited the ability to detect potential survival differences based on the detection of mutations in all genes of interest. However, at least MYC mutations [13] nor BCL2 mutations [14] have been clearly associated with inferior survival outcomes in patients with newly-diagnosed GCB DLBCL/HGBL treated with R-CHOP. Additionally, patients were not treated with a uniform first-line therapy, although the result of the CALGB/Alliance 50303 trial demonstrated no difference in survival for patients with newly diagnosed DLBCL/HGBL receiving R-CHOP as compared to dose-adjusted R-EPOCH [15].
In conclusion, CLMA performed on tumor biopsies from patients with newly-diagnosed GCB DLBCL/HGBL revealed frequent mutations in CREBBP which were predicted to result in loss of function as well as a significantly lower rate of estimated DFS at 2 years. These findings support efforts to confirm the predictive value of CREBBP mutations in a larger cohort of newly-diagnosed GCB DLBCL/HGBL patients as well investigation of agents such as histone deacetylase inhibitors which may overcome loss of histone acetyltransferase function in patients with CREBBP-mutated GCB DLBCL/HGBL. Furthermore, we have reported the first known attempt to translate findings from experimental molecular assays through CLMA in order to identify the frequency and predictive value of individual gene mutations within a specific DLBCL/HGBL patient population, and our results support future efforts of this type given their potential to practically inform DLBCL/HGBL patient risk stratification as well as the design of future clinical trials incorporating agents targeted against specific gene mutations.
Materials and Methods
Inclusion criteria for this analysis were diagnosis of GCB DLBCL/HGBL (either de novo or transformed indolent lymphoma but without receipt of prior cytotoxic chemotherapy), receipt of either R-CHOP or an intensive first-line immunochemotherapy and performance of mutation analysis with one of two lymphoma-specific gene sequencing panels at the Penn Center for Personalized Diagnostics at the University of Pennsylvania (Lymphoma Sequencing Panel [LSP] from 2018–20 and PennSeq™ Lymphoma Panel [PSLP] from 2020–22). Exclusion criteria included history of chronic lymphocytic leukemia, clinician or pathologist request for mutation analysis and lack of adequate follow-up.
LSP and PSL were designed to detect single nucleotide variants (SNVs), insertions and deletions (indels), with a minimum tumor percentage of 10% tumor cells. Acceptable specimens are blood, bone marrow, fine needle aspirations and formalin fixed paraffin embedded tissue. DNA was extracted using the Agencourt FormaPure kit (PET, Beckman Coulter, Brea, CA, USA) or the DSP Mini Kit or Gentra PureGene Blood Kit for low volume samples (blood, bone marrow, Qiagen, Germantown, MD, USA). LSP is an amplicon based Next Generation Sequencing (NGS) oncology panel designed to target genes recurrently mutated in lymphomas. A minimum input of 10 ng of DNA was required for the Lymphoma 40 Kit (Illumina, Carlsbad, CA, USA). Libraries were prepared based on manufacturers instruction to target 40 genes with SNVs and indels called at 5% VAF in the following genes: ATM, B2M, BIRC3, BRAF, BTK, CARD11, CD79A, CD79B, CIITA, CREBBP, CXCR4, EGR2, EZH2, GNA13, ID3, IDH2, JAK3, KLF2, MAP2K1, MYD88, NFKBIE, NOTCH1, NOTCH2, PLCG1, PLCG2, POT1, RHOA, RPS15, RRAGC, SF3B1, SOCS1, STAT3, STAT5B, TCF3, TET2, TNFAIP3, TNFRSF14, TP53, TRAF3, and XPO1. Sequencing of the genes in bold included the entire coding region. Sequencing of the pooled sequencing libraries took place on the MiSeq (Illumina, Carlsbad, CA, USA), with fastqs run through a custom bioinformatics pipeline, were reviewed and reported using HGVS nomenclature (http://varnomen.hgvs.org/). All variants are reported based on the hg19 genome build. PSLP is a custom hybrid-capture-based Next Generation Sequencing (NGS) oncology panel. A minimum input of 100 ng for DNA derived from non-FFPE or 200 ng of DNA derived from FFPE DNA is sheared and then prepped into a whole-genome library. The library is target-enriched using capture probes covering exonic regions of 502 gene targets, a 9,000 SNP backbone and additional probes for biomarker detection, with 116 genes reported for SNVs, indels and limited copy number calling: ABL1, ASXL1, ATM, B2M, BCL2, BCOR, BCORL1, BIRC3, BRAF, BRCA1, BRCA2, BRIP1, BRINP3, BTK, CALR, CARD11, CBL, CD79A, CD79B, CDKN2A, CEBPA, CIITA, CREBBP, CSF1R, CSF3R, CXCR4, DDX3X, DDX41, DICER1, DNMT3A, EGR2, ERCC4, ETV6, EZH2, FANCA, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCL, FANCM, FBXW7, FLT3, GATA2, GNA13, GNAS, HNRNPK, ID3, IDH1, IDH2, IKZF1, IL7R, JAK2, JAK3, KIT, KLF2, KLHL6, KRAS, MAP2K1, PAK1, RIP142, MPL, MYC, MYCN, MYD88, NF1, NFKBIE, NOTCH1, NOTCH2, NPM1, NRAS, PALB2, PDGFRA, PHF6, PLCG1, PLCG2, POT1, PRPF40B, PTEN, PTPN11, RAD21, RAD51, RAD51C, RHOA, RIT1, RPS15, RRAGC, RUNX1, SETBP1, SF1, SF3A1, SF3B1, SLX4, SMC1A, SOCS1, SRSF2, STAG2, STAT3, STAT5B, TBL1XR1, TCF3, TERT, TET2, TNFAIP3, TNFRSF14, TP53, TPMT, TRAF3, U2AF1, U2AF2, WT1, XPO1, XRCC2, ZMYM3, ZRSR2. Sequencing of the pooled sequencing libraries took place on the NovaSeq (Illumina, Carlsbad, CA, USA), with fastqs run through a custom bioinformatics pipeline, were reviewed and reported using HGVS nomenclature (http://varnomen.hgvs.org/). All variants are reported based on the hg38 genome build. Gene mutations were further characterized by structural type and effect on gene function was primarily determined through the OncoKB [16] and PolyPhen-2 [17] databases as well as primary literature when available as referenced for individual mutations [9, 18].
Based on the aforementioned publications, genetic mutations of interest were BCL2, CREBBP, DDX3X, EZH2, GNA13, KMT2D, MYC, PTEN TNFRSF14 and TP53; however, BCL2, DDX3X, MYC and PTEN were only available in the PSLP and KTM2D was not available in either panel. Institutional standards for pathologic evaluation of tumor biopsies included immunohistochemical staining (IHC) for CD10, BCL6, MUM1, MYC and BCL2 among other makers, with cell of origin assigned by Hans algorithm, [19] as well as fluorescence in situ hybridization (FISH) for MYC rearrangement with reflex testing for BCL2 and BCL6 rearrangement if positive. Therapy was given at the discretion of the treating physician. Disease free survival (DFS) was defined as the interval between diagnosis of DLBCL/HGBL to DLBCL/HGBL relapse or last follow-up in remission. Overall survival (OS) was defined as the interval between diagnosis and time of death or last follow-up while alive. Data were censored on 7/1/22. Disease response by computed tomography with or without positron emission tomography was determined by the Revised Response Criteria for Malignant Lymphoma.[20] Survival curves were plotted using Kaplan-Meier estimates, and survival analysis was performed using the log-rank test. Univariate analysis was performed using Cox proportional-hazards regression. Categorical variables were analyzed by Fisher’s exact test. Statistical significance was defined as a two-tailed P value < 0.05 unless otherwise specified. All statistical analyses were performed using Stata version 13 (StataCorp, College Station, TX, USA). This protocol was approved by the Institutional Review Board of the University of Pennsylvania.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
CONFLICTS OF INTEREST
DJL – Research Funding: Curis, Triphase; Advisory Board Member: ADC Therapeutics, Calithera, Epizyme, Morphosys; Independent Data Monitoring Committee Member: Karyopharm. JJDM: none. SJS – Research funding: Celgene, Genetech/Roche, Incyte, Novartis, Abbvie, Adaptive Biotechnologies, DTRM, Juno Therapeutics, Merck, Pharmacyclics, TG Therapeutics; Consultancy: Acerta, Beigene, Celgene, Genetech/Roche, Incyte, Janssen, Legend Biotech, Loxo, Morphosys, MustangBio, Nordic, Nanovector, Novartis, Regeneron; Advisory Board Member: Regeneron. SDN – Research funding: Pharmacyclics, Roche, Rafael, FortySeven/Gilead. JNG – Research Funding: Loxo Oncology; Consultancy: Genetech, Abbvie. SKB – Consultancy: Affimed, Daiichi Sankyo, Kyowa Kirin; Independent Data Monitoring Committee Member: Janssen; Honoraria: Kyowa Kirin, Seagen, Acrotech. JS – Research Funding: Adaptive, Astra Zeneca, BMS, Incyte, Merck, Pharmacyclics, Seagen, TG Therapeutics; Advisory Board Member: Adaptive, Astra Zeneca, Incyte; Consultancy: Atara, BMS, Genmab, Pharamcyclics, Seagen. EAC – Consultancy: Novartis, Beigene, KITE, Tessa, Juno/BMS. MSL – Honoraria: EUSA Pharma.
Ethical statement and consent
This protocol was approved by the institutional review board of the University of Pennsylvania and conducted in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with International Conference on Harmonization Good Clinical Practice guidelines. Waiver of informed consent obtained.
References
1. Lenz G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B, Goldschmidt N, Iqbal J, Vose J, Bast M, Fu K, et al, and Lymphoma/Leukemia Molecular Profiling Project. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008; 359:2313–23. https://doi.org/10.1056/NEJMoa0802885. [PubMed].
2. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, Staiger AM, Wala JA, Ducar MD, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018; 24:679–90. https://doi.org/10.1038/s41591-018-0016-8. [PubMed].
3. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, Hodson DJ, Xiao W, Yu X, et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med. 2018; 378:1396–407. https://doi.org/10.1056/NEJMoa1801445. [PubMed].
4. Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, Rushton C, Tang J, Thomas N, Slack GW, Farinha P, Takata K, Miyata-Takata T, et al. Double-Hit Gene Expression Signature Defines a Distinct Subgroup of Germinal Center B-Cell-Like Diffuse Large B-Cell Lymphoma. J Clin Oncol. 2019; 37:190–201. https://doi.org/10.1200/JCO.18.01583. [PubMed].
5. Sha C, Barrans S, Cucco F, Bentley MA, Care MA, Cummin T, Kennedy H, Thompson JS, Uddin R, Worrillow L, Chalkley R, van Hoppe M, Ahmed S, et al. Molecular High-Grade B-Cell Lymphoma: Defining a Poor-Risk Group That Requires Different Approaches to Therapy. J Clin Oncol. 2019; 37:202–12. https://doi.org/10.1200/JCO.18.01314. [PubMed].
6. Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011; 471:189–95. https://doi.org/10.1038/nature09730. [PubMed].
7. Juskevicius D, Jucker D, Klingbiel D, Mamot C, Dirnhofer S, Tzankov A. Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J Hematol Oncol. 2017; 10:70. https://doi.org/10.1186/s13045-017-0438-7. [PubMed].
8. Bolen CR, Klanova M, Trneny M, Sehn LH, He J, Tong J, Paulson JN, Kim E, Vitolo U, Di Rocco A, Fingerle-Rowson G, Nielsen T, Lenz G, Oestergaard MZ. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: data from the phase III GOYA study. Haematologica. 2020; 105:2298–307. https://doi.org/10.3324/haematol.2019.227892. [PubMed].
9. Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood. 2012; 120:3986–96. https://doi.org/10.1182/blood-2012-05-433334. [PubMed].
10. Sehn LH, Berry B, Chhanabhai M, Fitzgerald C, Gill K, Hoskins P, Klasa R, Savage KJ, Shenkier T, Sutherland J, Gascoyne RD, Connors JM. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007; 109:1857–61. https://doi.org/10.1182/blood-2006-08-038257. [PubMed].
11. Johnson NA, Slack GW, Savage KJ, Connors JM, Ben-Neriah S, Rogic S, Scott DW, Tan KL, Steidl C, Sehn LH, Chan WC, Iqbal J, Meyer PN, et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol. 2012; 30:3452–59. https://doi.org/10.1200/JCO.2011.41.0985. [PubMed].
12. Barrans S, Crouch S, Smith A, Turner K, Owen R, Patmore R, Roman E, Jack A. Rearrangement of MYC is associated with poor prognosis in patients with diffuse large B-cell lymphoma treated in the era of rituximab. J Clin Oncol. 2010; 28:3360–65. https://doi.org/10.1200/JCO.2009.26.3947. [PubMed].
13. Xu-Monette ZY, Deng Q, Manyam GC, Tzankov A, Li L, Xia Y, Wang XX, Zou D, Visco C, Dybkær K, Li J, Zhang L, Liang H, et al. Clinical and Biologic Significance of MYC Genetic Mutations in De Novo Diffuse Large B-cell Lymphoma. Clin Cancer Res. 2016; 22:3593–605. https://doi.org/10.1158/1078-0432.CCR-15-2296. [PubMed].
14. Schuetz JM, Johnson NA, Morin RD, Scott DW, Tan K, Ben-Nierah S, Boyle M, Slack GW, Marra MA, Connors JM, Brooks-Wilson AR, Gascoyne RD. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012; 26:1383–90. https://doi.org/10.1038/leu.2011.378. [PubMed].
15. Bartlett NL, Wilson WH, Jung SH, Hsi ED, Maurer MJ, Pederson LD, Polley MC, Pitcher BN, Cheson BD, Kahl BS, Friedberg JW, Staudt LM, Wagner-Johnston ND, et al. Dose-Adjusted EPOCH-R Compared With R-CHOP as Frontline Therapy for Diffuse Large B-Cell Lymphoma: Clinical Outcomes of the Phase III Intergroup Trial Alliance/CALGB 50303. J Clin Oncol. 2019; 37:1790–99. https://doi.org/10.1200/JCO.18.01994. [PubMed].
16. Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, Chang MT, Chandarlapaty S, Traina TA, et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol. 2017; 2017:1–16. https://doi.org/10.1200/PO.17.00011. [PubMed].
17. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–49. https://doi.org/10.1038/nmeth0410-248. [PubMed].
18. de Andrade KC, Lee EE, Tookmanian EM, Kesserwan CA, Manfredi JJ, Hatton JN, Loukissas JK, Zavadil J, Zhou L, Olivier M, Frone MN, Shahzada O, Longabaugh WJR, et al. The TP53 Database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ. 2022; 29:1071–73. https://doi.org/10.1038/s41418-022-00976-3. [PubMed].
19. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004; 103:275–82. https://doi.org/10.1182/blood-2003-05-1545. [PubMed].
20. Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, Coiffier B, Fisher RI, Hagenbeek A, Zucca E, Rosen ST, Stroobants S, Lister TA, et al, and International Harmonization Project on Lymphoma. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007; 25:579–86. https://doi.org/10.1200/JCO.2006.09.2403. [PubMed].