
Introduction
Gastrointestinal stromal tumor (GIST), the most common primary mesenchymal neoplasm of the gastrointestinal tract, accounts for 2–3% of all gastric malignancies [1, 2]. GISTs are characterized by variable behavior and differentiation towards the interstitial cells of Cajal [1, 2]. The tumors are generally immunopositive for CD117 (KIT) and DOG1 [3, 4].
Approximately 60–70% of GISTs occur in the stomach, 20–30% in the small intestine, 5% in the colon and rectum, and 1% in the esophagus [2]. On rare occasions, they form solitary masses (extragastrointestinal tumors) in the omentum or mesentery [2]. GISTs are mainly sporadic and occur in older adults (median age, 60–65 years) without gender predilection [2]. Complete resection is the treatment of choice in localized disease [5].
KIT proto-oncogene, receptor tyrosine kinase (KIT) and platelet derived growth factor receptor alpha (PDGFRA) oncogenic mutations have been detected in 80% and 10% of GISTs, respectively. They appear to be early events in GIST development [6, 7].
Additional genomic/chromosomal alterations are required for tumor progression. Such aberrations have been studied by different techniques: banding analysis, fluorescence in situ hybridization (FISH), comparative genomic hybridization (CGH), array CGH, and high-throughput sequencing [8–14].
A cytogenetic approach was, compared with other methods, infrequently applied to the study of genomic changes of GISTs, with karyotypes of less than 60 tumors having been reported [8]. This notwithstanding, cytogenetics provides a genome-wide overview of chromosome alterations while at the same time being informative about intratumor heterogeneity and the direction in which any clonal evolution may be proceeding. Furthermore, certain aberrations, such as balanced translocations, inversions, and telomeric associations, may not be detectable in studies using other methods.
Over more than two decades, we have as part of our diagnostic practice collected a consecutive series of GISTs analyzed cytogenetically and genotyped molecularly. Here, we report the accumulated chromosome and mutation data focusing on intratumor heterogeneity as well as cytogenetic evolution.
Results
The findings of cytogenetic analyses of 306 GIST samples from 291 patients together with molecular data on 254 tumors are presented in Supplementary Table 1.
Clonal chromosome aberrations were found in 248 samples and 237 cases (81% each). 220 (89%) of the karyotypically abnormal samples were near-diploid, 15 (6%) were near-triploid, 7 (3%) had both near-diploid and near-triploid/tetraploid cells, whereas 6 (2%) were in the 4n–6n ploidy range.
The set of primary tumors consisted of 226 abnormal cases. Their clinical and histopathological data are summarized in Table 1. Of the 226 GISTs, 149 (66%) showed one clone only, whereas the remaining tumors demonstrated clonal evolution. Specifically, 69 tumors (30%) displayed from 2 to 5 and 8 tumors (4%) from 6 to 23 karyotypically related clones. Unrelated clones (those with entirely disparate chromosome changes in cells from the same tumor) were found in a small proportion of cases (9%, 21/226).
Table 1: Clinical and histopathological data on 226 primary GISTs
| Clinical and histopathological data | Number of patients (%) |
|---|---|
| Age (years)a | 66 (23–93) |
| Sex | |
| Female | 105 (46) |
| Male | 121 (54) |
| Tumor location | |
| Esophagus | 3 (1) |
| Stomach | 181 (80) |
| Small intestine | 33 (15) |
| Rectum | 7 (3) |
| Extragastrointestinal | 2 (1) |
| Tumor size (cm)a | 5.0 (1.5–28.0) |
| Mitoses per 50 HPFa,b | 2 (0–178) |
| Tumor rupture | |
| Yes | 22 (10) |
| No | 196 (87) |
| Not determined | 8 (4) |
| Modified NIH risk criteriac | |
| Very low | 7 (3) |
| Low | 93 (41) |
| Intermediate | 44 (19) |
| High | 58 (26) |
| Metastatic | 19 (8) |
| Not able to classify | 5 (2) |
| Mutational analysis | |
| KIT exon 9 | 8 (4) |
| KIT exon 11 | 133 (59) |
| KIT exon 13 | 4 (2) |
| KIT exon 17 | 5 (2) |
| PDGFRA exon 12 | 3 (1) |
| PDGFRA exon 14 | 2 (1) |
| PDGFRA exon 18 | 31 (14) |
| No mutation detected | 11 (5) |
| Not done | 29 (13) |
| Preoperative systemic treatment | |
| No | 208 (92) |
| Yes | 18 (8) |
The number of chromosome aberrations per tumor/clone varied from 1 to over 50. 135 tumors (60%) displayed simple karyotypes (defined as ≤5 chromosomal changes) whereas 91 (40%) tumors had complex ones (>5 changes). A comparison of these two groups (Table 2) reveals that the karyotypically complex tumors were more often non-gastric (P < 0.001), larger (P < 0.001), more mitotically active (P = 0.009) and had a higher risk of tumor rupture (P < 0.001) and recurrence (P < 0.001). In contrast, no significant differences in mutation status between these two cytogenetic groups were found.
Table 2: Associations between karyotypic complexity and clinicopathological variables in 226 primary GISTs
| Clinicopathological variables | Karyotype | P value | |
|---|---|---|---|
| Simple (≤5 changes) | Complex (>5 changes) | ||
| Agea | 67 (25–87) | 66 (23–93) | 0.60 |
| Sex | 0.42 | ||
| Female | 66 | 39 | |
| Male | 69 | 52 | |
| Tumor location | <0.001 | ||
| Esophagus | 1 | 2 | |
| Stomach | 122 | 59 | |
| Small intestine | 11 | 22 | |
| Rectum | 1 | 6 | |
| Extragastrointestinal | 0 | 2 | |
| Tumor size (cm)a | 4.2 (1.5–24.0) | 7.0 (2.0–28.0) | <0.001 |
| Mitoses per 50 HPFa,b | 2 (0–53) | 3 (0–178) | 0.009 |
| Tumor rupture | <0.001 | ||
| Yes | 6 | 16 | |
| No | 128 | 68 | |
| Not determined | 1 | 7 | |
| Modified NIH risk criteriac | <0.001 | ||
| Very low | 7 | 0 | |
| Low | 66 | 27 | |
| Intermediate | 34 | 10 | |
| High | 20 | 38 | |
| Metastatic | 5 | 14 | |
| Not able to classify | 3 | 2 | |
| Mutational analysis | 0.31d | ||
| KIT exon 9 | 3 | 5 | |
| KIT exon 11 | 75 | 58 | |
| KIT exon 13 | 3 | 1 | |
| KIT exon 17 | 5 | 0 | |
| PDGFRA exon 12 | 3 | 0 | |
| PDGFRA exon 14 | 2 | 0 | |
| PDGFRA exon 18 | 18 | 13 | |
| No mutation detected | 4 | 7 | |
| Not done | 22 | 7 | |
To get more accurate and representative data on the cytogenetic profile and specific aberrations of GISTs, we excluded tumors with incomplete karyotypes (cases 19, 93, 201, and 207) from further analyses. We also excluded changes found in unrelated clones, the karyotypic relevance of which is uncertain. These near-diploid clones displayed simple changes, most frequently -Y and +7, or occasionally balanced aberrations. Tumors with only such aberrations (cases 9, 51, 59, 104, 132, and 226) were also left out. The remaining 216 cases comprised 173 gastric and 43 non-gastric GISTs.
The identified chromosome abnormalities were both numerical and structural. Among the latter, balanced translocations were uncommon. Recurrent structural aberrations were the dicentric chromosome dic(19;19)(q13;q13) (7 cases), the isochromosomes i(1)(q10), i(8)(q10), and i(17)(q10), and the ring chromosomes r(16)(p13q24) and r(19)(p13q13) (2–3 cases each). 25 GISTs demonstrated clonal telomeric associations with up to 16 different tas per tumor.
Losses of chromosomes, entire or partial, were more common than gains in the vast majority of the near-diploid tumors/clones whereas in polyploid clones, particularly in near-triploid ones, the proportion of gains increased and could be comparable to losses.
The frequencies of losses and gains in the 42 chromosome arms found in the 216 GISTs are summarized in Supplementary Table 2 and depicted in Figure 1A. In total, the most frequent losses affected chromosomes 14 (76%), 22 (44%), 1/1p (36%), and 15 (30%). In acrocentric chromosomes 14, 15, and 22, losses of the q-arm (p-arm changes were mostly untraceable) were partial only in less than 15% of cases, whereas whole chromosome losses predominated. In contrast, loss of an entire chromosome 1 was seen in less than 10% of tumors.
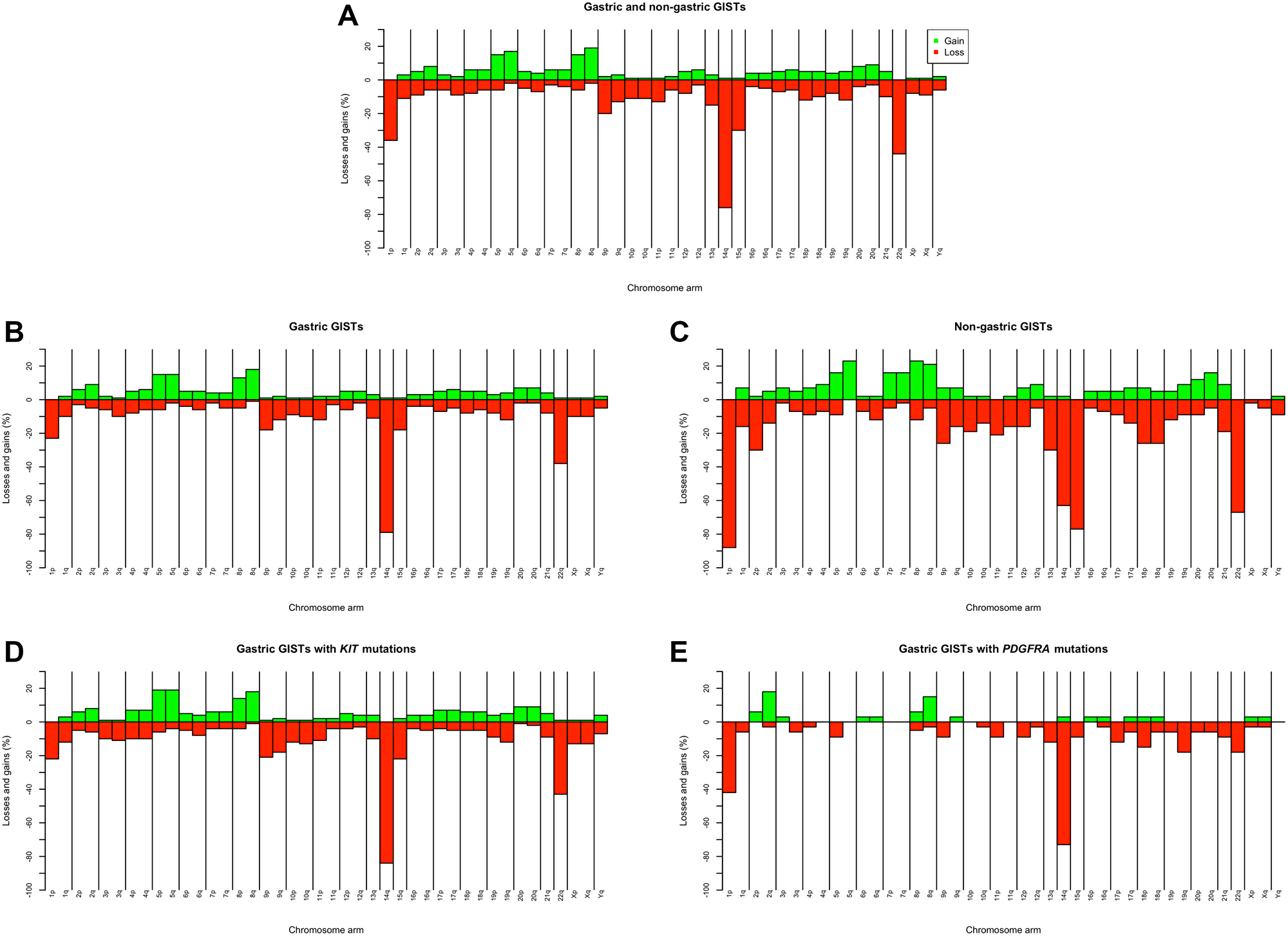
Figure 1: Chromosome imbalances identified by karyotyping in 216 primary both gastric and non-gastric GISTs (A); chromosome imbalances in 173 gastric (B) and 43 non-gastric (C) GISTs, and chromosome imbalances in 113 gastric GISTs with KIT mutations (D) and 33 gastric GISTs with PDGFRA mutations (E).
The most frequent loss, -14, was found in 29 GISTs as the sole abnormality. In addition, in 10 tumors displaying clonal evolution, a solitary -14 was present in the basic clone. The double loss -14 and -22 was the only change in 12 tumors, whereas in 6 cases such a clone was the basic one but was accompanied by karyotypic evolution.
Among whole chromosome gains, +5 (16%) and +8 (15%) were most common, followed by +2 and +20 (7% each) and +4 and +7 (6% each).
Four gastric (cases 31, 146, 178, and 282) and 2 esophageal (cases 57 and 288) GISTs demonstrated different karyotypic patterns; they were hyperdiploid (50–56 chromosomes) and displayed several (3 to 10) trisomies/tetrasomies alongside structural changes.
We further compared the cytogenetic profiles of gastric and non-gastric GISTs. The associations between tumor location and other clinicopathological variables are presented in Supplementary Table 3.
The patterns of imbalances in 42 chromosome arms of 173 gastric and 43 non-gastric GISTs are shown in Supplementary Table 4 and Figure 1B and 1C. Table 3 summarizes data on the associations between the four most frequent losses – of 1p, 14q, 15q, and 22q - on the one hand and tumor site on the other. Significant differences were found in the incidence of all these losses, with -14q being the most common in gastric and 1p- in non-gastric GISTs. Additionally, the non-gastric tumors showed statistically higher rates of the less frequent losses 2p- (30% vs. 3%; P < 0.001) and -13q (30% vs. 11%; P = 0.003).
Table 3: Associations between main chromosome losses, tumor location, karyotypic complexity, and genotype in GISTs
| Loss of chromosome arma | 1p | 14q | 15q | 22q | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | P value | n | % | P value | n | % | P value | n | % | P value | |
| Location Gastric (n = 173) Non-gastric (n = 43) | 40 38 | 23.1 88.4 | <0.001 | 137 27 | 79.2 62.8 | 0.029 | 31 33 | 17.9 76.7 | <0.001 | 65 29 | 37.6 67.4 | <0.001 |
| Karyotype (≤5 changes) Gastric (n = 137) Non-gastric (n = 16) | 16 14 | 11.9 87.5 | <0.001 | 112 8 | 81.8 50.0 | 0.007 | 9 12 | 6.6 75.0 | <0.001 | 42 8 | 30.9 50.0 | 0.16 |
| Genotype of gastric KIT (n = 113) PDGFRA (n = 33) | 25 14 | 22.1 42.4 | 0.026 | 95 24 | 84.1 72.7 | 0.20 | 25 3 | 22.1 9.1 | 0.13 | 48 6 | 42.5 18.2 | 0.013 |
To find out when in karyotypic evolution the said differences between gastric and non-gastric GISTs become evident, we selected for study tumors/clones with no more than 5 changes (i.e., with simple karyotypes). Altogether 55 chromosome imbalances were assessed in a total of 137 gastric and 16 non-gastric such tumors. As shown in Table 3, significant differences in the rate of losses of 1p, 14q, and 15q (but not 22q) were found between karyotypically simple gastric and non-gastric tumors/clones. Besides, non-gastric tumors with ≤5 changes had significantly higher incidence of 2p losses than did gastric tumors (20% vs. 0%; P < 0.001). For practically all the remaining imbalances (not shown), the values were below 10%, in fact 0% for half of them. Thus, the main differences in the cytogenetic profile between gastric and non-gastric GISTs emerge early in karyotypic evolution.
We further assessed a possible impact of mutation status on the cytogenetic profile of GISTs by comparing chromosome imbalances in the two main genotypic groups, KIT- and PDGFRA-mutated tumors. Since no PDGFRA mutations were identified in non-gastric GISTs in this study (Supplementary Table 3), and to avoid any site-specific effect in KIT-mutated tumors, only gastric tumors were compared.
Chromosome imbalances found in 42 chromosome arms of gastric 113 KIT- and 33 PDGFRA-mutated tumors are presented in Supplementary Table 5 and Figure 1D and 1E. Overall, PDGFRA-mutated tumors exhibited considerably fewer aberrations: in 46% (39/84) of all imbalances (losses or gains) the values were 0%, in contrast to only 2% (2/84) in KIT-mutated tumors. Table 3 shows that the frequencies of 14q and 15q losses did not differ significantly between KIT- and PDGFRA-mutated GISTs. In PDGFRA-mutated tumors, on the other hand, the rate of 1p losses was higher and 22q losses lower.
Altogether, 16 abnormal metastatic samples from 13 tumors (5 gastric and 8 non-gastric) were available for analyses. The majority of metastatic samples (11/16, 69%) were, like the primary tumor set, near-diploid, 4 (25%) were near-triploid, while one sample had 2n-4n clones. Half of the metastatic samples displayed clonal evolution in the form of 2–5 related clones whereas the clones in two samples were unrelated. All but one sample had complex karyotypes; however, that one simple karyotype carried a solitary +7 that probably was not representative of the tumor parenchyma.
On six occasions (cases 1a-c; 8a,b; 21b,c; 28a,b; 43a,b; and 78a-d) it was possible to compare primary tumors with their metastases. In general, metastatic samples resembled their matching primaries. Apart from that, the relationship between the genetic alterations of primary tumors and metastatic samples reflected three main evolutionary scenarios: First, the primary tumor and metastasis sometimes had practically identical or very similar aberrations (cases 43a,b and 28a,b). Second, the metastases could display a markedly higher degree of karyotypic complexity and/or heterogeneity than did the primary tumor (cases 1a-c and 78a-d). Third, and in contrast to the preceding scenario, the metastases sometimes exhibited less karyotypic complexity and/or heterogeneity (cases 8a,b and 21a,b). In three cases (1, 2, and 78), two metastatic samples were available for comparison; the synchronous metastases (cases 1b,c and 2a,b) showed closer karyotypic similarity to each other than did the consecutive samples (case 78c,d).
DISCUSSION
Published cytogenetic data on GIST include less than 60 tumors with chromosome aberrations [3, 8], in contrast to information on several large series (around 50–200 cases) of tumors investigated by CGH/array CGH or high-throughput sequencing. The present, largest by far, study adds nearly 240 abnormal tumor samples, both gastric and non-gastric, to the existing karyotypic database, coupled with mutation data for the majority of them. Besides obtaining more knowledge about the general pattern of chromosome aberrations in GISTs, we also aimed at investigating cytogenetic heterogeneity in this tumor type and a comparison of the cytogenetic pathways taken by gastric and non-gastric GISTs.
It is generally accepted that whereas oncogenic KIT and PDGFRA mutations are necessary for the neoplastic transformation of GIST, additional somatic genomic alterations are required for tumor progression. Banding cytogenetics is one of the techniques best suited to monitor the stepwise acquisition of chromosome aberrations, i.e., clonal evolution, characteristic of this process. Indeed, detailed analysis revealed extensive cytogenetic heterogeneity in the present series, much higher than reported previously [8]. Clonal evolution was found in nearly 34% of GISTs, generally in the form of 2–5 clones/subclones (up to 23).
The acquisition of new aberrations did not always proceed in a linear manner but rather led to a variety of changes upon which further selection might work. By way of example, case 116b showed gain of an extra 8q arm occurring in one subclone through formation of der(8;13)(q10;q10), whereas in another subclone the same imbalance was the result of a dic(8;15)(p11;p11). In case 181, the initial changes -14 and -22 were followed in one subclone by a der(1)t(1;2)(q44;q11) leading to gain of 2q, while another subclone instead had a der(1)t(1;8)(q44;q22) causing gain of 8q.
Clonal telomeric associations were found in 25, mostly gastric, GISTs. In spite of the fact that telomeric associations do not allegedly involve loss of chromosome material, they may be the precursors of dicentric chromosomes. The latter are known to be prone to further rearrangements because of additional risk of breakage during mitosis. The karyotypes of some tumors demonstrated a strong association between clonal telomeric fusions of particular chromosomes and clonal dicentrics or derivatives of the same chromosomes with variable breakpoints. Thus, coexistence of clones with tas(11;19)(p15;q13), dic(11;19)(p11;q13), and der(11;19)(q10;q10) was seen in case 266. A particular dicentric chromosome, dic(19;19)(q13;q13), was found recurrently; a dic(19;19), no breakpoints were given, was reported in GIST previously [15].
In agreement with the findings of CGH/array CGH studies [9–11], chromosome losses were more common than gains among primary GISTs also in the present series. The most frequent losses were of 14q (76%), 22q (44%), 1p (36%), and 15q (30%). These losses, albeit with somewhat variable rates, were reported also previously as the most common in GISTs [9, 11].
Monosomies for chromosomes 14 and 22 were cytogenetically detected early on [3, 16] and have since been accepted as highly characteristic aberrations for GISTs with both benign and malignant behavior. Yet, the pathogenetic roles of these losses, as well as of 1p and 15q, are not fully established [17–19]. Thus, it was reported [9, 11, 20, 21] that gastric and intestinal GISTs show site-dependent differences in the frequencies of the said losses. According to the proposed model, GISTs evolving along the -14q pathway are mainly gastric tumors with non-complex karyotypes and a more favorable clinical course, whereas losses of 1p and 15q are typical of intestinal tumors with more complex karyotypes and a tendency towards malignancy [11].
The present study also showed site-specific cytogenetic differences between gastric and non-gastric GISTs in the incidence of losses of 14q, 22q, 1p, 15q, and 13q. Our results are very similar to the corresponding frequencies found by CGH [11]. The higher rate we saw of the less common loss of 2p in non-gastric tumors, compared with gastric, was not reported earlier [11].
It appears that the main statistically significant differences in the cytogenetic profile between gastric and non-gastric GISTs become evident early during karyotypic evolution, when the karyotypes are still simple. In the present series, all GISTs with -14 as the sole aberration (n = 29) and tumors in which the -14 was the first event in clonal evolution (n = 10), as well as with double losses (-14, -22) (n = 12) and tumors in which these two changes were the first in clonal evolution (n = 6), were gastric. On the other hand, the high prevalence of 1p and 15q losses in non-gastric GISTs signifies their importance in site-specific, but non-gastric, clonal evolution. The evolutionary scenario in non-gastric GISTs is complicated by the fact that these tumors have no single loss, equivalent to -14, that is seen regularly and as the sole change. Nevertheless, one can hypothesize that loss of 1p plays a primary role in the tumorigenesis of non-gastric GIST, based on the data that this aberration is the most frequent and early event in karyotypic evolution. Furthermore, cases 126 and 238 of non-gastric tumors showed a single loss of 1p; sole 1p loss was also found in tumors of this location by CGH [11]. As to the next step, clones with the solitary double loss, 1p- and -15, were occasionally detected in non-gastric GISTs, not only in the present dataset (case 221) but also in reported cases [3, 11, 17]. Losses of 14q and 22q are seen in combination with 1p- and -15q at relatively high rates and also at early stages of cytogenetic evolution indicating that these four losses are nonrandomly involved in the development of non-gastric GISTs [9, 11, 20, 21].
Several CGH/array CGH studies [9, 10, 12] focused on the possible impact of mutation status - KIT vs. PDGFRA mutations - on the cytogenetic profile and progression pathways of GISTs. Among the gastric GISTs of the present series analyzed in detail, 65% carried KIT mutations whereas 19% had PDGFRA mutations. The PDGFRA-mutated GISTs displayed markedly fewer chromosome changes, in accordance with their generally more favorable outcome [22], even though three of them were metastatic. The frequencies of 14q and 15q losses in our study did not differ statistically between KIT- and PDGFRA-mutated gastric tumors although the rate of 22q loss in the latter was significantly lower while the rate of 1p losses was higher.
The reported data on the impact of the said mutations on the chromosome imbalance profile of the gastric GISTs are inconsistent. According to Silva et al. [10], tumors with PDGFRA mutations had the same overall pattern of alterations as those with KIT mutations but displayed less genomic complexity. Wozniak et al. [9] found no statistical difference in the frequency of any most common chromosome loss studied, such as -14q, -22q, -1p, and -15q, between KIT- and PDGFRA-mutant gastric tumors. In a systematic study of PDGFRA-mutated GISTs, Schaefer et al. [12] found significantly fewer chromosome aberrations in them than in KIT-mutated tumors. A comparison of GISTs of only gastric sites showed that losses of 14q and 22q occurred statistically less frequently in PDGFRA-mutated tumors.
In addition to the relative paucity of aberrations in GISTs with PDGFRA mutations seen in the present study, the finding of a lower incidence of 22q losses in gastric tumors with this genotype was also in accordance with published data [12]. Notably, the frequency of 22q losses (18%) in PDGFRA-mutated gastric GISTs found by us agrees well with the rate (15%) reported by Schaefer et al. [12]. It is of interest that in our series, none of the gastric tumors with either the double loss -14 and -22 as the only change, or this change in the basic clone, carried PDGFRA mutations. The importance of 22q losses in the progression of PDGFRA-mutated gastric GISTs is further corroborated by the fact that, in our series of totally six primary PDGFRA-mutated tumors with -22q, three (cases 215, 232, and 279) were high-risk and the other three (cases 37, 78a,b, and 190) were metastatic. Two of the three metastatic GISTs had nonetheless simple karyotypes and all three tumors shared losses of/in 1p, 19q, and 22q, whereas the three high-risk tumors with complex karyotypes had 1p- and -22 in common. The importance of these observations has to be evaluated on larger numbers of PDGFRA-mutated tumors.
Among the 33 karyotypically abnormal GISTs with PDGFRA mutation, two tumors had rare exon 14 and another two rare exon 12 mutations. All four had simple karyotypes. Chromosome changes in GISTs with exon 14 mutations were the same as in tumors with exon 18 mutations and typical for gastric GISTs, such as -14. Aberrations were less common in tumors with exon 12 mutations.
Among the karyotypically abnormal GISTs of the present series, KIT exon 9 mutations (n = 10) were seen mostly in non-gastric tumors, the majority of which had complex karyotypes or were metastatic. Also in another series [10], the highest cytogenetic complexity was seen in tumors bearing KIT exon 9 mutations. In contrast, all KIT exon 17 mutations (n = 5) were found in abnormal gastric GISTs. They displayed simple karyotypes, mainly with changes typical for this location (such as sole -14).
To our knowledge, no karyotypes of KIT/PDGFRA mutation-negative GISTs have been reported and existing CGH/array CGH array data on chromosome alterations in tumors with this genotype are partly inconsistent [10, 13, 23, 24]. There were 14 KIT/PDGFRA mutation-negative tumors in our dataset, three of which were karyotypically normal. The remaining 11 (6 gastric and 5 non-gastric) tumors comprised 5% of the 226 primary karyotypically abnormal GISTs. Three gastric GISTs (cases 39, 88, and 179) showed aberrations mainly consistent with this tumor site (such as a solitary -14), whereas the remainder displayed unusual patterns: a sole i(4)(q10) (case 228), a solitary +6 (case 226), or several polysomies (case 282). All 5 non-gastric tumors (cases 93, 94, 126, 208, and 281) had chromosome changes typical for this location. Hence, our series demonstrates a high proportion of karyotypically abnormal tumors with wild-type genotype. Some insights into the mechanisms of molecular heterogeneity of mutation-negative GISTs were provided recently [19, 25, 26].
The present study included 3 esophageal tumors (cases 57, 79, and 288). GISTs in this location are rare (1%) and, to the best of our knowledge, no karyotypic information about such tumors has been reported. The esophageal tumor of case 79 displayed changes observed in other non-gastric GISTs, such as partial deletions of 1p, 2p, 13q, and 22q. The two other tumors exhibited several trisomies, of which +4, +5, and +8 were common. The tumor of case 57 also showed concomitant losses of 1p and 2p in subclones. Surprisingly, both esophageal GISTs with trisomies (cases 57 and 288) showed the same KIT exon 13 mutation, Lys642Glu, whereas the tumor of case 79 had a KIT exon 11 mutation. The numbers are too small to say whether the karyotypic and molecular similarities between the two esophageal GISTs are coincidental or reflect something systematic.
In conclusion, the intratumor heterogeneity, including site-specific cytogenetic evolution patterns, identified in the tumors of the present series are consistent with the current concept of GISTs as a heterogeneous collection of molecular entities linked by a common histology and presumed cell of origin [27]. Based on the examination of such a large dataset, we were able to elucidate the roles of individual chromosomes in the pathogenesis of tumors with different genotypes and sites of origin, and provide further evidence that gastric and non-gastric GISTs develop via different cytogenetic pathways. Finally, karyotypically complex tumors had a more malignant behavior meaning that cytogenetic profiling could be explored as a prognostic marker, especially when applying statistical methods. A better understanding of the variable cytogenetic pathways during tumor progression may help to improve diagnostic, prognostic, and treatment decisions.
Materials and Methods
Ethics statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Data Protection Officer at Oslo University Hospital (approval nr 2016/16853 and 21/03190, dated Nov. 2016 and Feb. 2021, respectively). Informed consent was obtained from all subjects involved in the study. All information about patients has been de-identified.
Patients and tumor material
The present study comprises an unselected consecutive series of GIST samples from 291 patients received during 1998–2020. All patients were treated at Oslo University Hospital, Norway. There were 159 men and 132 women. Median age at diagnosis was 66 years (range 23–93 years). Samples from primary tumors were received from 281 patients whereas only metastases were available from 10 patients. Several samples (2 to 4) were received from 10 patients, raising the total number of samples studied to 306. 222 GISTs were located in the stomach, 48 in the small intestine, 14 in the rectum, 3 in the esophagus, and 4 were extragastrointestinal tumors. Nineteen patients (7%) had been treated with imatinib and one patient had received both imatinib and sunitinib before sampling of primary tumors; 10 patients had received imatinib before sampling of metastases (Supplementary Table 1). No treatment impact on the cytogenetic pattern was noticed.
For main analyses and correlations studies, only primary GISTs were selected. Tumors whose karyotype showed a solitary -Y (Supplementary Table 1, cases 32, 68, 86, and 272) were not included. From tumor cases 78 and 116, each represented by 2 samples, all aberrations were recorded only once.
Chromosome banding
Fresh tissue from a representative area of the resected tumor was received and analysed cytogenetically as previously described [28].
Mutation analyses
Genomic DNA was extracted from fresh frozen or paraffin-embedded tumor tissue. Exons 9, 11, 13, and 17 of KIT and exons 12, 14, and 18 of PDGFRA were analyzed by Sanger sequencing and categorized as described previously [29]. Tumors not analyzed in clinical routine were analyzed using AmpliSeq for Illumina Cancer Hotspot Panel version 2 as previously described [30]. The scoring was based on the sequence NM_000222.2 for KIT and NM_006206.4 for PDGFRA on the Human GRCh37/hg19 assembly [31].
Statistical analyses
Associations between variables were investigated with two-tailed Fisher’s exact test or Pearson’s χ2 test for categorical variables and independent Mann-Whitney U test or Kruskal-Wallis test for continuous variables. P < 0.05 was considered statistically significant. IBM SPSS Statistics for Windows version 25.0 (Armonk, NY, USA) was used.
Data availability
The data presented in this study are available on request from the corresponding authors. The data are not publicly available due to ethical issues.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This study was supported by the Radium Hospital Legacy and the South-East Norway Regional Health Authority (Grant Number 2019064).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors' response and revisions as well as expedited peer-review in Oncotarget.
References
1. Kang HC, Menias CO, Gaballah AH, Shroff S, Taggart MW, Garg N, Elsayes KM. Beyond the GIST: mesenchymal tumors of the stomach. Radiographics. 2013; 33:1673–90. https://doi.org/10.1148/rg.336135507. [PubMed].
2. Tos AD, Hornick J, Miettinen M, Wanless I, Waldermann E. Gastrointestinal stromal tumor. WHO Classification of Tumors - Soft Tissue and Bone Tumors. 2020; 216–21.
3. Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. desmoplastic small round-cell tumors. Cancer Genet Cytogenet. 2002; 138:1–10. https://doi.org/10.1016/s0165-4608(02)00680-5. [PubMed].
4. West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R, Goldblum JR, Brown PO, Heinrich MC, van de Rijn M. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004; 165:107–13. https://doi.org/10.1016/S0002-9440(10)63279-8. [PubMed].
5. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006; 130:1466–78. https://doi.org/10.5858/2006-130-1466-GSTROM. [PubMed].
6. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279:577–80. https://doi.org/10.1126/science.279.5350.577. [PubMed].
7. Liegl-Atzwanger B, Fletcher JA, Fletcher CD. Gastrointestinal stromal tumors. Virchows Arch. 2010; 456:111–27. https://doi.org/10.1007/s00428-010-0891-y. [PubMed].
8. Mitelman F, Johansson B, Mertens F. Mitelman database of chromosome aberrations in cancer. 2020. https://mitelmandatabase.isb-cgc.org.
9. Wozniak A, Sciot R, Guillou L, Pauwels P, Wasag B, Stul M, Vermeesch JR, Vandenberghe P, Limon J, Debiec-Rychter M. Array CGH analysis in primary gastrointestinal stromal tumors: cytogenetic profile correlates with anatomic site and tumor aggressiveness, irrespective of mutational status. Genes Chromosomes Cancer. 2007; 46:261–76. https://doi.org/10.1002/gcc.20408. [PubMed].
10. Silva M, Veiga I, Ribeiro FR, Vieira J, Pinto C, Pinheiro M, Mesquita B, Santos C, Soares M, Dinis J, Santos L, Lopes P, Afonso M, et al. Chromosome copy number changes carry prognostic information independent of KIT/PDGFRA point mutations in gastrointestinal stromal tumors. BMC Med. 2010; 8:26. https://doi.org/10.1186/1741-7015-8-26. [PubMed].
11. Gunawan B, von Heydebreck A, Sander B, Schulten HJ, Haller F, Langer C, Armbrust T, Bollmann M, Gasparov S, Kovac D, Füzesi L. An oncogenetic tree model in gastrointestinal stromal tumours (GISTs) identifies different pathways of cytogenetic evolution with prognostic implications. J Pathol. 2007; 211:463–70. https://doi.org/10.1002/path.2128. [PubMed].
12. Schaefer IM, Delfs C, Cameron S, Gunawan B, Agaimy A, Ghadimi BM, Haller F. Chromosomal aberrations in primary PDGFRA-mutated gastrointestinal stromal tumors. Hum Pathol. 2014; 45:85–97. https://doi.org/10.1016/j.humpath.2013.05.027. [PubMed].
13. Astolfi A, Nannini M, Pantaleo MA, Di Battista M, Heinrich MC, Santini D, Catena F, Corless CL, Maleddu A, Saponara M, Lolli C, Di Scioscio V, Formica S, Biasco G. A molecular portrait of gastrointestinal stromal tumors: an integrative analysis of gene expression profiling and high-resolution genomic copy number. Lab Invest. 2010; 90:1285–94. https://doi.org/10.1038/labinvest.2010.110. [PubMed].
14. Haefliger S, Marston K, Juskevicius D, Meyer-Schaller N, Forster A, Nicolet S, Komminoth P, Stauffer E, Cathomas G, Hoeller S, Tornillo L, Dirnhofer S, Terracciano LM, et al. Molecular Profile of Gastrointestinal Stromal Tumors in Sixty-Eight Patients from a Single Swiss Institution. Pathobiology. 2020; 87:171–78. https://doi.org/10.1159/000505407. [PubMed].
15. Gunawan B, Bergmann F, Höer J, Langer C, Schumpelick V, Becker H, Füzesi L. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Hum Pathol. 2002; 33:316–21. https://doi.org/10.1053/hupa.2002.32216. [PubMed].
16. Marci V, Casorzo L, Sarotto I, Dogliani N, Milazzo MG, Risio M. Gastrointestinal stromal tumor, uncommitted type, with monosomies 14 and 22 as the only chromosomal abnormalities. Cancer Genet Cytogenet. 1998; 102:135–38. https://doi.org/10.1016/s0165-4608(97)00319-1. [PubMed].
17. Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, Kordek R, Miettinen M. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet. 2001; 128:24–30. https://doi.org/10.1016/s0165-4608(01)00395-8. [PubMed].
18. El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000; 60:3899–903. [PubMed].
19. Schaefer IM, Mariño-Enríquez A, Fletcher JA. What is New in Gastrointestinal Stromal Tumor? Adv Anat Pathol. 2017; 24:259–67. https://doi.org/10.1097/PAP.0000000000000158. [PubMed].
20. Gunawan B, Schulten HJ, von Heydebreck A, Schmidt B, Enders C, Höer J, Langer C, Schüler P, Schindler CG, Kuhlgatz J, Füzesi L. Site-independent prognostic value of chromosome 9q loss in primary gastrointestinal stromal tumours. J Pathol. 2004; 202:421–29. https://doi.org/10.1002/path.1537. [PubMed].
21. Chen Y, Liou CP, Tseng HH, Jan YJ, Li CF, Tzeng CC. Deletions of chromosome 1p and 15q are associated with aggressiveness of gastrointestinal stromal tumors. J Formos Med Assoc. 2009; 108:28–37. https://doi.org/10.1016/S0929-6646(09)60029-2. [PubMed].
22. Lasota J, Dansonka-Mieszkowska A, Sobin LH, Miettinen M. A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Lab Invest. 2004; 84:874–83. https://doi.org/10.1038/labinvest.3700122. [PubMed].
23. Assämäki R, Sarlomo-Rikala M, Lopez-Guerrero JA, Lasota J, Andersson LC, Llombart-Bosch A, Miettinen M, Knuutila S. Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2007; 46:564–76. https://doi.org/10.1002/gcc.20439. [PubMed].
24. Belinsky MG, Skorobogatko YV, Rink L, Pei J, Cai KQ, Vanderveer LA, Riddell D, Merkel E, Tarn C, Eisenberg BL, von Mehren M, Testa JR, Godwin AK. High density DNA array analysis reveals distinct genomic profiles in a subset of gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2009; 48:886–96. https://doi.org/10.1002/gcc.20689. [PubMed].
25. Astolfi A, Indio V, Nannini M, Saponara M, Schipani A, De Leo A, Altimari A, Vincenzi B, Comandini D, Grignani G, Secchiero P, Urbini M, Pantaleo MA. Targeted Deep Sequencing Uncovers Cryptic KIT Mutations in KIT/PDGFRA/SDH/RAS-P Wild-Type GIST. Front Oncol. 2020; 10:504. https://doi.org/10.3389/fonc.2020.00504. [PubMed].
26. Astolfi A, Pantaleo MA, Indio V, Urbini M, Nannini M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int J Mol Sci. 2020; 21:3313. https://doi.org/10.3390/ijms21093313. [PubMed].
27. Barnett CM, Corless CL, Heinrich MC. Gastrointestinal stromal tumors: molecular markers and genetic subtypes. Hematol Oncol Clin North Am. 2013; 27:871–88. https://doi.org/10.1016/j.hoc.2013.07.003. [PubMed].
28. Gorunova L, Bjerkehagen B, Micci F, Heim S, Panagopoulos I. Cytogenetic and Molecular Study of an Adult Sclerosing Rhabdomyosarcoma of the Extremity: MYOD1-mutation and Clonal Evolution. Cancer Genomics Proteomics. 2020; 17:563–69. https://doi.org/10.21873/cgp.20212. [PubMed].
29. Boye K, Berner JM, Hompland I, Bruland ØS, Stoldt S, Sundby Hall K, Bjerkehagen B, Hølmebakk T. Genotype and risk of tumour rupture in gastrointestinal stromal tumour. Br J Surg. 2018; 105:e169–75. https://doi.org/10.1002/bjs.10743. [PubMed].
30. Hølmebakk T, Wiedswang AM, Meza-Zepeda LA, Hompland I, Lobmaier IVK, Berner JM, Stoldt S, Boye K. Integrating Anatomical, Molecular and Clinical Risk Factors in Gastrointestinal Stromal Tumor of the Stomach. Ann Surg Oncol. 2021; 28:6837–45. https://doi.org/10.1245/s10434-021-09605-8. [PubMed].
31. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. https://doi.org/10.1101/gr.229102. [PubMed].