
Introduction
Chlorpromazine (CPZ), a member of the thiazine-class of heterocyclic compounds known as phenothiazines, appeared in the repertoire of psychotherapeutic drugs in 1952, revolutionizing the treatment of psychiatric disorders [1, 2]. Before the discovery of CPZ, the treatments for psychiatric disorders were limited to either invasive therapies, such as insulin comas and electroconvulsive therapy, or non-specific and addictive pharmacological agents such as opium, morphine and cocaine [3, 4]. Although CPZ is a first-generation antipsychotic medication, it is still widely used and several recent studies found that the latest (second) generation antipsychotic drugs do not provide substantial advantages for patient treatment [5, 6].
Not long after CPZ entered clinical use as an antipsychotic, it was proposed that CPZ also possessed anticancer activity. Epidemiological studies conducted in Denmark from 1957 to 1980 suggested that psychiatric patients prescribed CPZ had decreased risk of developing cancer [7]. In 1972, a case report indicated a significant inhibition of tumor growth in a patient with squamous-cell carcinoma of the larynx after directly injecting CPZ into the tumor [8]. Anticancer effect of CPZ was also observed in animal studies. It was reported that CPZ and other structurally similar phenothiazines inhibited sarcoma tumor growth in mice [9–11]. Importantly, the antitumor potency of the antipsychotic drugs did not correlate with their antipsychotic activity, suggesting that their antitumor effect is due to cellular mechanisms independent from the regulation of neuronal excitability [10, 11]. The epidemiological data coupled with the animal studies brought a newfound attention to CPZ as a potential anti-tumor medication, leading to follow-up studies focused on CPZ’s antitumor mechanisms. Studies in the past few decades have found that CPZ inhibits cancer growth through multiple independent pathways, via remarkably diverse targets ranging from histone deacetylases to ion channels. Here, we review the proposed molecular mechanisms of CPZ’s anti-tumor activity and cancer type specificity, and discuss the clinical potential of CPZ for cancer treatment.
CHLORPROMAZINE AS AN ANTIPSYCHOTIC MEDICATION
CPZ, first synthesized by Paul Charpentier in 1951, was initially used as an anesthetic (structure shown in Figure 1A) [12]. CPZ’s psychiatric relevance was discovered in 1952 when its administration as an anesthetic during surgery resulted in ‘disinterest’ in patients [1]. Subsequent studies showed that CPZ can calm severely agitated psychotic patients, paving the way for its use as an antipsychotic medication worldwide [13]. Within the first decade of CPZ’s discovery, more than 50 million people worldwide had taken the drug [14]. CPZ remains one of the most common drugs used for treatment of psychiatric disorders and is designated as an essential drug for treatment of schizophrenia by the World Health Organization.
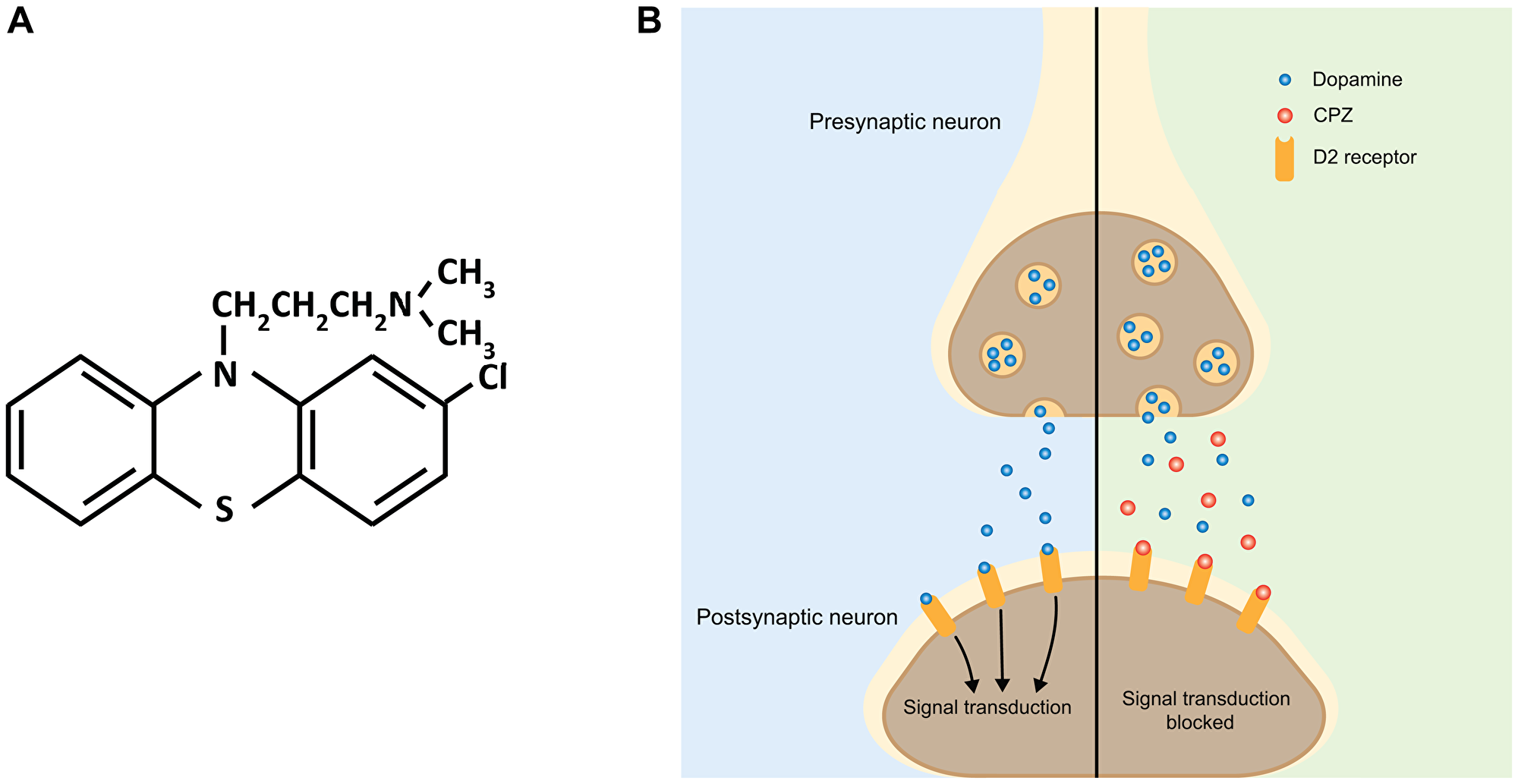
Figure 1: Schematic of the effect of CPZ on dopaminergic neurotransmission. (A) Chemical structure of CPZ. (B) In the absence of CPZ, release of dopamine from presynaptic terminals activates postsynaptic dopamine receptors, including D2, initiating downstream signal transduction (left side, blue). CPZ binds to D2 receptors without activating them. This prevents dopamine from binding to D2 receptors and blocks downstream signal transduction (right side, green).
As the use of CPZ for treatment of psychiatric disorders increased in popularity, identification of its pharmacological mechanisms gained interest as well. Initially, it was thought that the antipsychotic effect of CPZ was mediated through the inhibition of serotonin receptors [15]. However, subsequent studies established a model still accepted today, in which inhibition of D2 dopamine receptors is primarily responsible for CPZ’s antipsychotic effect (Figure 1B) [16–18]. CPZ shows high specificity for dopamine receptors with a binding affinity of 3–23 nM for D2 dopamine receptors [19, 20]. Subsequent studies showed that CPZ and related drugs prevent dopamine from binding and activating D2 dopamine receptors in the areas of the brain involved in the control of emotional behavior, such as the limbic system [21, 22]. The antagonistic action of CPZ on D2 receptors alleviates psychotic symptoms associated with increased dopamine signaling found in psychiatric disorders, including schizophrenia, bipolar disorder and psychotic disorders [23].
The effectiveness of CPZ as an antipsychotic drug stems from its ability to readily permeate the blood brain barrier (BBB) with an apparent permeability of 303 nm/s [24, 25]. The high BBB permeability of CPZ is conferred by its low molecular weight (318.86 g/mol), absence of hydrogen bond donors, and low flexibility of its chemical structure (presence of rotatable bonds) - properties typical of successful Central Nervous System (CNS) drugs [25]. CPZ’s effectiveness as a CNS drug is further potentiated by its ability to remain in the brain for sufficient periods to exert its intended effect, with a half-life of 8 to 35 hours [26]. Typical oral doses of CPZ range from 100 to 1000 mg/day, depending on the severity of the psychiatric ailment [27]. The plasma levels of CPZ normally observed in patients are in the range of 0.1 μM to 2 μM [28, 29]. However, due to the high membrane permeability and tissue accumulation of phenothiazines, the effective cellular concentration of CPZ is expected to be 10 to 1000-fold higher [30–32].
EARLY REPORTS OF CPZ’S CELLULAR ANTITUMORIGENIC MECHANISMS
The wide use of CPZ as an antipsychotic medication prompted studies on its effect on a wide range of cellular targets. It became clear that in addition to the neuronal receptors, CPZ also affects multiple seemingly unrelated targets and cellular processes, including inhibition of DNA synthesis [33], uncoupling of oxidative phosphorylation and inhibition of cytochrome oxidase [34, 35], suppression of adenosine triphosphatase (ATPase) enzymatic activity [34, 36], alteration of membrane permeability [37, 38] and inhibition of lipase activity [39]. Many of the identified cellular mechanisms were either initially or eventually linked to CPZ’s anticancer activity. These early studies indicated that while CPZ’s antipsychotic mechanism is relatively straightforward, its anticancer effect involves a diverse range of cellular pathways. During the last few decades, the number of targets and pathways implicated in CPZ’s anticancer activity has continued to grow, strengthening the potential of repurposing CPZ for cancer treatment. Below, we discuss some of CPZ’s most prominent cellular targets and mechanisms of action related to cancer.
EFFECT OF CPZ ON DNA SYNTHESIS
Cancer is characterized by an uncontrolled proliferation of cells. DNA synthesis is crucial to cell division, thus, inhibition of DNA synthesis may bridle cancer progression (Figure 2A). DNA synthesis inhibition was amongst the first identified CPZ’s anticancer cellular mechanisms. In 1965, it was shown that CPZ decreases H3-thymidine incorporation into human bone marrow cells during DNA synthesis, indicating that CPZ inhibits DNA synthesis (Figure 2B (iii)) [40, 41]. Similarly, inhibition of DNA synthesis was observed in Meth A sarcoma cells [42]. The inhibition of DNA synthesis by CPZ was reported in cell culture and cell-free systems [33, 42], suggesting that CPZ could be directly inhibiting enzymes involved in DNA synthesis. In addition to inhibiting DNA synthesis, CPZ caused DNA fragmentation in leukemic and mouse mastocytoma cells [43, 44], and in human oral cancer cells via the inhibition of Akt/mTOR phosphorylation (Figure 2B (iii) (see below) [45]. Importantly, CPZ decreased the viability of leukemic cells with minimal cytotoxicity and at clinically relevant doses (1–40 μM range) [43]. Similarly, while CPZ inhibited oral cancer cell proliferation with the IC50 of ~20 μM, it had little effect on healthy (noncancerous) oral cells [45]. CPZ has also been shown to inhibit the initial step of SV40 DNA replication in HeLa cell extract, specifically the pre-elongation step [46]. Taken together, these studies demonstrate that CPZ can inhibit cell proliferation in cancer by modulating DNA synthesis, although further studies are needed to tease out the direct mechanisms involved.
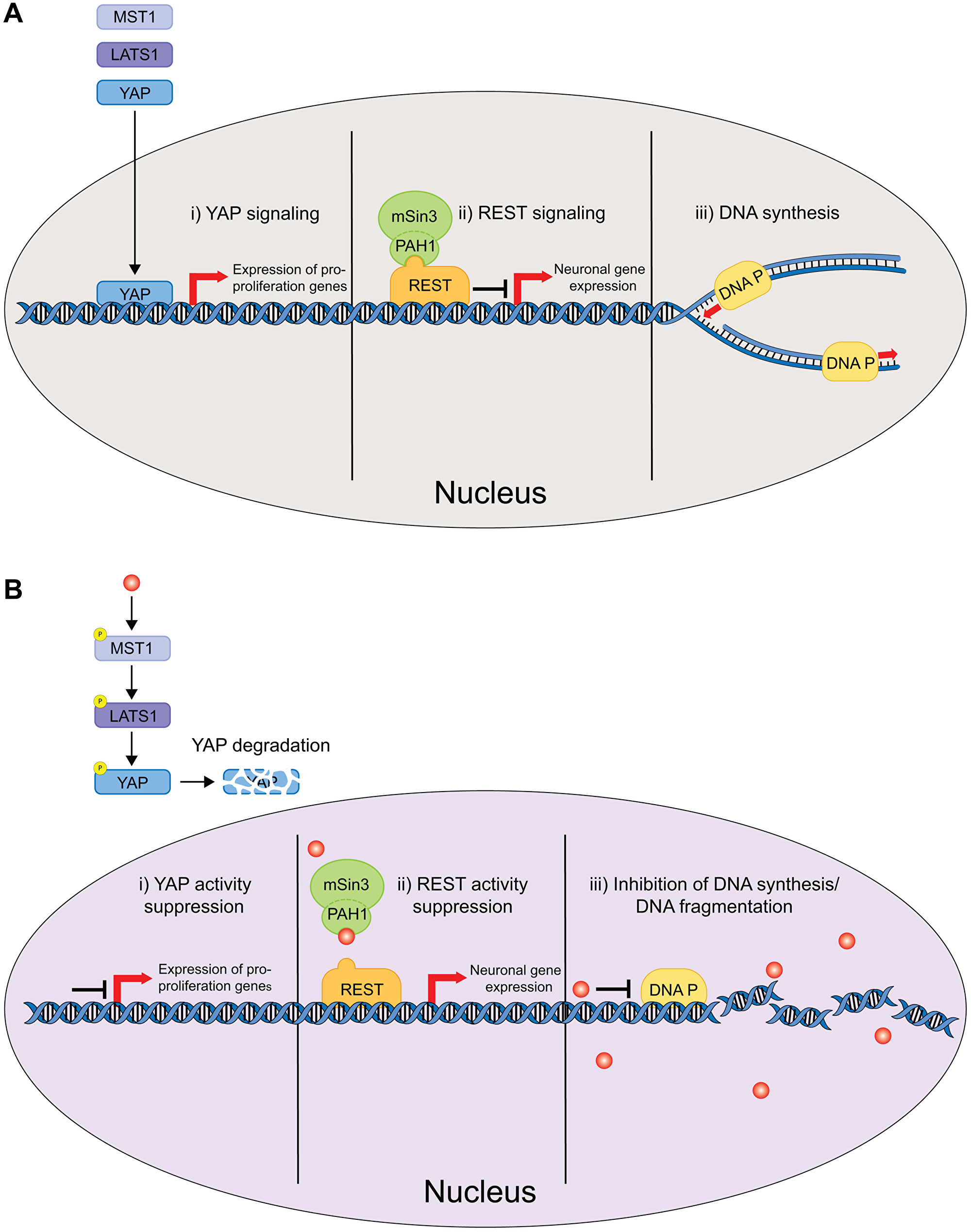
Figure 2: Schematic of the nucleus delimited effects of CPZ. (A) In the absence of CPZ, YAP signaling upregulates expression of genes involved in cell proliferation (i), REST signaling activates neuronal gene expression (ii) and DNA Polymerase carries out DNA replication necessary for cell division (iii). (B) CPZ inhibits YAP signaling by inducing YAP degradation (i), inhibits REST signaling most likely by binding to PAH1 and inhibiting mSin3/REST interactions (ii), and inhibits DNA Polymerase and causes DNA fragmentation (iii). These effects of CPZ inhibit cell proliferation and tumorigenesis.
EFFECT OF CPZ ON REST MEDIATED GENE TRANSCRIPTION
Repressor-element 1 silencing transcription factor (REST), also known as neural restrictive silencer factor (NRSF), represses neuronal gene transcription in non-neuronal cells, and regulates neuronal differentiation and gene expression (Figure 2A (ii)) [47–49]. REST represses gene expression by recruiting co-repressors mammalian Swi-independent 3 (mSin3) and CoREST1 [50–52]. The co-repressor mSin3 contains four paired amphipathic helical (PAH) domains, PAH1-PAH4, with PAH1 binding to REST [53, 54]. Importantly, upregulation of REST expression has been linked to several brain tumors, including medulloblastoma and glioblastoma [55–58].
In 2018, Kurita and coworkers identified CPZ as a small-molecule binder of mSin3 PAH1, using a ligand-based and structure based in-silico drug screening [59]. NMR titration experiments indicated that CPZ binds to the isolated PAH1 fused to Glutathione S-transferases (GST) with the binding affinity of 15 ± 4.2 μM. NMR data-guided docking of CPZ into PAH1 structure indicated CPZ-induced structural rearrangements. It has been proposed that by binding to PAH1 and inducing structural changes, CPZ could block mSin3-REST interactions, thereby reducing REST mediated transcription repression (Figure 2B (ii)). Consistent with this hypothesis, application of CPZ to human DAOY medulloblastoma cells decreased spheroid growth [59]. Although other mechanisms of the CPZ action have not been ruled out in the study, destabilization of mSin3-REST interactions could be yet another mechanism contributing to tumor growth inhibition by CPZ.
EFFECT OF CPZ ON YAP SIGNALING
Yes-associated protein (YAP) is a component of the Hippo signaling pathway and promotes cell proliferation and organ growth when it is not phosphorylated (Figure 2A (i)) [60–62]. Activation of the Hippo signaling cascade results in the phosphorylation of large tumor suppressor kinase 1/2 (LATS1/2) by the mammalian Ste2-like kinase (MST1/2). LATS1/2 in turn phosphorylates YAP, which leads to YAP proteasomal degradation. Recently, YAP overexpression was found to be associated with liver [63] and breast cancers [64, 65]. Moreover, increased YAP expression was shown to confer cancer cells with stem-like properties, including chemoresistance [66, 67]. Cancer stem cells (CSC) are one of the most intractable obstacles for cancer treatment due to their self-renewal properties and the ability to differentiate, leading to multidrug resistance and tumor recurrence. Importantly, YAP inhibitors have been shown to suppress CSC properties, such as drug resistance [67–69].
In 2019, Yang and coworkers found that CPZ suppresses YAP signaling in MCF7 and MDA-MB-231 breast cancer cells [70]. CPZ treatment induced a dose-dependent increase in the phosphorylation of YAP’s upstream regulators, LATS1 and MST1, thereby promoting YAP phosphorylation and subsequent proteasomal degradation, and decreased nuclear expression levels of YAP (Figure 2B (i)). Importantly, inhibition of YAP signaling by CPZ suppressed stemness in breast cancer cells, causing decrease in self-renewal and chemoresistance to doxorubicin and taxol, commonly used for breast cancer chemotherapy. In summary, the effect of CPZ on YAP and upstream Hippo signaling could be an important pathway for decreasing stemness and increasing the effectiveness of existing breast cancer chemotherapies.
EFFECT OF CPZ ON MITOTIC KINESIN
Mitosis is a crucial phase in the cell cycle and, therefore, is an important target for cancer therapies. Recent efforts to target mitosis for cancer treatment focus on suppressing cell division via affecting mitotic kinesins [71]. Mitotic kinesins are a family of mechanochemical enzymes that use ATP to traverse along or destabilize microtubules. These proteins are crucial for cell division and regulate the function of mitotic spindle (Figure 3, left) [72, 73]. One member of the mitotic kinesin family, KSP/Eg5, has been considered a promising oncogenic target. Substantial effort has been levied to identify KSP/Eg5 inhibitors for therapeutic use [74].
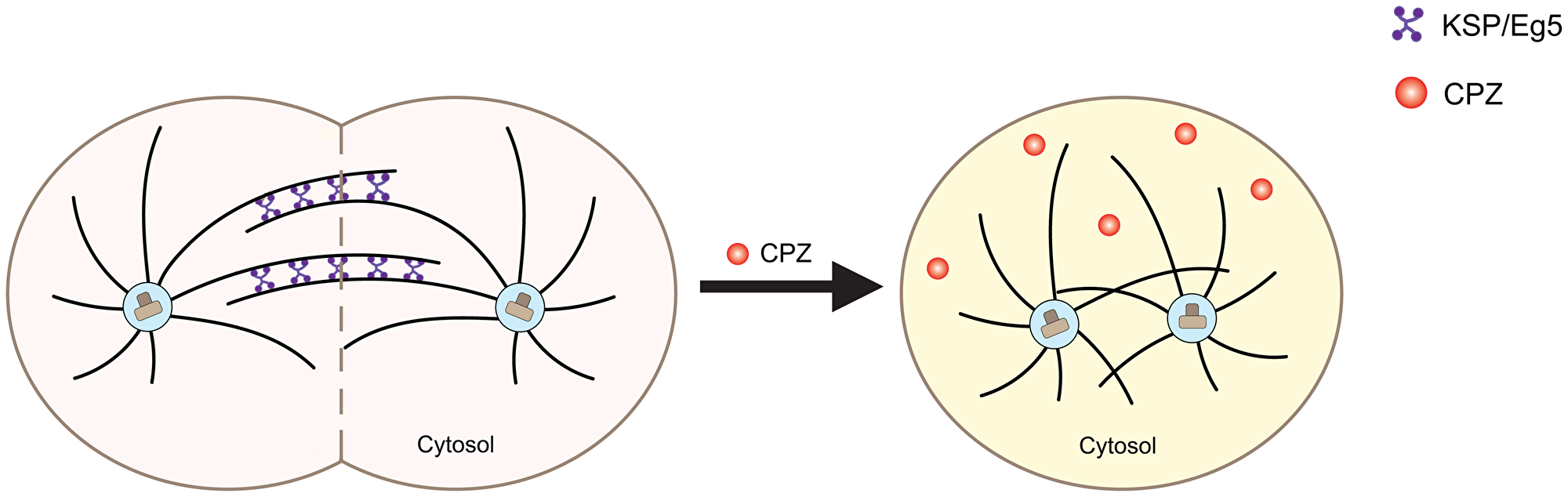
Figure 3: Schematic of the effect of CPZ on mitotic kinesins. Mitotic kinesins, such as KSP/Eg5, are crucial for mitotic spindle assembly for chromosome segregation during mitosis (left, pink). CPZ is thought to inhibit mitotic kinesins, hindering the spindle assembly and increasing the number of cells with monopolar spindles (right, yellow).
In 2007, Lee and coworkers showed that CPZ inhibited KSP/Eg5’s ability to hydrolyze ATP with an IC50 of 5 to 10 μM [75]. The effect was specific to KSP/Eg5, as no CPZ-dependent inhibition was observed for any other mitotic kinesins or chromokinesins, even at CPZ concentrations as high as 100 μM. Treatment with CPZ inhibited proliferation of HCT116 human colon carcinoma cell lines with an IC50 of 5 to 7 μM. Moreover, application of CPZ in combination with pentamidine, an antiparasitic agent, inhibited tumor growth in mice xenograft models implanted with A549, non–small cell lung cancer (NSCLC), or HCT116 colon cancer cells. At the cellular level, CPZ treatment increased the population of cells with monopolar spindle and the population of cells in the G2-M phase of cell cycle. Both accumulation of monopolar spindles and cell cycle arrest are characteristic of KSP/Eg5 inhibition, thereby suggesting that CPZ inhibits KSP/Eg5 activity, and this inhibition is involved in the observed antitumorigenic effect (Figure 3, right). Application of other phenothiazines with chemical structures similar to CPZ caused a similar antitumorigenic effect, although, with lower potency, further suggesting that the inhibition of KSP/EG5 activity is contributing to the reduction in cancer growth. Consistent with these results, Riffell and coworkers reported that CPZ treatment caused mitotic arrest in MCF-7mp53 breast cancer cells expressing dominant-negative p53, MDA-MB-231 breast cancer cells and T98G glioblastoma cells [76]. CPZ induces mitotic arrest at concentrations higher than 10 μM when used alone, however, it was effective at lower concentrations when administered in combination with 10–30 nM paclitaxel, a microtubule-targeting drug, suggesting the drugs work synergistically. Taken together, these studies indicate that inhibition of mitotic kinesin activity by CPZ can suppress tumor growth by inducing cell cycle arrest due to an accumulation of cells with monopolar spindle. Therefore, mitotic kinesin inhibition is yet another mechanism contributing to CPZ’s antitumorigenic effect.
EFFECT OF CPZ ON CYTOCHROME C OXIDASE
Cytochrome c oxidase (CcO) is the final enzyme of the mitochondrial electron transport chain (Figure 4A). CcO catalyzes the transfer of electrons from cytochrome c to oxygen (O2) molecules and subsequently converts O2 to water. In mammals, CcO is composed of thirteen subunits, many of which have multiple isoforms [77]. Several studies have identified links between CcO and tumor development and progression. It has been shown that the various CcO subunits are differentially expressed in certain tumors. For instance, expression of COX3 subunit of CcO is lower than normal in colon adenomas [78], while COX4 subunit isoform 1 (COX4-1) is overexpressed in glioblastoma (GBM) cells, and its upregulation is linked to the development of temozolomide (TMZ) chemoresistance in GBM cells [79]. Differential expression of COX5b subunit has been reported in colorectal carcinomas [80], and knockdown of the COX5a subunit by siRNA has been shown to reduce non-small cell lung cancer (NSCLC) cell migration and invasion [81].
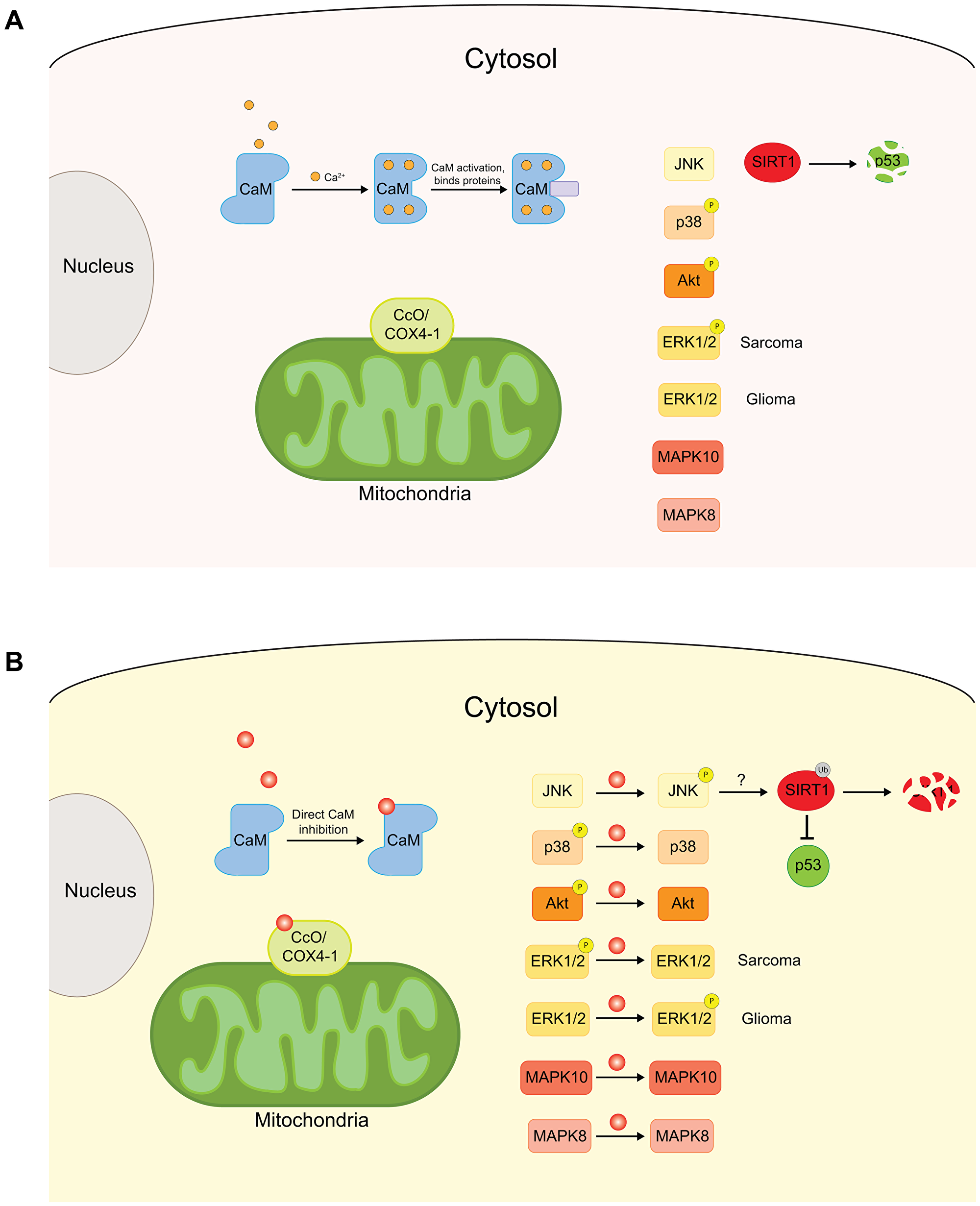
Figure 4: Schematic of the effects of CPZ on cytoplasmic and mitochondrial proteins. (A) Cytoplasmic proteins, CaM, kinases, including JNK, p38, Akt, ERK1/2 and MAPKs, and mitochondrial CcO complex are important proteins for normal cellular functions. Defects in these proteins can increase cell proliferation and drive tumorigenesis. (B) CPZ has been shown to inhibit CaM, affect the phosphorylation levels of the kinases, and preferentially bind to COX4-1 subunit, enriched in CcO complexes associated with cancer. Through these effects, CPZ inhibits cell proliferation and tumor progression.
Early studies of the effects of CPZ on CcO were conducted in mid 1950s on the rat brain and liver mitochondria [30, 32]. It was found that CPZ inhibits CcO activity by 50% at 50 μM concentration in rat brain mitochondria [34]. The direct link of CcO activity inhibition by CPZ to cancer was investigated in a recent study by Oliva and coworkers [82]. It was found that CPZ inhibits CcO activity in a dose-dependent manner in TMZ resistant U251 cell-derived UTMZ glioma cells while having no effect on TMZ-sensitive U251 cells. The specificity of the CcO inhibition was proposed to stem from a preferential binding of CPZ to a binding site at the interface of COX4-1 and two other subunits of the CcO complex (Figure 4B). Since the expression of COX4-1 subunits is increased in TMZ-resistant glioma cells, the preferential binding of CPZ to COX4-1 would explain the specificity of the CPZ effect for the TMZ-resistant U251 glioma cells [79]. Taken together with the previous reports, the study by Oliva and coworkers indicates that the inhibition of CcO activity by CPZ is one of the contributing factors to the CPZ’s antitumorigenic activity.
EFFECT OF CPZ ON KINASES
Mitogen-activated protein kinases (MAPK) are protein kinases that are involved in directing cellular responses to a diverse range of signals, including mitogens, osmotic stress, pro-inflammatory cytokines, and growth factors (Figure 4A) [83]. MAPK signaling pathways play an important role in cell proliferation and differentiation and are frequently dysregulated in cancer [83, 84]. Recent bioinformatics study based on regulation network analysis of protein interactions identified MAPK8 and MAPK10 as potential targets of CPZ (Figure 4B) [85]. Experimental findings further substantiated the possibility of MAPK contributing to the antitumor effects of CPZ. In 2010, Shin and coworkers showed that CPZ induced phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK), members of the MAPK family, in C6 glioma cells [86]. The increased phosphorylation of ERK1/2 and JNK kinases was linked to the induction of cyclin-dependent kinase inhibitor 1 (CDKN1), also known as p21 or p21Waf1/Cip1, by the transcription factor early growth response-1 (Egr-1), which itself is transcriptionally activated by another transcription factor, the ternary complex factor also known as Elk-1 [87]. CDKN1 promotes cell cycle arrest at the G1 and G2/M phases [88–90] and mediates the ability of the tumor suppressor p53 to arrest cell proliferation [91]. The upregulation of the JNK(ERK1/2)/Elk-1/Egr-1/p21 pathway contributed to the antitumorigenic effect of CPZ in C6 glioma cells, as summarized in Figure 4B) [86].
While CPZ increased JNK and ERK1/2 phosphorylation in C6 cells, the same study demonstrated that CPZ decreased p38 MAPK phosphorylation in the same cell line [86]. This suggests that the effect of CPZ on kinases is diverse and could be different even for the same kinases depending on the cell-type. In agreement with the varied effect of CPZ on MAPK phosphorylation, Martins and coworkers showed that CPZ decreases phosphorylation of ERK1/2 in Ewing sarcoma (ES) cells leading to reduction in cell proliferation and increase in the cell apoptosis levels [92], whereas Lee and coworkers showed that CPZ increased JNK phosphorylation in human colorectal cancer HCT116 cells, causing inhibition of the tumor cell growth and increased apoptosis [93]. Further investigation indicated that CPZ-induced JNK phosphorylation caused degradation of p53 inhibitor sirtuin 1 (SIRT1), either directly or indirectly, thereby inducing p53-dependent apoptosis of HCT116 cells (Figure 4B) [93]. Taken together, the reported effects of CPZ on the MAPK family are overwhelmingly antitumorigenic, however, the direction of the effect varied with tissue- and kinase-type.
Another kinase affected by CPZ is Akt, also known as protein kinase B, which in turn activates the mammalian target of rapamycin (mTOR). Akt/mTOR pathway regulates cell cycle and the induction of autophagy, and deficiencies in the pathway have been linked to cancer [94, 95]. Recently, Shin and coworkers found that CPZ, either directly or via intermediate players, inhibited mTOR by decreasing the levels of phosphorylated Akt (Figure 4B) [96]. CPZ inhibited the Akt/mTOR pathway in PTEN (phosphatase and tensin homolog)-null U-87MG glioma cells, with constitutively active Akt/mTOR pathway, causing cell cycle arrest and autophagic cell death. Similarly, Jhou and coworkers found that in oral cancer, CPZ reduced levels of phosphorylation of Akt and mTOR, leading to the cell cycle arrest in G2/M phase and inhibition of cell proliferation [45]. Despite the evidence that the kinases are involved in the effect of CPZ in cancer, experimental proof of direct binding of CPZ to kinases is lacking. Therefore, the molecular mechanisms of CPZ action on the kinases are not clear and require further investigation.
EFFECT OF CPZ ON CALMODULIN
Calmodulin (CaM) is a multifunctional calcium-binding protein ubiquitously expressed in eukaryotic cells (Figure 4A). CaM interacts with many other proteins to mediate a diverse range of cellular functions and signaling pathways. Amongst CaM’s main functions are the regulation of cell proliferation, division and differentiation [97], all of which contribute to its role in tumorigenesis. CaM was found to be upregulated in human primary lung cancer cells [98], and in rat fibroblasts transfected with oncogenes [99]. Moreover, CaM antagonists have been reported to decrease lung metastasis induced by implanting Lewis lung carcinomas in mice [100] and decrease tumor cell growth in different cancer types [101–103].
CPZ is known to inhibit CaM activity by direct binding to the protein in a calcium-dependent manner (Figure 4B) [104]. In 1982, Prozialeck and Weiss reported that the hydrophobicity of the phenothiazine ring of CPZ and other phenothiazine antipsychotics correlates well with their potency in inhibiting CaM activity, as reflected in the inhibition of CaM-induced activation of phosphodiesterases [105]. The authors proposed that CPZ binding to CaM is mediated by hydrophobic interactions between the phenothiazine ring and a nonpolar region of calmodulin, and by an electrostatic interaction between the positively charged amino group of CPZ and negatively charged residues on CaM protein. In the same study, the IC50 for CaM activity inhibition by CPZ was reported to be 40 μM.
Although several CaM inhibitors have successfully quelled cancer growth and metastasis, it is not clear if CaM inhibition by CPZ is therapeutically relevant. For instance, Zhong and coworkers reported that CPZ has antitumorigenic effect on small cell lung carcinoma (SCLC) [106]. However, application of CaM inhibitor W7 failed to reproduce the effect of CPZ, indicating that the CPZ’s antitumorigenic activity in SCLC is independent of CaM. Therefore, although it would be reasonable to speculate that inhibition of CaM activity by CPZ could be yet another mechanism contributing to CPZ’s antitumorigenic effect, evidence substantiating this hypothesis has yet to be found.
EFFECT OF CPZ ON MEMBRANES
Cellular membranes are vital for cellular integrity and contain numerous transmembrane proteins essential for cellular signaling (Figure 5A). An effect of CPZ on membranes was first reported in the 1960s when it was shown that CPZ inhibited the swelling of rat liver mitochondrial membranes in isotonic sucrose solution [37], reduced the rate of hypotonic hemolysis in human erythrocytes [107], and inhibited tissue uptake of radioactively labeled norepinephrine [108, 109]. It was suggested that these effects were due to the CPZ-induced decrease in membrane permeability, yet the exact mechanism was not elucidated. On the other hand, in vivo and in vitro studies of effects of CPZ on rat liver lysosomes suggested that at high concentrations (>100 μM) CPZ increases membrane permeability [38]. The discrepancy in the effect of CPZ on cellular membranes reported in the early studies could reflect the difference in the experimental systems used or could indicate that CPZ’s effect is dependent on membrane composition.
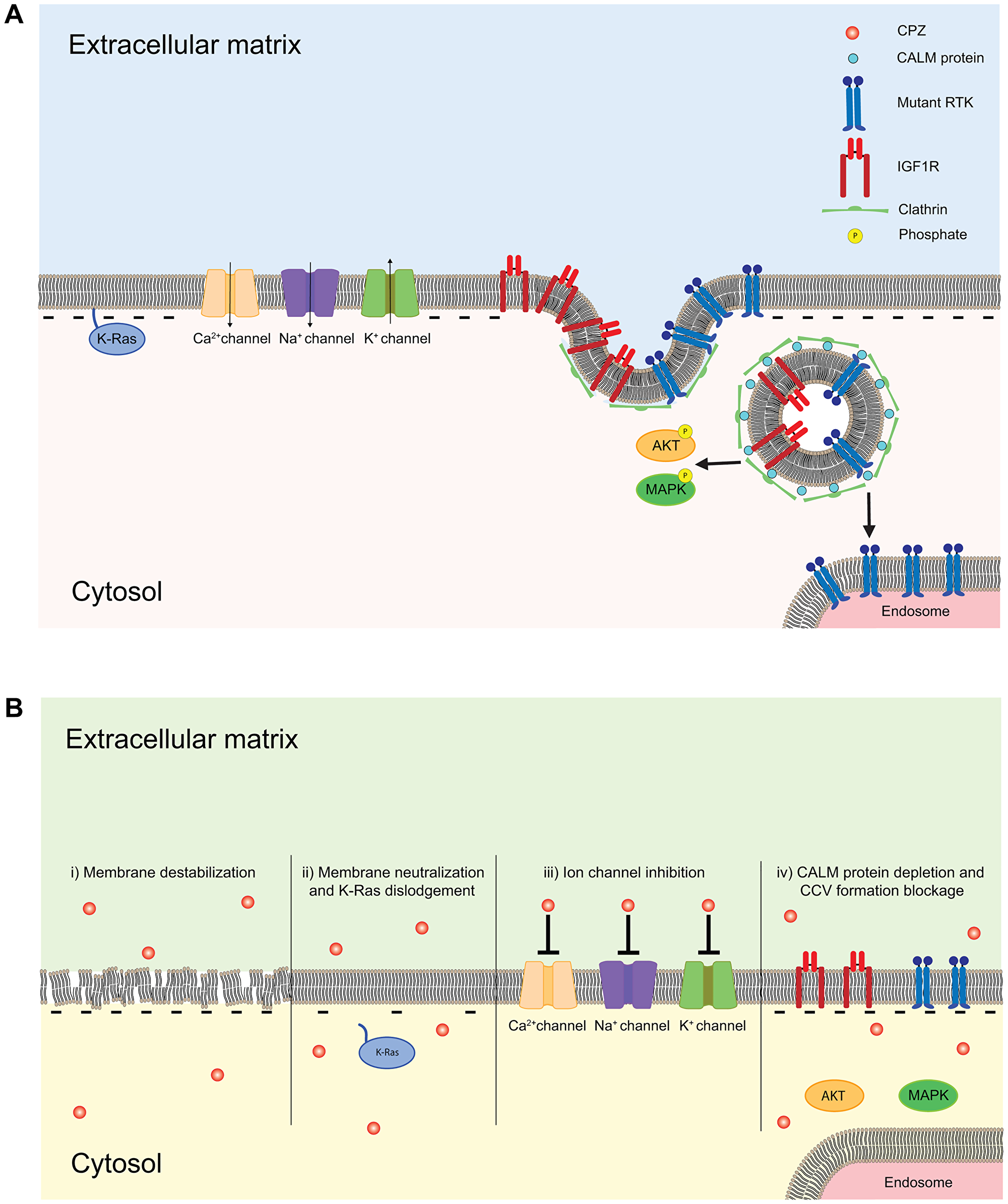
Figure 5: Schematic of the membrane delimited effects of CPZ. (A) Cellular surface membrane is formed by lipids, and contains transmembrane and membrane-associated proteins. Transmembrane proteins such as ion channels, including EAG, ERG and other K+ channels, Na+ channels and Ca2+ channels, receptors, including RTKs and IGF1Rs, and membrane-associated proteins, including Ras proteins, are key regulators of cell cycle and tumorigenesis. (B) CPZ directly affects membrane by destabilizing the lipid bilayer and increasing membrane permeability (i), decreasing membrane association of Ras protein by neutralizing the negative charge of phospholipids (ii), inhibiting various ion channels, including K+, Na+ and Ca2+ selective channels (iii) and altering RTK and IGF1R receptor internalization by suppressing clathrin mediated signaling (iv).
More recent studies showed that application of CPZ causes leakage of chromophore carboxyfluorescein from liposomes [110], and leakage of [18F]2-fluoro-2-deoxy-D-glucose-6-phosphate tracer and increased membrane fluidity in rat brain slices (Figure 5B (i)) [111]. The latter effect of CPZ was observed only at concentrations >100 μM, which are higher than the putative therapeutically relevant plasma levels of CPZ. However, due to the 20- to 30-fold accumulation of antipsychotic drugs in brain tissues [30, 31] and further accumulation in cellular membranes due to their high lipophilicity, the effect of CPZ on the membrane permeability and stability could be therapeutically important. In 2009, CPZ’s effect on membrane permeability was further characterized in membrane vesicles, where CPZ drastically increased membrane permeability as reflected in the CPZ concentration-dependent release of fluorophor calcein [112], and lipid monolayers, where CPZ increased the surface area of monolayers formed from acidic phospholipids while having no effect on monolayers formed from neutral lipids [113]. Investigation of interactions between CPZ and model membranes with the differential scanning calorimetry and isothermal calorimetry suggested that CPZ both interacts with negatively charged membrane phospholipids and also intercalates into the hydrophobic region of the membrane bilayer [112]. The interaction between CPZ and the membrane resulted in the increased membrane permeability and overall destabilization of the membrane, possibly by introducing membrane pores (Figure 5B (i)). Consistent with this hypothesis, CPZ-induced increase in membrane permeability resulted in increased MCF-7 human breast cancer cells sensitivity to tamoxifen, demonstrated by the prevention of cell growth and reduction of metabolic activity in the cells [112]. Therefore, CPZ-induced increase in membrane permeability facilitated increased accumulation of tamoxifen in the MCF-7 cancer cells, thereby increasing the drug’s efficacy. Taken together, these recent studies demonstrate that CPZ induces increased membrane permeability, thereby increasing intracellular accumulation of anticancer drugs and ultimately enhancing their effectiveness.
In addition to its effect on membrane permeability, CPZ has been shown to alter membrane polarity by interacting with negatively charged membrane phospholipids, in both live cells and phospholipid bilayers (Figure 5B (ii)). Confocal microscopy and fluorescence recovery after photobleaching (FRAP)-based studies found that CPZ facilitated the membrane dislodgment of oncogenic K-Ras4B(G12V), an isoform of Ras protein that anchors to the membrane via electrostatic interaction [114]. Ras proteins are membrane-associated GTPases, cycling between inactive GDP-bound and active GTP-bound states. Ras proteins are important regulators of cell growth and proliferation, and about 30% of human cancers are associated with defects in Ras protein function [115]. Proper anchorage of Ras proteins to the membrane is essential for their role in cellular signaling and tumorigenesis. CPZ significantly increased cytoplasmic and/or mitochondrial levels of K-Ras4B(G12V), while decreasing K-Ras4B(G12V) levels in the membrane. Importantly, CPZ did not have an effect on H-Ras, a Ras protein isoform which does not require electrostatic interaction with negatively charged phospholipids for its association with the membrane. This suggests that the CPZ dislodges K-Ras4B(G12V) by neutralizing negatively charged membrane phospholipids, thereby weakening the protein’s electrostatic interaction with the membrane (Figure 5B (ii)). Importantly, CPZ-induced membrane dislodgement of K-Ras4B(G12V) was associated with apoptosis of Rat-1 fibroblasts cell lines stably expressing GFP-K-Ras(G12V), and cell cycle arrest and suppressed wound healing in human pancreatic cancer PANC-1 cell lines expressing GFP-K-Ras(G12V) cells [114]. Consistent with CPZ-induced membrane dislodgment of K-Ras4B(G12V) via affecting membrane phospholipids, fluorescence spectroscopy studies indicated that CPZ increased disorder of the polar heads and acyl chain regions of the phospholipid bilayer, destabilizing the membrane [116]. In summary, the studies on the effects of CPZ on cellular membranes indicate that it affects both membrane permeability and polarity, and both effects contribute to CPZ’s antitumorigenic activity, as summarized in Figure 5B. The diverse range of CPZ’s effects on cellular membranes further underscores the complexity of its mechanism.
EFFECT OF CPZ ON ION CHANNELS
The initial reports of CPZ effects on ion channels came from studies of calcium channels. It was shown that CPZ and other antipsychotic drugs inhibit the binding of [3H]nitrendipine, a calcium channel antagonist, to calcium channels with an affinity comparable to CPZ’s affinity for dopamine receptors [117], causing the inhibition of smooth muscle contractions [117, 118]. Subsequent studies found that CPZ blocks a variety of sodium, potassium, and calcium channels (Figure 5B (iii)) [119–123].
More recently, it was shown that CPZ inhibits ether-a`-go-go (EAG) and EAG-related gene (ERG) channels, which belong to the KCNH family of potassium selective channels [124–126]. EAG and ERG are considered bona fide oncogenic channels [127, 128]. Upregulation of EAG and ERG channel activity has been implicated in the proliferation and progression of various cancers [129–137]. Additionally, the overexpression of EAG channels in human tumors is clinically used as tumor marker [129, 138, 139]. Inhibition of EAG and ERG channel activity, either with non-specific blockers or by siRNA, has suppressed cancer growth [137, 140–142]. It was shown that CPZ inhibits hERG channels in a concentration and voltage-dependent manner with the IC50 of 10.5 μM at -30 mV, and 4.9 μM at +30 mV, most likely by blocking channel pore [125, 126]. More recently, we found that CPZ also inhibits EAG channel activity [124], however, the mechanism of inhibition seems to be different from ERG channel inhibition. CPZ inhibited EAG currents in a concentration-dependent and voltage-independent manner with the IC50 of 3.7 μM [124]. Our study suggests that CPZ inhibits EAG channels by binding to the intracellular Per-Arnt-Sim (PAS) domain of the channels, as fluorescence-based and surface plasmon resonance-based assays showed direct binding of CPZ to the PAS domain and deletion of the PAS domain dramatically decreased the CPZ inhibition of EAG currents. Interestingly, we found that CPZ does not bind to the PAS domain of ERG channels. Taken together, the studies of CPZ’s effect on oncogenic EAG and ERG channels suggest that inhibition of these channels by CPZ likely contributes to its antitumorigenic effect.
EFFECT OF CPZ ON CALM PROTEIN
Clathrin assembly lymphoid myeloid leukemia (CALM) protein is crucial for the formation of clathrin coated vesicles for intracellular trafficking of receptor tyrosine kinases (RTKs) [143]. Mutations in RTKs are linked to various cancers, including lung cancer and acute myeloid leukemia (AML) [144]. While wild-type RTKs are continuously shuffled between the plasma membrane and endosomes via clathrin-mediated internalization and subsequent recycling, mutant RTKs are mistargeted to different intracellular compartments, such as endosomes, where they activate downstream targets and promote oncogenic activity [145, 146]. Recently, CPZ was shown to reduce the levels of CALM protein in a concentration-dependent manner, thereby altering the cellular localization of RTKs with AML-associated mutations, without disturbing wild-type RTKs (Figure 5B (iv)) [147]. Both treatment with CPZ and CALM knock-out resulted in the dislocation of the mutant RTKs and decreased levels of phosphorylation of downstream targets of RTKs. Also, both CPZ treatment and CALM knock-out inhibited the growth of AML cells with mutant RTKs. Importantly, CPZ treatment had no effect on the growth rate of AML cells with CALM knock-out, implicating suppression of CALM protein levels as the primary mechanism of CPZ’s anticancer effect in AML cells expressing mutant RTKs. In summary, the study provides evidence that CPZ is capable of blocking compartment-dependent oncogenic activity of mutant RTKs, via inhibition of the CALM protein.
ANTITUMORIGENIC ACTIVITY OF CPZ IN DIFFERENT CANCERS
In this section we list known antitumorigenic CPZ effects in different cancers and reported/proposed cellular mechanisms (Table 1). In breast cancer, CPZ exhibits an anti-proliferative effect, suppresses stemness, and increases cancer cell sensitivity to existing chemotherapies, thereby reversing drug resistance [70, 112]. The implicated mechanisms of CPZ action in breast cancer are suppression of YAP signaling [70] and increase in membrane permeability that promotes accumulation of chemotherapeutic agents, including doxorubicin and taxol [70]. In colorectal cancer, CPZ was shown to inhibit tumor growth and induce apoptosis [93]. The examined effect of CPZ in colorectal cancer was attributed to a down-regulation of p53 inhibitor, SIRT1. Interestingly, YAP levels promote colorectal tumor aggressiveness [148]. Therefore, similar to breast cancer, YAP suppression by CPZ in colorectal cancer could be in part responsible for its observed antioncogenic effect.
Table 1: Summary of antitumorigenic effects of CPZ in different cancers
| Cancer type | Effective concentration | Proposed Mechanism | Cell line (human, unless indicated) | Ref |
|---|---|---|---|---|
| Breast cancer | 10 μM | Increase in membrane permeability | MCF-7 | [112] |
| > 2 μM | Suppression of YAP signaling | MCF-7, MDA-MB-231 | [70] | |
| ≥6 μM | Inhibition of KSP/Eg5 | MCF-7mp53, MDA-MB-231 | [76] | |
| Colorectal cancer | 3–10 μM (IC50) | SIRT1 inhibition | HCT116 LoVo | [93] |
| 5–7 μM (IC50) | Inhibition of KSP/Eg5 | HCT116 | [75] | |
| Brain tumor | 4.5 μM (IC50) | Destabilization of REST-mSin3 interaction | DAOY (medulloblastoma) | [59] |
| 13 μM (IC50) | COX4-1 inhibition | TMZ-resistant U251 (glioblastoma) | [82] | |
| 20–40 μM | Upregulation of the JNK(ERK1/2)/Elk-1/Egr-1/p21 pathway | C6 (glioma) | [86] | |
| >20 μM | Inhibition of Akt/mTOR pathway | PTEN and U-87MG glioma cells | [96] | |
| ≥6 μM | Inhibition of KSP/Eg5 | T98G (glioblastoma) | [76] | |
| Not known | Not known | RG2 (rat glioblastoma) | [151] | |
| Skin cancer | 36.6 μM (IC50) | Not known | B16 mouse melanoma | [149, 150] |
| 12–19 mg/kg/day (drug dose used in mouse model) | Not known | Harding-Passey Melanoma mouse melanoma | [150] | |
| Leukemia | 12.04 μmol/l (IC50) | Inhibition of mitochondrial DNA polymerase and decreased ATP production | K-562 (chronic myelogenous leukemia) | [43] |
| 11.19 μmol/l (IC50) | Same as above | BALL-1 (B-acute lymphoblastic leukemia) | [43] | |
| 6.57 μmol/l (IC50) | Same as above | MOLT-4 (T-acute lymphoblastic leukemia) | [43] | |
| 11.81 μmol/l (IC50) | Same as above | CCRF-HSB-2 (T-acute lymphoblastic leukemia) | [43] | |
| 12.33 μmol/l (IC50) | Same as above | HPB-ALL (T-acute lymphoblastic leukemia) | [43] | |
| 6.940 μM (IC50) | Suppressed mutant RTK activity via CALM inhibition | Ba/F3/FLT3 ITD (modified murine pro-B cell line) | [147] | |
| 6.942 μM (IC50) | Same as above | Ba/F3/ KIT D814V (modified mouse pro-B cell line) | [147] | |
| Lymphoma | 6.95 μmol/l (IC50) | Inhibition of mitochondrial DNA polymerase and decreased ATP production | Raji (Burkitt’s lymphoma) | [43] |
| 14.89 μmol/l (IC50) | Same as above | Daudi (Burkitt’s lymphoma) | [43] | |
| Lung cancer | 10 μM | lysosomal dysfunction | H69, H82, H592 and U-1285 (small cell lung carcinoma cell lines) | [106] |
| Sarcoma | 25 μM (IC50) | Inhibition of DNA synthesis | Meth A cells | [42] |
| 10–15 μM (IC50) | Suppressed IGF1R internalization | A673, A4573, TC71 cells (ES cells) | [92] | |
| Mastocytoma | ~ 7 μM (IC50) | DNA fragmentation | PY815 (mouse mast cells) | [44] |
| Pancreatic cancer | 25 μM | Dislodging K-Ras from plasma membrane | PANC-1 (pancreatic carcinoma) | [114] |
| Lung cancer | CPZ + pentamidine | Inhibition of KSP/Eg5 | A549 (NSCLC) | [75] |
| Oral cancer | 26.65 ± 1.1 μM (IC50) | Inhibition of Akt and mTOR phosphorylation | HSC-3 | [45] |
| 23.49 ± 1.26 μM (IC50) | Same as above | Ca9-22 | [45] |
CPZ has been shown to exert antitumorigenic activity in several brain tumors. Medulloblastoma is the most common pediatric brain tumor frequently associated with overexpression of transcriptional repressor REST protein [55, 58]. CPZ inhibited growth of DAOY medulloblastoma cell spheroids, possibly by preventing interaction between REST and mSin3 necessary for REST mediated transcription suppression [59]. Another brain tumor with reported antitumorigenic effect of CPZ is GBM, one of the most lethal malignant brain tumors. A common chemotherapy for GBM involves treatment with temozolomide (TMZ). However, development of chemoresistance and CSC-promoted tumor recurrence present a substantial barrier for treatment of GBM. CPZ exhibited a potent cytotoxic effect on TMZ-resistant U251 human GBM cells with an IC50 of 13 μM, and also inhibited the cell neurosphere formation and anchorage-independent growth in soft agar [82]. This antitumorigenic effect of CPZ was specific to the TMZ-resistant GBM cells, as little growth inhibition was observed for TMZ sensitive U251 cells. The specificity was attributed to preferential binding of CPZ to the COX4-1 subunit of CcO enriched in the TMZ-resistant U251 cells, as discussed above. Notably, GBM has also been linked to the dysregulation of REST protein [56, 57]. Though it has not been examined, CPZ-induced suppression of REST signaling may be acting in parallel to the CcO inhibition shown to be responsible for the antitumorigenic effect of CPZ in GBM [82]. CPZ also inhibited growth of C6 glioma cells by upregulation of the JNK and ERK1/2 kinase phosphorylation [86].
CPZ suppresses proliferation and induces apoptosis in cultured lymphoma and leukemia cells via DNA fragmentation without affecting the viability of normal lymphocytes [43]. Inhibition of DNA polymerase and ATP production by CPZ was proposed as the most likely mechanism for this effect. CPZ also suppresses the growth of AML cells by inhibiting CALM protein involved in the formation of clathrin-coated vesicles and causing dislocation of the mutant RTKS, without disturbing wild-type RTKs [147]. Interestingly, suppression of clathrin-mediated endocytosis has been implicated in the antitumorigenic effect of CPZ in Ewing sarcoma (ES), a childhood cancer that occurs in bones or surrounding soft tissue, where CPZ was shown to disrupt clathrin-dependent internalization of the insulin-like growth factor 1 receptor (IGF1R), inhibiting AKT and MAPK phosphorylation and reducing ES cell proliferation [92]. Therefore, inhibition of clathrin-dependent signaling might be one of the conserved mechanisms of CPZ’s antitumorigenic effects.
CPZ decreased proliferation of oral cancer cells while showing low toxicity in normal oral cell lines [45]. This effect was attributed to the decreased phosphorylation of Akt and mTOR, as was the case in glioma cells [96]. CPZ inhibited proliferation of human colon carcinoma cells via inhibition of KSP/Eg5 mitotic kinesin [75], which was the same mechanisms responsible for the inhibition of MCF-7mp53 and MDA-MB-231 human breast cancer, and T98G glioblastoma cell lines [76]. Additionally, CPZ inhibited mouse mastocytoma cell growth by inducing DNA breakage [44], Meth A cell sarcoma growth via DNA synthesis inhibition [42], lung cancer growth by inducing lysosomal dysfunction [106], and growth of skin cancer in B16 mouse melanoma model [149, 150].
In summary, the antitumorigenic effect of CPZ in different cancers frequently involves common molecular/cellular targets that are known to be involved in cancer formation. At the same time, the CPZ’s diverse cellular targets and mechanisms uniquely confer the drug a multiscale antitumorigenic therapeutic potential which has not been fully harnessed yet.
EFFECT OF CPZ IN ANIMAL CANCER MODELS
Studies in animal models overwhelmingly recapitulate the antitumorigenic effect of CPZ that emerged from in vitro studies. One of the earliest studies of the effect of CPZ in vivo was conducted by Van Woert and Palmer in 1969 [150]. CPZ was injected intraperitoneally into mice transplanted with B-16 and Harding-Passey mouse melanomas. Examination of the tumors excised 18 days post-transplantation revealed about three-fold decreases in the weight of the tumors treated with CPZ in comparison with the CPZ-untreated controls. Shortly after this study, the anti-tumor effect of CPZ was tested in hamsters by Levij and Polliack in 1970 [152]. Tumor growth was induced by painting (topically applying) different chemical carcinogens to cheek pouches of hamsters in the absence and presence of CPZ. Histological examination of tissue cross-sections 9 and 12 weeks after the treatment indicated that animals painted with both carcinogens and CPZ showed drastically decreased levels of tumorigenesis compared to animals exposed only to carcinogens, including almost complete inhibition of carcinoma formation. These early studies on animal models yielded promising results and prompted many follow-up in vivo studies aimed to elucidate CPZ’s anticancer potential and mechanism.
Multiple studies further utilized mice xenograft models to investigate CPZ’s therapeutic potential for different cancer types, including sarcoma [42], colorectal cancer [93], glioma [82], leukemia [147], and oral cancer [45]. These studies showed that CPZ treatment in mice bearing various tumors significantly increased animal survival time [82, 147], quelled tumor growth [45, 93], and caused tumor regression [42]. Importantly, multiple studies reported no significant difference in mice weight between CPZ-treated and control groups, suggesting that CPZ treatment has no adverse effects on body weight [45, 82, 93]. Interestingly, Aas and coworkers found that in rats transplanted with RG2 neuroblastoma cells, CPZ enhanced effect of the anti-neoplastic drug, 1,3-bis(2-chloroethyl-l)-nitrosourea (BCNU), on inhibiting the transplanted tumor growth [151]. In addition, Lee and coworkers found that CPZ enhanced the anticancer effect of pentamidine, an antiparasitic agent, in mice transplanted with A549 lung cancer cells [75]. The synergistic effect of CPZ was also observed in several in vitro studies, as discussed above. Therefore, future studies should further explore CPZ’s anticancer potential as a stand-alone medication, as well as in combination with existing therapies.
In addition to the rodent xenograft models, CPZ’s antitumorigenic effect has also been demonstrated in zebrafish. Earlier this year, Jhou and coworkers found that CPZ treatment decreased tumor growth in zebrafish larvae grafted with oral cancer tumors [45]. Notably, CPZ treatment had no observable effect on larval development. Consistent with this report, an earlier study also reported no effect of CPZ on zebrafish development at clinically relevant levels [93]. Taken together, the studies in animal models strongly support antitumorigenic potential of CPZ demonstrated by in vitro studies. Furthermore, animal xenograft studies suggest that repurposing CPZ for cancer treatment as a stand-alone medication or in combination with other cancer drugs should be well tolerated and have low adverse effects.
POPULATION STUDIES OF THE EFFECT OF ANTIPSYCHOTIC MEDICATION ON CANCER RISK
Population studies on the cancer rate in psychiatric patients administered antipsychotics/antidepressants, including CPZ, are somewhat limited and yielded contradictory results. Additionally, most of the studies do not specify which antipsychotic medication was administered to the patients. In a systemic literature review conducted in 2012, Fond and coworkers concluded that overall the patients with schizophrenia may be less likely to develop cancer than the general population [153]. However, individual studies are controversial and frequently inconclusive. Moreover, the reported differences in the cancer rates between psychiatric patients and normal population are frequently cancer type- and gender-specific.
One of the first relevant population studies was conducted in Denmark based on the analysis of 6168 patients with schizophrenia followed from 1957 to 1984 [154]. The study found an overall decrease in the incidence of cancer in the patients, with especially marked decrease in the rate of respiratory system cancers, prostate cancer in male patients and uterine cancer in female patients. Another Danish study of 25,264 users of unspecified antipsychotic drugs monitored between 1989 and 2002, found decreased risk of rectal cancer in both female and male subjects, decreased risk in colon cancer in females and reduced risk of prostate cancer in male subjects [155]. In agreement with the protective effect of antipsychotics on colorectal cancer in Danish population, a study conducted in Taiwan in 2019, based on the analysis of 34,470 gastric cancer patients and 163,430 non-gastric cancer controls, also found a substantial decrease in the rate of gastric cancer in patients prescribed antipsychotic medication, including CPZ [156]. Another comprehensive study based on the analysis of 31,953 cancer cases in United Kingdom (UK), found a statistically significant dose- and time-dependent decrease in the incidences of colorectal cancer and glioma in the users of tricyclic antidepressants [157]. Unlike the above mentioned studies, a study of 26,996 patents with schizophrenia conducted from 1969 and 1991 in Finland found a slight increase in the overall cancer rate in the patients in comparison with the general population, especially marked increase in lung cancer in male patients [158]. However, the study also found a decrease in the incidence of rectal cancer in female patients, and interestingly, a substantial decrease in cancer incidents among non-schizophrenic siblings and parents of the patients. Similar to the other studies, the primary medication administered to the schizophrenic patients was not indicated.
Perhaps the most controversial is the effect of antidepressants/antipsychotics on the rate of breast cancer. A study of 29,641 female schizophrenia patients receiving antidepressants and 59,282 non-users selected based on Taiwan Insurance Claims Data from 1998 to 2008 found that the risk of breast cancer was almost 2-fold higher in the users of antidepressants [159]. This is by far the strongest report of adverse effects of antidepressants and is in agreement with an earlier study in Danish population that found a slight increase in the incidence of breast cancer in female patients [154]. However, several other studies report no dependence in the breast cancer rate and the use of antidepressant/antipsychotic drugs. For instance, a recently conducted study in Denmark on 4,951 antipsychotic users and 47,643 non-users found no clinically important association between the antipsychotic drug use and risk of breast cancer [160]. Two other earlier studies conducted in Denmark and UK, already mentioned in this section, also found that the rate of breast cancer is unaffected by the use of antidepressants [155, 157]. The cohort size for UK study is comparable to the cohort size of the Taiwanese study, yet they reached different conclusions, and the reasons for these differences are not clear. However, it should be noted that the interpretation of population studies on the effect of antipsychotic medications on cancer incidence is very sensitive to the study design and could be affected by a variety of factors, including genetic differences between populations, environmental influence and differences in lifestyle [161]. Therefore, the aforementioned studies and conclusions must be interpreted with caution. Studies on larger populations, based on robust models taking into account the multitude of cofactors and distinctions in the various antipsychotic drugs are necessary to dissect the link between cancer rates and the use of antipsychotics.
CLINICAL POTENTIAL OF CPZ FOR CANCER TREATMENT
As reviewed above, CPZ has a potent anti-proliferative effect on many cancer types achieved by affecting a variety of molecular targets and cellular pathways. CPZ also has an additional benefit due to the documented sedative effect that can reduce anxiety, insomnia and other symptoms related to the mental state of cancer patients [162]. Therefore, there is a strong rationale for repurposing CPZ for treatment of cancer. Development of new cancer drugs can take many years, and clinical testing can be halted at any stage in the event of unexpected toxicity and safety concerns, making a new drug development a risky and costly endeavor. As an FDA-approved drug that has been used for many decades to treat psychiatric patients, CPZ could be repurposed for the treatment of tumors where the molecular mechanisms of CPZ’s action are well understood, and preclinical and clinical studies support benefits of its administration.
Despite the potential for cancer treatment there are also known side-effects of CPZ that must be considered. CPZ can cause hypertension, bradycardia and ventricular arrhythmias [163]. Therefore, patients have to be monitored for the potential cardiac side-effects. Additionally, CPZ can cause weight gain [164], sexual dysfunction [165], movement disorders, including dystonia and tremors [166], and increase prolactin levels in females, leading to menstrual irregularities [167]. However, many of these symptoms are reversible once the treatment is discontinued [164–166]. The severity of the side-effects depends on the dose and the duration of the treatment. Therefore, fine-tuning CPZ’s dose and duration for optimal treatment of cancer patients, exploring combination therapy with CPZ and other existing therapies and cancer type-specific effects of CPZ should be the focus of future clinical studies. While this review is focused on CPZ, the founding member of the phenothiazines family of drugs with arguably the most studied effects in cancer, many other phenothiazines have been shown to possess anticancer properties as well [43, 168]. Consideration of other phenothiazines that may have a more favorable balance between their antitumorigenic activity and potential side-effects is also a valuable strategy for drug repurposing. Notably, a Phase II clinical trial involving a combination of CPZ and temozolomide for glioblastoma treatment was recently initiated [169]. This and other future clinical studies will shed light on the therapeutic potential of CPZ for cancer treatment.
Conclusions
Unequivocally, CPZ is a potent antitumorigenic agent acting through a diverse range of molecular targets and cellular pathways. The rationale for repurposing CPZ for cancer treatment is strong. However, further studies on the molecular and cellular mechanisms of CPZ’s action, and their interdependence are necessary to attain a comprehensive understanding of CPZ’s action in cancer. More refined population studies, specifically focused on the effect of CPZ on cancer patients will also shed light on the therapeutic potential of this drug for cancer therapy. Finally, clinical studies involving dose- and treatment duration-dependence of the CPZ’s effect in cancer are necessary to evaluate a therapeutic potential of repurposing CPZ for cancer treatment.
ACKNOWLEDGMENTS
We thank Drs. Eric Glasgow and Aykut Uren, and Stephanie Soohoo for comments on the manuscript. This work was supported by the NCI, National Institutes of Health Grant R01CA252969 (to T. I. B.).
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Laborit H, Huguenard P, Alluaume R. [A new vegetative stabilizer; 4560 R.P.]. Presse Med. 1952; 60:206–08. [PubMed].
2. Hamon P, Paraire, Velluz. [Effect of R. P. 4560 on maniacal agitation]. Ann Med Psychol (Paris). 1952; 110:331–35. [PubMed].
3. Rosenbloom M. Chlorpromazine and the psychopharmacologic revolution. JAMA. 2002; 287:1860–61. [PubMed].
4. López-Muñoz F, Alamo C, Cuenca E, Shen WW, Clervoy P, Rubio G. History of the discovery and clinical introduction of chlorpromazine. Ann Clin Psychiatry. 2005; 17:113–35. https://doi.org/10.1080/10401230591002002. [PubMed].
5. Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, Samara M, Barbui C, Engel RR, Geddes JR, Kissling W, Stapf MP, Lässig B, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013; 382:951–62. https://doi.org/10.1016/S0140-6736(13)60733-3. [PubMed].
6. Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology. 2008; 33:2061–79. https://doi.org/10.1038/sj.npp.1301639. [PubMed].
7. Mortensen PB. Neuroleptic treatment and other factors modifying cancer risk in schizophrenic patients. Acta Psychiatr Scand. 1987; 75:585–90. https://doi.org/10.1111/j.1600-0447.1987.tb02839.x. [PubMed].
8. Csatary LK. Chlorpromazines and cancer. Lancet. 1972; 2:338–39. https://doi.org/10.1016/s0140-6736(72)92955-8. [PubMed].
9. Belkin M, Hardy WG. Effect of reserpine and chlorpromazine on sarcoma 37. Science. 1957; 125:233–34. https://doi.org/10.1126/science.125.3241.233. [PubMed].
10. Kanzawa F, Hoshi A, Kuretani K. Relationship between antitumor activity and chemical structure in psychotropic agents. Gan. 1970; 61:529–34. [PubMed].
11. Hoshi A, Kanzawa F, Kuretani K. Antitumor activity of psychotropic drugs and their synergic action with cyclophosphamide. Chem Pharm Bull (Tokyo). 1969; 17:848–50. https://doi.org/10.1248/cpb.17.848. [PubMed].
12. Charpentier P, Gailliot P, Jacob R, Gaudechon J, Buisson P. Recherches sur les diméthylaminopropyl-N phénothiazines substituées. Comptes Rendus Hebdomadaires Des Seances De L Academie Des Sciences (Paris). 1952; 235:59–60.
13. Ban TA. Fifty years chlorpromazine: a historical perspective. Neuropsychiatr Dis Treat. 2007; 3:495–500. [PubMed].
14. Ayd FJ Jr. Chlorpromazine: ten years' experience. JAMA. 1963; 184:51–54. https://doi.org/10.1001/jama.1963.73700140004017. [PubMed].
15. Gyermek L, Lazar I, Csak AZ. The antiserotonin action of chlorpromazine and some other phenothiazine derivatives. Arch Int Pharmacodyn Ther. 1956; 107:62–74. [PubMed].
16. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on formation of 3methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh). 1963; 20:140–44. https://doi.org/10.1111/j.1600-0773.1963.tb01730.x. [PubMed].
17. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976; 192:481–83. https://doi.org/10.1126/science.3854. [PubMed].
18. Andén NE, Butcher SG, Corrodi H, Fuxe K, Ungerstedt U. Receptor activity and turnover of dopamine and noradrenaline after neuroleptics. Eur J Pharmacol. 1970; 11:303–14. https://doi.org/10.1016/0014-2999(70)90006-3. [PubMed].
19. Hals PA, Hall H, Dahl SG. Phenothiazine drug metabolites: dopamine D2 receptor, alpha 1- and alpha 2-adrenoceptor binding. Eur J Pharmacol. 1986; 125:373–81. https://doi.org/10.1016/0014-2999(86)90793-4. [PubMed].
20. Abbas AI, Hedlund PB, Huang XP, Tran TB, Meltzer HY, Roth BL. Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo. Psychopharmacology (Berl). 2009; 205:119–28. https://doi.org/10.1007/s00213-009-1521-8. [PubMed].
21. Crow TJ, Deakin JF, Longden A. The nucleus accumbens--possible site of antipsychotic action of neuroleptic drugs? Psychol Med. 1977; 7:213–21. https://doi.org/10.1017/s0033291700029287. [PubMed].
22. Bartholini G. Differential effect of neuroleptic drugs on dopamine turnover in the extrapyramidal and limbic system. J Pharm Pharmacol. 1976; 28:429–33. https://doi.org/10.1111/j.2042-7158.1976.tb04648.x. [PubMed].
23. Boyd KN, Mailman RB. Dopamine receptor signaling and current and future antipsychotic drugs. Handb Exp Pharmacol. 2012; 212:53–86. https://doi.org/10.1007/978-3-642-25761-2_3. [PubMed].
24. Jolliet P, Nion S, Allain-Veyrac G, Tilloy-Fenart L, Vanuxeem D, Berezowski V, Cecchelli R. Evidence of lowest brain penetration of an antiemetic drug, metopimazine, compared to domperidone, metoclopramide and chlorpromazine, using an in vitro model of the blood-brain barrier. Pharmacol Res. 2007; 56:11–17. https://doi.org/10.1016/j.phrs.2006.12.004. [PubMed].
25. Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002; 303:1029–37. https://doi.org/10.1124/jpet.102.039255. [PubMed].
26. Dollery C, Boobis AR. Therapeutic Drugs. Edinburgh; London: Churchill Livingstone, 1991.
27. Rey MJ, Schulz P, Costa C, Dick P, Tissot R. Guidelines for the dosage of neuroleptics. I: Chlorpromazine equivalents of orally administered neuroleptics. Int Clin Psychopharmacol. 1989; 4:95–104. https://doi.org/10.1097/00004850-198904000-00001. [PubMed].
28. Curry SH, Marshall JH, Davis JM, Janowsky DS. Chlorpromazine plasma levels and effects. Arch Gen Psychiatry. 1970; 22:289–96. https://doi.org/10.1001/archpsyc.1970.01740280001001. [PubMed].
29. Cooper TB, Simpson GM, Lee JH. Thymoleptic and neuroleptic drug plasma levels in psychiatry: current status. Int Rev Neurobiol. 1976; 19:269–309. https://doi.org/10.1016/s0074-7742(08)60706-0. [PubMed].
30. Baldessarini RJ, Centorrino F, Flood JG, Volpicelli SA, Huston-Lyons D, Cohen BM. Tissue concentrations of clozapine and its metabolites in the rat. Neuropsychopharmacology. 1993; 9:117–24. [PubMed].
31. Kornhuber J, Schultz A, Wiltfang J, Meineke I, Gleiter CH, Zöchling R, Boissl KW, Leblhuber F, Riederer P. Persistence of haloperidol in human brain tissue. Am J Psychiatry. 1999; 156:885–90. https://doi.org/10.1176/ajp.156.6.885. [PubMed].
32. Seeman P. Anti-schizophrenic drugs--membrane receptor sites of action. Biochem Pharmacol. 1977; 26:1741–48. https://doi.org/10.1016/0006-2952(77)90340-9. [PubMed].
33. Pisciotta AV, Ziebert AP, Hinz JE. Effect of chlorpromazine on DNA synthesis in a cell free system. Proc Soc Exp Biol Med. 1965; 120:305–08. https://doi.org/10.3181/00379727-120-30518. [PubMed].
34. Abood LG. Effect of chlorpromazine on phosphorylation of brain mitochondria. Proc Soc Exp Biol Med. 1955; 88:688–90. https://doi.org/10.3181/00379727-88-21695. [PubMed].
35. Dawkins MJ, Judah JD, Rees KR. The effect of chlorpromazine on the respiratory chain; cytochrome oxidase. Biochem J. 1959; 72:204–09. https://doi.org/10.1042/bj0720204. [PubMed].
36. Dawkins MJ, Judah JD, Rees KR. Action of chlorpromazine. 3. Mitochondrial adenosine triphosphatase and the adenosine triphosphate-adenosine diphosphate exchange. Biochem J. 1960; 76:200–05. https://doi.org/10.1042/bj0760200. [PubMed].
37. Spirtes MA, Guth PS. Effects of chlorpromazine on biological membranes--I. Chlorpromazine-induced changes in liver mitochondria. Biochem Pharmacol. 1963; 12:37–46. https://doi.org/10.1016/0006-2952(63)90007-8. [PubMed].
38. Guth PS, Amaro J, Sellinger OZ, Elmer L. Studies in vitro and in vivo of the effects of chlorpromazine on rat liver lysosomes. Biochem Pharmacol. 1965; 14:769–75. https://doi.org/10.1016/0006-2952(65)90095-x. [PubMed].
39. Huelsmann WC, Fabius AJ, De Ruiter H. Inhibition of lipase activity of lung and adipose tissue by phenothiazine derivatives. Nature. 1964; 202:1336–37. https://doi.org/10.1038/2021336a0. [PubMed].
40. Pisciotta AV, Kaldahl J. Studies on agranulocytosis. IV. Effects of chlorpromazine on nucleic acid synthesis of bone marrow cells in vitro. Blood. 1962; 20:364–76. [PubMed].
41. Pisciotta AV. Studies on agranulocytosis. VII. Limited proliferative potential of cpz-sensitive patients. J Lab Clin Med. 1965; 65:240–47. [PubMed].
42. Mizushima T, Natori S, Sekimizu K. Inhibition of DNA synthesis in Meth A cells by chlorpromazine. Biol Pharm Bull. 1993; 16:953–55. https://doi.org/10.1248/bpb.16.953. [PubMed].
43. Zhelev Z, Ohba H, Bakalova R, Hadjimitova V, Ishikawa M, Shinohara Y, Baba Y. Phenothiazines suppress proliferation and induce apoptosis in cultured leukemic cells without any influence on the viability of normal lymphocytes. Phenothiazines and leukemia. Cancer Chemother Pharmacol. 2004; 53:267–75. https://doi.org/10.1007/s00280-003-0738-1. [PubMed].
44. Darkin S, McQuillan J, Ralph RK. Chlorpromazine: a potential anticancer agent? Biochem Biophys Res Commun. 1984; 125:184–91. https://doi.org/10.1016/s0006-291x(84)80352-6. [PubMed].
45. Jhou AJ, Chang HC, Hung CC, Lin HC, Lee YC, Liu WT, Han KF, Lai YW, Lin MY, Lee CH. Chlorpromazine, an antipsychotic agent, induces G2/M phase arrest and apoptosis via regulation of the PI3K/AKT/mTOR-mediated autophagy pathways in human oral cancer. Biochem Pharmacol. 2021; 184:114403. https://doi.org/10.1016/j.bcp.2020.114403. [PubMed].
46. Hirai H, Takeda S, Natori S, Sekimizu K. Inhibition of SV40 DNA replication in vitro by chlorpromazine. Biol Pharm Bull. 1993; 16:565–67. https://doi.org/10.1248/bpb.16.565. [PubMed].
47. Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995; 267:1360–63. https://doi.org/10.1126/science.7871435. [PubMed].
48. Rodenas-Ruano A, Chávez AE, Cossio MJ, Castillo PE, Zukin RS. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci. 2012; 15:1382–90. https://doi.org/10.1038/nn.3214. [PubMed].
49. Gao Z, Ure K, Ding P, Nashaat M, Yuan L, Ma J, Hammer RE, Hsieh J. The master negative regulator REST/NRSF controls adult neurogenesis by restraining the neurogenic program in quiescent stem cells. J Neurosci. 2011; 31:9772–86. https://doi.org/10.1523/JNEUROSCI.1604-11.2011. [PubMed].
50. Naruse Y, Aoki T, Kojima T, Mori N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc Natl Acad Sci U S A. 1999; 96:13691–96. https://doi.org/10.1073/pnas.96.24.13691. [PubMed].
51. Andrés ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci U S A. 1999; 96:9873–78. https://doi.org/10.1073/pnas.96.17.9873. [PubMed].
52. Yu HB, Johnson R, Kunarso G, Stanton LW. Coassembly of REST and its cofactors at sites of gene repression in embryonic stem cells. Genome Res. 2011; 21:1284–93. https://doi.org/10.1101/gr.114488.110. [PubMed].
53. Nomura M, Uda-Tochio H, Murai K, Mori N, Nishimura Y. The neural repressor NRSF/REST binds the PAH1 domain of the Sin3 corepressor by using its distinct short hydrophobic helix. J Mol Biol. 2005; 354:903–15. https://doi.org/10.1016/j.jmb.2005.10.008. [PubMed].
54. Wang H, Clark I, Nicholson PR, Herskowitz I, Stillman DJ. The Saccharomyces cerevisiae SIN3 gene, a negative regulator of HO, contains four paired amphipathic helix motifs. Mol Cell Biol. 1990; 10:5927–36. https://doi.org/10.1128/mcb.10.11.5927-5936.1990. [PubMed].
55. Fuller GN, Su X, Price RE, Cohen ZR, Lang FF, Sawaya R, Majumder S. Many human medulloblastoma tumors overexpress repressor element-1 silencing transcription (REST)/neuron-restrictive silencer factor, which can be functionally countered by REST-VP16. Mol Cancer Ther. 2005; 4:343–49. https://doi.org/10.1158/1535-7163.MCT-04-0228. [PubMed].
56. Conti L, Crisafulli L, Caldera V, Tortoreto M, Brilli E, Conforti P, Zunino F, Magrassi L, Schiffer D, Cattaneo E. REST controls self-renewal and tumorigenic competence of human glioblastoma cells. PLoS One. 2012; 7:e38486. https://doi.org/10.1371/journal.pone.0038486. [PubMed].
57. Kamal MM, Sathyan P, Singh SK, Zinn PO, Marisetty AL, Liang S, Gumin J, El-Mesallamy HO, Suki D, Colman H, Fuller GN, Lang FF, Majumder S. REST regulates oncogenic properties of glioblastoma stem cells. Stem Cells. 2012; 30:405–14. https://doi.org/10.1002/stem.1020. [PubMed].
58. Lawinger P, Venugopal R, Guo ZS, Immaneni A, Sengupta D, Lu W, Rastelli L, Marin Dias Carneiro A, Levin V, Fuller GN, Echelard Y, Majumder S. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells. Nat Med. 2000; 6:826–31. https://doi.org/10.1038/77565. [PubMed].
59. Kurita JI, Hirao Y, Nakano H, Fukunishi Y, Nishimura Y. Sertraline, chlorprothixene, and chlorpromazine characteristically interact with the REST-binding site of the corepressor mSin3, showing medulloblastoma cell growth inhibitory activities. Sci Rep. 2018; 8:13763. https://doi.org/10.1038/s41598-018-31852-1. [PubMed].
60. Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014; 15:642–56. https://doi.org/10.15252/embr.201438638. [PubMed].
61. Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol. 2019; 7:49. https://doi.org/10.3389/fcell.2019.00049. [PubMed].
62. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008; 22:1962–71. https://doi.org/10.1101/gad.1664408. [PubMed].
63. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006; 125:1253–67. https://doi.org/10.1016/j.cell.2006.05.030. [PubMed].
64. Chang SS, Yamaguchi H, Xia W, Lim SO, Khotskaya Y, Wu Y, Chang WC, Liu Q, Hung MC. Aurora A kinase activates YAP signaling in triple-negative breast cancer. Oncogene. 2017; 36:1265–75. https://doi.org/10.1038/onc.2016.292. [PubMed].
65. Kim T, Yang SJ, Hwang D, Song J, Kim M, Kyum Kim S, Kang K, Ahn J, Lee D, Kim MY, Kim S, Seung Koo J, Seok Koh S, et al. A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun. 2015; 6:10186. https://doi.org/10.1038/ncomms10186. [PubMed].
66. Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, Ishimoto T, Kuroki H, Nitta H, Hashimoto D, Chikamoto A, Oki E, Beppu T, Baba H. An Imbalance in TAZ and YAP Expression in Hepatocellular Carcinoma Confers Cancer Stem Cell-like Behaviors Contributing to Disease Progression. Cancer Res. 2015; 75:4985–97. https://doi.org/10.1158/0008-5472.CAN-15-0291. [PubMed].
67. Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW, Heallen TR, Xiao L, Hofstetter WL, Weston B, Lee JH, Wadhwa R, Sudo K, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014; 74:4170–82. https://doi.org/10.1158/0008-5472.CAN-13-3569. [PubMed].
68. Fisher ML, Grun D, Adhikary G, Xu W, Eckert RL. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget. 2017; 8:110257–72. https://doi.org/10.18632/oncotarget.22628. [PubMed].
69. Li YW, Xu J, Zhu GY, Huang ZJ, Lu Y, Li XQ, Wang N, Zhang FX. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018; 4:105. https://doi.org/10.1038/s41420-018-0124-8. [PubMed].
70. Yang CE, Lee WY, Cheng HW, Chung CH, Mi FL, Lin CW. The antipsychotic chlorpromazine suppresses YAP signaling, stemness properties, and drug resistance in breast cancer cells. Chem Biol Interact. 2019; 302:28–35. https://doi.org/10.1016/j.cbi.2019.01.033. [PubMed].
71. Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat Rev Cancer. 2007; 7:107–17. https://doi.org/10.1038/nrc2049. [PubMed].
72. Wordeman L. How kinesin motor proteins drive mitotic spindle function: Lessons from molecular assays. Semin Cell Dev Biol. 2010; 21:260–68. https://doi.org/10.1016/j.semcdb.2010.01.018. [PubMed].
73. Bergnes G, Brejc K, Belmont L. Mitotic kinesins: prospects for antimitotic drug discovery. Curr Top Med Chem. 2005; 5:127–45. https://doi.org/10.2174/1568026053507697. [PubMed].
74. Knight SD, Parrish CA. Recent progress in the identification and clinical evaluation of inhibitors of the mitotic kinesin KSP. Curr Top Med Chem. 2008; 8:888–904. https://doi.org/10.2174/156802608784911626. [PubMed].
75. Lee MS, Johansen L, Zhang Y, Wilson A, Keegan M, Avery W, Elliott P, Borisy AA, Keith CT. The novel combination of chlorpromazine and pentamidine exerts synergistic antiproliferative effects through dual mitotic action. Cancer Res. 2007; 67:11359–67. https://doi.org/10.1158/0008-5472.CAN-07-2235. [PubMed].
76. Riffell JL, Zimmerman C, Khong A, McHardy LM, Roberge M. Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle. 2009; 8:3025–38. [PubMed].
77. Kadenbach B, Hüttemann M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion. 2015; 24:64–76. https://doi.org/10.1016/j.mito.2015.07.002. [PubMed].
78. Heerdt BG, Halsey HK, Lipkin M, Augenlicht LH. Expression of mitochondrial cytochrome c oxidase in human colonic cell differentiation, transformation, and risk for colonic cancer. Cancer Res. 1990; 50:1596–600. [PubMed].
79. Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, Griguer CE. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem. 2010; 285:39759–67. https://doi.org/10.1074/jbc.M110.147504. [PubMed].
80. Liang L, Qu L, Ding Y. Protein and mRNA characterization in human colorectal carcinoma cell lines with different metastatic potentials. Cancer Invest. 2007; 25:427–34. https://doi.org/10.1080/07357900701512258. [PubMed].
81. Chen WL, Kuo KT, Chou TY, Chen CL, Wang CH, Wei YH, Wang LS. The role of cytochrome c oxidase subunit Va in non-small cell lung carcinoma cells: association with migration, invasion and prediction of distant metastasis. BMC Cancer. 2012; 12:273. https://doi.org/10.1186/1471-2407-12-273. [PubMed].
82. Oliva CR, Zhang W, Langford C, Suto MJ, Griguer CE. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget. 2017; 8:37568–83. https://doi.org/10.18632/oncotarget.17247. [PubMed].
83. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011; 75:50–83. https://doi.org/10.1128/MMBR.00031-10. [PubMed].
84. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002; 12:9–18. https://doi.org/10.1038/sj.cr.7290105. [PubMed].
85. Qi L, Ding Y. Potential antitumor mechanisms of phenothiazine drugs. Sci China Life Sci. 2013; 56:1020–27. https://doi.org/10.1007/s11427-013-4561-6. [PubMed].
86. Shin SY, Kim CG, Kim SH, Kim YS, Lim Y, Lee YH. Chlorpromazine activates p21Waf1/Cip1 gene transcription via early growth response-1 (Egr-1) in C6 glioma cells. Exp Mol Med. 2010; 42:395–405. https://doi.org/10.3858/emm.2010.42.5.041. [PubMed].
87. Choi BH, Kim CG, Bae YS, Lim Y, Lee YH, Shin SY. p21 Waf1/Cip1 expression by curcumin in U-87MG human glioma cells: role of early growth response-1 expression. Cancer Res. 2008; 68:1369–77. https://doi.org/10.1158/0008-5472.CAN-07-5222. [PubMed].
88. Chan TA, Hwang PM, Hermeking H, Kinzler KW, Vogelstein B. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev. 2000; 14:1584–88. [PubMed].
89. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995; 82:675–84. https://doi.org/10.1016/0092-8674(95)90039-x. [PubMed].
90. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995; 377:552–57. https://doi.org/10.1038/377552a0. [PubMed].
91. Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001; 61:6234–38. [PubMed].
92. Martins AS, Ordóñez JL, Amaral AT, Prins F, Floris G, Debiec-Rychter M, Hogendoorn PC, de Alava E. IGF1R signaling in Ewing sarcoma is shaped by clathrin-/caveolin-dependent endocytosis. PLoS One. 2011; 6:e19846. https://doi.org/10.1371/journal.pone.0019846. [PubMed].
93. Lee WY, Lee WT, Cheng CH, Chen KC, Chou CM, Chung CH, Sun MS, Cheng HW, Ho MN, Lin CW. Repositioning antipsychotic chlorpromazine for treating colorectal cancer by inhibiting sirtuin 1. Oncotarget. 2015; 6:27580–95. https://doi.org/10.18632/oncotarget.4768. [PubMed].
94. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005; 24:7455–64. https://doi.org/10.1038/sj.onc.1209085. [PubMed].
95. Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014; 26:2694–701. https://doi.org/10.1016/j.cellsig.2014.08.019. [PubMed].
96. Shin SY, Lee KS, Choi YK, Lim HJ, Lee HG, Lim Y, Lee YH. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis. 2013; 34:2080–89. https://doi.org/10.1093/carcin/bgt169. [PubMed].
97. Berchtold MW, Villalobo A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim Biophys Acta. 2014; 1843:398–435. https://doi.org/10.1016/j.bbamcr.2013.10.021. [PubMed].
98. Liu GX, Sheng HF, Wu S. A study on the levels of calmodulin and DNA in human lung cancer cells. Br J Cancer. 1996; 73:899–901. https://doi.org/10.1038/bjc.1996.160. [PubMed].
99. Ye Q, Wei Y, Fischer R, Borner C, Berchtold MW. Expression of calmodulin and calmodulin binding proteins in rat fibroblasts stably transfected with protein kinase C and oncogenes. Biochim Biophys Acta. 1997; 1359:89–96. https://doi.org/10.1016/s0167-4889(97)00086-4. [PubMed].
100. Ito H, Wang JZ, Shimura K. Inhibition of lung metastasis by a calmodulin antagonist, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W-7), in mice bearing Lewis lung carcinoma. Anticancer Res. 1991; 11:249–52. [PubMed].
101. Gulino A, Barrera G, Vacca A, Farina A, Ferretti C, Screpanti I, Dianzani MU, Frati L. Calmodulin antagonism and growth-inhibiting activity of triphenylethylene antiestrogens in MCF-7 human breast cancer cells. Cancer Res. 1986; 46:6274–78. [PubMed].
102. Shim JS, Lee J, Kim KN, Kwon HJ. Development of a new Ca2+/calmodulin antagonist and its anti-proliferative activity against colorectal cancer cells. Biochem Biophys Res Commun. 2007; 359:747–51. https://doi.org/10.1016/j.bbrc.2007.05.174. [PubMed].
103. Mac Neil S, Walker SW, Senior HJ, Bleehen SS, Tomlinson S. Effects of extracellular calmodulin and calmodulin antagonists on B16 melanoma cell growth. J Invest Dermatol. 1984; 83:15–19. https://doi.org/10.1111/1523-1747.ep12261637. [PubMed].
104. Weiss B, Prozialeck W, Cimino M, Barnette MS, Wallace TL. Pharmacological regulation of calmodulin. Ann N Y Acad Sci. 1980; 356:319–45. https://doi.org/10.1111/j.1749-6632.1980.tb29621.x. [PubMed].
105. Prozialeck WC, Weiss B. Inhibition of calmodulin by phenothiazines and related drugs: structure-activity relationships. J Pharmacol Exp Ther. 1982; 222:509–16. [PubMed].
106. Zong D, Zielinska-Chomej K, Juntti T, Mörk B, Lewensohn R, Hååg P, Viktorsson K. Harnessing the lysosome-dependent antitumor activity of phenothiazines in human small cell lung cancer. Cell Death Dis. 2014; 5:e1111. https://doi.org/10.1038/cddis.2014.56. [PubMed].
107. Freeman AR, Spirtes MA. Effects of chlorpromazine on biological membranes-II. Chlorpromazine-induced changes in human erythrocytes. Biochem Pharmacol. 1963; 12:47–53. https://doi.org/10.1016/0006-2952(63)90008-x. [PubMed].
108. Axelrod J, Whitby LG, Hertting G. Effect of psychotropic drugs on the uptake of H3-norepinephrine by tissues. Science. 1961; 133:383–84. https://doi.org/10.1126/science.133.3450.383. [PubMed].
109. Dengler HJ, Spiegel HE, Titus EO. Effects of drugs on uptake of isotopic norepinephrine by cat tissues. Nature. 1961; 191:816–17. https://doi.org/10.1038/191816a0. [PubMed].
110. Drori S, Eytan GD, Assaraf YG. Potentiation of anticancer-drug cytotoxicity by multidrug-resistance chemosensitizers involves alterations in membrane fluidity leading to increased membrane permeability. Eur J Biochem. 1995; 228:1020–29. https://doi.org/10.1111/j.1432-1033.1995.tb20352.x. [PubMed].
111. Maruoka N, Murata T, Omata N, Takashima Y, Tanii H, Yonekura Y, Fujibayashi Y, Wada Y. Effects of chlorpromazine on plasma membrane permeability and fluidity in the rat brain: a dynamic positron autoradiography and fluorescence polarization study. Prog Neuropsychopharmacol Biol Psychiatry. 2007; 31:178–86. https://doi.org/10.1016/j.pnpbp.2006.08.019. [PubMed].
112. Yde CW, Clausen MP, Bennetzen MV, Lykkesfeldt AE, Mouritsen OG, Guerra B. The antipsychotic drug chlorpromazine enhances the cytotoxic effect of tamoxifen in tamoxifen-sensitive and tamoxifen-resistant human breast cancer cells. Anticancer Drugs. 2009; 20:723–35. https://doi.org/10.1097/CAD.0b013e32832ec041. [PubMed].
113. Agasøsler AV, Tungodden LM, Cejka D, Bakstad E, Sydnes LK, Holmsen H. Chlorpromazine-induced increase in dipalmitoylphosphatidylserine surface area in monolayers at room temperature. Biochem Pharmacol. 2001; 61:817–25. https://doi.org/10.1016/s0006-2952(01)00542-1. [PubMed].
114. Eisenberg S, Giehl K, Henis YI, Ehrlich M. Differential interference of chlorpromazine with the membrane interactions of oncogenic K-Ras and its effects on cell growth. J Biol Chem. 2008; 283:27279–88. https://doi.org/10.1074/jbc.M804589200. [PubMed].
115. Lu S, Jang H, Gu S, Zhang J, Nussinov R. Drugging Ras GTPase: a comprehensive mechanistic and signaling structural view. Chem Soc Rev. 2016; 45:4929–52. https://doi.org/10.1039/c5cs00911a. [PubMed].
116. Suwalsky M, Villena F, Sotomayor CP, Bolognin S, Zatta P. Human cells and cell membrane molecular models are affected in vitro by chlorpromazine. Biophys Chem. 2008; 135:7–13. https://doi.org/10.1016/j.bpc.2008.02.014. [PubMed].
117. Gould RJ, Murphy KM, Reynolds IJ, Snyder SH. Antischizophrenic drugs of the diphenylbutylpiperidine type act as calcium channel antagonists. Proc Natl Acad Sci U S A. 1983; 80:5122–25. https://doi.org/10.1073/pnas.80.16.5122. [PubMed].
118. Flaim SF, Brannan MD, Swigart SC, Gleason MM, Muschek LD. Neuroleptic drugs attenuate calcium influx and tension development in rabbit thoracic aorta: effects of pimozide, penfluridol, chlorpromazine, and haloperidol. Proc Natl Acad Sci U S A. 1985; 82:1237–41. https://doi.org/10.1073/pnas.82.4.1237. [PubMed].
119. Ogata N, Yoshii M, Narahashi T. Psychotropic drugs block voltage-gated ion channels in neuroblastoma cells. Brain Res. 1989; 476:140–44. https://doi.org/10.1016/0006-8993(89)91546-1. [PubMed].
120. Kon K, Krause E, Gögelein H. Inhibition of K+ channels by chlorpromazine in rat ventricular myocytes. J Pharmacol Exp Ther. 1994; 271:632–37. [PubMed].
121. McCann JD, Welsh MJ. Neuroleptics antagonize a calcium-activated potassium channel in airway smooth muscle. J Gen Physiol. 1987; 89:339–52. https://doi.org/10.1085/jgp.89.2.339. [PubMed].
122. Ogata N, Tatebayashi H. Differential inhibition of a transient K+ current by chlorpromazine and 4-aminopyridine in neurones of the rat dorsal root ganglia. Br J Pharmacol. 1993; 109:1239–46. https://doi.org/10.1111/j.1476-5381.1993.tb13755.x. [PubMed].
123. Müller M, De Weille JR, Lazdunski M. Chlorpromazine and related phenothiazines inhibit the ATP-sensitive K+ channel. Eur J Pharmacol. 1991; 198:101–04. https://doi.org/10.1016/0014-2999(91)90569-c. [PubMed].
124. Wang ZJ, Soohoo SM, Tiwari PB, Piszczek G, Brelidze TI. Chlorpromazine binding to the PAS domains uncovers the effect of ligand modulation on EAG channel activity. J Biol Chem. 2020; 295:4114–23. https://doi.org/10.1074/jbc.RA119.012377. [PubMed].
125. Lee SY, Choi SY, Youm JB, Ho WK, Earm YE, Lee CO, Jo SH. Block of HERG human K(+) channel and IKr of guinea pig cardiomyocytes by chlorpromazine. J Cardiovasc Pharmacol. 2004; 43:706–14. https://doi.org/10.1097/00005344-200405000-00014. [PubMed].
126. Thomas D, Wu K, Kathöfer S, Katus HA, Schoels W, Kiehn J, Karle CA. The antipsychotic drug chlorpromazine inhibits HERG potassium channels. Br J Pharmacol. 2003; 139:567–74. https://doi.org/10.1038/sj.bjp.0705283. [PubMed].
127. Asher V, Sowter H, Shaw R, Bali A, Khan R. Eag and HERG potassium channels as novel therapeutic targets in cancer. World J Surg Oncol. 2010; 8:113. https://doi.org/10.1186/1477-7819-8-113. [PubMed].
128. Pardo LA, Sühmer W. Eag1 as a cancer target. Expert Opin Ther Targets. 2008; 12:837–43. https://doi.org/10.1517/14728222.12.7.837. [PubMed].
129. Hemmerlein B, Weseloh RM, Mello de Queiroz F, Knötgen H, Sánchez A, Rubio ME, Martin S, Schliephacke T, Jenke M, Heinz-Joachim-Radzun, Stühmer W, Pardo LA. Overexpression of Eag1 potassium channels in clinical tumours. Mol Cancer. 2006; 5:41. https://doi.org/10.1186/1476-4598-5-41. [PubMed].
130. Ousingsawat J, Spitzner M, Puntheeranurak S, Terracciano L, Tornillo L, Bubendorf L, Kunzelmann K, Schreiber R. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin Cancer Res. 2007; 13:824–31. https://doi.org/10.1158/1078-0432.CCR-06-1940. [PubMed].
131. Farias LM, Ocaña DB, Díaz L, Larrea F, Avila-Chávez E, Cadena A, Hinojosa LM, Lara G, Villanueva LA, Vargas C, Hernández-Gallegos E, Camacho-Arroyo I, Dueñas-González A, et al. Ether a go-go potassium channels as human cervical cancer markers. Cancer Res. 2004; 64:6996–7001. https://doi.org/10.1158/0008-5472.CAN-04-1204. [PubMed].
132. Meyer R, Schönherr R, Gavrilova-Ruch O, Wohlrab W, Heinemann SH. Identification of ether à go-go and calcium-activated potassium channels in human melanoma cells. J Membr Biol. 1999; 171:107–15. https://doi.org/10.1007/s002329900563. [PubMed].
133. Badaoui M, Mimsy-Julienne C, Saby C, Van Gulick L, Peretti M, Jeannesson P, Morjani H, Ouadid-Ahidouch H. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget. 2018; 9:24653–71. https://doi.org/10.18632/oncotarget.19065. [PubMed].
134. Valdés-Abadía B, Morán-Zendejas R, Rangel-Flores JM, Rodríguez-Menchaca AA. Chloroquine inhibits tumor-related Kv10.1 channel and decreases migration of MDA-MB-231 breast cancer cells in vitro. Eur J Pharmacol. 2019; 855:262–66. https://doi.org/10.1016/j.ejphar.2019.05.017. [PubMed].
135. Pardo LA, del Camino D, Sánchez A, Alves F, Brüggemann A, Beckh S, Stühmer W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999; 18:5540–47. https://doi.org/10.1093/emboj/18.20.5540. [PubMed].
136. Asher V, Warren A, Shaw R, Sowter H, Bali A, Khan R. The role of Eag and HERG channels in cell proliferation and apoptotic cell death in SK-OV-3 ovarian cancer cell line. Cancer Cell Int. 2011; 11:6. https://doi.org/10.1186/1475-2867-11-6. [PubMed].
137. Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, Yang H, Wang TA, Stehbens SJ, Younger S, Barshow S, Zhu S, Cooper MK, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015; 18:1236–46. https://doi.org/10.1038/nn.4088. [PubMed].
138. Stühmer W, Alves F, Hartung F, Zientkowska M, Pardo LA. Potassium channels as tumour markers. FEBS Lett. 2006; 580:2850–52. https://doi.org/10.1016/j.febslet.2006.03.062. [PubMed].
139. Chávez-López MG, Zúñiga-García V, Pérez-Carreón JI, Avalos-Fuentes A, Escobar Y, Camacho J. Eag1 channels as potential early-stage biomarkers of hepatocellular carcinoma. Biologics. 2016; 10:139–48. https://doi.org/10.2147/BTT.S87402. [PubMed].
140. Hartung F, Stühmer W, Pardo LA. Tumor cell-selective apoptosis induction through targeting of K(V)10.1 via bifunctional TRAIL antibody. Mol Cancer. 2011; 10:109. https://doi.org/10.1186/1476-4598-10-109. [PubMed].
141. Weber C, de Queiroz FM, Downie BR, Suckow A, Stühmer W, Pardo LA. Silencing the activity and proliferative properties of the human EagI Potassium Channel by RNA Interference. J Biol Chem. 2006; 281:13030–37. https://doi.org/10.1074/jbc.M600883200. [PubMed].
142. Glassmeier G, Hempel K, Wulfsen I, Bauer CK, Schumacher U, Schwarz JR. Inhibition of HERG1 K+ channel protein expression decreases cell proliferation of human small cell lung cancer cells. Pflugers Arch. 2012; 463:365–76. https://doi.org/10.1007/s00424-011-1045-z. [PubMed].
143. Rai S, Tanaka H, Suzuki M, Ogoh H, Taniguchi Y, Morita Y, Shimada T, Tanimura A, Matsui K, Yokota T, Oritani K, Tanabe K, Watanabe T, et al. Clathrin assembly protein CALM plays a critical role in KIT signaling by regulating its cellular transport from early to late endosomes in hematopoietic cells. PLoS One. 2014; 9:e109441. https://doi.org/10.1371/journal.pone.0109441. [PubMed].
144. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010; 141:1117–34. https://doi.org/10.1016/j.cell.2010.06.011. [PubMed].
145. Obata Y, Toyoshima S, Wakamatsu E, Suzuki S, Ogawa S, Esumi H, Abe R. Oncogenic Kit signals on endolysosomes and endoplasmic reticulum are essential for neoplastic mast cell proliferation. Nat Commun. 2014; 5:5715. https://doi.org/10.1038/ncomms6715. [PubMed].
146. Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Böhmer FD, Gerke V, Schmidt-Arras DE, Berdel WE, Müller-Tidow C, Mann M, Serve H. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009; 36:326–39. https://doi.org/10.1016/j.molcel.2009.09.019. [PubMed].
147. Rai S, Tanaka H, Suzuki M, Espinoza JL, Kumode T, Tanimura A, Yokota T, Oritani K, Watanabe T, Kanakura Y, Matsumura I. Chlorpromazine eliminates acute myeloid leukemia cells by perturbing subcellular localization of FLT3-ITD and KIT-D816V. Nat Commun. 2020; 11:4147. https://doi.org/10.1038/s41467-020-17666-8. [PubMed].
148. Ling HH, Kuo CC, Lin BX, Huang YH, Lin CW. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp Cell Res. 2017; 350:218–25. https://doi.org/10.1016/j.yexcr.2016.11.024. [PubMed].
149. Gil-Ad I, Shtaif B, Levkovitz Y, Nordenberg J, Taler M, Korov I, Weizman A. Phenothiazines induce apoptosis in a B16 mouse melanoma cell line and attenuate in vivo melanoma tumor growth. Oncol Rep. 2006; 15:107–12. [PubMed].
150. Van Woert MH, Palmer SH. Inhibition of the growth of mouse melanoma by chlorpromazine. Cancer Res. 1969; 29:1952–55. [PubMed].
151. Aas AT, Brun A, Pero RW, Salford LG. Chlorpromazine in combination with nitrosourea inhibits experimental glioma growth. Br J Neurosurg. 1994; 8:187–92. https://doi.org/10.3109/02688699409027965. [PubMed].
152. Levij IS, Polliack A. Inhibition of chemical carcinogenesis in the hamster cheek pouch by topical chlorpromazine. Nature. 1970; 228:1096–97. https://doi.org/10.1038/2281096a0. [PubMed].
153. Fond G, Macgregor A, Attal J, Larue A, Brittner M, Ducasse D, Capdevielle D. Antipsychotic drugs: pro-cancer or anti-cancer? A systematic review. Med Hypotheses. 2012; 79:38–42. https://doi.org/10.1016/j.mehy.2012.03.026. [PubMed].
154. Mortensen PB. The incidence of cancer in schizophrenic patients. J Epidemiol Community Health. 1989; 43:43–47. https://doi.org/10.1136/jech.43.1.43. [PubMed].
155. Dalton SO, Johansen C, Poulsen AH, Nørgaard M, Sørensen HT, McLaughlin JK, Mortensen PB, Friis S. Cancer risk among users of neuroleptic medication: a population-based cohort study. Br J Cancer. 2006; 95:934–39. https://doi.org/10.1038/sj.bjc.6603259. [PubMed].
156. Hsieh YH, Chan HL, Lin CF, Liang SH, Lu ML, McIntyre RS, Lee Y, Lin TC, Chiu WC, Chen VC. Antipsychotic use is inversely associated with gastric cancer risk: A nationwide population-based nested case-control study. Cancer Med. 2019; 8:4484–96. https://doi.org/10.1002/cam4.2329. [PubMed].
157. Walker AJ, Card T, Bates TE, Muir K. Tricyclic antidepressants and the incidence of certain cancers: a study using the GPRD. Br J Cancer. 2011; 104:193–97. https://doi.org/10.1038/sj.bjc.6605996. [PubMed].
158. Lichtermann D, Ekelund J, Pukkala E, Tanskanen A, Lönnqvist J. Incidence of cancer among persons with schizophrenia and their relatives. Arch Gen Psychiatry. 2001; 58:573–78. https://doi.org/10.1001/archpsyc.58.6.573. [PubMed].
159. Wu Chou AI, Wang YC, Lin CL, Kao CH. Female schizophrenia patients and risk of breast cancer: A population-based cohort study. Schizophr Res. 2017; 188:165–71. https://doi.org/10.1016/j.schres.2017.01.019. [PubMed].
160. Pottegård A, Lash TL, Cronin-Fenton D, Ahern TP, Damkier P. Use of antipsychotics and risk of breast cancer: a Danish nationwide case-control study. Br J Clin Pharmacol. 2018; 84:2152–61. https://doi.org/10.1111/bcp.13661. [PubMed].
161. Kisely S, Campbell LA, Cox M. The effect of study design on the reporting of mortality due to colorectal cancer in adults with mental illness in Nova Scotia. Can J Psychiatry. 2012; 57:389–94. https://doi.org/10.1177/070674371205700609. [PubMed].
162. Mehta RD, Roth AJ. Psychiatric considerations in the oncology setting. CA Cancer J Clin. 2015; 65:300–14. https://doi.org/10.3322/caac.21285. [PubMed].
163. Lathers CM, Lipka LJ. Cardiac arrhythmia, sudden death, and psychoactive agents. J Clin Pharmacol. 1987; 27:1–14. https://doi.org/10.1177/009127008702700101. [PubMed].
164. Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, Weiden PJ. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999; 156:1686–96. https://doi.org/10.1176/ajp.156.11.1686. [PubMed].
165. Greenberg HR. Inhibition of ejaculation by chlorpromazine. J Nerv Ment Dis. 1971; 152:364–66. https://doi.org/10.1097/00005053-197105000-00008. [PubMed].
166. Martino D, Karnik V, Osland S, Barnes TRE, Pringsheim TM. Movement Disorders Associated With Antipsychotic Medication in People With Schizophrenia: An Overview of Cochrane Reviews and Meta-Analysis. Can J Psychiatry. 2018; 63:706743718777392. https://doi.org/10.1177/0706743718777392. [PubMed].
167. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009; 29:64–73. https://doi.org/10.1592/phco.29.1.64. [PubMed].
168. Otręba M, Kośmider L. In vitro anticancer activity of fluphenazine, perphenazine and prochlorperazine. A review. J Appl Toxicol. 2021; 41:82–94. https://doi.org/10.1002/jat.4046. [PubMed].
169. Abbruzzese C, Matteoni S, Persico M, Villani V, Paggi MG. Repurposing chlorpromazine in the treatment of glioblastoma multiforme: analysis of literature and forthcoming steps. J Exp Clin Cancer Res. 2020; 39:26. https://doi.org/10.1186/s13046-020-1534-z. [PubMed].