
Introduction
Spiced up with words like “emerging” and “promising” [1–4], numerous excellent reviews on senolytics can be friendly parodied in one sentence: ‘New promising strategies to fight devastating diseases are rapidly emerging, fueling new hopes and promising healthier lifespan with potential benefits to win the war on aging by using emergent senomorphics and promising senolytics’.
Despite these promises, only two studies showed lifespan extension by senolytics in mammals. Namely, fisetin extended lifespan in a small mouse study [5]. A combination of Dasatinib plus Quercetin (D+Q) increased median lifespan from 937 days to 996 days (by 6.3%) in mice (see Figure 6I in ref. [6]). As we will discuss, this modest increase in lifespan can be explained not only by killing of senescent cells, but also by off-target effects such as mTOR inhibition. These senolytics are available for human use and, for reasons discussed elsewhere [7], can be used for life extension in humans without the need for lifelong clinical trials.
Senolytics
The term senolytics, drugs that selectively kill senescent cells, was introduced by Kirkland and Tchkonia in 2015 [8]. Senolytics must extend lifespan by killing senescent cells, not by off-target mechanisms [8]. Kirkland and co-workers attempted to develop senolytics using bioinformatics followed by screening for siRNAs that kill senescent cells, followed by screening of potential drugs that may target these pathways [8]. They hypothesized that senescent cells can be selectively targeted, because they express pro-survival pathways, making them resistant to death [8–10]. While it seems paradoxical to kill cells, because they are resistant to killing there is a relevant analogy in oncology known as oncogene-addiction.
Crossroad of oncology and geroscience
The field of senolytics is at a crossroads of two disciplines: oncology and gerontology. Development of drugs that kills senescent cells selectively is an oncology-like task. All potential senolytics are either approved for cancer therapy (dasatinib, venetoclax) or experimental anti-cancer drugs (Fisetin and Quercetin), including failed drugs (the Hsp-90 inhibitor geldanamycin). But whether killing of senescent cells is the goal of anti-aging therapy is the realm of gerontology.
Lessons from cancer therapy
If cancer cells could be killed selectively without killing normal cells, then cancer would be curable. For almost a century, millions of scientists worldwide have worked on the cure for cancer, spending hundreds of billions of research dollars. Still, most common cancers remain incurable by chemotherapy. So, similarly, we cannot expect miracle from senolytics in such short time. Especially given that the cancer cell is an easier target than the senescent cell. In cancer therapy, some selectivity can be achieved by targeting cell proliferation. For example, microtubule active drugs such as paclitaxel and vinblastine kill cells entering mitosis. But targeting proliferation cannot be possibly exploited for killing senescent cells.
A second way to achieve selectivity in cancer therapy is targeting the tissue of cancer origin [11]. For example, targeting all prostate cells (normal and cancer) by anti-androgen deprivation or breast epithelial cells by anti-estrogens. This approach is not applicable for anti-aging therapy.
The third approach is targeting oncogenes that support the survival of cancer cells. For example, the Bcr-Abl oncoprotein, an anti-apoptotic kinase, drives chronic myelogenous leukemia [12]. Dasatinib, an inhibitor of Bcr-Abl, is approved for treatment of the BCR-ABL-driven leukemias [13].
Oncogene addiction and matching targets (technical description)
Inhibitors of Bcr-Abl (imatinib and dasatinib) induce apoptosis in Bcr-Abl-expressing cells [12]. The paradox is that Bcr-Abl is not necessary for cell survival, if cells do not have it, but it becomes necessary, if cells do have it. Normally, no cell has Bcr-Abl. For example, HL60 leukemia cells do not have and do not need Bcr-Abl. Inhibitors of Bcr-Abl exert no effect on HL60 cells [14]. But once HL60 cells are transfected with Bcr-Abl, they become Bcr-Abl-addicted. Inhibitors of Bcr-Abl induce apoptosis in BCR-Abl-transfected HL60 cells, while they have no effect on parental HL60 cells [14]. And this is even more surprising because Bcr-Abl renders HL60 resistant to standard chemotherapy. Oncogene addiction can be explained by the dam model [15]. Because Bcr-Abl blocks the apoptotic cascade, another pro-survival mechanism (for example, Bcl-2) may become dispensable. Specifically, whereas parental HL60 cells express high levels of Bcl-2, Bcr-Abl-expressing cells have no Bcl-2 [16]. Due to loss of Bcl-2, caspase-9 is activated (Figure 2 in ref. [17]). However, this activation does not cause apoptosis due to the Bcr-Abl dam. When the Bcr-Abl dam is inactivated by dasatinib or degraded by geldanamycin, then the stream overflows, killing the cell [15]. Combined targeting of BCL-2 and BCR-ABL eradicates chronic myeloid leukemia stem cells [18].
Noticeably, these anti-cancer drugs developed for oncogene-addicted cancers were re-discovered as senolytics: the Bcr-Abl kinase inhibitor dasatinib, the Bcl-2/BclxL inhibitors Venetoclax (ABT-199) and Navitoclax (ABT-263) and Hsp-90 inhibitors (geldanamycin).
Another side of the same coin is synthetic lethality [19, 20]. In 1997, Synthetic lethality was defined as a condition when “the loss of either of two genes is viable for the cell, but the simultaneous inactivation of both genes is lethal” [21]. In other words, loss of one target renders cell sensitive to inhibition of its matching target.
Let us take this one step further: combinations aimed at both targets (Figure 1). Matching drug combinations can selectively kill cells with known genetic/epigenetic background, while sparing other cells [22, 23]. I discussed anti-cancer combinations previously [23, 24]. And it is remarkable that out of two senolytic modalities that extend lifespan in mice, one is an empirical drug combination. Also, remarkably, one drug in this combination is the Bcr-Abl inhibitor that is used for oncogene-addicted leukemias. The next step would be designing mechanism-based combinations aimed toward matching and well-defined targets.
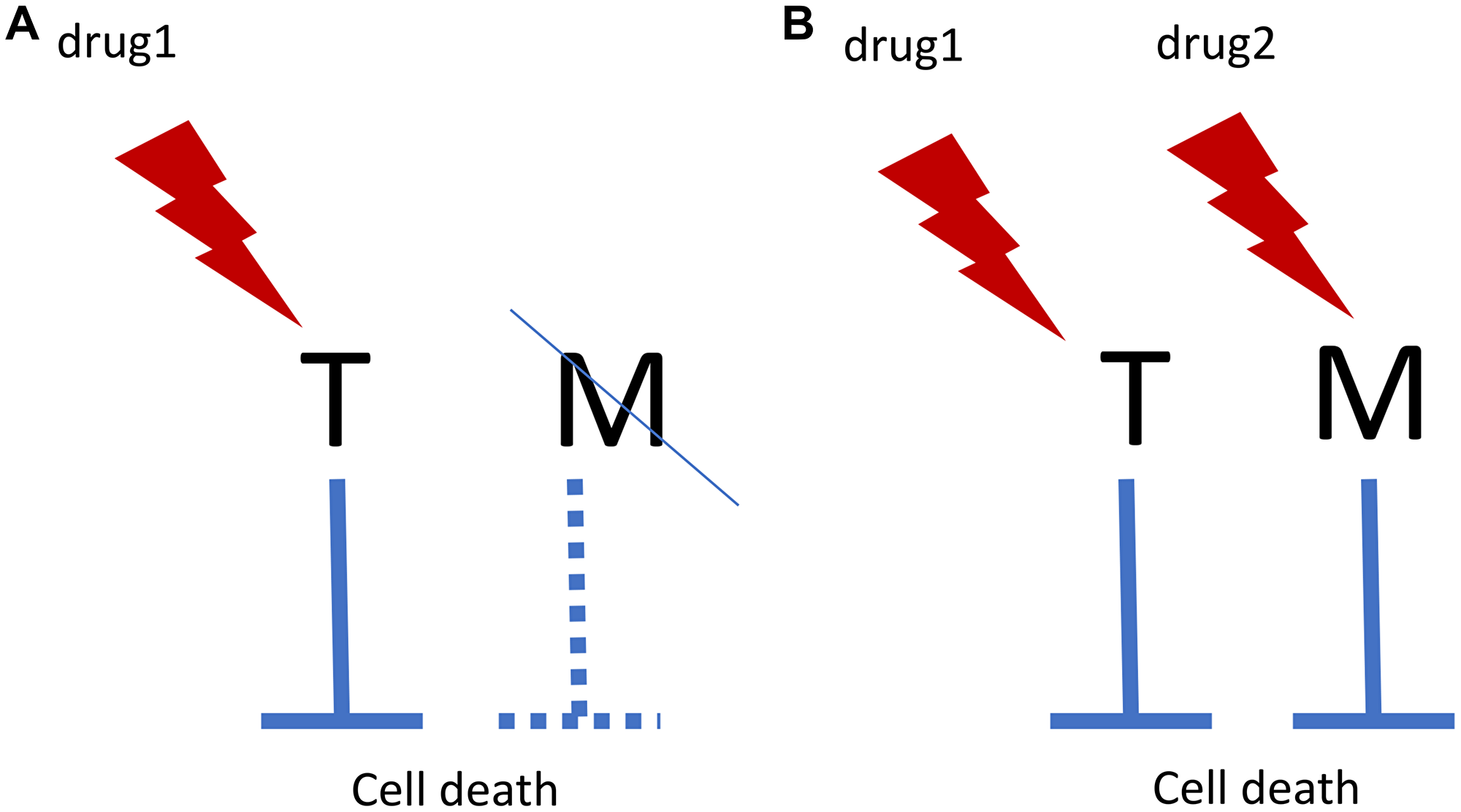
Figure 1: Oncogene addiction/synthetic lethality and matching targets. (A) In oncogene addiction, expression of an anti-apoptotic oncoprotein (Target, T) eventually leads to deactivation of matching (M) survival pathway. For example, T is Bcr-Abl and M is Bcl-2. Drug 1 kills such cells selectively. In synthetic lethality, loss of M renders cells sensitive to drug 1. (B) Matching drug combination. Targeting M by drug 2 renders cells sensitive to drug 1. And vice versa.
Senolytics: from oncology back to gerontology
The main problem in cancer therapy is how to kill cells selectively. Senolytics face a similar problem. Venetoclax (ABT-199) and Navitoclax (ABT-263), inhibitors of Bcl-2 and BclxL, are approved as anti-leukemia drugs [25]. These drugs have serious side effects due to damage of neutrophils and blood platelets. HSP-90 inhibitors (e.g., geldanamycin), which target multiple oncogene-addiction [14, 26] were tested for cancer treatment but have not be approved because of their toxicity even at intermittent doses typical for cancer therapy.
But toxicity is not the only problem. In oncology, a cancer cell is the undisputed target, which must be killed or at least permanently arrested. But is a senescent cell the correct target to slow down organismal aging? [27, 28]. Do senescent cells drive aging, or they are just markers of aging? Is it feasible to kill senescent cells instead of rejuvenating them? And more fundamental questions: What is cellular senescence? Is it a loss of function? If yes, why then do we choose to decrease their functioning further by killing these cells? Or in contrast, is senescence a hyperfunction, such as the Senescence-Associated Secretory Phenotype (SASP), in which case it makes sense to kill these cells? Is senescence caused by damage? And if yes, some senolytics are damaging drugs and may cause senescence by themselves [29]. Or senescence is not functional decline due to accumulation of molecular damage? Then what causes cellular senescence and organismal aging?
Senescence in cell culture
The program of cellular senescence consists of two steps: cell cycle arrest followed by gerogenic conversion from initially reversible arrest to senescence (geroconversion) [30, 31]. The cell cycle arrest can be induced by a variety of means: DNA damaging and anticancer drugs, telomere shortening, hyperactivation of oncogenic pathways (Ras, Raf, Akt) and ectopic expression of p21 and p16. In all these cases, arrest is ultimately mediated by p21 and p16, which inhibit CDK [30, 31].
When the cell cycle is arrested by p16 or p21, then growth-promoting pathways such as mTOR and MAPK convert this arrest to irreversible senescence (geroconversion). Cellular senescence is caused by geroconversion, not by cell cycle arrest. Geroconversion is a continuation of cellular growth, when actual growth is limited because of the cell cycle arrest [32]. Geroconversion is associated with the proliferation-like activity of mTOR and MAPK pathways. Geroconversion is a proliferative state of non-proliferating cells [30, 31]. Hyperfunctional growth-promoting pathways lead to cellular hypertrophy (large flat morphology), hyper-secretion (senescence-associated secretory phenotype, SASP) and lysosomal hyperfunction (senescence associated beta-galactosidase, SA-β-gal), accumulation of lipids (red-O-staining), overexpression of cyclin D1, hyperproduction of lactate, as well as secondary growth factor- and insulin-resistance [30, 31]. These are hallmarks of cellular senescence, predictable by the model that cellular senescence is a continuation of cellular growth [33]. When the cell gets arrested in the presence of rapamycin, geroconversion is decelerated [34]. Rapamycin maintain reversible quiescence (or G0), by delaying senescence. Rapamycin inhibits cellular growth in size and thus slows down geroconversion, which is a continuation of growth [30, 31].
Geroconversion in vivo
In G0/quiescent cells, mTOR is inactive. Then activation of mTOR leads either to proliferation or to geroconversion [35]. In the organism, mTOR activation may lead to partial geroconversion such as the transition of stem cells from G0 to GAlert, associated with cell size growth [36]. Prolonged GAlert leads to stem cell depletion [37]. Alternatively, quiescent stem cells may undergo geroconversion to senescence [38, 39]. mTOR is involved in stem cell senescence, and inhibition of mTOR maintains stem cell quiescence [37, 40–42].
Senescent and gerogenic cells in the organism
According to mainstream theories of aging, cellular senescence is a permanent growth arrest caused by DNA damage and other stresses. SASP promotes organismal aging and its diseases (Figure 2A). By killing senescent cells, senolytics delay diseases and/or aging [43–45].
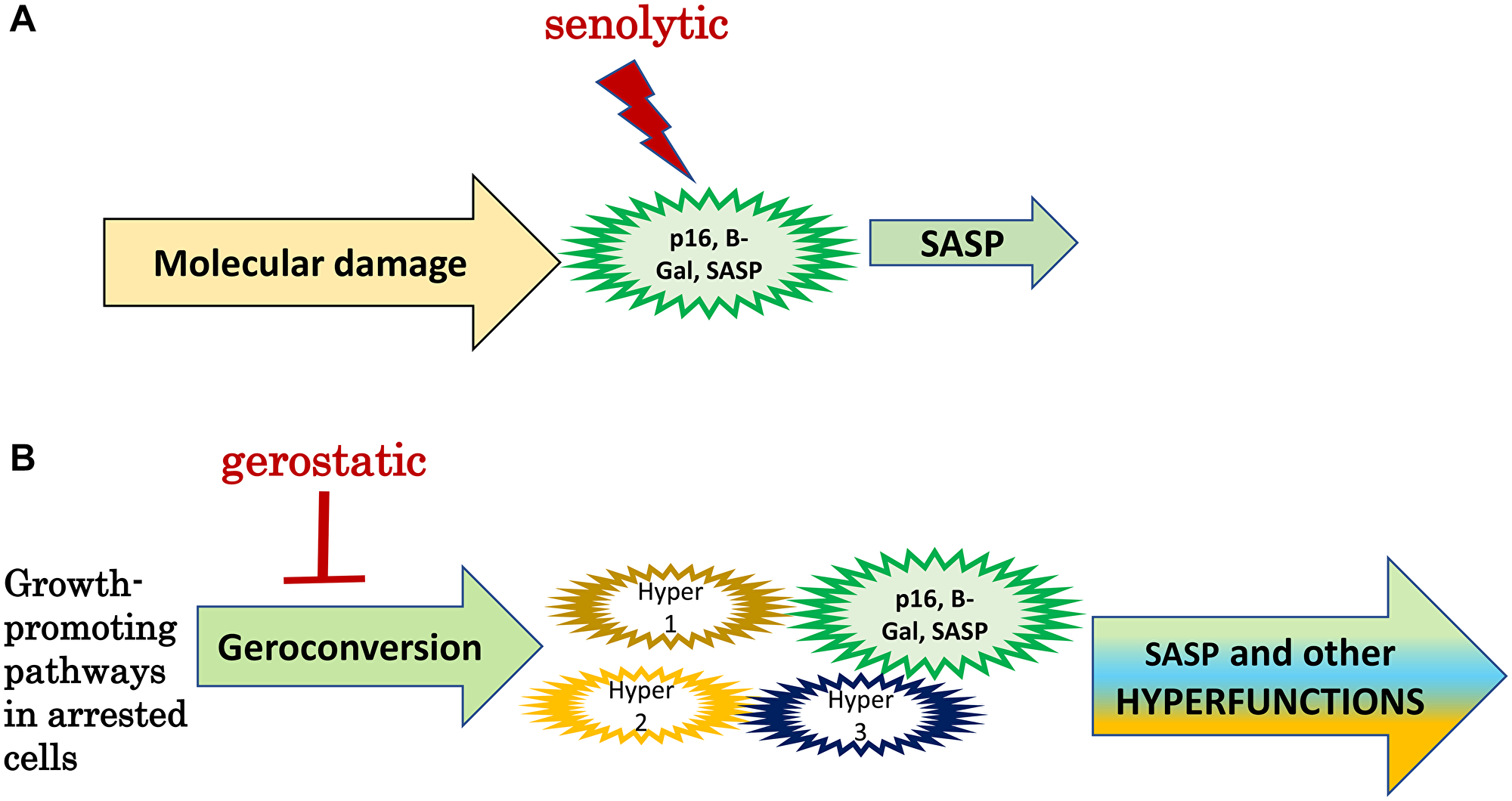
Figure 2: Senolytics versus gerostatics. (A) Senolytics: Standard model. Molecular damage causes functional decline associated with p16 expression, SA-β-gal-staining and SASP (a large green cell). SASP is involved in some diseases of aging. Senolytics kill senescent cells. (B) Gerostatics: simplified hyperfunction model. In arrested cells, growth-promoting and nutrient-sensing signaling pathways drive geroconversion instead of growth, rendering them gerogenic. Only a few cells (green) become phenotypically senescent. Most gerogenic cells are just slightly hyperfunctional (hyper 1, 2, 3). SASP is one of numerous hyperfunctions. Activated p16-positive macrophages are an example of gerogenic cells. Hyperfunctional cells drive age-related disease and aging is a sum of all diseases.
According to hyperfunction theory, cellular senescence is a continuation of cellular growth and cellular functions, leading to hyperfunctions [46]. SASP is only one of numerous hyperfunctions, which are tissue-specific (cells of different tissues have different functions). Although noticeable, fully senescent cells are rare in the organism. According to hyperfunction theory (Figure 2B), most cells undergo partial geroconversion, but only some cells (mostly of connective tissue and macrophages) acquire classically senescent morphology. Most cells undergo partial geroconversion (or no geroconversion at all). According to hyperfunction theory, the key feature of senescent cells is hyperfunction caused by higher than optimal activity of signaling pathways such as mTOR. These pathways drive development and growth but are not deactivated enough in post-development [46]. Hyperfunctional cells are gerogenic, producing age-related diseases. Senescent cells with p16 and SA-β-gal expression are a subgroup of gerogenic cells.
P16 is a marker of cell cycle arrest, but cell cycle arrest is not yet senescence. SA-β-gal is a hallmark of hyperfunctional lysosomes [47–49]. Cells arrested by serum-starvation and by contact-inhibition are also SA-β-gal-positive [47], (Figure 4 in [50]).
Hyperfunction theory is based on the cell culture model of proliferation-like level of signaling pathways in non-proliferating cells. This is the simplest hyperfunction. Quasi-programmed nature of aging is not an absolutely essential element of hyperfunction theory.
Non-senescent cells in organismal aging
According to hyperfunction theory, phenotypically-senescent cells are a subgroup of gerogenic cells. The fully senescent phenotype develops when growth-promoting pathways (for example, mTOR, MAPK) are active in acutely arrested (by DNA damage, for instance) cells [31]. Some other gerogenic cells are the product of partial geroconversion. And some gerogenic cells are not necessarily different from young, normal cells; it is enough that their function is not sufficiently decreased, when it becomes unnecessary in post-development. For example, cells that facilitate collagen cross-linking (an important function in development), should not do that in post-development (except in special cases, such as wound healing [51]. Or, the nematode Caenorhabditis elegans senesces without senescent cells. Simply, cells continue their developmental and reproductive functions in post-development and thus drive quasi-programmed (age-related) diseases [52, 53]. For example, they continue to produce yolk when it is not needed anymore, leading to intestinal atrophy and ectopic yolk deposition [54]. As another example, teratoma-like tumors develop from unfertilized oocytes which enter the uterus and become hypertrophic after exhaustion of sperm stocks [55, 56].
I believe that phenotypically-senescent cells contribute to some age-related diseases in some (but not all) organisms. Aging is driven by all gerogenic cells combined (Figure 2B).
Gerostatics in life extension
A decade ago, I introduced the term gerostatic or gero-suppressant (see for references [30, 31]). The immuno-suppressant rapamycin is a prototypical gero-suppresssant (gerostatic). The term gerostatic emphasizes static effects of rapamycin on both proliferation and geroconversion. At low doses, inhibitors of the mTOR kinase [57–59], PI3K and MEK [60, 61], S6K [61], PDK1 [62] and mdm-2, such as nutlin-3B [63, 64] are gerostatics. Deep hypoxia [65] and contact inhibition [50] are physiological gerostatics. In contrast, metformin is not gerostatic because it does not affect geroconversion and cellular senescence. Gerostatics should not be confused with senostatics. The term gerostatic has precise meaning: a drug that slows down geroconversion.
Like cellular senescence is a continuation of cellular mass growth [46], organismal aging is a continuation of developmental growth, driven in part by growth-promoting pathways [46, 52, 53, 55, 56, 66, 67]. Signal pathways that drive geroconversion in cell culture also promote aging in animals. Inhibition of the IGF-1/PI3K/mTOR/S6K pathway delays senescence and increases lifespan in animals including mammals. For example, mice with reduced mTOR expression [68], low mTORC1 activity due to growth hormone resistance [69, 70] are small and live longer [68–70].
As a gerostatic, rapamycin suppresses growth and senescence in yeast [71] and mammalian cells [32, 40–42, 72–82]. Rapamycin slows aging, stem cell exhaustion and extends lifespan in the simplest organism: Hydra [83]. Rapamycin extends lifespan in C. elegans [84] and Drosophila [85, 86]. Rapamycin increases lifespan and healthspan in mice [42, 87–121].
Fisetin inhibits the PI3K/mTOR pathway
Fisetin (3, 7, 3′ ,4′-tetrahydroxyflavone) inhibits multiple signaling kinases, including the PI3K/mTOR pathway and is considered a natural dual inhibitor of PI3K/Akt and mTOR signaling [122–131]. Fisetin inhibits the mTOR pathway both indirectly and directly by binding to mTOR and its downstream target, p70S6K [129]. Fisetin causes death of cancer cells, which is associated with mTOR inhibition [124–129]. Fisetin exerts multiple rapamycin-like effects in animals. It prevents cardiac hypertrophy by inhibiting mTOR [131]. Fisetin inhibits Akt, S6K1 and mTORC1, S6K1 in adipose tissue and prevents adipocyte differentiation and obesity in HFD-fed mice [130].
Quercetin inhibits multiple kinases
In numerous studies, quercetin inhibited the PI3K/Akt/mTOR pathway by multiple mechanisms in cell culture and animals [132–143]. At concentrations that also inhibit the PI3K/Akt/mTOR-signaling pathway, quercetin suppresses cancer cell growth [137–138]. Quercetin inhibits multiple kinases including ABL1, Aurora-A, -B, -C, CLK1, FLT3, JAK3, MET, NEK4, NEK9, PAK3, PIM1, RET, FGF-R2, PDGF and may kill cells in mitosis [144]. Inhibition of multiple targets, when only one is an intended target, may increase side effects without increasing therapeutic effect.
Dasatinib and Quercetin (D+Q) combination
Quercetin alone does not extend lifespan in mice [145], but a Dasatinib and Quercetin (D+Q) combination extended lifespan. The first empirical senolytic combination includes D, originally developed to target oncogene-addiction in leukemia, and Q, which inhibits the mTOR pathway, among numerous others. Dasatinib is an inhibitor of multiple tyrosine kinases including Bcr-Abl, ABL, SRC, c-KIT, PDGFR and ephrin receptor. Due to its inhibition of multiple kinases, it suppresses bone marrow, resulting in pancytopenia [13] and causing pulmonary endothelial cell apoptosis, lung vascular toxicity, pleural effusions and predisposition to pulmonary hypertension [146]. As a long-term side effect, Dasatinib increases mortality from ischemic heart disease [147, 148].
In humans, D 100 mg and Q 1000 mg given for three days decreased the number of p16- and SA-β-gal-positive cells in adipose tissue [9]. In patients with idiopathic pulmonary fibrosis, the senolytics effect of this treatment on relevant markers was inconclusive [149].
However, Kovacovicova et al. found that D+Q was ineffective in clearing chemotherapy-induced senescent cells. Furthermore, D+Q exerted acute pro-tumorigenic effects [150]. And furthermore, dasatinib plus quercetin treatment led to exacerbation of obesity- and age-dependent liver disease progression [151].
Do senolytics exist?
By the strict definition given by Kirkland [8], the existence of senolytics has not yet been proven. Although F and D+Q decrease the number of SA-β-gal and p16-positive cells in some tissues, there is no proof that this decrease is due to the killing of senescent cells in the organism. It could be due to reduction of these markers per cell, or even cell rejuvenation. In fact, rapamycin, which does not kill senescent cells, decreases expression of SA-β-gal and p16 [73, 74, 152]. In the organism, low doses of rapamycin decrease levels of p16 and tend to decrease SA-β-gal activity [153]. Given that current senolytics (F, D+Q) can inhibit mTOR, this scenario is possible. In order to demonstrate that senolytics work as senolytics, it is necessary to detect dead and apoptotic senescent cells, rather than only a decrease in SA-β-gal and p16. This is exactly how cytotoxic therapy is validated in oncology [154–156].
One may argue that because senolytics can be administered intermittently—a ‘hit-and-run’ approach, rather than continuously (daily)—this proves that they kill cells. This argument is not compelling. For example, rapamycin (a gerostatic, which does not kill cells) nevertheless can be given intermittently and transiently to extends lifespan and prevent cancer [88, 104, 111, 112, 157–160]. Even a single dose has long lasting effects. For example, a single administration decreases weight gain for at least 10 weeks, by shifting the set point long-term [161]. Rapamycin treatment for 2 weeks in young mice results in long-term preservation of primordial follicles and prolongation of ovarian lifespan in old mice [162].
Hyperfunctional senescent cells over-secrete cytokines and growth factors that may drive senescence of other cells and make them hyperfunctional too. Mutual overstimulation establishes self-maintained positive feedback loops. I suggest that disruption of such loops, even by a single (but high) dose of rapamycin, can have prolonged effects without killing cells.
SA-β-gal-positive/p16-expressing cells are not always senescent [163–166]. SA-β-gal and p16 can be reversibly induced in macrophages by physiological stimuli [163–166]. In groundbreaking studies, Gudkov and co-workers found that “significant proportion of p16/βGal-positive cells in aging mice are activated macrophages” [163–165]. Given that activated (hyperfunctional) macrophages and macrophage-derived foam cells are involved in age-related diseases, this may explain why the elimination of p16/ SA-β-gal-positive positive cells can be beneficial.
Although hyperfunction is a characteristic of senescent phenotype, p16/ SA-β-gal-positive macrophages are different from senescent cells used to screen for senolytics in cell culture [163–165].
Activated macrophages are gerogenic. Oxidized Low-Density Lipoprotein (ox-LDL) activates macrophages and induces formation of foam senescent cells characterized with SA-β-gal and p16 expression [167]. Remarkably, Quercetin [167] and Fisetin [168] inhibit formation of foam cells, prevent SA-β-gal and p16 induction and delay senescence [168].
Given that current senolytics may work as gerostatics, the significance of killing of senescent cells is unclear, even if it occurs (Figure 3). Is it the mechanism of life-extension or an unwanted side effect? Detrimental killing of senescent cells has been discussed in ref. [169].
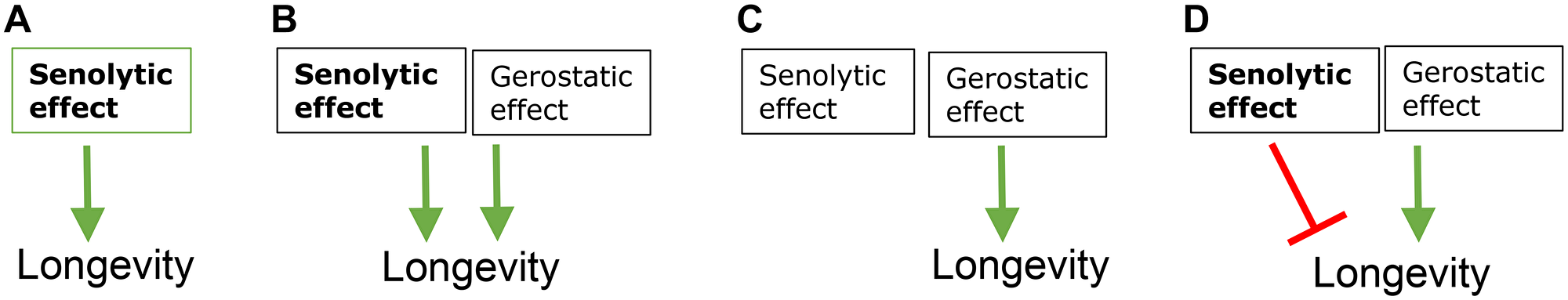
Figure 3: Potential mechanisms of life-extensions by fisetin and D+Q. (A) Life extension is purely through their senolytic effects. (B) Senolytic and gerostatic (off target) effects are additive (C) Senolytic effect is either absent or irrelevant. Life extension is purely through gerostatic (off target) mechanism. (D) Senolytic effect is detrimental and antagonizes life extension. Green arrows - stimulation; red symbol - inhibition.
Two gerostatics as one senolytic
At low concentrations, inhibitors of MEK, PI3K, the mTOR kinase are gerostatics. At high concentrations, they may become cytotoxic, probably due to inhibition of multiple kinases (an off-target effect). (In contrast, rapamycin and other rapalogs are not cytotoxic at any achievable doses. Still, everolimus and rapamycin potentiate cytotoxicity of dasatinib against cancer cells [170, 171]). A combination of two gerostatics can act as a senolytic. For example, MEK inhibitors especially combined with pan-mTOR inhibitors are cytotoxic to some senescent cells [172, 173]. It would be important to investigate life-extension in mice by combinations of MEK and pan-TOR inhibitors, pan-mTOR inhibitors and rapamycin, MEK inhibitors and rapamycin.
Conclusions
Rapamycin and other gerostatics do not kill senescent cells but slow down cell growth, gerogenesis and oncogenesis. Gerostatics mostly act on non-senescent cells, decreasing their hyperfunction and decelerating their geroconversion to senescence. Rapamycin robustly extends lifespan and tumor-free survival in mice. It is also effective, when used intermittently and transiently. In theory, inhibition of the mTOR pathway can explain life extension by current senolytics such as F, D+Q. However, it is not clear whether these senolytics inhibit mTOR sufficiently to slow aging at doses that are achievable in humans.
It is expected that rapamycin-like effect may be responsible for the therapeutic effects of senolytics in disease. Some senolytics are investigated for treatment of diseases such as idiopathic pulmonary fibrosis [10, 45, 149, 174]. Although the treatment of specific diseases is very important, it is a different story entirely. For example, DNA damaging drugs such as doxorubicin are successfully used for cancer therapy; insulin is a life-saving drug in terminal diabetes; glucocorticoids are useful for arthritis; antibiotics cure bacterial infections common in the elderly. And these conditions are common age-related diseases. But doxorubicin, insulin, corticosteroids and penicillin are not anti-aging drugs. And they do not extend lifespan in mice. Unless drugs extend lifespan, they are not drugs to treat aging as a common cause of age-related diseases. Life extension in mice by D+Q and F was shown in one study for each of these modalities [5, 6]. It is desirably to reproduce these results, preferably in a variety of mouse models, in order to advocate their use (alone or in combination with rapamycin) in humans for longer and healthier life. Given that these senolytics are available for human use and well-tolerated, they could be used under doctor supervision without life-long clinical trials [7]. But first it must be shown reproducibly that they extend lifespan consistently in animals.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Martel J, Ojcius DM, Wu CY, Peng HH, Voisin L, Perfettini JL, Ko YF, Young JD. Emerging use of senolytics and senomorphics against aging and chronic diseases. Med Res Rev. 2020; 40:2114–31. https://doi.org/10.1002/med.21702. [PubMed].
2. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017; 16:718–35. https://doi.org/10.1038/nrd.2017.116. [PubMed].
3. Shetty AK, Kodali M, Upadhya R, Madhu LN. Emerging Anti-Aging Strategies - Scientific Basis and Efficacy. Aging Dis. 2018; 9:1165–84. https://doi.org/10.14336/AD.2018.1026. [PubMed].
4. Prašnikar E, Borišek J, Perdih A. Senescent cells as promising targets to tackle age-related diseases. Ageing Res Rev. 2021; 66:101251. https://doi.org/10.1016/j.arr.2020.101251. [PubMed].
5. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018; 36:18–28. https://doi.org/10.1016/j.ebiom.2018.09.015. [PubMed].
6. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9. [PubMed].
7. Blagosklonny MV. The goal of geroscience is life extension. Oncotarget. 2021; 12:131–44. https://doi.org/10.18632/oncotarget.27882. [PubMed].
8. Kirkland JL, Tchkonia T. Clinical strategies and animal models for developing senolytic agents. Exp Gerontol. 2015; 68:19–25. https://doi.org/10.1016/j.exger.2014.10.012. [PubMed].
9. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019; 47:446–56.
10. Kirkland JL, Tchkonia T. Senolytic drugs: from discovery to translation. J Intern Med. 2020; 288:518–36. https://doi.org/10.1111/joim.13141. [PubMed].
11. Blagosklonny MV. Tissue-selective therapy of cancer. Br J Cancer. 2003; 89:1147–51. https://doi.org/10.1038/sj.bjc.6601256. [PubMed].
12. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996; 2:561–66. https://doi.org/10.1038/nm0596-561. [PubMed].
13. Lindauer M, Hochhaus A. Dasatinib. Recent Results Cancer Res. 2018; 212:29–68. https://doi.org/10.1007/978-3-319-91439-8_2. [PubMed].
14. Blagosklonny MV, Fojo T, Bhalla KN, Kim JS, Trepel JB, Figg WD, Rivera Y, Neckers LM. The Hsp90 inhibitor geldanamycin selectively sensitizes Bcr-Abl-expressing leukemia cells to cytotoxic chemotherapy. Leukemia. 2001; 15:1537–43. https://doi.org/10.1038/sj.leu.2402257. [PubMed].
15. Blagosklonny MV. Do cells need CDK2 and ... Bcr-Abl? Cell Death Differ. 2004; 11:249–51. https://doi.org/10.1038/sj.cdd.4401361. [PubMed].
16. Amarante-Mendes GP, Naekyung Kim C, Liu L, Huang Y, Perkins CL, Green DR, Bhalla K. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood. 1998; 91:1700–05. https://doi.org/10.1182/blood.V91.5.1700. [PubMed].
17. Demidenko ZN, An WG, Lee JT, Romanova LY, McCubrey JA, Blagosklonny MV. Kinase-addiction and bi-phasic sensitivity-resistance of Bcr-Abl- and Raf-1-expressing cells to imatinib and geldanamycin. Cancer Biol Ther. 2005; 4:484–90. https://doi.org/10.4161/cbt.4.4.1702. [PubMed].
18. Carter BZ, Mak PY, Mu H, Zhou H, Mak DH, Schober W, Leverson JD, Zhang B, Bhatia R, Huang X, Cortes J, Kantarjian H, Konopleva M, Andreeff M. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016; 8:355ra117. https://doi.org/10.1126/scitranslmed.aag1180. [PubMed].
19. Singh A, Settleman J. Oncogenic K-ras "addiction" and synthetic lethality. Cell Cycle. 2009; 8:2676–77. https://doi.org/10.4161/cc.8.17.9336. [PubMed].
20. Zhao D, DePinho RA. Synthetic essentiality: Targeting tumor suppressor deficiencies in cancer. BioEssays. 2017; 39:1700076. https://doi.org/10.1002/bies.201700076. [PubMed].
21. Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997; 278:1064–68. https://doi.org/10.1126/science.278.5340.1064. [PubMed].
22. Blagosklonny MV. Sequential activation and inactivation of G2 checkpoints for selective killing of p53-deficient cells by microtubule-active drugs. Oncogene. 2002; 21:6249–54. https://doi.org/10.1038/sj.onc.1205793. [PubMed].
23. Blagosklonny MV. The power of chemotherapeutic engineering: arresting cell cycle and suppressing senescence to protect from mitotic inhibitors. Cell Cycle. 2011; 10:2295–98. https://doi.org/10.4161/cc.10.14.16819. [PubMed].
24. Blagosklonny MV. Matching targets for selective cancer therapy. Drug Discov Today. 2003; 8:1104–07. https://doi.org/10.1016/s1359-6446(03)02806-x. [PubMed].
25. Lafontaine J, Cardin GB, Malaquin N, Boisvert JS, Rodier F, Wong P. Senolytic Targeting of Bcl-2 Anti-Apoptotic Family Increases Cell Death in Irradiated Sarcoma Cells. Cancers (Basel). 2021; 13:386. https://doi.org/10.3390/cancers13030386. [PubMed].
26. Blagosklonny MV. Hsp-90-associated oncoproteins: multiple targets of geldanamycin and its analogs. Leukemia. 2002; 16:455–62. https://doi.org/10.1038/sj.leu.2402415. [PubMed].
27. Martin N, Huna A, Bernard D. Elimination of Senescent Endothelial Cells: Good or Bad Idea? Trends Cell Biol. 2021; 31:327–30. https://doi.org/10.1016/j.tcb.2021.02.009. [PubMed].
28. Carpenter VJ, Saleh T, Gewirtz DA. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers (Basel). 2021; 13:723. https://doi.org/10.3390/cancers13040723. [PubMed].
29. Restall IJ, Lorimer IA. Induction of premature senescence by hsp90 inhibition in small cell lung cancer. PLoS One. 2010; 5:e11076. https://doi.org/10.1371/journal.pone.0011076. [PubMed].
30. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014; 13:3628–35. https://doi.org/10.4161/15384101.2014.985507. [PubMed].
31. Blagosklonny MV. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle. 2018; 17:2655–65. https://doi.org/10.1080/15384101.2018.1554781. [PubMed].
32. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7:3355–61. https://doi.org/10.4161/cc.7.21.6919. [PubMed].
33. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4:358–62. https://doi.org/10.1038/sj.embor.embor806. [PubMed].
34. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle. 2009; 8:1888–95. https://doi.org/10.4161/cc.8.12.8606. [PubMed].
35. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2:924–35. https://doi.org/10.18632/aging.100265. [PubMed].
36. Rodgers JT, King KY, Brett JO, Cromie MJ, Charville GW, Maguire KK, Brunson C, Mastey N, Liu L, Tsai CR, Goodell MA, Rando TA. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature. 2014; 510:393–96. https://doi.org/10.1038/nature13255. [PubMed].
37. Yue F, Bi P, Wang C, Li J, Liu X, Kuang S. Conditional Loss of Pten in Myogenic Progenitors Leads to Postnatal Skeletal Muscle Hypertrophy but Age-Dependent Exhaustion of Satellite Cells. Cell Rep. 2016; 17:2340–53. https://doi.org/10.1016/j.celrep.2016.11.002. [PubMed].
38. Sousa-Victor P, Perdiguero E, Muñoz-Cánoves P. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle. 2014; 13:3183–90. https://doi.org/10.4161/15384101.2014.965072. [PubMed].
39. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506:316–21. https://doi.org/10.1038/nature13013. [PubMed].
40. Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Liu Y, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008; 205:2397–408. https://doi.org/10.1084/jem.20081297. [PubMed].
41. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97:20–29. https://doi.org/10.1097/TP.0b013e3182a7fcf8. [PubMed].
42. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal. 2009; 2:ra75. https://doi.org/10.1126/scisignal.2000559. [PubMed].
43. Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol. 2014; 39:45–61. https://doi.org/10.1159/000358899. [PubMed].
44. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021; 22:75–95. https://doi.org/10.1038/s41580-020-00314-w. [PubMed].
45. Song S, Tchkonia T, Jiang J, Kirkland JL, Sun Y. Targeting Senescent Cells for a Healthier Aging: Challenges and Opportunities. Adv Sci (Weinh). 2020; 7:2002611. https://doi.org/10.1002/advs.202002611. [PubMed].
46. Blagosklonny MV. Aging and immortality: quasi-programmed senescence and its pharmacologic inhibition. Cell Cycle. 2006; 5:2087–102. https://doi.org/10.4161/cc.5.18.3288. [PubMed].
47. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92:9363–67. https://doi.org/10.1073/pnas.92.20.9363. [PubMed].
48. Severino J, Allen RG, Balin S, Balin A, Cristofalo VJ. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp Cell Res. 2000; 257:162–71. https://doi.org/10.1006/excr.2000.4875. [PubMed].
49. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5:187–95. https://doi.org/10.1111/j.1474-9726.2006.00199.x. [PubMed].
50. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014; 111:8832–37. https://doi.org/10.1073/pnas.1405723111. [PubMed].
51. Cai L, Xiong X, Kong X, Xie J. The Role of the Lysyl Oxidases in Tissue Repair and Remodeling: A Concise Review. Tissue Eng Regen Med. 2017; 14:15–30. https://doi.org/10.1007/s13770-016-0007-0. [PubMed].
52. Gems D, de la Guardia Y. Alternative Perspectives on Aging in Caenorhabditis elegans: Reactive Oxygen Species or Hyperfunction? Antioxid Redox Signal. 2013; 19:321–29. https://doi.org/10.1089/ars.2012.4840. [PubMed].
53. Gems D, Partridge L. Genetics of longevity in model organisms: debates and paradigm shifts. Annu Rev Physiol. 2013; 75:621–44. https://doi.org/10.1146/annurev-physiol-030212-183712. [PubMed].
54. Ezcurra M, Benedetto A, Sornda T, Gilliat AF, Au C, Zhang Q, van Schelt S, Petrache AL, Wang H, de la Guardia Y, Bar-Nun S, Tyler E, Wakelam MJ, Gems D. C. elegans Eats Its Own Intestine to Make Yolk Leading to Multiple Senescent Pathologies. Curr Biol. 2018; 28:2544–56.e5. https://doi.org/10.1016/j.cub.2018.06.035. [PubMed].
55. Wang H, Zhang Z, Gems D. Monsters in the uterus: teratoma-like tumors in senescent C. elegans result from a parthenogenetic quasi-program. Aging (Albany NY). 2018; 10:1188–89. https://doi.org/10.18632/aging.101486. [PubMed].
56. Wang H, Zhao Y, Ezcurra M, Benedetto A, Gilliat AF, Hellberg J, Ren Z, Galimov ER, Athigapanich T, Girstmair J, Telford MJ, Dolphin CT, Zhang Z, Gems D. A parthenogenetic quasi-program causes teratoma-like tumors during aging in wild-type C. elegans. NPJ Aging Mech Dis. 2018; 4:6. https://doi.org/10.1038/s41514-018-0025-3. [PubMed].
57. Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016; 8:3535–51. https://doi.org/10.18632/aging.101155. [PubMed].
58. Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016; 8:231–44. https://doi.org/10.18632/aging.100872. [PubMed].
59. Walters HE, Cox LS. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int J Mol Sci. 2018; 19:2325. https://doi.org/10.3390/ijms19082325. [PubMed].
60. Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009; 8:1896–900. https://doi.org/10.4161/cc.8.12.8809. [PubMed].
61. Leontieva OV, Demidenko ZN, Blagosklonny MV. S6K in geroconversion. Cell Cycle. 2013; 12:3249–52. https://doi.org/10.4161/cc.26248. [PubMed].
62. An S, Cho SY, Kang J, Lee S, Kim HS, Min DJ, Son E, Cho KH. Inhibition of 3-phosphoinositide-dependent protein kinase 1 (PDK1) can revert cellular senescence in human dermal fibroblasts. Proc Natl Acad Sci U S A. 2020; 117:31535–46. https://doi.org/10.1073/pnas.1920338117. [PubMed].
63. Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, Desprez PY, Benz C, Davalos AR, Campisi J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep. 2018; 8:2410. https://doi.org/10.1038/s41598-018-20000-4. [PubMed].
64. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107:9660–64. https://doi.org/10.1073/pnas.1002298107. [PubMed].
65. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109:13314–18. https://doi.org/10.1073/pnas.1205690109. [PubMed].
66. Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013; 12:3736–42.
67. de la Guardia Y, Gilliat AF, Hellberg J, Rennert P, Cabreiro F, Gems D. Run-on of germline apoptosis promotes gonad senescence in C. elegans. Oncotarget. 2016; 7:39082–96. https://doi.org/10.18632/oncotarget.9681. [PubMed].
68. Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, Fergusson MM, Rovira II, Allen M, Springer DA, Lago CU, Zhang S, DuBois W, et al. Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 2013; 4:913–20. https://doi.org/10.1016/j.celrep.2013.07.030. [PubMed].
69. Dominick G, Berryman DE, List EO, Kopchick JJ, Li X, Miller RA, Garcia GG. Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology. 2015; 156:565–75. https://doi.org/10.1210/en.2014-1690. [PubMed].
70. Bartke A, Sun LY, Longo V. Somatotropic signaling: trade-offs between growth, reproductive development, and longevity. Physiol Rev. 2013; 93:571–98. https://doi.org/10.1152/physrev.00006.2012. [PubMed].
71. Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20:174–84. https://doi.org/10.1101/gad.1381406. [PubMed].
72. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, Limbad C, Demaria M, Li P, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015; 17:1049–61. https://doi.org/10.1038/ncb3195. [PubMed].
73. Antonioli E, Torres N, Ferretti M, Piccinato CA, Sertie AL. Individual response to mTOR inhibition in delaying replicative senescence of mesenchymal stromal cells. PLoS One. 2019; 14:e0204784. https://doi.org/10.1371/journal.pone.0204784. [PubMed].
74. Nie D, Zhang J, Zhou Y, Sun J, Wang W, Wang JH. Rapamycin Treatment of Tendon Stem/Progenitor Cells Reduces Cellular Senescence by Upregulating Autophagy. Stem Cells Int. 2021; 2021:6638249. https://doi.org/10.1155/2021/6638249. [PubMed].
75. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, Georgilis A, Montoya A, Wolter K, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015; 17:1205–17. https://doi.org/10.1038/ncb3225. [PubMed].
76. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, Guo G, Xia Y, Zhu X, Shi G, Cheng C. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016; 8:1102–14. https://doi.org/10.18632/aging.100925. [PubMed].
77. Hinojosa CA, Mgbemena V, Van Roekel S, Austad SN, Miller RA, Bose S, Orihuela CJ. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012; 47:958–65. https://doi.org/10.1016/j.exger.2012.08.013. [PubMed].
78. Wang R, Sunchu B, Perez VI. Rapamycin and the inhibition of the secretory phenotype. Exp Gerontol. 2017; 94:89–92. https://doi.org/10.1016/j.exger.2017.01.026. [PubMed].
79. Gao C, Ning B, Sang C, Zhang Y. Rapamycin prevents the intervertebral disc degeneration via inhibiting differentiation and senescence of annulus fibrosus cells. Aging (Albany NY). 2018; 10:131–43. https://doi.org/10.18632/aging.101364. [PubMed].
80. Houssaini A, Breau M, Kebe K, Abid S, Marcos E, Lipskaia L, Rideau D, Parpaleix A, Huang J, Amsellem V, Vienney N, Validire P, Maitre B, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018; 3:e93203. https://doi.org/10.1172/jci.insight.93203. [PubMed].
81. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012; 11:401–14. https://doi.org/10.1016/j.stem.2012.06.007. [PubMed].
82. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181:278–93. https://doi.org/10.1016/j.ajpath.2012.03.017. [PubMed].
83. Tomczyk S, Suknovic N, Schenkelaars Q, Wenger Y, Ekundayo K, Buzgariu W, Bauer C, Fischer K, Austad S, Galliot B. Deficient autophagy in epithelial stem cells drives aging in the freshwater cnidarian Hydra. Development. 2020; 147:dev177840. https://doi.org/10.1242/dev.177840. [PubMed].
84. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713–24. https://doi.org/10.1016/j.cmet.2012.04.007. [PubMed].
85. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010; 11:35–46. https://doi.org/10.1016/j.cmet.2009.11.010. [PubMed].
86. Wang A, Mouser J, Pitt J, Promislow D, Kaeberlein M. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget. 2016; 7:80131–39. https://doi.org/10.18632/oncotarget.12560. [PubMed].
87. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392–95. https://doi.org/10.1038/nature08221. [PubMed].
88. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176:2092–97. https://doi.org/10.2353/ajpath.2010.091050. [PubMed].
89. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10:4230–36. https://doi.org/10.4161/cc.10.24.18486. [PubMed].
90. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66:191–201. https://doi.org/10.1093/gerona/glq178. [PubMed].
91. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4:715–22. https://doi.org/10.18632/aging.100496. [PubMed].
92. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/– mice. Aging (Albany NY). 2012; 4:709–14. https://doi.org/10.18632/aging.100498. [PubMed].
93. Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012; 4:144ra103. https://doi.org/10.1126/scitranslmed.3003802. [PubMed].
94. Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, Woodward MA, Miller RA. Rapamycin slows aging in mice. Aging Cell. 2012; 11:675–82. https://doi.org/10.1111/j.1474-9726.2012.00832.x. [PubMed].
95. Fang Y, Westbrook R, Hill C, Boparai RK, Arum O, Spong A, Wang F, Javors MA, Chen J, Sun LY, Bartke A. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013; 17:456–62. https://doi.org/10.1016/j.cmet.2013.02.008. [PubMed].
96. Flynn JM, O'Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, Kapahi P, Nelson MD, Kennedy BK, Melov S. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. 2013; 12:851–62. https://doi.org/10.1111/acel.12109. [PubMed].
97. Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, Oh K, Wasko BM, Ramos FJ, et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013; 342:1524–28. https://doi.org/10.1126/science.1244360. [PubMed].
98. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P, Sharp ZD. Rapamycin extends life span of Rb1+/– mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5:100–10. https://doi.org/10.18632/aging.100533. [PubMed].
99. Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schröder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013; 123:3272–91. https://doi.org/10.1172/JCI67674. [PubMed].
100. Ye L, Widlund AL, Sims CA, Lamming DW, Guan Y, Davis JG, Sabatini DM, Harrison DE, Vang O, Baur JA. Rapamycin doses sufficient to extend lifespan do not compromise muscle mitochondrial content or endurance. Aging (Albany NY). 2013; 5:539–50. https://doi.org/10.18632/aging.100576. [PubMed].
101. Fok WC, Chen Y, Bokov A, Zhang Y, Salmon AB, Diaz V, Javors M, Wood WH 3rd, Zhang Y, Becker KG, Pérez VI, Richardson A. Mice fed rapamycin have an increase in lifespan associated with major changes in the liver transcriptome. PLoS One. 2014; 9:e83988. https://doi.org/10.1371/journal.pone.0083988. [PubMed].
102. Hasty P, Livi CB, Dodds SG, Jones D, Strong R, Javors M, Fischer KE, Sloane L, Murthy K, Hubbard G, Sun L, Hurez V, Curiel TJ, Sharp ZD. eRapa restores a normal life span in a FAP mouse model. Cancer Prev Res (Phila). 2014; 7:169–78. https://doi.org/10.1158/1940-6207.CAPR-13-0299. [PubMed].
103. Khapre RV, Kondratova AA, Patel S, Dubrovsky Y, Wrobel M, Antoch MP, Kondratov RV. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging (Albany NY). 2014; 6:48–57. https://doi.org/10.18632/aging.100633. [PubMed].
104. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Weekly administration of rapamycin improves survival and biomarkers in obese male mice on high-fat diet. Aging Cell. 2014; 13:616–22. https://doi.org/10.1111/acel.12211. [PubMed].
105. Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JF, Pletcher S, Salmon AB, Sharp ZD, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014; 13:468–77. https://doi.org/10.1111/acel.12194. [PubMed].
106. Popovich IG, Anisimov VN, Zabezhinski MA, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol Ther. 2014; 15:586–92. https://doi.org/10.4161/cbt.28164. [PubMed].
107. Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, Diaz V, Sloane L, Maslin K, Treaster S, Réndon S, van Remmen H, Ward W, et al. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014; 69:119–30. https://doi.org/10.1093/gerona/glt056. [PubMed].
108. Hurez V, Dao V, Liu A, Pandeswara S, Gelfond J, Sun L, Bergman M, Orihuela CJ, Galvan V, Padrón Á, Drerup J, Liu Y, Hasty P, et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell. 2015; 14:945–56. https://doi.org/10.1111/acel.12380. [PubMed].
109. Johnson SC, Yanos ME, Bitto A, Castanza A, Gagnidze A, Gonzalez B, Gupta K, Hui J, Jarvie C, Johnson BM, Letexier N, McCanta L, Sangesland M, et al. Dose-dependent effects of mTOR inhibition on weight and mitochondrial disease in mice. Front Genet. 2015; 6:247. https://doi.org/10.3389/fgene.2015.00247. [PubMed].
110. Karunadharma PP, Basisty N, Dai DF, Chiao YA, Quarles EK, Hsieh EJ, Crispin D, Bielas JH, Ericson NG, Beyer RP, MacKay VL, MacCoss MJ, Rabinovitch PS. Subacute calorie restriction and rapamycin discordantly alter mouse liver proteome homeostasis and reverse aging effects. Aging Cell. 2015; 14:547–57. https://doi.org/10.1111/acel.12317. [PubMed].
111. Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE, Lamming DW. Intermittent Administration of Rapamycin Extends the Life Span of Female C57BL/6J Mice. J Gerontol A Biol Sci Med Sci. 2016; 71:876–81. https://doi.org/10.1093/gerona/glw064. [PubMed].
112. Bitto A, Ito TK, Pineda VV, LeTexier NJ, Huang HZ, Sutlief E, Tung H, Vizzini N, Chen B, Smith K, Meza D, Yajima M, Beyer RP, et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife. 2016; 5:e16351. https://doi.org/10.7554/eLife.16351. [PubMed].
113. Liao CY, Anderson SS, Chicoine NH, Mayfield JR, Academia EC, Wilson JA, Pongkietisak C, Thompson MA, Lagmay EP, Miller DM, Hsu YM, McCormick MA, O'Leary MN, Kennedy BK. Rapamycin Reverses Metabolic Deficits in Lamin A/C-Deficient Mice. Cell Rep. 2016; 17:2542–52. https://doi.org/10.1016/j.celrep.2016.10.040. [PubMed].
114. Felici R, Buonvicino D, Muzzi M, Cavone L, Guasti D, Lapucci A, Pratesi S, De Cesaris F, Luceri F, Chiarugi A. Post onset, oral rapamycin treatment delays development of mitochondrial encephalopathy only at supramaximal doses. Neuropharmacology. 2017; 117:74–84. https://doi.org/10.1016/j.neuropharm.2017.01.039. [PubMed].
115. Siegmund SE, Yang H, Sharma R, Javors M, Skinner O, Mootha V, Hirano M, Schon EA. Low-dose rapamycin extends lifespan in a mouse model of mtDNA depletion syndrome. Hum Mol Genet. 2017; 26:4588–605. https://doi.org/10.1093/hmg/ddx341. [PubMed].
116. Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017; 18:57. https://doi.org/10.1186/s13059-017-1186-2. [PubMed].
117. Bielas J, Herbst A, Widjaja K, Hui J, Aiken JM, McKenzie D, Miller RA, Brooks SV, Wanagat J. Long term rapamycin treatment improves mitochondrial DNA quality in aging mice. Exp Gerontol. 2018; 106:125–31. https://doi.org/10.1016/j.exger.2018.02.021. [PubMed].
118. Weiss R, Fernandez E, Liu Y, Strong R, Salmon AB. Metformin reduces glucose intolerance caused by rapamycin treatment in genetically heterogeneous female mice. Aging (Albany NY). 2018; 10:386–401. https://doi.org/10.18632/aging.101401. [PubMed].
119. Ferrara-Romeo I, Martinez P, Saraswati S, Whittemore K, Graña-Castro O, Thelma Poluha L, Serrano R, Hernandez-Encinas E, Blanco-Aparicio C, Maria Flores J, Blasco MA. The mTOR pathway is necessary for survival of mice with short telomeres. Nat Commun. 2020; 11:1168. https://doi.org/10.1038/s41467-020-14962-1. [PubMed].
120. Parihar M, Dodds SG, Hubbard G, Javors MA, Strong R, Hasty P, Sharp ZD. Rapamycin Extends Life Span in ApcMin/+ Colon Cancer FAP Model. Clin Colorectal Cancer. 2021; 20:e61–70. https://doi.org/10.1016/j.clcc.2020.08.006. [PubMed].
121. Strong R, Miller RA, Bogue M, Fernandez E, Javors MA, Libert S, Marinez PA, Murphy MP, Musi N, Nelson JF, Petrascheck M, Reifsnyder P, Richardson A, et al. Rapamycin-mediated mouse lifespan extension: Late-life dosage regimes with sex-specific effects. Aging Cell. 2020; 19:e13269. https://doi.org/10.1111/acel.13269. [PubMed].
122. Chamcheu JC, Esnault S, Adhami VM, Noll AL, Banang-Mbeumi S, Roy T, Singh SS, Huang S, Kousoulas KG, Mukhtar H. Fisetin, a 3,7,3',4'-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells. 2019; 8:1089. https://doi.org/10.3390/cells8091089. [PubMed].
123. Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012; 84:1277–81. https://doi.org/10.1016/j.bcp.2012.07.012. [PubMed].
124. Lim JY, Lee JY, Byun BJ, Kim SH. Fisetin targets phosphatidylinositol-3-kinase and induces apoptosis of human B lymphoma Raji cells. Toxicol Rep. 2015; 2:984–89. https://doi.org/10.1016/j.toxrep.2015.07.004. [PubMed].
125. Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, Mukhtar H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis. 2010; 31:1424–33. https://doi.org/10.1093/carcin/bgq115. [PubMed].
126. Watanabe M, Hisatake M, Fujimori K. Fisetin Suppresses Lipid Accumulation in Mouse Adipocytic 3T3-L1 Cells by Repressing GLUT4-Mediated Glucose Uptake through Inhibition of mTOR-C/EBPα Signaling. J Agric Food Chem. 2015; 63:4979–87. https://doi.org/10.1021/acs.jafc.5b00821. [PubMed].
127. Sun X, Ma X, Li Q, Yang Y, Xu X, Sun J, Yu M, Cao K, Yang L, Yang G, Zhang G, Wang X. Anti-cancer effects of fisetin on mammary carcinoma cells via regulation of the PI3K/Akt/mTOR pathway: In vitro and in vivo studies. Int J Mol Med. 2018; 42:811–20. https://doi.org/10.3892/ijmm.2018.3654. [PubMed].
128. Sundarraj K, Raghunath A, Panneerselvam L, Perumal E. Fisetin Inhibits Autophagy in HepG2 Cells via PI3K/Akt/mTOR and AMPK Pathway. Nutr Cancer. 2020 Oct 21. https://doi.org/10.1080/01635581.2020.1836241. [Epub ahead of print]. [PubMed]
129. Syed DN, Chamcheu JC, Khan MI, Sechi M, Lall RK, Adhami VM, Mukhtar H. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling. Biochem Pharmacol. 2014; 89:349–60. https://doi.org/10.1016/j.bcp.2014.03.007. [PubMed].
130. Jung CH, Kim H, Ahn J, Jeon TI, Lee DH, Ha TY. Fisetin regulates obesity by targeting mTORC1 signaling. J Nutr Biochem. 2013; 24:1547–54. https://doi.org/10.1016/j.jnutbio.2013.01.003. [PubMed].
131. Dong B, Liu C, Xue R, Wang Y, Sun Y, Liang Z, Fan W, Jiang J, Zhao J, Su Q, Dai G, Dong Y, Huang H. Fisetin inhibits cardiac hypertrophy by suppressing oxidative stress. J Nutr Biochem. 2018; 62:221–29. https://doi.org/10.1016/j.jnutbio.2018.08.010. [PubMed].
132. He L, Zhang N, Wang L, Du L, Li C, Li Y, Li X, Zhu X, Lu Q, Yin X. Quercetin inhibits AQP1 translocation in high-glucose-cultured SRA01/04 cells through PI3K/Akt/mTOR Pathway. Curr Mol Pharmacol. 2020 Sep 8. https://doi.org/10.2174/1874467213666200908120501. [Epub ahead of print]. [PubMed]
133. Bruning A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anticancer Agents Med Chem. 2013; 13:1025–31. https://doi.org/10.2174/18715206113139990114. [PubMed].
134. Xiao Y, Zhou L, Zhang T, Qin C, Wei P, Luo L, Luo L, Huang G, Chen A, Liu G. Anti-fibrosis activity of quercetin attenuates rabbit tracheal stenosis via the TGF-β/AKT/mTOR signaling pathway. Life Sci. 2020; 250:117552. https://doi.org/10.1016/j.lfs.2020.117552. [PubMed].
135. Wang R, Qiu Z, Wang G, Hu Q, Shi N, Zhang Z, Wu Y, Zhou C. Quercetin attenuates diabetic neuropathic pain by inhibiting mTOR/p70S6K pathway-mediated changes of synaptic morphology and synaptic protein levels in spinal dorsal horn of db/db mice. Eur J Pharmacol. 2020; 882:173266. https://doi.org/10.1016/j.ejphar.2020.173266. [PubMed].
136. Rivera Rivera A, Castillo-Pichardo L, Gerena Y, Dharmawardhane S. Anti-Breast Cancer Potential of Quercetin via the Akt/AMPK/Mammalian Target of Rapamycin (mTOR) Signaling Cascade. PLoS One. 2016; 11:e0157251. https://doi.org/10.1371/journal.pone.0157251. [PubMed].
137. Li X, Zhou N, Wang J, Liu Z, Wang X, Zhang Q, Liu Q, Gao L, Wang R. Quercetin suppresses breast cancer stem cells (CD44+/CD24−) by inhibiting the PI3K/Akt/mTOR-signaling pathway. Life Sci. 2018; 196:56–62. https://doi.org/10.1016/j.lfs.2018.01.014. [PubMed].
138. Jia L, Huang S, Yin X, Zan Y, Guo Y, Han L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018; 208:123–30. https://doi.org/10.1016/j.lfs.2018.07.027. [PubMed].
139. Granato M, Rizzello C, Gilardini Montani MS, Cuomo L, Vitillo M, Santarelli R, Gonnella R, D'Orazi G, Faggioni A, Cirone M. Quercetin induces apoptosis and autophagy in primary effusion lymphoma cells by inhibiting PI3K/AKT/mTOR and STAT3 signaling pathways. J Nutr Biochem. 2017; 41:124–36. https://doi.org/10.1016/j.jnutbio.2016.12.011. [PubMed].
140. Huang Y, Chen Y, Shaw AM, Goldfine H, Tian J, Cai J. Enhancing TFEB-Mediated Cellular Degradation Pathways by the mTORC1 Inhibitor Quercetin. Oxid Med Cell Longev. 2018; 2018:5073420. https://doi.org/10.1155/2018/5073420. [PubMed].
141. Ren J, Li J, Liu X, Feng Y, Gui Y, Yang J, He W, Dai C. Quercetin Inhibits Fibroblast Activation and Kidney Fibrosis Involving the Suppression of Mammalian Target of Rapamycin and β-catenin Signaling. Sci Rep. 2016; 6:23968. https://doi.org/10.1038/srep23968. [PubMed].
142. Pratheeshkumar P, Budhraja A, Son YO, Wang X, Zhang Z, Ding S, Wang L, Hitron A, Lee JC, Xu M, Chen G, Luo J, Shi X. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR- 2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS One. 2012; 7:e47516. https://doi.org/10.1371/journal.pone.0047516. [PubMed].
143. Lu Q, Ji XJ, Zhou YX, Yao XQ, Liu YQ, Zhang F, Yin XX. Quercetin inhibits the mTORC1/p70S6K signaling-mediated renal tubular epithelial-mesenchymal transition and renal fibrosis in diabetic nephropathy. Pharmacol Res. 2015; 99:237–47. https://doi.org/10.1016/j.phrs.2015.06.006. [PubMed].
144. Boly R, Gras T, Lamkami T, Guissou P, Serteyn D, Kiss R, Dubois J. Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int J Oncol. 2011; 38:833–42. https://doi.org/10.3892/ijo.2010.890. [PubMed].
145. Spindler SR, Mote PL, Flegal JM, Teter B. Influence on longevity of blueberry, cinnamon, green and black tea, pomegranate, sesame, curcumin, morin, pycnogenol, quercetin, and taxifolin fed iso-calorically to long-lived, F1 hybrid mice. Rejuvenation Res. 2013; 16:143–51. https://doi.org/10.1089/rej.2012.1386. [PubMed].
146. Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant EM, Chaumais MC, Bouchet S, Manéglier B, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016; 126:3207–18. https://doi.org/10.1172/JCI86249. [PubMed].
147. Caocci G, Mulas O, Annunziata M, Luciano L, Abruzzese E, Bonifacio M, Orlandi EM, Albano F, Galimberti S, Iurlo A, Pregno P, Sgherza N, Martino B, et al. Long-term mortality rate for cardiovascular disease in 656 chronic myeloid leukaemia patients treated with second- and third-generation tyrosine kinase inhibitors. Int J Cardiol. 2020; 301:163–66. https://doi.org/10.1016/j.ijcard.2019.10.036. [PubMed].
148. Moslehi JJ, Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J Clin Oncol. 2015; 33:4210–18. https://doi.org/10.1200/JCO.2015.62.4718. [PubMed].
149. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, Kirkland JL. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. 2019; 40:554–63. https://doi.org/10.1016/j.ebiom.2018.12.052. [PubMed].
150. Kovacovicova K, Skolnaja M, Heinmaa M, Mistrik M, Pata P, Pata I, Bartek J, Vinciguerra M. Senolytic Cocktail Dasatinib+Quercetin (D+Q) Does Not Enhance the Efficacy of Senescence-Inducing Chemotherapy in Liver Cancer. Front Oncol. 2018; 8:459. https://doi.org/10.3389/fonc.2018.00459. [PubMed].
151. Raffaele M, Kovacovicova K, Frohlich J, Lo Re O, Giallongo S, Oben JA, Faldyna M, Leva L, Giannone AG, Cabibi D, Vinciguerra M. Mild exacerbation of obesity- and age-dependent liver disease progression by senolytic cocktail dasatinib + quercetin. Cell Commun Signal. 2021; 19:44. https://doi.org/10.1186/s12964-021-00731-0. [PubMed].
152. Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012; 11:2391–401. https://doi.org/10.4161/cc.20683. [PubMed].
153. Singh M, Jensen MD, Lerman A, Kushwaha S, Rihal CS, Gersh BJ, Behfar A, Tchkonia T, Thomas RJ, Lennon RJ, Keenan LR, Moore AG, Kirkland JL. Effect of Low-Dose Rapamycin on Senescence Markers and Physical Functioning in Older Adults with Coronary Artery Disease: Results of a Pilot Study. J Frailty Aging. 2016; 5:204–07. https://doi.org/10.14283/jfa.2016.112. [PubMed].
154. Burcombe R, Wilson GD, Dowsett M, Khan I, Richman PI, Daley F, Detre S, Makris A. Evaluation of Ki-67 proliferation and apoptotic index before, during and after neoadjuvant chemotherapy for primary breast cancer. Breast Cancer Res. 2006; 8:R31. https://doi.org/10.1186/bcr1508. [PubMed].
155. Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, Yee HT, McMullen H, Oratz R, Klein P, Formenti SC, Muggia F. Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res. 2000; 6:4610–17. [PubMed].
156. Farczádi E, Szántó J, Kaszás I, Benyó I, Bodnár Z, Szlobodnyik J, Szende B. Changes in apoptotic and mitotic activity in rectal carcinoma after short-term cytostatic therapy as possible predictive factors. Neoplasma. 1999; 46:219–23. [PubMed].
157. Leontieva OV, Paszkiewicz GM, Blagosklonny MV. Comparison of rapamycin schedules in mice on high-fat diet. Cell Cycle. 2014; 13:3350–56. https://doi.org/10.4161/15384101.2014.970491. [PubMed].
158. Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, Djukovic D, Beyer RP, Raftery D, MacCoss M, Tian R, Rabinovitch PS. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016; 8:314–27. https://doi.org/10.18632/aging.100881. [PubMed].
159. Bhatt KH, Rudraraju R, Brooks JF, Jung JW, Galea R, Wells JW, Steptoe RJ. Short-course rapamycin treatment enables engraftment of immunogenic gene-engineered bone marrow under low-dose irradiation to permit long-term immunological tolerance. Stem Cell Res Ther. 2017; 8:57. https://doi.org/10.1186/s13287-017-0508-3. [PubMed].
160. Longo VD, Fontana L. Intermittent supplementation with rapamycin as a dietary restriction mimetic. Aging (Albany NY). 2011; 3:1039–40. https://doi.org/10.18632/aging.100401. [PubMed].
161. Hebert M, Licursi M, Jensen B, Baker A, Milway S, Malsbury C, Grant VL, Adamec R, Hirasawa M, Blundell J. Single rapamycin administration induces prolonged downward shift in defended body weight in rats. PLoS One. 2014; 9:e93691. https://doi.org/10.1371/journal.pone.0093691. [PubMed].
162. Dou X, Sun Y, Li J, Zhang J, Hao D, Liu W, Wu R, Kong F, Peng X, Li J. Short-term rapamycin treatment increases ovarian lifespan in young and middle-aged female mice. Aging Cell. 2017; 16:825–36. https://doi.org/10.1111/acel.12617. [PubMed].
163. Frescas D, Hall BM, Strom E, Virtuoso LP, Gupta M, Gleiberman AS, Rydkina E, Balan V, Vujcic S, Chernova OB, Gudkov AV. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle. 2017; 16:1526–33. https://doi.org/10.1080/15384101.2017.1339850. [PubMed].
164. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). 2016; 8:1294–315. https://doi.org/10.18632/aging.100991. [PubMed].
165. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, Leonova KI, Consiglio CR, Gollnick SO, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017; 9:1867–84. https://doi.org/10.18632/aging.101268. [PubMed].
166. Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, Sharpless NE. Cells exhibiting strong p16INK4a promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A. 2019; 116:2603–11. https://doi.org/10.1073/pnas.1818313116. [PubMed].
167. Cao H, Jia Q, Yan L, Chen C, Xing S, Shen D. Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells. Int J Mol Sci. 2019; 20:6093. https://doi.org/10.3390/ijms20236093. [PubMed].
168. Jia Q, Cao H, Shen D, Yan L, Chen C, Xing S. Fisetin, via CKIP-1/REGγ, limits oxidized LDL-induced lipid accumulation and senescence in RAW264.7 macrophage-derived foam cells. Eur J Pharmacol. 2019; 865:172748. https://doi.org/10.1016/j.ejphar.2019.172748. [PubMed].
169. Grosse L, Wagner N, Emelyanov A, Molina C, Lacas-Gervais S, Wagner KD, Bulavin DV. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020; 32:87–99.e6. https://doi.org/10.1016/j.cmet.2020.05.002. [PubMed].
170. Miklja Z, Yadav VN, Cartaxo RT, Siada R, Thomas CC, Cummings JR, Mullan B, Stallard S, Paul A, Bruzek AK, Wierzbicki K, Yang T, Garcia T, et al. Everolimus improves the efficacy of dasatinib in PDGFRα-driven glioma. J Clin Invest. 2020; 130:5313–25. https://doi.org/10.1172/JCI133310. [PubMed].
171. Chen B, Xu X, Luo J, Wang H, Zhou S. Rapamycin Enhances the Anti-Cancer Effect of Dasatinib by Suppressing Src/PI3K/mTOR Pathway in NSCLC Cells. PLoS One. 2015; 10:e0129663. https://doi.org/10.1371/journal.pone.0129663. [PubMed].
172. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Targeted elimination of senescent Ras-transformed cells by suppression of MEK/ERK pathway. Aging (Albany NY). 2017; 9:2352–75. https://doi.org/10.18632/aging.101325. [PubMed].
173. Kochetkova EY, Blinova GI, Bystrova OA, Martynova MG, Pospelov VA, Pospelova TV. Suppression of mTORC1 activity in senescent Ras-transformed cells neither restores autophagy nor abrogates apoptotic death caused by inhibition of MEK/ERK kinases. Aging (Albany NY). 2018; 10:3574–89. https://doi.org/10.18632/aging.101686. [PubMed].
174. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016; 15:973–77. https://doi.org/10.1111/acel.12458. [PubMed].