
Introduction
Renal cell carcinoma (RCC) is the third most common type of urinary cancer in the world, and the sixth most common cancer type in Europe. Approximately 403 000 new cases and 175 000 deaths were registered worldwide in 2018 [1, 2]. Although most patients have localized disease at presentation, 20–40% experience either local or distant relapse, requiring systemic treatment. In addition, approximately 25% of all kidney cancer patients are presented with metastatic disease already at the time of diagnosis [3].
RCC is a malignancy comprising different histological subtypes with distinctive genetic and molecular alterations [4]. The three major histologic subtypes are clear cell renal carcinoma (ccRCC), which accounts for ~75% cases, papillary renal cell carcinoma (pRCC), which accounts for 15–20%, and chromophobe renal cell carcinoma (ChRCC), representing ~5% of all RCC cases. Principally, metastatic RCC (mRCC) is incurable and affected patients require systemic treatment which only confers palliative management.
The tumor development and cellular proliferation that take place in RCC may be accredited to changes in intracellular activity, involving many growth factors and growth factor receptors. The proangiogenic growth factors such as vascular endothelial growth factor (VEGF), VEGF receptor (VEGFR), platelet derived growth factor receptor (PDGFR), and basic fibroblast growth factor (bFGF) are specifically overexpressed in RCC, which indicates that the tumor is in a highly vascular state [5]. As a result, a range of therapies have been developed targeting these growth factor receptors via the phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathways, including TKIs (sorafenib, sunitinib, pazopanib), VEGF antibodies (bevacizumab) and mTOR inhibitors (everolimus, temsirolimus) [6–8]. In addition, immune checkpoint inhibitors (pembrolizumab, nivolumab, avelumab and ipilimumab) are available for systemic treatment of mRCC, with promising results [9]. Combination treatment with checkpoint-inhibitors and TKIs are the new standard treatment in the first-line setting, as well as the combination of different checkpoint inhibitors (immunotherapy doublet) for some subgroups of patients. Except for the immunotherapy doublet, virtually all lines of systemic RCC treatment recommended by clinical practice guidelines consist of TKIs. Despite a number of new treatment options improving RCC patients’ disease control rates and survival, the lack of useful biomarkers remains a major clinical concern. Cancer progression during TKI therapy in mRCC is inevitable and subsequent treatment lines with TKIs usually yield ever-shortening progression free survival (PFS) as RCC clones resistant to the applied TKIs are positively selected, rendering the RCC increasingly therapy resistant. Of note, both TKI-induced benefits and toxicity display remarkable inter-individual variation. Where some patients respond well for several months and even years from one TKI without noteworthy toxicity, others may have lower quality of life due to fatigue, diarrhea, hypertension, sore hands and feet, and oropharyngeal ulcers without affecting their progression to mRCC. Clinical parameters and predefined clinical risk groups are established for first line treatment [10, 11], but do not represent a substitute for tumor- and patient- specific biomarkers predicting the likelihood of clinical benefit for distinct TKIs.
Thus, the aim of this study was to expand the understanding of the tyrosine kinome of human kidney cancer before and during TKI therapy to identify possible biomarkers of clinical relevance that might help to select the optimal patient groups for therapeutic interventions involving targeting compounds. In the present study, we focused primarily on the overall tyrosine kinase activity patterns in RCC and the effects of ex vivo TKIs. At the same time, we were looking for particularly important pathways and potential novel therapeutic targets.
Results
Tyrosine kinase activity profiles in cancer and normal kidney tissue
Unsupervised heatmap analysis of kinase substrate phosphorylation profiles showed distinct differences in phosphorylation patterns between normal and cancerous kidney tissue using log2-transformed values (Figure 1). We identified 36 kinase substrates that had significantly different phosphorylation profiles (False discovery rate (FDR) < 0.05) in normal and cancer tissue (Table 1). Twenty substrates showed higher kinase activity in cancer, whereas 16 substrates showed significantly lower kinase activity in cancer compared to normal kidney tissue (FDR < 0.05). Through pathway analysis we identified that most kinase substrates that exhibited high kinase activity were part of the PI3K-akt pathway (p.value = 3.1E-3, Benjamini = 9.0E-2), whereas kinase substrates within the Rap1 signaling pathway (p.value = 1.7E-9, Benjamini= 1.9E-7) showed lower kinase activity in cancer, and higher in normal kidney tissue. We could not identify any significant differences in phosphorylation profiles between different histological groups, or other clinical parameters (age, gender, risk profile etc.).
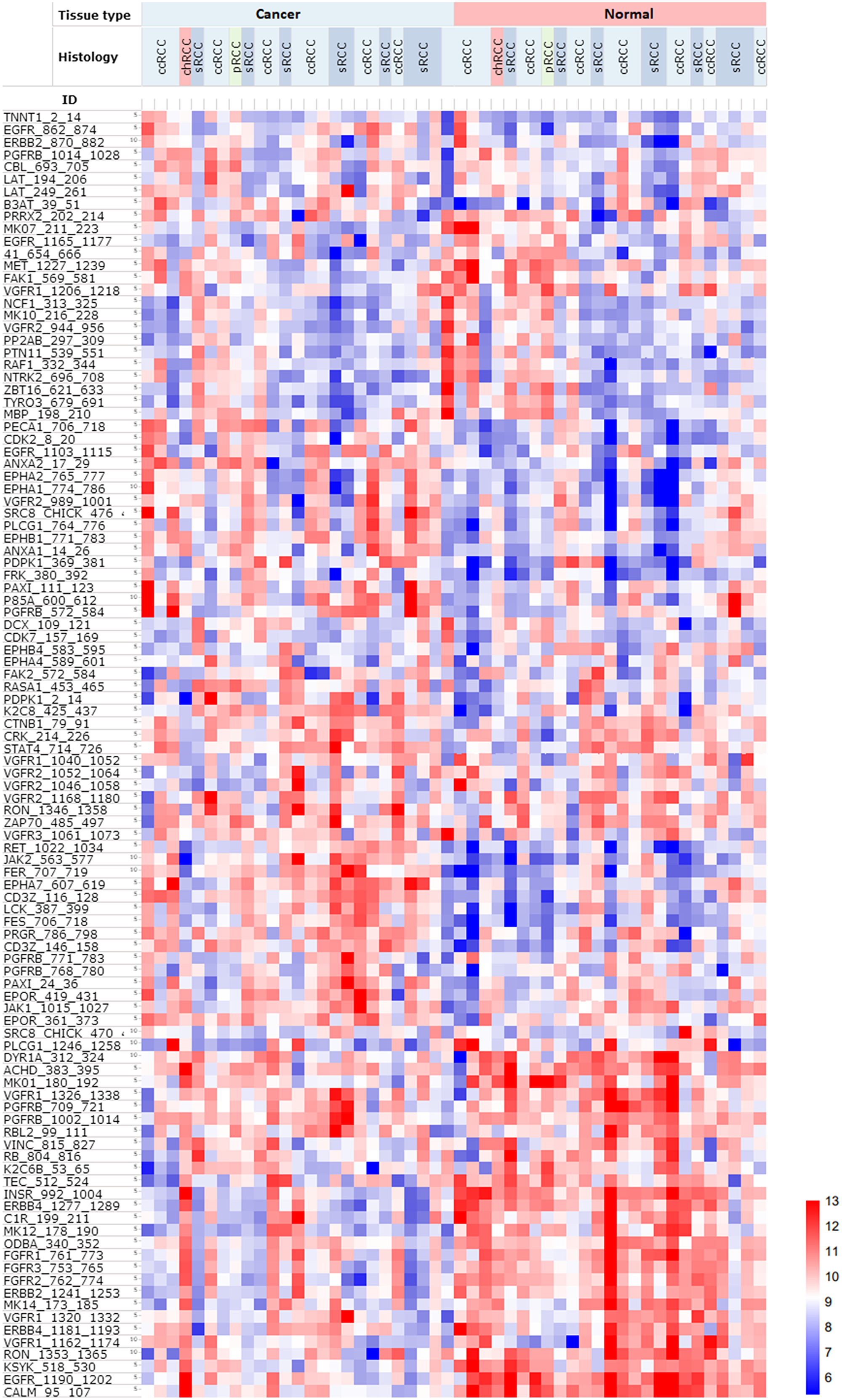
Figure 1: Heatmap of PTK phosphorylation profiles of 25 RCC patients, including malignant and matched normal tissue from the same patient (n = 50). There is a clear separation of normal and cancer kidney tissue, but no significant differences amongst the different histological types. Red indicate higher phosphorylation, whereas blue indicate lower phosphorylation of PTK. Samples are displayed on the horizontal axis, whereas kinase substrates are displayed on the vertical axis. Abbreviations: ccRCC: clear cell carcinoma; chRCC: chromophobe renal cell carcinoma; pRCC: papillary renal cell carcinoma; sRCC: renal cell carcinoma with sarcomatoid features.
Table 1: Significant kinase substrates (FDR < 0.05) between normal and RCC tissue
| Substrate ID | Description | p.value | FDR | Delta |
|---|---|---|---|---|
| CD3Z_116_128 | T-cell surface glycoprotein CD3 zeta chain precursor | 1,9E-04 | 1,5E-03 | 0,296 |
| CD3Z_146_158 | T-cell surface glycoprotein CD3 zeta chain precursor | 4,0E-04 | 2,6E-03 | 0,278 |
| ANXA2_17_29 | Annexin A2 | 1,2E-03 | 6,6E-03 | 0,255 |
| EPHA1_774_786 | Ephrin type-A receptor 1 precursor | 1,0E-02 | 3,4E-02 | 0,241 |
| EPHA7_607_619 | Ephrin type-A receptor 7 precursor | 3,6E-04 | 2,5E-03 | 0,232 |
| FES_706_718 | Proto-oncogene tyrosine-protein kinase Fes/Fps | 6,1E-05 | 9,0E-04 | 0,219 |
| JAK2_563_577 | Tyrosine-protein kinase JAK2 | 5,8E-05 | 9,0E-04 | 0,209 |
| EPHA2_765_777 | Ephrin type-A receptor 2 precursor | 1,6E-02 | 4,7E-02 | 0,203 |
| CDK2_8_20 | Cell division protein kinase 2 | 1,8E-03 | 8,3E-03 | 0,200 |
| RET_1022_1034 | Proto-oncogene tyrosine-protein kinase receptor ret precursor | 1,4E-04 | 1,4E-03 | 0,182 |
| PECA1_706_718 | Platelet endothelial cell adhesion molecule precursor | 2,9E-03 | 1,2E-02 | 0,178 |
| FER_707_719 | Proto-oncogene tyrosine-protein kinase FER | 1,6E-04 | 1,4E-03 | 0,174 |
| PDPK1_369_381 | 3-phosphoinositide-dependent protein kinase 1 | 8,6E-03 | 2,9E-02 | 0,174 |
| LCK_387_399 | Proto-oncogene tyrosine-protein kinase LCK | 1,2E-03 | 6,6E-03 | 0,146 |
| K2C8_425_437 | Keratin, type II cytoskeletal 8 | 2,2E-03 | 9,6E-03 | 0,143 |
| FRK_380_392 | Tyrosine-protein kinase FRK | 7,0E-03 | 2,5E-02 | 0,131 |
| ANXA1_14_26 | Annexin A1 | 1,4E-02 | 4,3E-02 | 0,119 |
| EPHB1_771_783 | Ephrin type-B receptor 1 precursor | 3,2E-03 | 1,2E-02 | 0,113 |
| B3AT_39_51 | Band 3 anion transport protein | 1,3E-02 | 3,9E-02 | 0,106 |
| EPOR_361_373 | Erythropoietin receptor precursor | 1,1E-02 | 3,5E-02 | 0,094 |
| TEC_512_524 | Tyrosine-protein kinase Tec | 8,7E-03 | 2,9E-02 | −0,076 |
| MK14_173_185 | Mitogen-activated protein kinase 14 | 1,6E-03 | 8,0E-03 | −0,153 |
| RON_1353_1365 | Macrophage-stimulating protein receptor precursor | 1,5E-03 | 7,7E-03 | −0,163 |
| MK01_180_192 | Mitogen-activated protein kinase 1 | 3,2E-03 | 1,2E-02 | −0,187 |
| MK12_178_190 | Mitogen-activated protein kinase 12 | 1,4E-04 | 1,4E-03 | −0,192 |
| ERBB2_1241_1253 | Receptor tyrosine-protein kinase erbB-2 precursor | 3,0E-04 | 2,2E-03 | −0,226 |
| FGFR3_753_765 | Fibroblast growth factor receptor 3 precursor | 8,6E-05 | 1,1E-03 | −0,247 |
| ODBA_340_352 | 2-oxoisovalerate dehydrogenase subunit alpha, mitochondrial precursor | 8,8E-04 | 5,1E-03 | −0,252 |
| KSYK_518_530 | Tyrosine-protein kinase SYK | 6,2E-06 | 1,3E-04 | −0,253 |
| C1R_199_211 | Complement C1r subcomponent precursor | 3,4E-03 | 1,2E-02 | −0,261 |
| FGFR2_762_774 | Fibroblast growth factor receptor 2 precursor | 1,2E-04 | 1,4E-03 | −0,291 |
| FGFR1_761_773 | Basic fibroblast growth factor receptor 1 precursor | 7,8E-04 | 4,8E-03 | −0,308 |
| ERBB4_1277_1289 | Receptor tyrosine-protein kinase erbB-4 precursor | 1,5E-06 | 4,0E-05 | −0,520 |
| EGFR_1190_1202 | Epidermal growth factor receptor precursor | 1,2E-06 | 4,0E-05 | −0,521 |
| CALM_93_105 | Calmodulin | 9,5E-08 | 4,9E-06 | −0,573 |
| INSR_992_1004 | Insulin receptor precursor | 8,2E-08 | 4,9E-06 | −0,676 |
Upstream kinases that might be responsible for differences observed between normal and cancerous tissue
In order to identify the kinases potentially responsible for the observed differences in kinase activity between normal and cancerous tissue, we performed upstream kinase analysis. Through this analysis we identified a list of predicted kinases, with several of them showing higher kinase activity in cancerous compared to normal kidney tissue (Figure 2). Amongst them were members of the Src family kinases such as Fyn, BLK, Src, LCK, Yes, HCK, Lyn and Fgr. Some kinases identified had lower kinase activity in cancer compared to normal tissue, amongst them Lmr1, EPHA7 and JAK1.
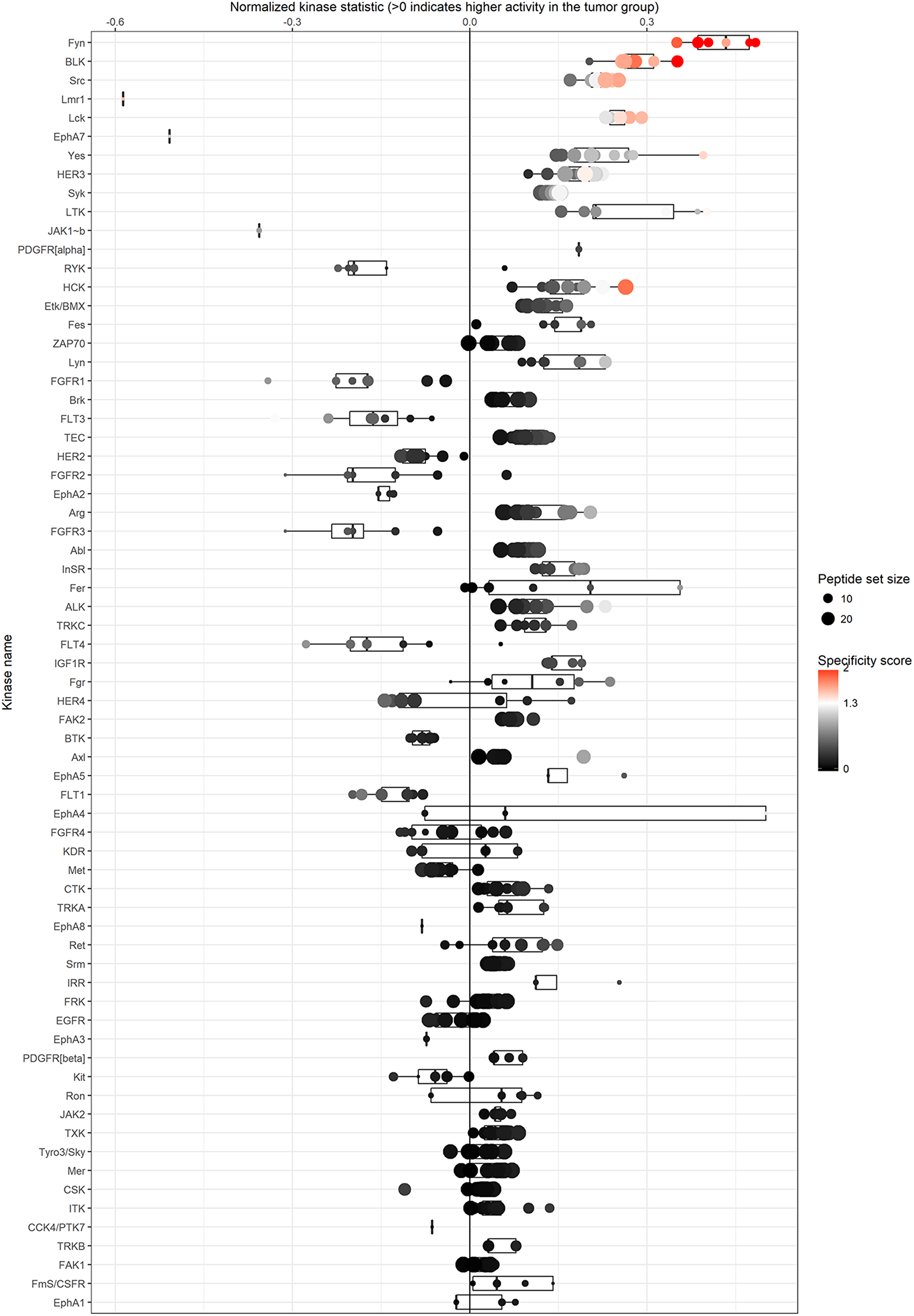
Figure 2: Upstream kinase analysis identifies kinases that might be responsible for the differences in phosphorylation profiles between normal and cancer tissue. Kinases at the top are the ones that are most likely to be involved compared to the ones at the bottom of the list. Src-kinase family members seems to be dominating at the top.
Tyrosine kinase profiles upon ex vivo inhibition with TKIs and analysis of affected pathways
The inhibition profiles obtained during ex-vivo exposure of cancer tissue lysates to four different TKIs (pazopanib, tivozanib, sunitinib and cabozantinib) showed altered kinase activity (Figure 3A). The heatmap shows the log fold change (LFC) values between control and treated RCC. Due to the low number of samples of pRCC and chRCC, we could not get a clear indication of the differences between histological kidney cancer types, and they were therefore excluded from the analysis with TKIs. From our results, tivozanib clearly exhibited stronger inhibitory potential compared to the other TKIs (Supplementary File 1).
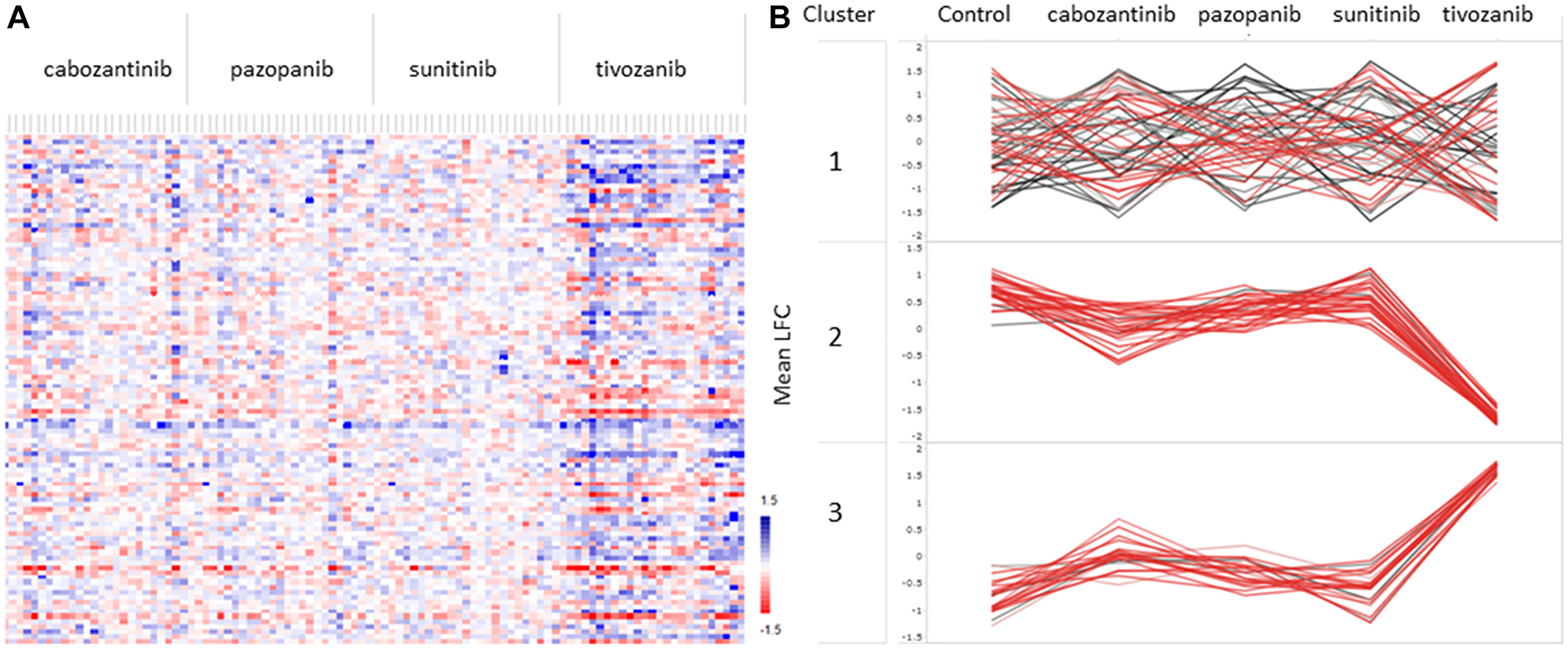
Figure 3: A Mean log fold change (LFC) values between RCC tissue without treatment (control) and with ex vivo treatment with four different tyrosine kinase inhibitors (TKIs): sunitinib, pazopanib, cabozantinib, and tivozanib. (A) Heatmap of mean LFC values of each kinase substrate in all samples treated with different TKIs. Red indicates positive LFC between control and TKI, whereas blue indicates negative LFC between control and TKI. (B) Kinase substrates were clustered in three groups, cluster 1, 2 and 3, based on PTK activity profiles in treated versus untreated (control) RCC samples. Cluster 2 and cluster 3 showed similar trends upon treatment with TKIs, whereas substrates in cluster 1 were affected differently by all TKIs in different samples. Values indicate the mean LFC between treated and untreated samples.
Further, we identified three distinct clusters of kinase substrates that were differently affected by the TKIs (Figure 3B, Supplementary File 2). Cluster 1 contained 54 kinase substrates that were differently affected by all four TKIs. Cluster 2 consisted of 29 kinase substrates showing reduced kinase activity upon treatment with all four of the TKIs. Pazopanib and sunitinib exhibited little inhibitory effects on most samples, whereas cabozantinib and tivozanib clearly showed inhibition of kinase substrates in cluster 2, exhibiting greater potency. Interestingly, 21 kinase substrates in cluster 3 showed increased kinase activity upon treatment with TKIs, with tivozanib showing greater potency in activating these kinases. Supplementary File 3 contains information on P-values and LFC values of each kinase substrate upon treatment with the different TKIs, and the cluster to which each kinase substrate belongs.
Pathway analysis was performed using UNIPROT accession number of each kinase substrate in cluster 2 and cluster 3 in order to identify the pathways affected by the TKIs (Supplementary File 2). Here, the results show that the PI3K pathway is overrepresented, especially in cluster 2. Kinase substrates encoding for endothelial growth factor receptor (EGFR), Src substrate protein p85 (p80) and VEGFR1 and VEGFR2 are both in cluster 2 and cluster 3, but the phosphorylation sites of these kinases are different in the two clusters. In addition to PI3K, kinases within the Rap1 and Ras signaling pathways are highly affected by the TKIs in cluster 2, whereas kinases within the neurotrophin and Ras signaling pathway are activated by the TKIs (Table 2).
Table 2: Selected kinase substrates in cluster 2 and 3 involved in different pathways
| Kinase substrate ID | UNIPROT Accession | Pathway | Cluster |
|---|---|---|---|
| EGFR_1190_1202 | P00533 | Rap1 /Ras/PI3K | 2 |
| FGFR1_761_773 | P11362 | Rap1 /Ras/PI3K | |
| FGFR3_753_765 | P22607 | Rap1 /Ras/PI3K | |
| PGFRB_1002_1014 | P09619 | Rap1 /Ras/PI3K | |
| PGFRB_709_721 | P09619 | Rap1 /Ras/PI3K | |
| VGFR1_1326_1338 | P17948 | Rap1/Ras/PI3K | |
| CALM_95_107 | P62158 | Rap1 / Ras | |
| VGFR2_1052_1064 | P35968 | Ras /PI3K | |
| EPOR_361_373 | P19235 | PI3K | |
| JAK1_1015_1027 | P23458 | PI3K | |
| RBL2_99_111 | Q08999 | PI3K | |
| CRK_214_226 | P46108 | Rap1 | |
| CTNB1_79_91 | P35222 | Rap1 | |
| VGFR2_1168_1180 | P35968 | Rap1 | |
| RASA1_453_465 | P20936 | Ras | |
| ZAP70_485_497 | P43403 | Ras | |
| MK07_211_223 | Q13164 | Neurotrophin | 3 |
| MK12_178_190 | P53778 | Neurotrophin | |
| NTRK2_696_708 | Q16620 | Neurotrophin | |
| RAF1_332_344 | P04049 | Neurotrophin / Ras | |
| MK10_216_228 | P53779 | Neurotrophin /Ras | |
| PLCG1_1246_1258 | P19174 | Neurotrophin /Ras | |
| PTN11_539_551 | Q06124 | Neurotrophin /Ras | |
| EGFR_1165_1177 | P00533 | Ras | |
| LAT_194_206 | O43561 | Ras | |
| VGFR1_1206_1218 | P17948 | Ras | |
| VGFR2_1046_1058 | P35968 | Ras | |
| VGFR2_944_956 | P35968 | Ras |
DISCUSSION
We identified significant differences in the overall PTK activity when comparing normal and cancer kidney tissue samples from patients diagnosed with RCC. Especially kinases within the PI3K pathway had higher activity in cancer compared to normal kidney tissue, but the PI3K pathway was also most affected by the TKIs in our in-vitro experiments. On the contrary, kinases within the Rap1 pathway were noted to be less active in cancer and higher in normal kidney tissue, but after ex vivo TKI treatment of cancer tissue, the Rap1 signaling pathway seemed to be the most affected. This indicates that these pathways play an important role in RCC patients and treatment response.
A recent study by Anderson et al. assessed the kinase activity profiles of ccRCC patients, and identified a number of kinase substrates different between normal and cancer kidney tissue [12]. However, although they had a smaller dataset of matched normal and cancerous tissue samples (n = 12), and their findings predominantly included serine/threonine kinases (STKs), our findings support that the kinase substrate encoding for the erythropoietin-producing hepatoma B1 (EPHB1) protein is higher phosphorylated in cancer tissue compared to normal tissue (Table 3). EPH proteins are involved in cell processes such as cell growth and differentiation, and have long been suggested as potential targets in cancer treatment as overexpression of EPH proteins have been found in several types of cancer, including kidney cancer [13]. High EPH-ephrin signaling has been shown to be involved in the pathogenesis and progression of ccRCC, with worse prognosis associated with higher expression of EPHA1, EPHA2 and EPHA7 [14, 15]. In our dataset, in addition to EPHB1, we also identified EPHA1, EPHA2 and EPHA7 that were highly phosphorylated in cancer compared to normal tissue, making these proteins as potential therapeutic targets. In addition, upstream kinase analysis confirmed by both our study and that of Anderson et al. [12], that members of the Src family kinase proteins are significantly different in normal and kidney cancer tissue, including Fyn, Src and Lyn, exhibiting higher kinase activity levels in cancer. Research involving RCC has shown that Src is the most highly expressed gene, followed by Lyn, Hck, Fgr and Fyn [16], which fits well with our observations. Interestingly, the over-expression of Src family members has also been observed to have important roles in other types of malignancy, including prostate, breast, colon, and lung cancer [17–20]. In breast cancer especially, Src overexpression has been correlated to poor survival [19] and resistance to therapy [21]. Furthermore, interaction between EPHB1 and Src has been shown to activate Ras/Raf/MAPK pathway [22], and is concordant with the results we have obtained throughout our analysis. Previous research involving analysis on several “omic” levels on a large number of ccRCC patients revealed that several genes within the PI3K/AKT pathway were often mutated (28% of cases) [23], suggesting that kinases within this pathway are important potential therapeutic targets. Our study had a limited number of patients with different histological subtypes, and therefore our findings did not show major differences between different subtypes, i.e. pRCC versus ccRCC, nor did we find any significant associations between PTK profiles and clinical parameters. For future research, larger number of patients should be included, including different histological types.
Table 3: Patient characteristics
| Characteristics | Number (n =) |
|---|---|
| Gender | |
| Female | 9 |
| Male | 16 |
| Age group | |
| Median (range) | 65 (43–79) |
| Histology | |
| Clear cell | 23 |
| Papillary | 1 |
| Chromophobe | 1 |
| IDMC/ MSKCC risk group | |
| Good | 10 |
| Intermediate | 11 |
| Poor | 1 |
| Non-metastatic disease | 3 |
In this study, we also explored the effects of four different TKIs on kinase activity in RCC that are commonly used in clinical settings, such as pazopanib, sunitinib, cabozantinib and tivozanib. Our analysis showed that both cabozantinib and tivozanib exhibited greater potency with regards to decreasing phosphorylation levels, but they also showed greater activation of certain kinases. Sunitinib and pazopanib on the other hand, did not show the same potency in decreasing phosphorylation levels in RCC samples. Based on our results, tivozanib exhibited the best inhibitory effect on PTKs. Tivozanib is a VEGF-inhibitor that has been extensively studied in the context of solid tumors and in advanced RCC through preclinical data and clinical trials but has only recently (late 2017) been approved as therapy for mRCC patients [24, 25]. Previous studies have shown that tivozanib is effective for patients that have previously received TKI treatment and is superior to other TKIs as it prolongs response of mRCC patients substantially [26]. For future studies, there should be research studying whether tivozanib given as a first line drug might improve lifespan of RCC patients, as it shows greater inhibitory potential, although a very low concentration of the drug was used for the in vitro experiments.
Furthermore, we identified two clusters of kinase substrates that were either negatively affected (cluster 2), or positively affected by all four TKIs (cluster 3). Our findings suggest that while some kinases are targeted specifically by the TKIs and are therefore inhibited, others in the same pathway will compensate and increase their activity. We have observed a similar phenomenon in our previous study with malignant melanoma and treatment with ex vivo BRAF-inhibitor, with some kinases being inhibited, whereas others being activated upon TKI treatment [27], revealing the complexity of the use of TKIs in the clinics. Furthermore, we identified that kinases within the Rap1 and Ras pathway were significantly affected by TKIs. Ras and Rap1, play critical roles in regulating T- cell proliferative responses. Ras plays an essential role in transmitting signals from the T-cell receptor (TCR) to activation of the Raf-1/ERK signaling cascade, which is required for T- cell proliferation, IL-2 production, and thymic maturation [28]. Rap1, is also activated in T-lymphocytes following TCR stimulation, and is a known suppressor of Ras-dependent transformation [29]. Our study also showed that kinases within the neurotrophin and Ras signaling pathway were activated upon TKI treatment. Activation of the neurotrophin pathway can stimulate the Ras, PI3K, phospholipase C-γ1 signaling pathways controlled through these proteins, including the MAP kinases [30]. The neurotrophin pathway has also been previously implied in immunity. In fact, neurotrophins and their receptors are key molecules in survival and functions of cells of both the innate and adaptive immune system [31]. This can possibly explain why combinations of TKI and immune checkpoint inhibitors work better as first line therapy for mRCC patients [32]. Furthermore, the tumor microenvironment (TME) has been suggested as an additional major player influencing on the effect and response of TKIs. Studies have shown that stromal cells in the TME exposed to TKIs produce cytokines, hormones, or growth factors that modulate the response of the tumor to TKIs [33, 34]. Interestingly, stromal cells can also secrete growth factors activating the MAPK and Pi3K/AKT/mTOR, leading to resistance of cancer cells [35, 36]. The role of TME in resistance to TKI treatment in RCC patients is however not fully explored and is an area that needs further investigations.
Materials and Methods
Patient material
In total, 324 nephrectomy samples were consecutively collected at the Akershus University Hospital from 2013 to 2019. From these, we selected 25 RCC patients suitable for analysis as we had matching normal kidney tissue available. Twenty-two nephrectomy samples were available from patients with either primary or recurrent metastatic disease, and three with non-metastatic disease (see Table 3 for patient characteristics and Supplementary File 3 for extensive information). Twenty-three patients were classified with ccRCC histology, whereas one patient with chRCC, and one with pRCC. An experienced pathologist assessed tumor and normal tissue collected from each patient.
Sample preparation
Tissue specimens from normal and cancer kidney tissue (n = 50) were sectioned with a cryostat (at –35°C) into 10 μm thick coupes, to a total volume of ∼15 mm3 for each sample. The number of coupes needed to get 15 mm3 for the experiment was calculated based on the surface area of the tissue specimen. Tissue samples were kept frozen at all times during the procedure, and to avoid contamination between tumor and normal tissue, all tumor tissue and normal tissue were sliced separately. The sectioned tissue was with 100–200 μl of mammalian protein extraction reagent (M-PER) buffer (Pierce Biotechnology, Inc., Rockford, IL), supplemented with phosphatase and protease inhibitors (Pierce Biotechnology, Inc.) The protein concentration of lysates was determined using the BCA assay (Pierce Biotechnology, Inc.).
Tyrosine kinase activity profiling
PTK profiling was performed using the Protein Tyrosine Kinase PamChip® Array for Pamstation®12 (PamGene International B.V., ‘s-Hertogenbosch, The Netherlands) at Akershus University Hospital, according to manufacturer’s protocol. Briefly, the assay contains 144 kinase substrates, representing approximately 100 different kinases. For each experimental run, 5 μg of protein sample lysate was added to the reaction mixture containing 1 × PK buffer, 10 mM DTT, 400 μM ATP, 1 × PTK additive (PamGene), 1:400 Halt Phosphatase Inhibitors (Thermo Fisher Scientific), 0.01% BSA (Pamgene), and fluorescein isothiocyanate-labeled antiphosphotyrosine antibody (PamGene), as previously described [27]. Based on pilot experiments of increasing concentrations of the different TKIs added ex vivo to kidney cancer tissue lysates, concentrations that resulted in ~50% inhibition of most kinase substrates were chosen for the main experiments. As a result, the PTK profiles of kidney cancer samples were assessed with and without the presence of four TKIs with following concentrations; 2.5 μM cabozantinib (XL184, BMS-907351) (Selleck Chemicals), 2.5 μM sunitinib (Sigma Aldrich), 10 μM pazopanib (Sigma Aldrich) and 2 μM tivozanib (AV-951) (Selleck Chemicals). Kidney cancer samples and normal tissue samples that were not treated with inhibitors (control) contained 2% Dimethyl Sulfoxide (DMSO) instead of TKIs. Initially, experiments were run in triplicates, but after confirmation of insignificant variation between technical replicates (coefficient of variation < 9%), most of our samples were not run in replicates to allow more individual samples to be used. However, for quality controls, some samples were run in duplicates or triplicates, where necessary.
Data analysis
All data was processed and analyzed in Bionavigator v.6 (PamGene) interfaced to the statistical program R 3.3.1 (R-project, https://www.rproject.org/). Analysis performed included quality check, log2 transformation of data and averaging of the kinase signal replicates. During quality check, we excluded arrays that showed clear defects (broken array etc.). Signal-positive spots had to show a positive trend in the phosphorylation time course. Substrates in which we could not detect a positive trend in >75% of the samples were excluded from further analysis, resulting in 104/144 kinase substrates eligible for further analysis. For samples in which we had several replicates, the values from each replicate were averaged to contain only a single value per sample per kinase substrate. To identify significant differences in kinase activity between groups, student’s T-test or Mann–Whitney U test was used wherever appropriate to obtain P-values and false discovery rate (FDR). False discovery rate (FDR) < 0.05 was considered significant.
Upstream kinase analysis
BioNavigator software v.6 (PamGene Inc.) was used to perform upstream kinase analysis in order to identify kinases that might be responsible for the differences in kinase activity observed between two groups, normal and cancer tissue. The method uses in silico predictions to identify upstream kinases through Kinexus Kinase Predictor (http://www.phosphonet.ca/), as previously described [37]. The method calculates a significance score or specificity score of a kinase Q = −10log [max (m/M, 1/M)], where m is the number of times out of M permutations that | τp | >| τ |, where τp is the value of the statistical difference obtained after permutation of the sample or peptide labels, respectively. Kinases are then ranked based on the sum of both scores. The kinases that rank on top, are the ones that are most likely to drive the differences between two groups.
Pathway analysis
For kinase substrates that were significantly different between normal and cancer tissue, pathway analysis was performed using the UNIPROT accession number for each kinase substrate involved, using DAVIDS bioinformatics [38] and reactome (https://reactome.org/cite) online software tools.
Conclusions
The results of our study contribute to better understanding of the changes in kinase activity in RCC tumor cells involved in fundamental oncogenic cellular processes and the ex vivo effect of TKIs. We found tivozanib and cabozantinib to be more potent TKIs in RCC samples than sunitinib or pazopanib. The next step will be to correlate the efficacy and toxicity in individual patients with their respective kinase activity of normal and malignant kidney tissue. Thus, the presented findings might provide options to select the most promising TKI for individual RCC patients prior to initiation of TKI-therapies.
Abbreviations
ccRCC: clear cell renal cell carcinoma; ChRCC: Chromophobe Renal Carcinoma; chRCC: chromophobe renal cell carcinoma; DMSO: Dimethyl Sulfoxide; FDR: false discovery rate; LFC: log fold change; mRCC: metastatic renal cell carcinoma; mTOR: mammalian target of rapamycin; PDGFR: platelet derived growth factor receptor; PI3K: Phosphoinositide 3- Kinase; pRCC: papillary Renal Carcinoma; pRCC: papillary renal cell carcinoma; PTK: protein tyrosine kinase; RCC: Renal cell carcinoma; sRCC: renal cell carcinoma with sarcomatoid features; TKI: Tyrosine kinase inhibitor; TME: tumor microenvironment; VEFR: vascular endothelial growth factor receptor; VEGF: vascular endothelial growth factor.
Data availability
Data is available online as supplementary material. The datasets include log2 values from the tyrosine kinase profiling of normal and tumor tissue (Normal_tumor_dataset 1, see Supplementary Dataset 1) and tumor tissue treated ex vivo with four different inhibitors, tivozanib, sunitinib, pazopanib, and cabozantinib (Control and compound_dataset 2, see Supplementary Dataset 2). In addition, the enrichment file for the datasets with information for each sample used for the different tyrosine kinase profiling runs is provided (Enrichmentfile dataset 3, see Supplementary Dataset 3). Raw data is submitted to the online database Array Express (https://www.ebi.ac.uk/arrayexpress/).
CONFLICTS OF INTEREST
Faris Naji worked for Pamgene during this study, and has declared no competing interests.
FUNDING
This project has been funded by Medical Division, Akershus University Hospital.
References
1. Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D, Bray F. Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer. 2013; 49:1374–403. https://doi.org/10.1016/j.ejca.2012.12.027. [PubMed].
2. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, Znaor A, Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019; 144:1941–53. https://doi.org/10.1002/ijc.31937. [PubMed].
3. Dabestani S, Thorstenson A, Lindblad P, Harmenberg U, Ljungberg B, Lundstam S. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016; 34:1081–86. https://doi.org/10.1007/s00345-016-1773-y. [PubMed].
4. Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, Gibb EA, Akbani R, Beroukhim R, Bottaro DP, Choueiri TK, Gibbs RA, et al, and Cancer Genome Atlas Research Network. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018; 23:3698. https://doi.org/10.1016/j.celrep.2018.06.032. [PubMed].
5. Finley DS, Pantuck AJ, Belldegrun AS. Tumor biology and prognostic factors in renal cell carcinoma. Oncologist. 2011; 16:4–13. https://doi.org/10.1634/theoncologist.2011-S2-04. [PubMed].
6. Dabney R, Devine R, Sein N, George B. New agents in renal cell carcinoma. Target Oncol. 2014; 9:183–93. https://doi.org/10.1007/s11523-013-0303-8. [PubMed].
7. Rassy E, Flippot R, Albiges L. Tyrosine kinase inhibitors and immunotherapy combinations in renal cell carcinoma. Ther Adv Med Oncol. 2020; 12:1758835920907504. https://doi.org/10.1177/1758835920907504. [PubMed].
8. Sinha S, Cao Y, Dutta S, Wang E, Mukhopadhyay D. VEGF neutralizing antibody increases the therapeutic efficacy of vinorelbine for renal cell carcinoma. J Cell Mol Med. 2010; 14:647–58. https://doi.org/10.1111/j.1582-4934.2008.00578.x. [PubMed].
9. Parikh M, Bajwa P. Immune Checkpoint Inhibitors in the Treatment of Renal Cell Carcinoma. Semin Nephrol. 2020; 40:76–85. https://doi.org/10.1016/j.semnephrol.2019.12.009. [PubMed].
10. Ko JJ, Xie W, Kroeger N, Lee JL, Rini BI, Knox JJ, Bjarnason GA, Srinivas S, Pal SK, Yuasa T, Smoragiewicz M, Donskov F, Kanesvaran R, et al. The International Metastatic Renal Cell Carcinoma Database Consortium model as a prognostic tool in patients with metastatic renal cell carcinoma previously treated with first-line targeted therapy: a population-based study. Lancet Oncol. 2015; 16:293–300. https://doi.org/10.1016/S1470-2045(14)71222-7. [PubMed].
11. Motzer RJ, Escudier B, Bukowski R, Rini BI, Hutson TE, Barrios CH, Lin X, Fly K, Matczak E, Gore ME. Prognostic factors for survival in 1059 patients treated with sunitinib for metastatic renal cell carcinoma. Br J Cancer. 2013; 108:2470–77. https://doi.org/10.1038/bjc.2013.236. [PubMed].
12. Anderson JC, Willey CD, Mehta A, Welaya K, Chen D, Duarte CW, Ghatalia P, Arafat W, Madan A, Sudarshan S, Naik G, Grizzle WE, Choueiri TK, Sonpavde G. High Throughput Kinomic Profiling of Human Clear Cell Renal Cell Carcinoma Identifies Kinase Activity Dependent Molecular Subtypes. PLoS One. 2015; 10:e0139267. https://doi.org/10.1371/journal.pone.0139267. [PubMed].
13. Phan NN, Liu S, Wang CY, Hsu HP, Lai MD, Li CY, Chen CF, Chiao CC, Yen MC, Sun Z, Jiang JZ. Overexpressed gene signature of EPH receptor A/B family in cancer patients-comprehensive analyses from the public high-throughput database. Int J Clin Exp Pathol. 2020; 13:1220–42. [PubMed].
14. Jung M, Lee S, Moon KC. c-Met and EPHA7 Receptor Tyrosine Kinases Are Related to Prognosis in Clear Cell Renal Cell Carcinoma: Focusing on the Association with Myoferlin Expression. Cancers (Basel). 2022; 14:1095. https://doi.org/10.3390/cancers14041095. [PubMed].
15. Toma MI, Erdmann K, Diezel M, Meinhardt M, Zastrow S, Fuessel S, Wirth MP, Baretton GB. Lack of ephrin receptor A1 is a favorable independent prognostic factor in clear cell renal cell carcinoma. PLoS One. 2014; 9:e102262. https://doi.org/10.1371/journal.pone.0102262. [PubMed].
16. Qayyum T, McArdle PA, Lamb GW, Jordan F, Orange C, Seywright M, Horgan PG, Jones RJ, Oades G, Aitchison MA, Edwards J. Expression and prognostic significance of Src family members in renal clear cell carcinoma. Br J Cancer. 2012; 107:856–63. https://doi.org/10.1038/bjc.2012.314. [PubMed].
17. Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci U S A. 1990; 87:558–62. https://doi.org/10.1073/pnas.87.2.558. [PubMed].
18. Masaki T, Igarashi K, Tokuda M, Yukimasa S, Han F, Jin YJ, Li JQ, Yoneyama H, Uchida N, Fujita J, Yoshiji H, Watanabe S, Kurokohchi K, Kuriyama S. pp60c-src activation in lung adenocarcinoma. Eur J Cancer. 2003; 39:1447–55. https://doi.org/10.1016/s0959-8049(03)00276-4. [PubMed].
19. Elsberger B, Fullerton R, Zino S, Jordan F, Mitchell TJ, Brunton VG, Mallon EA, Shiels PG, Edwards J. Breast cancer patients’ clinical outcome measures are associated with Src kinase family member expression. Br J Cancer. 2010; 103:899–909. https://doi.org/10.1038/sj.bjc.6605829. [PubMed].
20. Gelman IH, Peresie J, Eng KH, Foster BA. Differential requirement for Src family tyrosine kinases in the initiation, progression, and metastasis of prostate cancer. Mol Cancer Res. 2014; 12:1470–79. https://doi.org/10.1158/1541-7786.MCR-13-0490-T. [PubMed].
21. Zhou J, Xu M, Le K, Ming J, Guo H, Ruan S, Huang T. SRC Promotes Tamoxifen Resistance in Breast Cancer via Up-Regulating SIRT1. Onco Targets Ther. 2020; 13:4635–47. https://doi.org/10.2147/OTT.S245749. [PubMed].
22. Vindis C, Cerretti DP, Daniel TO, Huynh-Do U. EphB1 recruits c-Src and p52Shc to activate MAPK/ERK and promote chemotaxis. J Cell Biol. 2003; 162:661–71. https://doi.org/10.1083/jcb.200302073. [PubMed].
23. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49. https://doi.org/10.1038/nature12222. [PubMed].
24. Saes L, Eskens FALM. Tivozanib: a new treatment option for renal cell carcinoma. Drugs Today (Barc). 2017; 53:609–18. https://doi.org/10.1358/dot.2017.53.11.2724804. [PubMed].
25. Kim ES. Tivozanib: First Global Approval. Drugs. 2017; 77:1917–23. https://doi.org/10.1007/s40265-017-0825-y. [PubMed].
26. Verzoni E, Escudier B, Hutson TE, McDermott DF, Pal SK, Porta C, Rini BI, Needle MN, Atkins MB. TIVO-3: Durability of response and updated overall survival of tivozanib versus sorafenib in metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2021; 39:4546. https://doi.org/10.1200/JCO.2021.39.15_suppl.4546.
27. Tahiri A, Røe K, Ree AH, de Wijn R, Risberg K, Busch C, Lønning PE, Kristensen V, Geisler J. Differential inhibition of ex-vivo tumor kinase activity by vemurafenib in BRAF(V600E) and BRAF wild-type metastatic malignant melanoma. PLoS One. 2013; 8:e72692. https://doi.org/10.1371/journal.pone.0072692. [PubMed].
28. Cantrell DA. GTPases and T cell activation. Immunol Rev. 2003; 192:122–30. https://doi.org/10.1034/j.1600-065x.2003.00028.x. [PubMed].
29. Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras-related gene with transformation suppressor activity. Cell. 1989; 56:77–84. https://doi.org/10.1016/0092-8674(89)90985-9. [PubMed].
30. Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006; 361:1545–64. https://doi.org/10.1098/rstb.2006.1894. [PubMed].
31. Triaca V, Carito V, Fico E, Rosso P, Fiore M, Ralli M, Lambiase A, Greco A, Tirassa P. Cancer stem cells-driven tumor growth and immune escape: the Janus face of neurotrophins. Aging (Albany NY). 2019; 11:11770–92. https://doi.org/10.18632/aging.102499. [PubMed].
32. Stühler V, Rausch S, Maas JM, Stenzl A, Bedke J. Combination of immune checkpoint inhibitors and tyrosine kinase inhibitors for the treatment of renal cell carcinoma. Expert Opin Biol Ther. 2021; 21:1215–26. https://doi.org/10.1080/14712598.2021.1890713. [PubMed].
33. Tan HY, Wang N, Lam W, Guo W, Feng Y, Cheng YC. Targeting tumour microenvironment by tyrosine kinase inhibitor. Mol Cancer. 2018; 17:43. https://doi.org/10.1186/s12943-018-0800-6. [PubMed].
34. Katoh M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int J Mol Med. 2016; 38:3–15. https://doi.org/10.3892/ijmm.2016.2620. [PubMed].
35. Chan XY, Singh A, Osman N, Piva TJ. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int J Mol Sci. 2017; 18:1527. https://doi.org/10.3390/ijms18071527. [PubMed].
36. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012; 487:500–4. https://doi.org/10.1038/nature11183. [PubMed].
37. Tahiri A, Tekpli X, Satheesh SV, DeWijn R, Lüders T, Bukholm IR, Hurtado A, Geisler J, Kristensen VN. Loss of progesterone receptor is associated with distinct tyrosine kinase profiles in breast cancer. Breast Cancer Res Treat. 2020; 183:585–98. https://doi.org/10.1007/s10549-020-05763-7. [PubMed].
38. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. https://doi.org/10.1038/nprot.2008.211. [PubMed].