
Introduction
Pancreatic cancer accounts for only 3% of all cancers diagnosed in the United States but is responsible for approximately 7% of all cancer-related deaths. Indeed, although relatively rare, pancreatic cancer represents the third most common cause of cancer-related death nationwide (https://www.cdc.gov/cancer/dcpc/research/update-on-cancer-deaths/index.htm) [1]. Incidence is increasing; it is projected that pancreatic cancer will become the second leading cause of cancer-related death by 2030 [2–4]. The reported frequency of early onset pancreatic cancer (EOPC) – defined here as pancreatic cancer diagnosed in patients under the age of 50 years – varies from 4% to 18% [5, 6], the majority of which are pancreatic ductal adenocarcinoma [7]. Although EOPC is less common than later-onset pancreatic cancer (LOPC), it contributes to a disproportionately high societal burden of potential years of life lost (PYLL), which has been estimated to be 25% in the United States and 40% in Europe [8]. Survival in patients with EOPC has improved over time but remains dismal [9].
There are no established data that definitively distinguish the tumor biology of EOPC from (LOPC). Previous studies have identified many genetic mutations that are shared between EOPC and LOPC [10–12]. However, genetic features that are unique to EOPC have also been described, including a low rate of KRAS and high rate of SMAD4 mutations [13]. Based on the limited available data, the biology and behavior of EOPC may be significantly different from that of LOPC.
Similar to early-onset lung and breast cancer, EOPC is associated with inferior survival relative to LOPC [5]. Although late presentation may contribute to poor survival in EOPC, the role of genetic factors remains incompletely understood.
The purpose of this review is to identify genetic mutations that are more prevalent in EOPC as compared to LOPC and discuss the potential impact of these mutations on tumor biology, treatment, and survival.
Risk factors, clinical presentation, and diagnosis of EOPC
The median age of diagnosis of pancreatic cancer in the United States is 71 years [14]. Risk factors for EOPC include smoking, obesity, diabetes mellitus, chronic pancreatitis, and a family history of pancreatic cancer [8, 15–17]. The effect of alcohol on pancreatic cancer risk varies based on age. However, it has been shown that excess alcohol use in individuals ≤45 years is associated with a markedly increased risk of both EOPC and LOPC [15].
The clinical presentation of EOPC and LOPC is identical and characterized by weight loss, jaundice, pruritus, nausea, and vomiting (Figure 1). EOPC has a predilection for the head of the pancreas while LOPC predominantly affects the tail [12]. Individuals with EOPC are more likely to present with advanced disease and are less likely to have multiple comorbidities as compared to their older counterparts [5, 6] (Table 1).
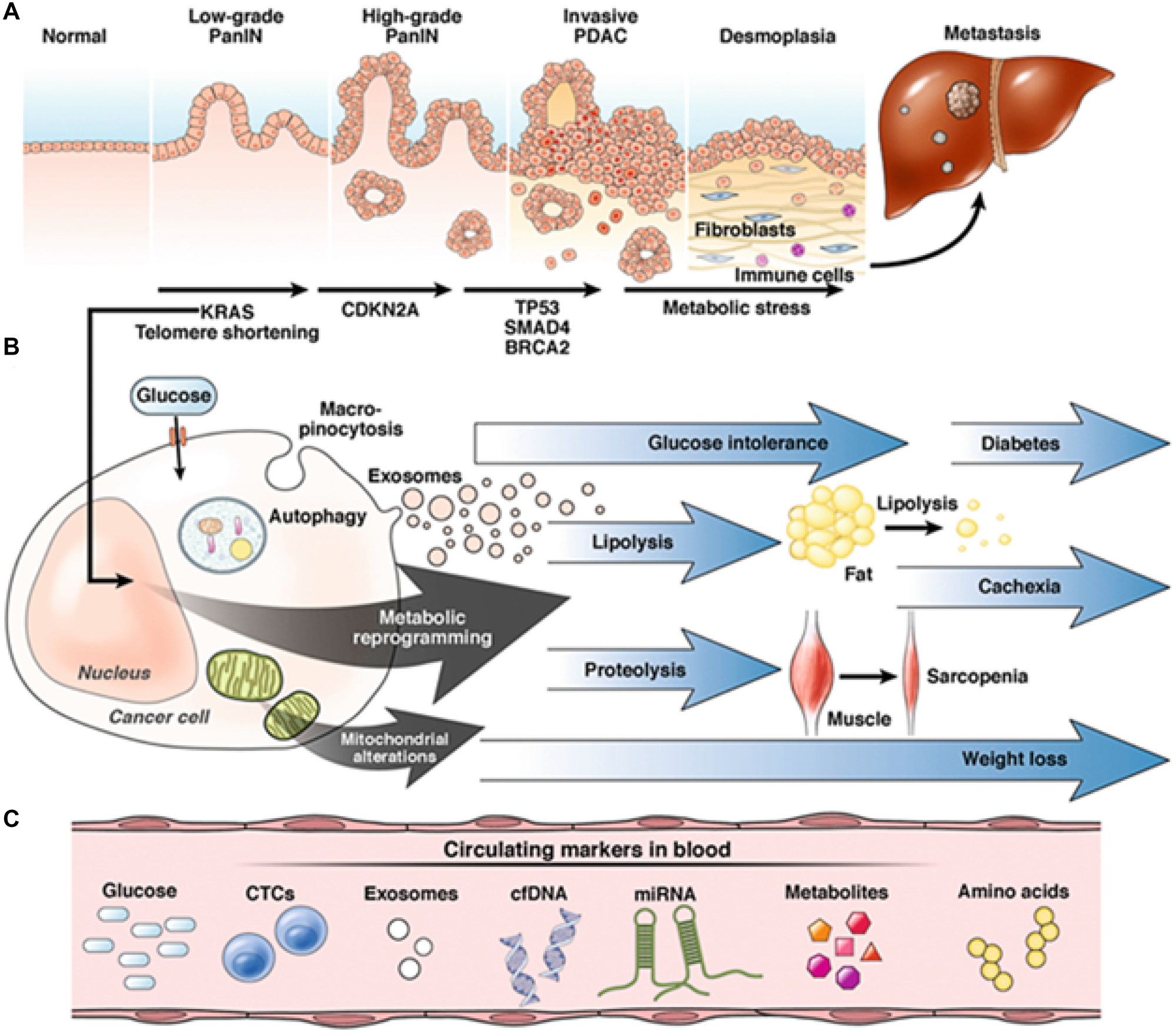
Figure 1: Progression of pancreatic adenocarcinoma (PDAC). (A) The progression of a normal pancreatic cell to PDAC begins with low-grade pancreatic intraepithelial neoplasia (PanIN); further mutagenesis – mediated by KRAS, CDKN2A, TP53, SMAD4, and BRCA2 – leads to high-grade PanIN and subsequently invasive PDAC. PDAC is characterized by an exuberant desmoplastic reaction and signaling between fibroblasts and immune cells, which promotes epithelial-mesenchymal transition and ultimately metastasis. (B) KRAS-mediated metabolic reprogramming and mitochondrial alterations leads to a cascade of downstream events, manifesting with the triad of diabetes, cachexia, and sarcopenia classically associated with PDAC. (C) Cell-free DNA (cfDNA) may be used to identify metabolic aberrations during the initial stages of carcinogenesis and facilitate early diagnosis (Abbreviations: CTC: circulating tumor cells; miRNA: microRNA). Reprinted with permission from Søreide et al. [110].
Table 1: Comparison of EOPC and LOPC
| Variable | EOPC | LOPC |
|---|---|---|
| Clinical presentation | More advanced at time of diagnosis, present with more aggressive disease | Likely to present with early-staged disease |
| Median age of diagnosis (years) | 46 | 71 |
| Gender difference | ||
| Male | More likely to be male | More common (1.3:1) |
| Female | Less likely to be female | Less likely to be female |
| Risk factors | Tobacco, alcohol, chronic pancreatitis, Hereditary pancreatitis, diabetes (type 1 and 2), BMI >40kg/m2, history of radiation therapy, cholecystectomy, gastrectomy | Tobacco, alcohol, chronic pancreatitis, diabetes (type 1 and 2), BMI >40kg/m2, history of radiation therapy, cholecystectomy, gastrectomy |
| Common genomic abnormality | BRCA1/2, SMAD4, KRAS, FOXC2, PI3KCA, CFTR, CDKN2a, c-MYC, MICROSATELLITE INSTABILITY (MSI) | BRCA1/2, SMAD4, KRAS, FOXC2, PI3KCA, CFTR, CDKN2a, c-MYC, MICROSATELLITE INSTABILITY (MSI) |
| Response to chemotherapy | May tolerate better and receive more aggressive therapy | Usually, poor candidate |
| Survival outcomes | Improves with more aggressive therapy, Median overall survival 8.6 months | Poor prognosis and less tolerant of surgery and systemic therapy, Median overall survival 8.0 months |
Next generation sequencing (NGS) is a “massive-parallel” or “high-throughput” sequencing technique used to determine the precise order of nucleotides within a strand of DNA/RNA. The first generation sequencing was developed by Frederick Sangers [18], who used two-dimensional chromatography to radiolabel base paired nucleotides. NGS represents a quantum leap in genetic analysis, facilitating millions of sequencing reactions run in parallel to allow for multiplexing of patient samples and thus simultaneous sequencing and detection of genetic mutations [19, 20]. NGS is now used in the analysis of both familial and sporadic pancreatic cancers [21].
Genomic basis of EOPC
Although numerous studies have examined modifiable risk factors associated with EOPC [22], only recently with the widespread adoption of genome sequencing have the lesser-known genetic factors been investigated. Much of the recent research has focused on inheritable causes of EOPC, which have proven to be significant given that 10% of patients with these tumors have some genetic risk factor [23, 24]. Numerous systemic conditions and genetic mutations have also been shown to increase the risk of pancreatic cancer, including Peutz-Jeghers syndrome (LKB1/STK11), Li-Fraumeni syndrome (TP53), familial atypical mole-multiple melanoma (FAMMM) syndrome (CDKN2A), Fanconi anemia, and mutations in BRCA1/2, CFTR, KRAS, SMAD4, and FOXC2 [6, 8, 25–27]. Notably, many of these genetic mutations are also associated with an increased risk of pancreatitis compared to the general population [28]. Several genetic mutations, including BRCA1/2, may be influenced by modifiable risk factors such as smoking and should be thought of in the context of the multistage theory of carcinogenesis.
There are only a few genetic mutations that have been linked specifically to EOPC in population studies, including CDKN2A, FOXC2, SMAD4, and PI3KCA [29–30]. Limited existing data are controversial but suggest that many of the genetic mutations associated with pancreatic cancer are shared between EOPC and LOPC. In a recent genomic and transcriptomic study by Raffenne et al., investigators compared mutational features of key genes and global methylation profiles between EOPC (defined as pancreatic cancer diagnosed at 55 years of age or younger) and late-onset pancreatic cancer (defined as pancreatic cancer diagnosed at age 70 or older). Authors concluded that the tumors in each group shared similar genetic and molecular features [31]. However, in another study by Bannon et al., investigators found a higher prevalence of germline mutations in EOPC. Individuals with EOPC also had better outcomes than those with LOPC, which was hypothesized to be related to superior DNA repair mechanisms and increased sensitivity to chemotherapy among younger patients [32].
The mechanism of pancreatic carcinogenesis involves a complex interplay of multiple related genetic and molecular factors (Figure 2). Other genetic mutations are discussed below.
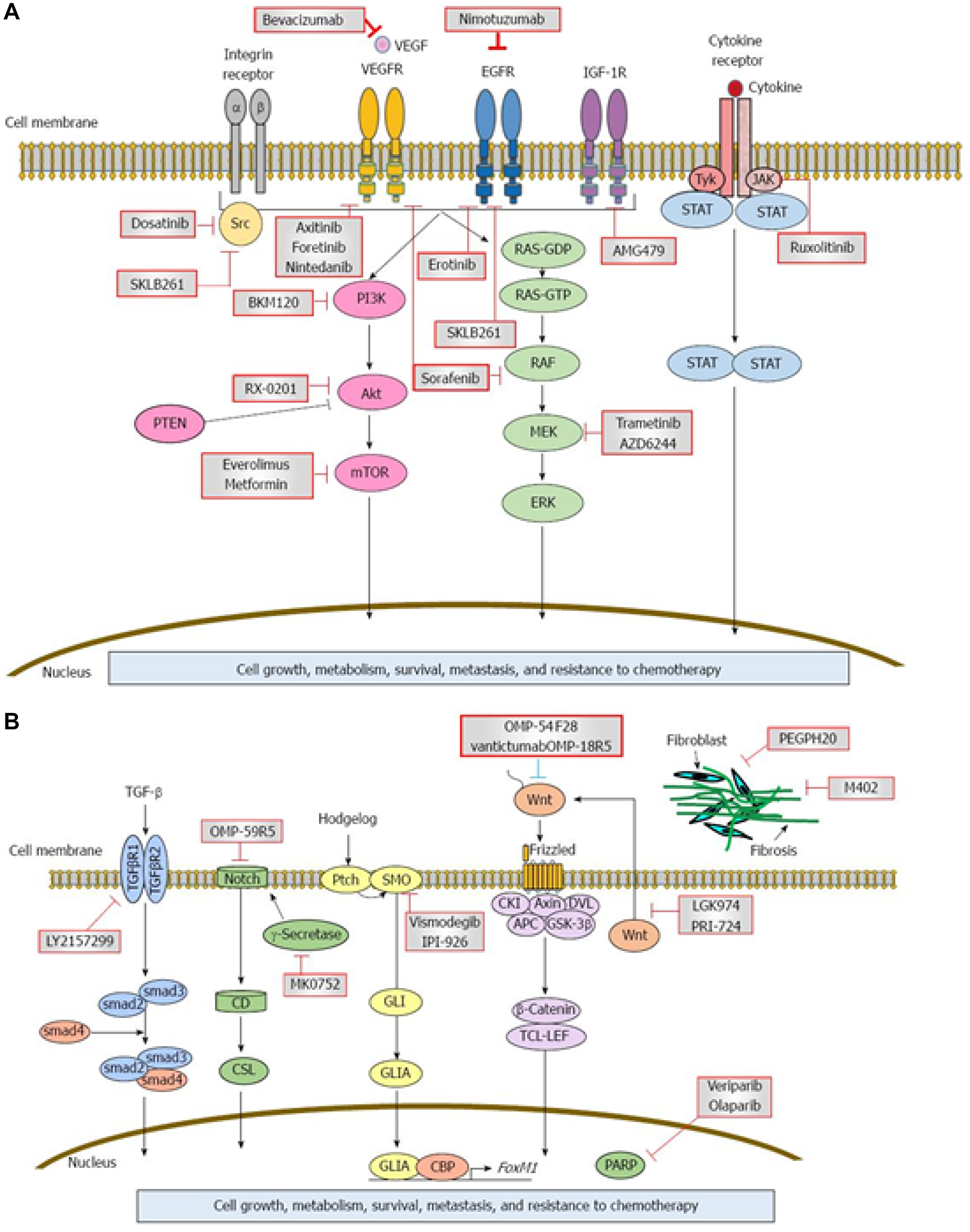
Figure 2: Signaling cascades and therapeutic inhibitors in pancreatic adenocarcinoma (PDAC). (A and B) Aberrant expression of multiple growth factors and growth factor receptors as well as intracellular, extracellular, and intranuclear proteins is implicated in the pathogenesis of PDAC. Examples include vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR), insulin-like growth factor-1 receptor (IGF-1R), gamma-secretase, and poly (ADP-ribose polymerase (PARP). Each abnormal protein may serve as a target for monoclonal antibodies or chemotherapeutic agents, which inhibit abnormal protein activity and can potentially reduce the rate of disease progression. Circles indicate critical signaling pathways and red squares indicate potential targets. Reprinted with permission from Matsuoka et al. [109].
Kirsten rat sarcoma virus (KRAS) mutation
KRAS mutations are present in approximately 90% of pancreatic ductal adenocarcinoma [33, 34]. However, KRAS mutations are relatively rare in EOPC. In one recent study, authors found that only 42.8% of patients with EOPC expressed a KRAS mutation [10]. The common types of KRAS are on exon 2 codons 12 and 13 with relative frequency of 71–80% [35, 36] and mostly located at G12C, G12D and G12R in pancreatic cancer [37, 38]. There is no data on frequency in EOPC verses LOPC.
Interestingly, mathematical models have shown that it takes nearly twelve years from the oncogenic event that initiates pancreatic carcinogenesis until the development of the parental clone and an additional seven years for the development of metastatic subclones within the primary cancer [39, 40]. Therefore, most KRAS mutant pancreatic cancers could be considered EOPC if detected via early screening. Screening for early detection in high-risk individuals such as familial pancreatic cancer could potentially identify more patients with pancreatic cancer at an early stage and age [41]. The mechanism of KRAS mutation-associated carcinogenesis is illustrated in Figure 3.
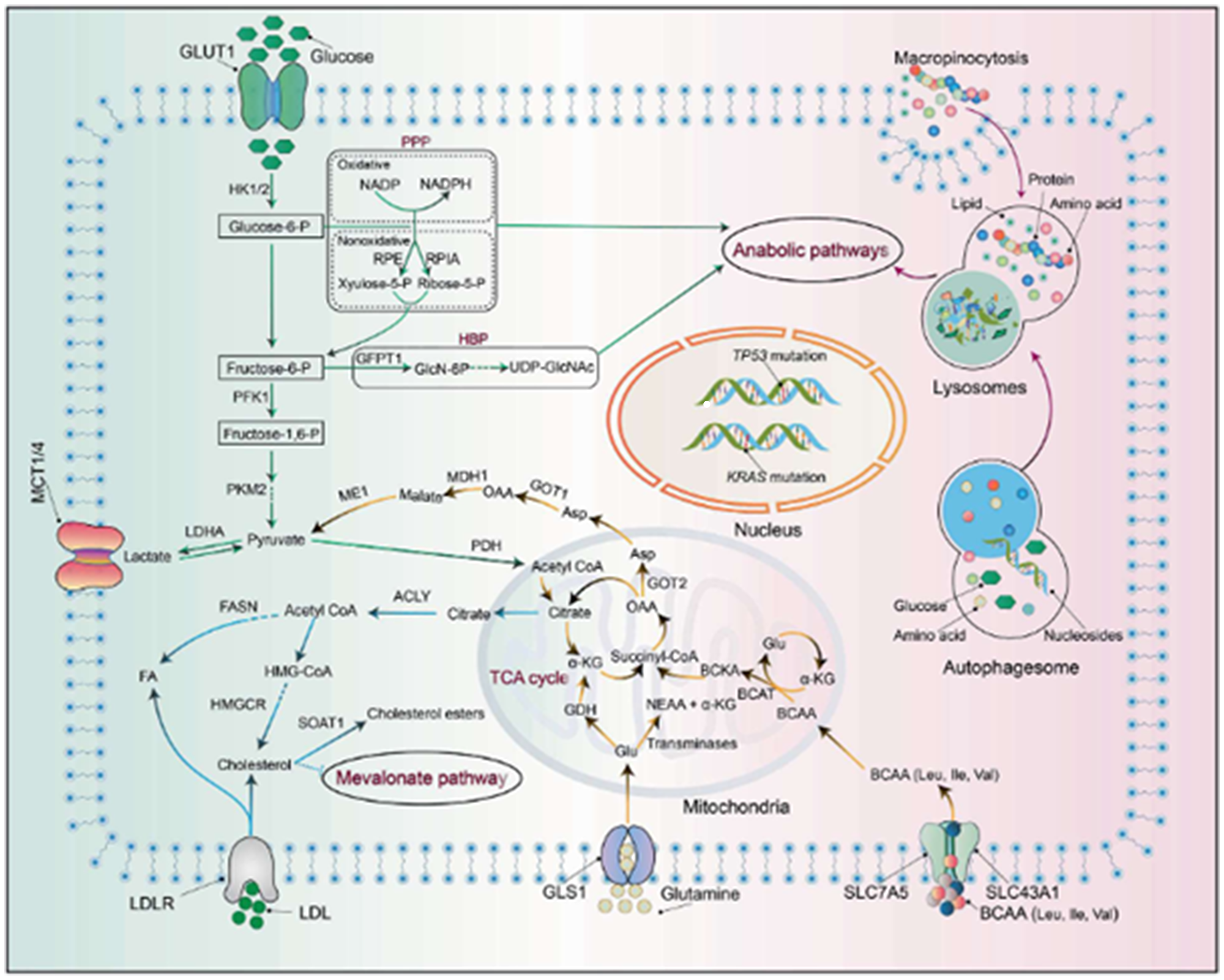
Figure 3: Metabolic reprogramming in pancreatic adenocarcinoma (PDAC). Activation of KRAS and mutations of TP53 results in a cascade of downstream effects that generates biosynthetic precursors to drive various anabolic pathways, including the non-oxidative arm of the pentose phosphate pathway (PPP) and the hexosamine biosynthesis pathway (HBP). In addition, KRAS activation affects glutamine metabolism, increasing the NADPH/NADP+ ratio and increasing turnover of glutathione (GSH) via reduction of its oxidized form. Branched chain amino acid (BCAA) catabolism is also catalyzed in the setting of KRAS activation via increased BCAT2 (branched chain amino acid transaminase 2) activity. The ultimate outcome of these aberrant pathways is enhanced nutrient salvaging and further promotion of neoplastic processes. Reprinted with permission from Wang et al. [110].
Microsatellite instability (MSI)
Mutations or epigenetic changes of mismatch repair genes such as MLH1, MSH2, MSH3, MSH6, PMS1, and PMS2, which repair DNA replication errors, result in MSI [42]. The presence of MSI is associated with a favorable prognosis in colorectal cancer [43], gastric cancer [44], and cancer of papilla of vater [45]. However, MSI-positivity is associated with a poor prognosis in breast cancer [46, 47] and non-small cell lung cancer [48]. The prevalence of MSI- positivity ranges from 3–67% in pancreatic cancer [49–53]; similar to other gastrointestinal malignancies, MSI-positivity is associated with a favorable prognosis [49, 54]. There is a paucity of data on the prevalence of MSI in EOPC. Bergmann et al. assessed molecular characteristics of 7 patients under 40 years and found mismatch repair gene products MLH1, MSH2 and MSH6 to be present in all patients [10]. There are no data specific to the prevalence of MSI positivity and survival among patients with EOPC.
Cyclin dependent kinase inhibitor 2A(CDKN2A)
One of the recently studied genes associated with EOPC is CKDN2A, which normally functions as a tumor suppressor gene involved in the pathway that inhibits the cell cycle at the G1 checkpoint. Mutations in CDKN2A are found in 95% of pancreatic cancers [55, 56]. CKDN2A encodes the protein p16inka, which acts as a cell cycle regulator. Previous studies have shown that loss of the p16inka protein may increase progression of cells that have a pre-existing KRAS mutation [57]. Notably, however, KRAS mutations prevalence appear to be less common in EOPC as compared to LOPC [29, 30]. Tsang and colleagues found that a biallelic CDKN2A mutation was significantly increased in EOPC patients [30]. Targeting different proteins in this pathway has been investigated in a few prior studies with limited clinical efficacy to date.
Forkhead box protein C2 (FOXC2)
FOXC2 is an oncogenic transcription factor associated with many different cancers, including hepatocellular carcinoma and breast cancer. The true prevalence of FOXC2 mutations in pancreatic cancer remains to be established. It is upregulated in pancreatic ductal adenocarcinoma and enhances growth and migration of cancer cells [58]. It interacts with beta-catenin and promote cell growth by activating beta-catenin/T-cell factor 4 signaling. The beta-catenin/T-cell factor 4 interaction may serve as a therapeutic target for future drug development [58, 59]. Tsang and colleagues showed that FOXC2 upregulation was associated with EOPC and potentially early epithelial-to-mesenchymal transition [30].
SMAD4
SMAD4 is an important tumor suppressor gene that is found to be inactivated in approximately 50% of pancreatic cancers and has been associated with a more aggressive clinical course [5, 10, 60, 61]. In addition to its tumor suppressor function, it also acts in part of the transforming growth factor beta (TGF-β) pathway. In the setting of EOPC, SMAD4 serves as part of this pathway, where it forms as complex with specific sequences of DNA in the cell nucleus after it is activated by a TGF-β protein. When SMAD4 is mutated, the cell proliferation process is left unchecked and rapid cell growth follows. Previous analyses have demonstrated that patients with EOPC had higher mutations rates of SMAD4 than those with LOPC [29].
PI3KCA
PI3KCA is a gene that encodes the p110 alpha protein which is involved in the phosphatidylinositol 3-kinase (PI3K) pathway. Mutations are present in 3–5% of patients with pancreatic cancers [62–64]. This frequently mutated oncogene has been previously studied and implicated in a number of cancers [65]. However, it is only recently that it has been linked to EOPC. Ben-Aharon et al. found that this gene was more often implicated in EOPC than in LOPC [29]. Furthermore, this pathway has been studied with regards to the PTEN tumor suppressor gene that normally inhibits this pathway. Previous studies have shown mutations to PTEN result in accelerated mutated pancreatic cell growth but have so far not been linked to EOPC [66].
CFTR
Cystic fibrosis transmembrane conductance regulator (CFTR) is a transmembrane chloride channel that helps maintain fluid homeostasis in the body. Mutations in the form of phenylalanine 508 deletion (ΔF508) are the most commonly seen alteration. The prevalence of CFTR mutations in pancreatic cancer is estimated range between 5 and 8% [67, 68]. A potential link between CFTR mutations and EOPC was proposed over a decade ago. In 2005, McWilliams and colleagues found that 8.4% of EOPC patients had CFTR mutations as compared to 4.1% in the control group [68]. However, this study was limited by its very small size (n = 33) with only two patients found to have a CFTR mutation; these patients were 42 and 50 years of age. A follow up study by the McWilliams group again found a significant increase in pancreatic cancer in carriers of the CFTR gene (5.3% versus 3.8%) along with a younger average onset (62 versus 67 years) [67]. Other studies have described variable findings on the influence of CFTR mutations in early pancreatic cancer, including a recent analysis in which investigators found no significant difference in the rate of EPOC among patients with the most common CFTR variants (ΔF508 mutation and 5T allele) as compared to a control group [69].
BRCA1/2
BRCA1/2 are tumor suppressor genes that are well known for their role in breast and ovarian cancers. They have also been described in pancreatic cancers. Pathogenic BRCA2 and BRCA1 mutations are found in approximately 2%, and ≤1% of pancreatic cancers, respectively [70–72]. In a 450 patient cohort of patients ≤50 years of age, 114 of whom underwent germline testing, 29% had pathologic germline alterations and nearly 16% of those patients demonstrated BRCA1/2 mutations [73]. However, other studies have found no significant increase in BRCA1/2 mutations among EOPC cohorts when compared to those with LOPC [29].
Treatment and survival of EOPC
Surgical treatment
Surgical treatment of pancreatic cancer is the only potentially curative modality when diagnosed at early stage of disease. Unfortunately, however, pancreatic cancer is most often diagnosed at an advanced stage [5]. Contraindications to surgery include metastasis to liver, omentum, peritoneum, or any other extra-abdominal sites, occlusion of the superior mesenteric artery or encasement of more than half of vessel circumference, and involvement of the inferior vena cava [74]. Stage II pancreatic cancer – defined as local tumor that has grown outside the margins of the pancreas but without vascular invasion – may be amenable to resection [75]. Individuals presenting with later stage cancers are typically treated with chemotherapy.
Medical treatment
Limited data suggest that EOPC is more aggressive and is associated with poorer survival than LOPC [5, 6, 76]. However, as of this writing, there are no large-scale studies comparing the outcomes between EOPC and LOPC; available studies are small and predominantly retrospective.
The prognosis of pancreatic cancer is dismal, with a 5-year survival rate of 8%, but modest gains have been made over the last decade with a gradual increase in median survival [77]. Treatment of early-staged pancreatic cancer is surgical resection [78, 79]. Improvements in survival have largely been attributable to novel targeted therapies in advanced and metastatic setting (Figure 2). Furthermore, increasing the use of germline testing, utilizing molecular tumor analysis, and employing adjuvant therapy with modified FOLFIRINOX (leucovorin, 5- fluorouracil, irinotecan, oxaliplatin) have all contributed to prolonged survival for individuals with pancreatic cancer [80].
As compared to patients with LOPC, individuals with EOPC are more likely to present with advanced stage disease. Notably, although patients with EOPC are more likely to undergo chemo- and radiation therapy and surgery as compared to their counterparts with LOPC, several studies suggest they nevertheless face poorer outcomes [5, 6]. Other studies have reported opposite or neutral findings regarding survival [76, 81–83].
Unfortunately, there are no randomized controlled trials (RCTs) examining management strategies and survival differences between EOPC and LOPC; data from existing studies can only be extrapolated to provide insight into the potential effect of available therapeutics on EOPC. For example, gemcitabine was compared to FOLFIRINOX in one RCT involving 342 patients aged 65 years or younger with metastatic pancreatic cancer and good performance status [84]. The FOLFIRINOX group was noted to have a significant survival advantage, albeit with increased toxicity, over the gemcitabine group. The hazard of death was 0.61 (95% CI; 0.46–0.82) for those ≤65 years and 0.48 (95% CI; 0.30–0.77) for individuals older than 65 years. In another RCT involving 493 patients [85] with pancreatic cancer status-post resection, and had no evidence of metastatic disease, malignant ascites, or pleural effusion were eligible for inclusion. In this study, 59.2% were ≤65 years. Patients who received a modified FOLFIRINOX regimen showed superior disease free and overall survival as compared to those treated with gemcitabine. In subgroup analysis, there was no significant difference in outcomes based on age. Given the relatively high rate of grade 3 and 4 adverse effects, FOLFIRINOX is currently recommended only for patients with good performance status. Patients with EOPC typically have fewer comorbidities than those with LOPC and are therefore more often deemed candidates for FOLFIRINOX therapy. Moreover, EOPC may demonstrate biology and behavior that is distinct from that of LOPC. Consequently, the response to available therapies may differ between the two clinical entities [60].
Approximately 10% of pancreatic cancers have familial inheritance [83, 86]. Screening first-degree relatives of individuals with multiple family members affected by pancreatic cancer can help identify disease precursors, as there are numerous syndromes associated with pancreatic cancer, including Peutz–Jeghers syndrome, p16, BRCA and hereditary non-polyposis colorectal cancer (HNPCC) [41, 57, 87]. The recent POLO trial investigated the effect of maintenance Olaparib therapy on individuals with germline BRCA-mutated metastatic pancreatic cancer that had not progressed during platinum-based chemotherapy. Subjects included in the study were relatively young, with median age of 57 years. Authors concluded that maintenance therapy with Olaparaib increased progression-free survival as compared to placebo [88]. Moreover, there was no statistically significant difference in the incidence of grade 3 or higher adverse events (40% in the Olaparib group versus 23% in the placebo group). In a prespecified subgroup analysis of progression-free survival, investigators demonstrated that outcomes were most improved among the youngest patients included in the cohort. The effect of poly ADP ribose polymerase (PARP) inhibitors in BRCA-mutated EOPC as a first-line treatment for metastatic disease warrants further exploration.
The response to treatment may vary in patients with both a germline BRCA1/2 and PALB2 (gBRCA/PALB2+) mutation. In a phase II trial comparing cisplatin and gemcitabine with or without veliparib in gBRCA/PALB2+ in patients with stage III or stage IV pancreatic adenocarcinoma, cisplatin and gemcitabine were shown to be the more effective regimen and concurrent veliparib did not improve response rate [89]. However, the median age of subjects was 64 years; it is conceivable that individuals with EOPC which are likely to have some genetic mutation [90–92] may likely benefit from the addition of veliparib. Similarly, in patients with metastatic pancreatic adenocarcinoma, nab-paclitaxel plus gemcitabine significantly improved overall survival, progression-free survival, and response rate compared with gemcitabine alone [93]. In subgroup analysis of same study, overall survival in patients <65 years old (majority; 57.6%) overall survival was superior (Hazard ratio; 0.65, 95% CI:0.53–0.79) versus older patients (Hazard ratio; 0.81, 95% CI:0.63–1.03).
Molecular targets, such as human epidermal growth factor receptor type 1 (HER1/EGFR), which are overexpressed in many pancreatic cancers have also been targeted clinically with mixed outcomes. Moore et al. first demonstrated a statistically significantly improvement in survival in advanced pancreatic cancer by adding erlotinib to gemcitabine [94]. In the Moore group study, the hazard ratio for survival was significant for patients ≤65 years (HR = 0.75 (0.58 to 0.96), but not for those > 65 years old. However, no difference was noted on stratifying by EGFR status. However, Hammel et al. conducted a phase 3 trial involving patients with locally advanced pancreatic cancer with disease controlled and did not find difference in overall survival with gemcitabine when compared with gemcitabine plus erlotinib as maintenance therapy [95]. The median age in the Hammel group study was older at 63 years (interquartile range of 58–71 years). Similarly, in a phase II trial by Abrams et al. showed addition of adjuvant erlotinib to gemcitabine did not show any clinical benefit [96]. Clinical trials to specifically look at effects of specific actionable molecular targets in EOPC is warranted.
Circulating tumor DNA (ctDNA) can be used to determine pancreatic adenocarcinoma prognosis and can conceivably help select targeted therapeutic agents [97]. Indeed, in a 2018 study by Riviere et al., investigators used ctDNA to identify patients with genomic alterations that were potentially actionable by experimental or approved drugs [98]. Furthermore, ctDNA can be used to identify germline mutations and may serve as an adjunct to traditional hereditary cancer gene testing [99]. The use of ctDNA in pancreatic adenocarcinoma diagnosis, prognosis, and treatment is rapidly expanding. Future large-scale analyses of ctDNA may be used to identify distinct genomic alterations and therapeutic targets among patients with EOPC.
Immune checkpoint inhibitors (ICIs) have historically been ineffective for the management of pancreatic adenocarcinoma. However, emerging evidence suggests that ICIs may play an important role in the management of both EOPC and LOPC. In a 2021 study by Lenzo et al., authors described several subsets of patients who may benefit from ICI therapy, including those with a PD-L1-driven phenotype or PD-1 axis-driven immunosuppression [100]. A subsequent study by Botta et al. showed that a small subset of patients with genomic alterations in SWItch/Sucrose Nonfermentable (SWI/SNF) chromatin remodeling genes may be responsive to ICIs; authors postulated that patients with SWI/SNF mutations demonstrate increased sensitivity to T cell-mediated cytotoxicity [101]. Future prospective studies may be of value to determine which patient populations are most likely to benefit from ICI therapy.
Summary of treatment recommendations for EOPC
There are no established treatment guidelines tailored toward EOPC. In advanced or metastatic disease, treatment for EOPC and LOPC is similar and based predominantly on performance status and comorbidities [102]. The first-line systemic treatment options are FOLFIRINOX or gemcitabine/nab-paclitaxel [84, 93]. A direct comparison between the two regimens reveals that FOLFIRINOX offers prolonged overall and progression-free survival as compared to gemcitabine/nab-paclitaxel. However, while there are no data on quality of life (QoL) for gemcitabine/nab-paclitaxel, data showed FOLFIRINOX showed a delay of deterioration of health status [103]. There are no clinical trials which directly compare FOLFIRINOX(FFN), nab-paclitaxel plus gemcitabine in unresectable tumor setting. In a study using real word experience, 225 patients received either FOLFIRINOX, gemcitabine plus nab-paclitaxel, or gemcitabine alone. Those who received FFN were more likely to be younger, and additionally, those receiving FFN or gemcitabine plus nab-paclitaxel had better overall survival than gemcitabine alone [104]. The second line options include gemcitabine, gemcitabine plus cisplatin, and gemcitabine/nab-paclitaxel [105].
Challenges in medical management of pancreatic cancer are vast and are related to metabolism of therapeutic agents [106], management of the inflammatory response [107], and the complex immunological milieu which leads to fibrotic reaction that promotes blockade of active immune cells [108]. Given the poor prognosis of pancreatic cancer, it is more pressing to examine unique features of EOPC that may lead to the identification of new molecular targets and the development of new therapeutics.
Conclusions
Pancreatic cancer is increasing in incidence and will become second leading cause of cancer death by 2030. Unfortunately, the prognosis of pancreatic cancer remains dismal. Individuals with EOPC tend to have fewer comorbidities and are more likely to undergo chemo- and radiation therapy as compared to their counterparts with LOPC, but nevertheless face a worse prognosis. Poor outcomes in EOPC patients may be multifactorial and related to advanced stage of presentation at diagnosis and unique tumor biology. The genomic basis of EOPC warrants further investigation and consideration as a distinct clinical entity. Future studies and trials may lead to the identification of genetic mutations distinct to EOPC and the development of new targeted interventions.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Lisa C, Richardson ND, Henley J. An Update on Cancer Deaths in the United States; 1999-2019, G.U.D.o.H.a.H.S. Centers for Disease Control and Prevention. Atlanta, Centers for Disease Control and Prevention, Division of Cancer Prevention and Control, Editor. 2021.
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913–21. https://doi.org/10.1158/0008-5472.CAN-14-0155. [PubMed].
3. Rahib L, Wehner MR, Matrisian LM, Nead KT. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw Open. 2021; 4:e214708. https://doi.org/10.1001/jamanetworkopen.2021.4708. [PubMed].
4. Gordon-Dseagu VL, Devesa SS, Goggins M, Stolzenberg-Solomon R. Pancreatic cancer incidence trends: evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int J Epidemiol. 2018; 47:427–39. https://doi.org/10.1093/ije/dyx232. [PubMed].
5. Ansari D, Althini C, Ohlsson H, Andersson R. Early-onset pancreatic cancer: a population-based study using the SEER registry. Langenbecks Arch Surg. 2019; 404:565–71. https://doi.org/10.1007/s00423-019-01810-0. [PubMed].
6. Tingstedt B, Weitkämper C, Andersson R. Early onset pancreatic cancer: a controlled trial. Ann Gastroenterol. 2011; 24:206–12. [PubMed].
7. Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, Cooc J, Weinkle J, Kim GE, Jakkula L, Feiler HS, Ko AH, Olshen AB, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011; 17:500–3. https://doi.org/10.1038/nm.2344. [PubMed].
8. Raimondi S, Maisonneuve P, Löhr JM, Lowenfels AB. Early onset pancreatic cancer: evidence of a major role for smoking and genetic factors. Cancer Epidemiol Biomarkers Prev. 2007; 16:1894–97. https://doi.org/10.1158/1055-9965.EPI-07-0341. [PubMed].
9. Alese OB, Jiang R, Shaib W, Wu C, Akce M, Gaines T, Ni L, Behera M, El-Rayes BF. Young Adults With Pancreatic Cancer: National Trends in Treatment and Outcomes. Pancreas. 2020; 49:341–54. https://doi.org/10.1097/MPA.0000000000001502. [PubMed].
10. Bergmann F, Aulmann S, Wente MN, Penzel R, Esposito I, Kleeff J, Friess H, Schirmacher P. Molecular characterisation of pancreatic ductal adenocarcinoma in patients under 40. J Clin Pathol. 2006; 59:580–84. https://doi.org/10.1136/jcp.2005.027292. [PubMed].
11. Lüttges J, Stigge C, Pacena M, Klöppel G. Rare ductal adenocarcinoma of the pancreas in patients younger than age 40 years. Cancer. 2004; 100:173–82. https://doi.org/10.1002/cncr.11860. [PubMed].
12. Ohmoto A, Yachida S, Kubo E, Takai E, Suzuki M, Shimada K, Okusaka T, Morizane C. Clinicopathologic Features and Germline Sequence Variants in Young Patients (≤40 Years Old) With Pancreatic Ductal Adenocarcinoma. Pancreas. 2016; 45:1056–61. https://doi.org/10.1097/MPA.0000000000000574. [PubMed].
13. Luo J, Xiao L, Wu C, Zheng Y, Zhao N. The incidence and survival rate of population-based pancreatic cancer patients: Shanghai Cancer Registry 2004-2009. PLoS One. 2013; 8:e76052. https://doi.org/10.1371/journal.pone.0076052. [PubMed].
14. Sites A. SEER Cancer Statistics review 1975-2010. Bethesda, MD: National Cancer Institute; 2013. Available from http://seer.cancer.gov/csr.
15. McWilliams RR, Maisonneuve P, Bamlet WR, Petersen GM, Li D, Risch HA, Yu H, Fontham ET, Luckett B, Bosetti C, Negri E, La Vecchia C, Talamini R, et al. Risk Factors for Early-Onset and Very-Early-Onset Pancreatic Adenocarcinoma: A Pancreatic Cancer Case-Control Consortium (PanC4) Analysis. Pancreas. 2016; 45:311–16. https://doi.org/10.1097/MPA.0000000000000392. [PubMed].
16. Lin JC, Chan DC, Chen PJ, Chu HC, Chueh TH, Huang HH, Chang PY, Yu CP, Chang WK, Hsieh TY. Clinical characteristics of early onset pancreatic adenocarcinoma: a medical center experience and review of the literature. Pancreas. 2011; 40:638–39. https://doi.org/10.1097/MPA.0b013e318214fe56. [PubMed].
17. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011; 378:607–20. https://doi.org/10.1016/S0140-6736(10)62307-0. [PubMed].
18. Sanger F. Sequences, sequences, and sequences. Annu Rev Biochem. 1988; 57:1–28. https://doi.org/10.1146/annurev.bi.57.070188.000245. [PubMed].
19. Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008; 26:1135–45. https://doi.org/10.1038/nbt1486. [PubMed].
20. Buchholz M, Gress TM. Molecular changes in pancreatic cancer. Expert Rev Anticancer Ther. 2009; 9:1487–97. https://doi.org/10.1586/era.09.107. [PubMed].
21. Shen GQ, Aleassa EM, Walsh RM, Morris-Stiff G. Next-Generation Sequencing in Pancreatic Cancer. Pancreas. 2019; 48:739–48. https://doi.org/10.1097/MPA.0000000000001324. [PubMed].
22. Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006; 20:197–209. https://doi.org/10.1016/j.bpg.2005.10.001. [PubMed].
23. Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol. 2019; 10:10–27. https://doi.org/10.14740/wjon1166. [PubMed].
24. Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med. 2009; 133:365–74. https://doi.org/10.5858/133.3.365. [PubMed].
25. Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. 2012; 51:14–24. https://doi.org/10.1002/mc.20855. [PubMed].
26. Ghadirian P, Lynch HT, Krewski D. Epidemiology of pancreatic cancer: an overview. Cancer Detect Prev. 2003; 27:87–93. https://doi.org/10.1016/s0361-090x(03)00002-3. [PubMed].
27. Goldstein AM, Fraser MC, Struewing JP, Hussussian CJ, Ranade K, Zametkin DP, Fontaine LS, Organic SM, Dracopoli NC, Clark WH Jr. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med. 1995; 333:970–74. https://doi.org/10.1056/NEJM199510123331504. [PubMed].
28. Bansal P, Sonnenberg A. Pancreatitis is a risk factor for pancreatic cancer. Gastroenterology. 1995; 109:247–51. https://doi.org/10.1016/0016-5085(95)90291-0. [PubMed].
29. Ben-Aharon I, Elkabets M, Pelossof R, Yu KH, Iacubuzio-Donahue CA, Leach SD, Lowery MA, Goodman KA, O'Reilly EM. Genomic Landscape of Pancreatic Adenocarcinoma in Younger versus Older Patients: Does Age Matter? Clin Cancer Res. 2019; 25:2185–93. https://doi.org/10.1158/1078-0432.CCR-18-3042. [PubMed].
30. Tsang ES, Topham JT, Karasinska JM, Lee MKC, Williamson LM, Mendis S, Denroche RE, Jang GH, Kalloger SE, Moore RA, Mungall AJ, Bathe OF, Tang PA, et al. Delving into Early-onset Pancreatic Ductal Adenocarcinoma: How Does Age Fit In? Clin Cancer Res. 2021; 27:246–54. https://doi.org/10.1158/1078-0432.CCR-20-1042. [PubMed].
31. Raffenne J, Martin FA, Nicolle R, Konta M, Blum Y, Torrisani J, Puleo F, Bachet JB, Svrcek M, Bardier-Dupas A, Emile JF, Demetter P, Radman M, et al. Pancreatic Ductal Adenocarcinoma Arising in Young and Old Patients Displays Similar Molecular Features. Cancers (Basel). 2021; 13:1234. https://doi.org/10.3390/cancers13061234. [PubMed].
32. Bannon SA, Montiel MF, Goldstein JB, Dong W, Mork ME, Borras E, Hasanov M, Varadhachary GR, Maitra A, Katz MH, Feng L, Futreal A, Fogelman DR, et al. High Prevalence of Hereditary Cancer Syndromes and Outcomes in Adults with Early-Onset Pancreatic Cancer. Cancer Prev Res (Phila). 2018; 11:679–86. https://doi.org/10.1158/1940-6207.capr-18-0014. [PubMed].
33. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, Quinn MC, Robertson AJ, Fadlullah MZ, et al, and Australian Pancreatic Cancer Genome Initiative. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015; 518:495–501. https://doi.org/10.1038/nature14169. [PubMed].
34. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, et al, and Australian Pancreatic Cancer Genome Initiative. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016; 531:47–52. https://doi.org/10.1038/nature16965. [PubMed].
35. Windon AL, Loaiza-Bonilla A, Jensen CE, Randall M, Morrissette JJD, Shroff SG. A KRAS wild type mutational status confers a survival advantage in pancreatic ductal adenocarcinoma. J Gastrointest Oncol. 2018; 9:1–10. https://doi.org/10.21037/jgo.2017.10.14. [PubMed].
36. AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017; 7:818–31. https://doi.org/10.1158/2159-8290.CD-17-0151. [PubMed].
37. Cefalì M, Epistolio S, Palmarocchi MC, Frattini M, De Dosso S. Research progress on KRAS mutations in colorectal cancer. J Cancer Metastasis Treat. 2021; 7:26. https://doi.org/10.20517/2394-4722.2021.61.
38. Li J, Gan S, Blair A, Min K, Rehage T, Hoeppner C, Halait H, Brophy VH. A Highly Verified Assay for KRAS Mutation Detection in Tissue and Plasma of Lung, Colorectal, and Pancreatic Cancer. Arch Pathol Lab Med. 2019; 143:183–89. https://doi.org/10.5858/arpa.2017-0471-OA. [PubMed].
39. Yachida S, Iacobuzio-Donahue CA. Evolution and dynamics of pancreatic cancer progression. Oncogene. 2013; 32:5253–60. https://doi.org/10.1038/onc.2013.29. [PubMed].
40. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467:1114–17. https://doi.org/10.1038/nature09515. [PubMed].
41. Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, Nio Y, Schulick RS, Bassi C, Kluijt I, Levy MJ, Chak A, Fockens P, et al, and International Cancer of Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013; 62:339–47. https://doi.org/10.1136/gutjnl-2012-303108. [PubMed].
42. Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996; 65:101–33. https://doi.org/10.1146/annurev.bi.65.070196.000533. [PubMed].
43. Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000; 342:69–77. https://doi.org/10.1056/NEJM200001133420201. [PubMed].
44. dos Santos NR, Seruca R, Constância M, Seixas M, Sobrinho-Simões M. Microsatellite instability at multiple loci in gastric carcinoma: clinicopathologic implications and prognosis. Gastroenterology. 1996; 110:38–44. https://doi.org/10.1053/gast.1996.v110.pm8536886. [PubMed].
45. Achille A, Biasi MO, Zamboni G, Bogina G, Iacono C, Talamini G, Capella G, Scarpa A. Cancers of the papilla of vater: mutator phenotype is associated with good prognosis. Clin Cancer Res. 1997; 3:1841–47. [PubMed].
46. Paulson TG, Wright FA, Parker BA, Russack V, Wahl GM. Microsatellite instability correlates with reduced survival and poor disease prognosis in breast cancer. Cancer Res. 1996; 56:4021–26. [PubMed].
47. Tomita S, Deguchi S, Miyaguni T, Muto Y, Tamamoto T, Toda T. Analyses of microsatellite instability and the transforming growth factor-beta receptor type II gene mutation in sporadic breast cancer and their correlation with clinicopathological features. Breast Cancer Res Treat. 1999; 53:33–39. https://doi.org/10.1023/a:1006167210269. [PubMed].
48. Zhou X, Kemp BL, Khuri FR, Liu D, Lee JJ, Wu W, Hong WK, Mao L. Prognostic implication of microsatellite alteration profiles in early-stage non-small cell lung cancer. Clin Cancer Res. 2000; 6:559–65. [PubMed].
49. Nakata B, Wang YQ, Yashiro M, Nishioka N, Tanaka H, Ohira M, Ishikawa T, Nishino H, Hirakawa K. Prognostic value of microsatellite instability in resectable pancreatic cancer. Clin Cancer Res. 2002; 8:2536–40. [PubMed].
50. Eatrides JM, Coppola D, Al Diffalha S, Kim RD, Springett GM, Mahipal A. Microsatellite instability in pancreatic cancer. J Clin Oncol. 2016; 34:e15753. https://doi.org/10.1200/JCO.2016.34.15_suppl.e15753.
51. Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, Imai K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001; 61:3139–44. [PubMed].
52. Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998; 152:1501–7. [PubMed].
53. Venkatasubbarao K, Ahmed MM, Swiderski C, Harp C, Lee EY, McGrath P, Mohiuddin M, Strodel W, Freeman JW. Novel mutations in the polyadenine tract of the transforming growth factor beta type II receptor gene are found in a subpopulation of human pancreatic adenocarcinomas. Genes Chromosomes Cancer. 1998; 22:138–44. https://doi.org/10.1002/(sici)1098-2264(199806)22:2<138::aid-gcc8>3.0.co;2-y. [PubMed].
54. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, Geva R, Gottfried M, Penel N, Hansen AR, Piha-Paul SA, Doi T, Gao B, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020; 38:1–10. https://doi.org/10.1200/JCO.19.02105. [PubMed].
55. Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, Baylin SB, Kern SE, Herman JG. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997; 57:3126–30. [PubMed].
56. McWilliams RR, Wieben ED, Rabe KG, Pedersen KS, Wu Y, Sicotte H, Petersen GM. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. Eur J Hum Genet. 2011; 19:472–78. https://doi.org/10.1038/ejhg.2010.198. [PubMed].
57. Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016; 388:73–85. https://doi.org/10.1016/S0140-6736(16)00141-0. [PubMed].
58. Cui L, Dang S, Qu J, Mao Z, Wang X, Zhang J, Chen J. FOXC2 is up-regulated in pancreatic ductal adenocarcinoma and promotes the growth and migration of cancer cells. Tumour Biol. 2016; 37:8579–85. https://doi.org/10.1007/s13277-015-4607-4. [PubMed].
59. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012; 149:1192–205. https://doi.org/10.1016/j.cell.2012.05.012. [PubMed].
60. Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Eshleman JR, Goggins M, Jaffee EM, Iacobuzio-Donahue CA, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009; 15:4674–79. https://doi.org/10.1158/1078-0432.CCR-09-0227. [PubMed].
61. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996; 271:350–53. https://doi.org/10.1126/science.271.5247.350. [PubMed].
62. Heestand GM, Kurzrock R. Molecular landscape of pancreatic cancer: implications for current clinical trials. Oncotarget. 2015; 6:4553–61. https://doi.org/10.18632/oncotarget.2972. [PubMed].
63. Weiss GA, Rossi MR, Khushalani NI, Lo K, Gibbs JF, Bharthuar A, Cowell JK, Iyer R. Evaluation of phosphatidylinositol-3-kinase catalytic subunit (PIK3CA) and epidermal growth factor receptor (EGFR) gene mutations in pancreaticobiliary adenocarcinoma. J Gastrointest Oncol. 2013; 4:20–29. https://doi.org/10.3978/j.issn.2078-6891.2012.012. [PubMed].
64. Schönleben F, Qiu W, Remotti HE, Hohenberger W, Su GH. PIK3CA, KRAS, and BRAF mutations in intraductal papillary mucinous neoplasm/carcinoma (IPMN/C) of the pancreas. Langenbecks Arch Surg. 2008; 393:289–96. https://doi.org/10.1007/s00423-008-0285-7. [PubMed].
65. Benvenuti S, Frattini M, Arena S, Zanon C, Cappelletti V, Coradini D, Daidone MG, Pilotti S, Pierotti MA, Bardelli A. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat. 2008; 29:284–88. https://doi.org/10.1002/humu.20648. [PubMed].
66. Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, Dry S, Wu H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010; 70:7114–24. https://doi.org/10.1158/0008-5472.CAN-10-1649. [PubMed].
67. McWilliams RR, Petersen GM, Rabe KG, Holtegaard LM, Lynch PJ, Bishop MD, Highsmith WE Jr. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations and risk for pancreatic adenocarcinoma. Cancer. 2010; 116:203–9. https://doi.org/10.1002/cncr.24697. [PubMed].
68. McWilliams R, Highsmith WE, Rabe KG, de Andrade M, Tordsen LA, Holtegaard LM, Petersen GM. Cystic fibrosis transmembrane regulator gene carrier status is a risk factor for young onset pancreatic adenocarcinoma. Gut. 2005; 54:1661–62. https://doi.org/10.1136/gut.2005.074534. [PubMed].
69. Malats N, Casals T, Porta M, Guarner L, Estivill X, Real FX. Cystic fibrosis transmembrane regulator (CFTR) DeltaF508 mutation and 5T allele in patients with chronic pancreatitis and exocrine pancreatic cancer. PANKRAS II Study Group. Gut. 2001; 48:70–74. https://doi.org/10.1136/gut.48.1.70. [PubMed].
70. Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, Siddiqui A, Witmer PD, Tamura K, Song TJ, Navarro Almario JA, Brant A, Borges M, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol. 2017; 35:3382–90. https://doi.org/10.1200/JCO.2017.72.3502. [PubMed].
71. Young EL, Thompson BA, Neklason DW, Firpo MA, Werner T, Bell R, Berger J, Fraser A, Gammon A, Koptiuch C, Kohlmann WK, Neumayer L, Goldgar DE, et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer. 2018; 18:697. https://doi.org/10.1186/s12885-018-4573-5. [PubMed].
72. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, Lilyquist J, Na J, Moore R, Antwi SO, Bamlet WR, Chaffee KG, DiCarlo J, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA. 2018; 319:2401–9. https://doi.org/10.1001/jama.2018.6228. [PubMed].
73. Varghese AM, Singh I, Singh RR, Capanu M, Chou JF, Wong W, Stadler ZK, Salo-Mullen EE, Iacobuzio-Donahue CA, Kelsen DP, Park W, Yu KH, O'Reilly EM. Young-onset pancreas cancer (PC) in patients less than or equal to 50 years old at Memorial Sloan Kettering (MSK): Descriptors, genomics, and outcomes. J Clin Oncol. 2020; 38:774. https://doi.org/10.1200/JCO.2020.38.4_suppl.774.
74. Ferrone CR, Marchegiani G, Hong TS, Ryan DP, Deshpande V, McDonnell EI, Sabbatino F, Santos DD, Allen JN, Blaszkowsky LS, Clark JW, Faris JE, Goyal L, et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann Surg. 2015; 261:12–17. https://doi.org/10.1097/SLA.0000000000000867. [PubMed].
75. van Roessel S, Kasumova GG, Verheij J, Najarian RM, Maggino L, de Pastena M, Malleo G, Marchegiani G, Salvia R, Ng SC, de Geus SW, Lof S, Giovinazzo F, et al. International Validation of the Eighth Edition of the American Joint Committee on Cancer (AJCC) TNM Staging System in Patients With Resected Pancreatic Cancer. JAMA Surg. 2018; 153:e183617. https://doi.org/10.1001/jamasurg.2018.3617. [PubMed].
76. Piciucchi M, Capurso G, Valente R, Larghi A, Archibugi L, Signoretti M, Stigliano S, Zerboni G, Barucca V, La Torre M, Cavallini M, Costamagna G, Marchetti P, et al. Early onset pancreatic cancer: risk factors, presentation and outcome. Pancreatology. 2015; 15:151–55. https://doi.org/10.1016/j.pan.2015.01.013. [PubMed].
77. Gupta R, Amanam I, Chung V. Current and future therapies for advanced pancreatic cancer. J Surg Oncol. 2017; 116:25–34. https://doi.org/10.1002/jso.24623. [PubMed].
78. Miyasaka Y, Ohtsuka T, Nakamura M. Minimally invasive surgery for pancreatic cancer. Surg Today. 2021; 51:194–203. https://doi.org/10.1007/s00595-020-02120-5. [PubMed].
79. Clancy TE. Surgery for Pancreatic Cancer. Hematol Oncol Clin North Am. 2015; 29:701–16. https://doi.org/10.1016/j.hoc.2015.04.001. [PubMed].
80. Tempero MA. NCCN Guidelines Updates: Pancreatic Cancer. J Natl Compr Canc Netw. 2019; 17:603–5. https://doi.org/10.6004/jnccn.2019.5007. [PubMed].
81. Ntala C, Debernardi S, Feakins RM, Crnogorac-Jurcevic T. Demographic, clinical, and pathological features of early onset pancreatic cancer patients. BMC Gastroenterol. 2018; 18:139. https://doi.org/10.1186/s12876-018-0866-z. [PubMed].
82. Raissouni S, Rais G, Mrabti H, Raissouni F, Mouzount H, Aitelhaj M, El Khoyaali S, Mohtaram A, Errihani H. Pancreatic adenocarcinoma in young adults in a moroccan population. J Gastrointest Cancer. 2012; 43:607–11. https://doi.org/10.1007/s12029-012-9407-0. [PubMed].
83. Duffy A, Capanu M, Allen P, Kurtz R, Olson SH, Ludwig E, Klimstra DS, O’Reilly EM. Pancreatic adenocarcinoma in a young patient population--12-year experience at Memorial Sloan Kettering Cancer Center. J Surg Oncol. 2009; 100:8–12. https://doi.org/10.1002/jso.21292. [PubMed].
84. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, Bennouna J, Bachet JB, Khemissa-Akouz F, et al, and Groupe Tumeurs Digestives of Unicancer, and PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817–25. https://doi.org/10.1056/NEJMoa1011923. [PubMed].
85. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, Choné L, Francois E, Artru P, Biagi JJ, Lecomte T, Assenat E, Faroux R, et al, and Canadian Cancer Trials Group and the Unicancer-GI–PRODIGE Group. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med. 2018; 379:2395–406. https://doi.org/10.1056/NEJMoa1809775. [PubMed].
86. Petersen GM. Familial pancreatic cancer. Semin Oncol. 2016; 43:548–53. https://doi.org/10.1053/j.seminoncol.2016.09.002. [PubMed].
87. Ghiorzo P. Genetic predisposition to pancreatic cancer. World J Gastroenterol. 2014; 20:10778–89. https://doi.org/10.3748/wjg.v20.i31.10778. [PubMed].
88. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, Reinacher-Schick A, Tortora G, Algül H, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med. 2019; 381:317–27. https://doi.org/10.1056/NEJMoa1903387. [PubMed].
89. O’Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, Tahover E, Lowery MA, Chou JF, Sahai V, Brenner R, Kindler HL, Yu KH, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J Clin Oncol. 2020; 38:1378–88. https://doi.org/10.1200/JCO.19.02931. [PubMed].
90. Lynch HT, Deters CA, Snyder CL, Lynch JF, Villeneuve P, Silberstein J, Martin H, Narod SA, Brand RE. BRCA1 and pancreatic cancer: pedigree findings and their causal relationships. Cancer Genet Cytogenet. 2005; 158:119–25. https://doi.org/10.1016/j.cancergencyto.2004.01.032. [PubMed].
91. Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015; 17:251–64. https://doi.org/10.1016/j.jmoldx.2014.12.006. [PubMed].
92. Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, Jayakumaran G, Middha S, Zehir A, Donoghue MTA, You D, Viale A, Kemeny N, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018; 33:125–36.e3. https://doi.org/doi:10.1016/j.ccell.2017.12.004. [PubMed].
93. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691–703. https://doi.org/10.1056/NEJMoa1304369. [PubMed].
94. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, Campos D, Lim R, Ding K, et al, and National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007; 25:1960–66. https://doi.org/10.1200/JCO.2006.07.9525. [PubMed].
95. Hammel P, Huguet F, van Laethem JL, Goldstein D, Glimelius B, Artru P, Borbath I, Bouché O, Shannon J, André T, Mineur L, Chibaudel B, Bonnetain F, Louvet C, and LAP07 Trial Group. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA. 2016; 315:1844–53. https://doi.org/10.1001/jama.2016.4324. [PubMed].
96. Abrams RA, Winter KA, Safran H, Goodman KA, Regine WF, Berger AC, Gillin MT, Philip PA, Lowy AM, Wu A, DiPetrillo TA, Corn BW, Seaward SA, et al. Results of the NRG Oncology/RTOG 0848 Adjuvant Chemotherapy Question-Erlotinib+Gemcitabine for Resected Cancer of the Pancreatic Head: A Phase II Randomized Clinical Trial. Am J Clin Oncol. 2020; 43:173–79. https://doi.org/10.1097/COC.0000000000000633. [PubMed].
97. Patel H, Okamura R, Fanta P, Patel C, Lanman RB, Raymond VM, Kato S, Kurzrock R. Clinical correlates of blood-derived circulating tumor DNA in pancreatic cancer. J Hematol Oncol. 2019; 12:130. https://doi.org/10.1186/s13045-019-0824-4. [PubMed].
98. Riviere P, Fanta PT, Ikeda S, Baumgartner J, Heestand GM, Kurzrock R. The Mutational Landscape of Gastrointestinal Malignancies as Reflected by Circulating Tumor DNA. Mol Cancer Ther. 2018; 17:297–305. https://doi.org/10.1158/1535-7163.MCT-17-0360. [PubMed].
99. Slavin TP, Banks KC, Chudova D, Oxnard GR, Odegaard JI, Nagy RJ, Tsang KWK, Neuhausen SL, Gray SW, Cristofanilli M, Rodriguez AA, Bardia A, Leyland-Jones B, et al. Identification of Incidental Germline Mutations in Patients With Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J Clin Oncol. 2018; 36:JCO1800328. https://doi.org/10.1200/JCO.18.00328. [PubMed].
100. Lenzo FL, Kato S, Pabla S, DePietro P, Nesline MK, Conroy JM, Burgher B, Glenn ST, Kuvshinoff B, Kurzrock R, Morrison C. Immune profiling and immunotherapeutic targets in pancreatic cancer. Ann Transl Med. 2021; 9:119. https://doi.org/10.21037/atm-20-1076. [PubMed].
101. Botta GP, Kato S, Patel H, Fanta P, Lee S, Okamura R, Kurzrock R. SWI/SNF complex alterations as a biomarker of immunotherapy efficacy in pancreatic cancer. JCI Insight. 2021; 6:e150453. https://doi.org/10.1172/jci.insight.150453. [PubMed].
102. Tempero MA, Malafa MP, Al-Hawary M, Behrman SW, Benson AB, Cardin DB, Chiorean EG, Chung V, Czito B, Del Chiaro M, Dillhoff M, Donahue TR, Dotan E, et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2021; 19:439–57. https://doi.org/10.6004/jnccn.2021.0017. [PubMed].
103. Gourgou-Bourgade S, Bascoul-Mollevi C, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Boige V, Bérille J, Conroy T. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: results from the PRODIGE 4/ACCORD 11 randomized trial. J Clin Oncol. 2013; 31:23–29. https://doi.org/10.1200/JCO.2012.44.4869. [PubMed].
104. Wang Y, Camateros P, Cheung WY. A Real-World Comparison of FOLFIRINOX, Gemcitabine Plus nab-Paclitaxel, and Gemcitabine in Advanced Pancreatic Cancers. J Gastrointest Cancer. 2019; 50:62–68. https://doi.org/10.1007/s12029-017-0028-5. [PubMed].
105. Ducreux M, Seufferlein T, Van Laethem JL, Laurent-Puig P, Smolenschi C, Malka D, Boige V, Hollebecque A, Conroy T. Systemic treatment of pancreatic cancer revisited. Semin Oncol. 2019; 46:28–38. https://doi.org/10.1053/j.seminoncol.2018.12.003. [PubMed].
106. de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol. 2014; 741:8–16. https://doi.org/10.1016/j.ejphar.2014.07.041. [PubMed].
107. Babic A, Schnure N, Neupane NP, Zaman MM, Rifai N, Welch MW, Brais LK, Rubinson DA, Morales-Oyarvide V, Yuan C, Zhang S, Poole EM, Wolpin BM, et al. Plasma inflammatory cytokines and survival of pancreatic cancer patients. Clin Transl Gastroenterol. 2018; 9:145. https://doi.org/10.1038/s41424-018-0008-5. [PubMed].
108. Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galbán CJ, Galbán S, di Magliano MP. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012; 7:e49707. https://doi.org/10.1371/journal.pone.0049707. [PubMed].
109. Matsuoka T, Yashiro M. Molecular targets for the treatment of pancreatic cancer: Clinical and experimental studies. World J Gastroenterol. 2016; 22:776–89. https://doi.org/10.3748/wjg.v22.i2.776. [PubMed].
110. Wang S, Zheng Y, Yang F, Zhu L, Zhu XQ, Wang ZF, Wu XL, Zhou CH, Yan JY, Hu BY, Kong B, Fu DL, Bruns C, et al. The molecular biology of pancreatic adenocarcinoma: translational challenges and clinical perspectives. Signal Transduct Target Ther. 2021; 6:249. https://doi.org/10.1038/s41392-021-00659-4. [PubMed].