
Introduction
In recent years, new mechanisms have been discovered and elucidated that link carcinogenesis with cellular metabolism in colon cancer. Despite these progresses, 25% of CRC patients are diagnosed at an advanced stage with a 5-year survival < 20%, and only limited targeted therapies are available [1]. For these reasons, it is necessary to understand comprehensively the underlying mechanisms that promote CRC progression. In particular, the importance of the action of AGEs on their cell receptors here is highlighted to suggest potential new interesting therapeutic targets.
MOLECULAR AND METABOLIC CHARACTERISTICS OF COLORECTAL CANCER
1-MAPK pathway
Most colorectal cancers result from the transformation and progression of precursors such as adenoma. From a molecular point of view, three main pathways of carcinogenesis have been identified: chromosomal instability (CIN), microsatellite instability (MSI) and the CpG island methylator phenotype (CIMP). 85% of CRCs develop from the CIN pathway and have alterations in structure and number of the chromosomes, leading to abnormalities such as aneuploidy, chromosomal rearrangement and loss of heterozygosity at the suppressor gene site. Tumours of the CIN pathway also acquire mutations in oncogenes and tumour suppressor genes including Adenomatous Polyposis Coli (APC), KRAS Proto-Oncogene, BRAF Proto-Oncogene and Tumour suppressor TP53. According to the model of Fearon and Vogelstein.
[2] APC inactivation occurs first in the proliferating epithelium. The most relevant pathways involved in CIN tumours are the Wnt and MAPK pathways [3]. The MAPK pathway is activated by a receptor tyrosine kinase. Activating mutations of molecules, such as RAS, RAF or ERK, related to these signalling pathways, can produce tumour cell proliferation. In CRC, MAPK can be constitutively overactive. This type of hyperactivation can occur through three main signal transduction pathways: usually overexpressed MAPK/ERK pathway, also known as the RAS/RAF/MEK/ERK pathway; MAPK14 (p38 MAPK) pathway; activation of the stress-activated protein kinases/c-Jun Nh(2)-terminal kinase (SAPK/JNK) signalling pathway. The overactive state in the MAPK signalling cascade is determinant to influence the responsivity degree of the patient treated with EGFR inhibitors [4]. Moreover, p38MAPK signaling enhances glycolysis in cancer cells through the up-regulation of the glucose transporters [5] that can trigger cellular stress responses. Dysfunctions of the metabolic signaling pathways in the colonic epithelium and local immune cells are highly intertwined with the the gene/pheno-type dysfunction responsible of tumour progression and relatively antitumour immunity [6].
2-Warburg effect
In the colon, insulin resistance, hyperglycaemia and chronic inflammation reinforce the metabolic dysregulation associated with cancer development and promote the progression [7]. In fact, in this phase, the tumour cells proliferate rapidly by increasing their absorption of nutrients to satisfy their bioenergetic demands, developing a metabolism oriented towards anabolic pathways. Moreover, the nutrient-poor tumour microenvironment can influence the phenotype of macrophages that control both the innate and adaptive immunity response. In fact, a metabolic adaptation of tumour-associated macrophages (TAM) is triggered through the upregulation of the glycolytic genes linked to lactate derived from neoplasia which contributes to the polarization of tissue-associated macrophages (TAM) M2 which have an anti-inflammatory and pro-tumoural effect also in CRC [8, 9].
On the other hand, this reprogrammed metabolism, documented in many types of cancers is considered a hallmark of cancer cells and is called “Warburg effect”. The Warburg effect (WE) results in a glycolytic switch associated with oncogene activation, resulting in increased cytoplasmic production of ATP and lactate and disturbances in mitochondrial energy production [10, 11]. It is well known that CRC demonstrate the Warburg metabolic phenotype [12]. It is now evident that insulin resistance, hyperglycaemia and chronic inflammation lead to colon carcinogenesis through interaction with molecular pathways. The WE clearly suggests that the link between glucose metabolism, protein protonation state and the microenvironment of neoplasms, as resulting from interactions with the inflammatory infiltrate and related cytokine production, is a major actor of the development and progression of tumours [13, 14].
WE as the main characteristics of cancer can thus be triggered by infectious agents, followed by consequent effects such as genomic mutations [15], possibly switching benign adenomas to malignant carcinomas in the case of colorectal cancers. Inhibitors for mitochondrial ATP synthesis are being developed for colon cancer [16] in which the uptake of glucose is overregulated [17]. Moreover, tumor suppressor protein p53 was demonstrated negatively influences the oncogenic metabolic adaptation of cancer cells reverting WE [18].
3-Oxidative stress
Recently, it was also shown that some cancer cells use mitochondria, not glycolysis to generate ATP [19]. Such a cancer phenotype has been classified as the reverse Warburg effect (RWE). The underlying mechanism is based on a pathological collaborative interaction in tissue [20]. In particular, loss of stromal Caveoline-1 (Cav-1) expression results in increased production of nitric oxide, increased reactive oxygen species (ROS) production, increased oxidative stress, and mitochondrial dysfunction occurring in fibroblasts (supporting cells) which supply energy-rich metabolites to a cancer cell with fully functional mitochondria. Extensive morphological changes occur, these tumours are highly aggressive, fast growing, with a strong metastatic potential and poor outcomes for a patient. The reverse Warburg effect may exist in many different types of epithelial cancers [21] and the loss of stromal Cav-1 thus may be involved mechanistically in all the different phases of epithelial tumorigenesis.
CAV1 is frequently overexpressed in advanced colorectal tumors and it is implicated in enhanced aerobic glycolysis of tumor cells. Elevated CAV1 increases glucose uptake and ATP production by stimulating transcription of the glucose transporter GLUT3 via a high mobility group A1 (HMGA1)-binding site within the promoter [22]. HMGA1 chromatin remodeling protein is known as an important factor required by cancer cells for tumor progression and acquisition of a stem-like state [23, 24]. However, HMGA1 is also a ligand of RAGE (Receptor for advanced glycation end products) with direct implications in metabolic and molecular signaling dysfunction in the colon cancer microenvironment [25].
AGES AND RAGES IN COLORECTAL CANCER
1-AGEs
The methylglyoxal (MG) produced through the glycolysis, is a highly reactive dicarbonyl compound and a major precursor of the cell advanced glycation end products (AGE) [26]. The AGE are highly reactive molecules, formed endogenously or exogenously during metabolic oxidative stress and accumulate in tissues normally with age [27]. Advanced glycation endproducts (AGEs) derive nonenzymatically from metabolism through glycation reaction between proteins, lipids, or nucleic acid with free reducing sugars [28].
AGEs can result from Maillard reaction or from glycolysis and oxidative pathways through the reaction between the electrophilic carbonyl group of reducing sugars with free amino group of proteins. Such reactions can generate unstable Schif’s bases that may form stable Amadori products after further rearrangements. Maillard in 1912 described the formation of melanoidins, a class of heterogeneous polymers that are formed when sugars and amino acids combine. In fact, Maillard products are synthesized trough a reaction between free amino groups of proteins, mainly arginine and lysine, and carbohydrates. This reaction was furtherly described by Amadori and is known as Amadori Heyns rear-rangement. In turn, the stable Amadori products undergo further modifcations, such as oxidation, dehydration or polymerization in the presence of transition metals to form more stable AGEs [29].
Many AGEs have been identifed such as the so-called GOLD (glyoxal lysine dimer), MOLD (methylglyoxallysine dimer), CEL (Nε-carboxyethyl-lysine), CML (pentosidine, and non-cross linking Nε- carboxymethyl-lysine).
The cellular functions of AGEs are mediated through binding to the receptor for advanced glycation end products (RAGE), a transmembrane molecule considered a pattern recognition receptor (PRR). It is a member of the immunoglobulin superfamily and a multi-ligand receptor that interacts with various ligands RAGE is thought to be associated with colon cancer metastasis and poor prognosis.
2-RAGE
RAGE is a multi-ligand signaling system highly expressed on the cell surface membrane of immune, endothelial or tumour cells when they are in the active phase. During embryonic development, there is a high RAGE expression especially in brain tissue. Its expression diminishes with the development and is almost absent in healthy adults. Only in healthy lung epithelium, does RAGE expression remain constitutive and relate to elevated levels of a unique isoform not found in other adult epithelia or cell types
RAGE consists in a 45–50 kDa molecule that binds to AGEs and other danger signaling molecules (DAMPs) to exert their pathophysiological roles in multiple disorders. These DAMPs include, among others, High-mobility group box proteins (HMGB1), S100 calgranulins and amphoterin.
Binding of RAGE with AGEs activates many signaling pathways also active in CRC, such as MAPK, NF-κB, PI3K, Akt, ERK1/2 and JAK-STAT, involved in inflammation, cell survival and cancer progression [30].
Hypoxia in cancer cells increases RAGEs expression and in cancer the involvement of the AGE-RAGE axis promotes the autophagic activity via the activation of autophagic proteins such as Beclin-1 with inhibition of apoptotic signaling. AGE-RAGE signaling also causes oxidative stress producing oxygen-free radicals, and activates NF-κB that causes secretion of pro-infammatory cytokines, growth factors, and adhesion molecules [31].
Studies conducted on knockout mouse models to understand the functions of RAGE, suggest that this receptor plays a regulatory role in sepsis [32]. In particular, it affects the homeostatic regulation of innate immunity, which in turn, can play a pivotal role in cancerogenesis [33].
When receptor ligand binding occurs, the resulting complex does not constitute a clear negative feedback signal aimed at inducing and regulating the clearance and degradation of the RAGE receptor. In fact, it has been shown that the receptor ligand complex instead functions almost as a positive feedback leading to a prolonged period of its expression and activation with consequent prolonged inflammation (Figure 1) [34]. This redundant positive effect created by the receptor ligand binding, results in an exasperated downstream molecular activation. This behaviour justifies why the pathways RAGE-dependent are involved in the pathogenesis of many simple and complex diseases, where inflammation is the least common. Currently, clinically relevant studies for RAGE associate its elevated plasma levels in inflammatory disease and brain injury with disease severity and functional outcome [35].
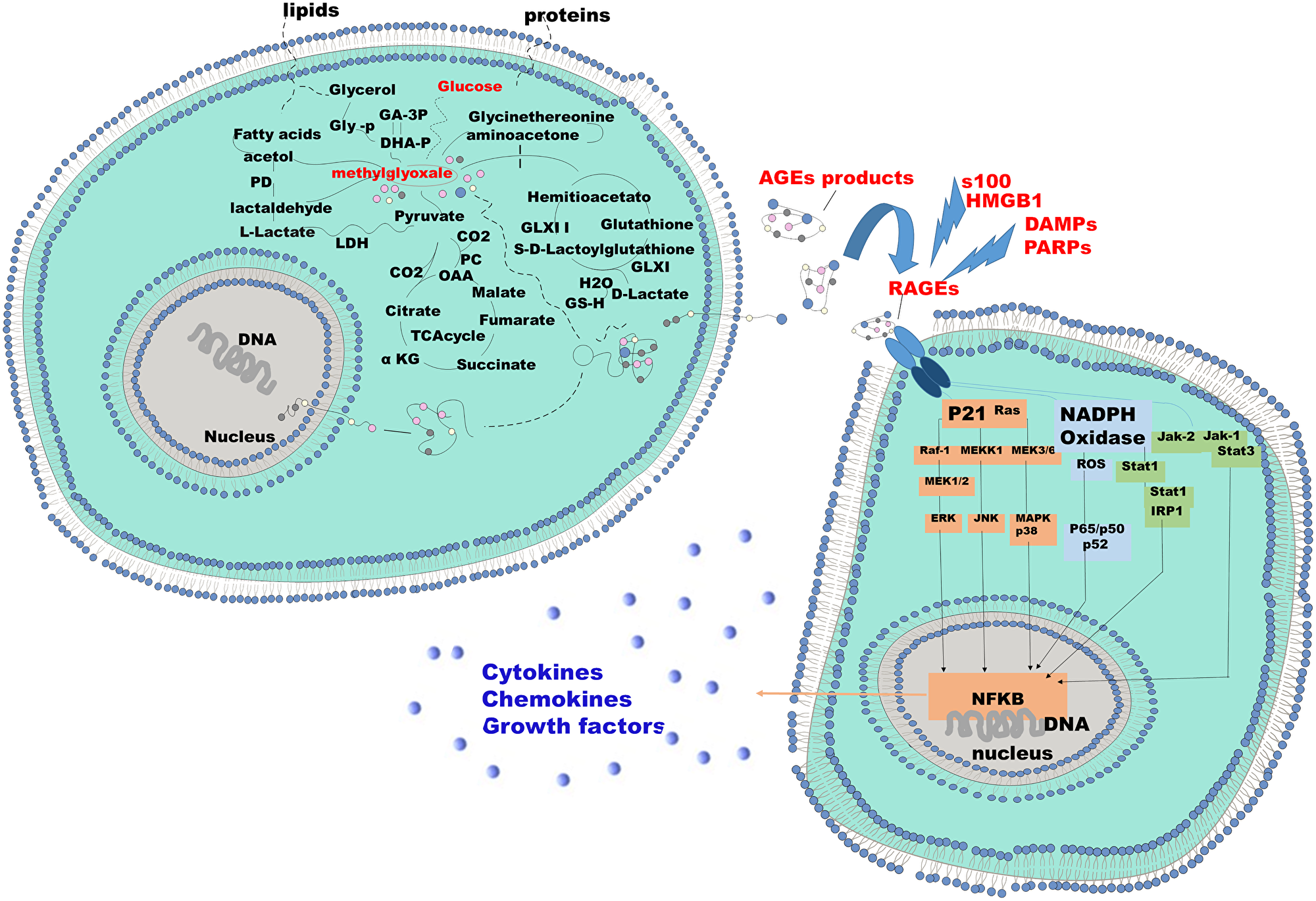
Figure 1: Mechanism of endogenous AGE formation and pathobiological actions of RAGE receptor ligands. The two cells represented here can be identified as a tumor cell and the stromal counterpart or vice versa. The energy demand of the tumor cell aimed at supporting the high proliferative standard that induces a proportional increase in the glyco-lytic pathway. The increased glycolysis results in an increased production of lactate and aldehydes such as glioxal and methylglioxal. These two aldehydes bind, by Maillards reaction, to the free radicals of the macromolecules, protein and non-protein present in the intracellular microenviroment resulting in adducts of Methylglyoxal, or advanced gly-cated adduct AGEs. These molecules gain extracellular space and bind to their receptors, RAGE. RAGE's expression is induced by the presence of the AGEs. RAGE activation results in the activation of downstream signaling pathways responsible for the release of cytokines, chemokines and growth factors. S-100 proteins DAMP and PAMP and HMBG in response to specific conditions share analogous ability to induce and bind RAGE.
Among the countless diseases in which RAGE is involved there is also the onset and progression of colon cancer [36] (Table 1).
Table 1: Principal RAGE-ligands and activated pathways involved in the pathogenesis and development of colon cancer
| Axis RAGE-ligand | Downstream Activated Pathways | Cancer phases | Pleiotropic effects | Study |
|---|---|---|---|---|
| RAGE-AGEs | NFkB, ERK, MAPK | Tumorigenesis and progression of colon cancer | cell proliferation, inflammation, cancer progression | Malara N, et al. [14] Liliensiek B [39] |
| RAGE-S-100 | NFkB, MAPK, IL-6, TNFα, TIL | Advanced colon cancer | cancer progression | Onyeagucha BC, et al. [37] Sun X, et al. [53] Wang HY, et al. [55] |
| RAGE-HMGB1 | NFkB, TIL, PD-1 | Resistant colon cancer | inflammation, cancer progression, immunosuppresion | Yao X, et al. [71] Huang CY, [65] |
| RAGE-DAMPs/PAMPs | SAPK/JNK, CdC42/Rac, p38MAPK, ERK1, CXCL2, IL-1, TNFα, | Tumorigenesis and progression of colon cancer | inflammation, cancer progression | Escamilla-Tilch M, et al. [75] Stephens M, et al. [76] |
3-AGEs-RAGE axis in vitro
Studies in human colon cancer cell lines have shown how the AGEs-RAGE axis induces the progression of cancer cells through the regulation of specific pathways, such as upregulation of carbohydrate response element binding protein (ChREBP) in the cell line HCT 116 (Human, colon, carcinoma). This protein, ChREBP, by promoting the enhancement of anaerobic glucose metabolism, drives the metabolic switch and suppression of p53. These actions combine to promote the growth and progression of cancer cells [37, 38]. Furthermore, in human colon cancer cell lines, Caco-2 and COLO320, the AGEs-RAGE axis increases ERK and MAPK and NF-ΚB respectively [39, 40]. The expression of Sp1 is also regulated by the binding of RAGE, with the AGEs through the activation of the ERK pathway. Sp1 is a key regulator of the molecular process that drives metastasis. This molecular actor intervenes in the modification of the stroma by promoting the degradation of type IV collagen with activation of metalloproteinases (MMPs). The prolonged NF-ΚB activation promotes tumour progression in colon cells through the inhibition of proapototic pathways that inhibit caspase activity [41–43].
RAGE LIGANDS IN CRC
S100 and RAGEs in colonrectal cancer
The S100 family of proteins is an important group that is part of the RAGE ligands. S100 proteins bind Ca2 + and their function is to regulate the levels of intracellular Ca2 + and consequently also of numerous Ca2 + signaling pathways. Unlike calmodulin and troponin-C, proteins linked to calcium metabolism and whose activities are limited to the intracellular environment, several proteins of the S100 family act as intracellular regulators but also as extracellular signaling proteins. They can be secreted and/or released to regulate the activities of target cells. Within the cell, S100 proteins exhibit a somewhat specific distribution. In the intracellular context, S100 proteins are involved in the regulation of proliferation, differentiation, apoptosis, Ca2 + homeostasis, energy metabolism, inflammation and migration/invasion. They interact with a variety of target proteins including enzymes, cytoskeletal subunits, receptors, transcription factors and nucleic acids. As extracellular signaling molecules, S100 proteins regulate cell proliferation, differentiation, survival and migration in normal and pathological conditions, inflammation and tissue repair and/or exert antimicrobial activity [43, 44]. Indeed, at the gene level, numerous S100 genes are specifically inducible by appropriate growth factors, cytokines and toll-like receptor (TLR) ligands. To fulfill the function of alarmins, or cell signaling molecules, they are secreted and may function as damage-associated molecular pattern factors that primarily mediate the functions of the innate and adaptive immune systems. Operationally, they stimulate the locomotion of tumour cells and/or participate in tissue repair. The S100B protein is expressed in astrocytes, some neuronal populations, Schwann cells, melanocytes, chondrocytes, adipocytes, skeletal myofibers and associated satellite cells, some dendritic cell and lymphocyte populations and a few other cell types. This protein acts as a stimulator of cell proliferation and migration and as an inhibitor of apoptosis and differentiation [45].
Cancer cells also exhibit a distinctive S100 protein profile which can be both stage-specific and subtype specific. In gliomas, S100B expression correlates positively with proneuronal, neuronal and classical subtypes, but not mesenchymal, whereas S100A8 and S100A9 expression correlates positively with mesenchymal subtypes [46, 47].
Extracellular protein S100A9, but not S100A8, binds to the EMMPRIN receptor and requires TNF receptor-associated factor 2 of adapter protein (TRAF2) to upregulate the expression of TNFα, IL-1, IL-6 and other factors [48]. Another example is given by the S10012 protein which mediates pro-inflammatory effects through binding to RAGE and the toll-like receptor such as TLR-4. The duple stimulation of RAGE and TLR-4s leads to the activation of the transcription nuclear factor NF-κB. Nuclear transfer of NF-κB induces inflammatory response and leukocyte recruitment. [47].
Furthermore, through RAGE receptors, under stress conditions, S100B, triggers NF-ΚB and MAPK signaling and stimulates the release of pro-inflammatory cytokines, such as IL6, TNFα and IL-1β [49, 50].
S100A8, S100A9 and S100P are other S100 proteins related to AGEs/RAGE progression mechanism of colorectalcarcinoma. They trigger many of the pathways above cited such as ERK, NF-kB [51, 52].
Interestingly, the membrane protein S100A16 and its main partner S100A14 and S100A4 are expressed by tumour cells in CRC and is associated with prognosis, indicating that the expression of these S100 proteins is a favorable prognostic biomarker and a therapeutic target.
The S100A4 protein also exhibits both intra- and extracellular activities. The expression of S100A4 in cells causes apoptosis, cell migration and maintenance of stem cells and in the extracellular space activates various processes by stimulating pro-inflammatory pathways and the expression of various molecules, such as cytokines [53]. In S100A4 knock out (S100A4 -/-) mice the expression of inflammatory cytokines and the recruitment of macrophages and neutrophils decreased significantly. On the other hand in wild type (WT) mice the effects, favouring colitis development, promoted by S100A4 could be abolished by a receptor for advanced glycation end products (RAGE)-specific inhibitor (FPS-ZM1) [54].
In summary, some effects of S100 proteins in CRC may be mediated by the regulation of TIL. Tumour-infiltrating lymphocytes (TILs) are associated with the host's immune status and are an important prognostic factor in many malignancies. Many reports have shown that an elevated level of TIL is a favorable biomarker in the prognosis of colorectal cancer. RAGE and its ligands are of fundamental importance in the modulation of TILs. RAGE and its ligands can play a fundamental role in the regulation of the lymphocyte infiltrate in CRC [55] by modulating its intensity and persistence in the tumour microenvironment (Table 1).
HMGB1 and RAGEs in colonrectal cancer
HMGB1 is a chromatin binding factor in the nuclei of cancerous and normal cells, it plays a role in DNA repair, transcription, differentiation, extracellular signaling and somatic recombination [56]. The function of HMGB1 takes place by binding non-specifically to a smaller groove of the DNA and thus modulates the interaction of the DNA with transcription factors [57]. It enhances the activity of p53, p73, the retinoblastoma protein, transcription factors such as the Rel/NF-κB family and the estrogen receptor [58]. ERK/MAPK pathway is upregulated by HMGB1 in colon carcinoma [59]. In cell culture studies HMGB1 may show DNA damage caused by chemotherapy [60]. In colon cancer, HMGB1 and its receptor, are indicators of progression [61]. In an immunohistochemical study, HMGB1 overexpression was determined as 55.6% in relation to tumour invasion, lymph node status, distant metastasis and stage of colon cancer disease [62]. c-IAP2 is an antiapoptotic protein which may be upregulated through NFk-B activation via HMGB1. There is a strong correlation between upregulation of the apoptosis repressing HMGB1 and c-IAP2 proteins in the pathogenesis of colon carcinoma [63]. Upregulation of MMPs with HMGB1 has been associated with cancer cell proliferation of colon through the RAGE/Snail/NF-κB signalling pathways accompanied by the activation of MMP-7 [64].
HMGB1 expression was also correlated with overall survival and proved to be an independent predictor of worse prognosis in CRC cases [65] (Table 1). High expression of HMGB1 in the cytoplasm of breast cancer cells has been associated with high histological grade, pT stage and abundant TIL [66]. In patients with locally advanced rectal cancer and with elevated cyto-HMGB1 and PD-1 + TIL they showed better results. The best outcome is probably due to the release of HMGB1, which stimulates the maturation of dendritic cells (DCs) through the activation of TLR4, and the subsequent recruitment of TIL into the tumour [67]. Another explanation of the best outcome related the elevated cyto-HMG is due the inverted relationship with the CAV1 expression. In colon calcer cells CAV1 depleted cytoplasmic levels of HMGA1 are concomitantly increased. Increased expression of CAV1 stimulates HMGA1 interaction with the HMGA1-binding site in the SLC2A3 promoter at nuclear levels to improve the GLUT3 expression [68]. When GLUT3 is highly expressed in colorectal cancer (CRC) it is negatively linked to CRC patient outcome. GLUT3 expression protects CRC cells by energy stress in the tumour microenvironment to withstand nutrient scarcity and to exacerbate the malignancy of CRC cells [69].
PAMPs/DAMPs and RAGEs in colonrectal cancer
The community of the microbiota in the lumen of the colon requires a careful system of surveillance of the intestinal immune system. In fact, the intestinal microbiota is a crucial biomodulator of the development and function of immune cells. Gut bacteria are directly involved in homeostatic regulation, function and differentiation of T cells. This is a saprophytic and mutually convenient contribution to both parties. In fact, while the microbes obtain a habitat and nourishment from the host, they return the favor by regulating the alimentary digestion and the protective immunity against the pathogens of the host [70]. Not surprisingly, in the early stages of the neoplastic transformation of the colonic epithelium there are phenomena of dysbiosis of the intestinal microbiota and increased intestinal permeability associated with inflammation. These same phenomena also underlie the tumour progression of CRC. Gut microbe-derived signals have been recognized to tune immune cells interacting with key players mediating host and microbe communication, known as pattern recognition receptors (PRRs). These signalling molecules, therefore considered to be part of the alarmine’s system, are expressed by innate immune cells such as dendritic cells (DC), monocytes/macrophages and natural killer (NK) cells [71]. RAGE, as an important pattern recognition receptor (PRR), may play a fundamental role in maintaining microbiological homeostasis within the colonic epithelium and how it can be considered as a perfect sensor for environmental stimuli. RAGE, as described above, is expressed on macrophages, smooth muscle cells, endothelial cells, epithelial cells. In addition to PAMP, RAGE also recognizes the molecular patterns associated with damage (DAMP) that induce inflammation [72]. The detection of PAMP and DAMP by RAGE drives a cascade of signals that converge to the nuclear factor κB (NF-κB) [73]. The interaction of ligands with RAGE activates a positive feedback loop by upregulating multiple signalling pathways such as SAPK/JNK (stress-activated protein kinase/c-jun-NH2-terminal kinase), Cdc42/Rac, p38 MAPK (protein kinase activated by mitogen) and ERK1/2 (Ras-extracellular kinase regulated by signal 1/2), which results, once again, in the increase of the transcription factor NF-KB and consequent production of cytokines, adhesive molecules and MMP (metalloproteinase of the matrix) [74]. Cytokines and chemokines that act as growth factors and promote angiogenesis have been associated with the development of polyps. In mice with defects in colon barrier integrity, bacterial invasion and increased expression of several inflammatory factors such as IL-17, Cxcl2, TNF-α and IL-1 can be observed [75, 76] (Table 1). In these proinflammatory responses, the mitochondrial dysfunction within the cancer cells, favors the excess reactive oxygen species release that can exacerbate the production of AGEs fueling the fire of inflammation [77].
RAGE/AGES CROSS-TALK IN IMMUNE STATE AND IMMUNOTHERAPY OF COLON CANCER
K-Ras is a small nucleotide-binding molecule of guanine that cycles between the active (GTP-bound) and inactive (GDP-bound) forms [78]. Mutations of the oncogene in K-Ras cancel its interactions with GTPase activating proteins (GAP) with a consequent decrease in the hydrolysis rate of GTP. Furthermore, the oncogenic mutation confers constitutive activity to K-Ras leading to prolonged but not permanent activity which returns to baseline in the absence of continuous external stimulation [79]. It has been shown that during the initial stages of neoplastic transformation of the colon the oncogen Kras requires not only external stimuli but also for the continuous maintenance of the signaling necessary to support the growth of tumour cells [80]. The high levels of K-Ras activity generate inflammatory stimuli and oxidative stress, NFκB activation, COX2 expression and DNA damage leading to the release of HMGB1 and nucleic acids. These events form a Ras/inflammation feed-forward loop.
In this scenario RAGE is a central key player involved in maintaining the Kras/inflammation feed-forward loop. The role in chronic colon inflammation and tumour promotion of upregulation dependent on RAGE and its potential ligands S100a8 and S100a9 [81, 82]. Furthermore, bone marrow chimera experiments revealed that RAGE expression on immune cells maintains the inflammatory reaction during tumour promotion [83]. However, the importance of RAGE in tumours expressing the Kras oncogene may be even stronger. Animals expressing oncogenic Kras but deficient in RAGE expression showed a delay in cancer development. Mice deficient in the RAGE ligand, S100A9, show a significant reduction in the incidence and burden of colorectal cancers associated with colitis [84].
Colon cancer is known to generate a variety of RAGE ligands, including HMGB1 and S100 molecules, suggesting that while oncogenic K-Ras itself may be “non-pharmacological,” it may be possible to inhibit or modulate its role in cancer by blocking the critical feed-forward mechanism in which RAGE plays an important role. In addition to its previously described intra-nuclear role, HMGB1 functions as an extracellular signaling molecule by mediating different responses and binding to different receptors, including RAGE and toll-like receptors (TLRs) −2 and 4. Consequent are pleiotropic effects, such as proliferation cellular, differentiation, death, inflammation and immunity. When HMGB1 is passively released from dying tumour cells following chemotherapy in colorectal cancer, HMGB1 facilitates autophagy following cytotoxic insults for chemoresistance via its RAGE receptor via the MEK/ERK signaling pathway. ERK mediated phosphorylation of Drp1 is necessary to resist chemotherapy cytotoxicity as it promotes the induction of autophagy on surviving tumour cells to promote regrowth. Patients with highly phosphorylated Drp1 proteins are associated with an increased risk of developing relapse lesions after neoadjuvant chemoradiotherapy treatment in locally advanced rectal cancer [85].
Furthermore, there are cumulative points of evidence towards a cooperative interaction in the host immune response between RAGE and members of the Toll-like receptor (TLR) family. The question relating the possible physical association between RAGE and TLR remains to be address. RAGE and TLR shared three ligands, HMGB, S100 and bacterial cell wall component, LPS. The mechanism or the physicochemical conditions underlying the choice of ligands to bind preferentially RAGE or TLR is not well understood. Increasing evidence in literature support their potential synergism on down-stream signaling pathways and the outcome of these interactions on the inflammatory response [86].
TLRs are found on the cell surface or intracellular in endosomes. They have well-established roles in the host immune response to infections and their activation normally contributes to the induction of protective immune responses [87].
TLR can be recognized by exogenous and endogenous ligands (DAMPs), as low-molecular-weight hyaluronic acid, fibrinogen, fibronectin, heparan sulfate peptidoglycans, and AGEs. AGE/TLR interaction determines the expression of NFκB with an increase in adhesion molecules (ICAM and VCAM, which lead to the recruitment of macrophages and neutrophils) and production of TNF, IL-1α, IL-4, IL -10, Cytokines IL-12 MCP-1, PAI-1 and tPA. [88]. TLRs represent a molecular link between the phenotype of inflamed tumour cells, the regulation of T cell activation and antitumour immunity and can modulate tumour progression and chemoresistance. The experimentally induced absence of the three receptor forms of TLR (TLR 3,7,9) results in tumour regression dependent on both CD4 and CD8 T cells and protects mice from subsequent tumour provocation. [89].
The CRC can evade immune surveillance in various ways, for example through the recruitment of immunosuppressive cells such as regulatory T cells (Treg), the secretion of factors such as TGF-β or through the activation of Immune checkpoints. On the other hand, these tumors can activate, through the secretion of pro-inflammatory cytokines, cellular factors promoting carcinogenesis such as STAT3. Among the immunotherapies used against CRC are recombinant cytokines, agents modulating the immune microenvironment, oncolytic viral therapy, small molecules, autologous T cells, vaccines, antibodies against specific antigens and immune checkpoint inhibitors. Combination of immunotherapies for CRC are also tested.
Imumune checkpoint inhibitors are used in order to treat CRC because they provide a prolonged disease control and exhibit an acceptable toxicity degree. Patients with MSI have higher responses to PD-1 treatment [90].
In the interaction between cancer cell and the immune system, a central role has recently been attributed to the immunosuppressive action of PD-L1. Cancer cells can locally produce PD-L1 to inhibit immune cell surveillance. In this regard, it has been shown that PD-L1 is expressed on the surface of exosomes secreted by tumour cells, which are found in the bloodstream with the function of inhibiting the anti-tumour activity of T cells. It has been shown that there are numerous transcriptional regulators. which can induce the expression of PD-L1. In the tumour microenvironment, PD-L1 can be modulated by inflammatory cytokines such as IFNγ, which induces activation of STAT1 and increased transcription of PD-L1 [91, 92].
HIF1, Myc, STAT3, AP-1, and NF-κB have also been shown to increase PD-L1 transcription in response to a variety of upstream signals. PD-L1 expression is significantly elevated in colon cancer tissues. During chemotherapy HMGB1 is passively released and through its RAGE receptor, upregulating the MEK/ERK signaling pathway increases NF-ΚB transcription and indirectly stabilizes PD-L1 [93]. Indeed, NF-ΚB, by upregulating the CSN5 deubiquitinase, inhibits the ubiquitination and proteasomal degradation of PD-L1 in tumour cells [94, 95]. Intriguing is the consideration that the RAGE-ligand axis can promote an immunosuppressive microenvironment by activating the PD-1/PD-L1 checkpoint, together with other previously reported immunosuppressive cytokines, such as IL4, IL10, PGE2 and TGFβ [96]. In this scenario, it is possible to imagine that possibly significantly blocking PD-1/PD-L1 could enhance CD8 + T-cell-mediated antitumour immunity against cancer cells. Therefore, a treatment with anti-PD-1 blocking antibodies to increase the number of CD8 + memory T cells in the tumour microenvironment may be useful as an immune prevention approach to antagonize the immune evasion of tumour cells [97–99].
Conclusions
The RAGE-ligand axis intervenes and supports the tumour progression of the CRC by inducing and activating molecular pathways promoting chemoresistance and immuno-escape phenomena. In particular, endogenous ligands as molecules produced and present in the intracellular microenvironment, which, once released in the extracellular space, become signalling molecules aimed at the endogenous activation of the RAGE. These molecules, AGEs, S100, HMGB1, are all connected to pro-cancerogenic molecular pathways of the colon and are also produced during chemotherapy. Their presence and concentration in the extracellular space induces and influences the activity of the RAGE receptor on cellular surface membrane of tumour cells, stromal cells and of immune cells. Moreover, exogenous microbial agents, relative to disturbance phenomena on colon microbiome, releasing DAMPs and PAMPs free to interact with RAGE, contributed to its constitutional state of activity. Metabolism and metabolites conditioned the microenvironment by modulating the immune response and on the other hand, the concomitant activation of RAGE, by inducing the indirect increasing of the transcriptional activity of NFKB, affects the production and release of cytokines and chemokines. Cytokines and chemokines are essential actors in the inflammatory scenario that implies the progressive evolution of the chemo resistant CRCs (Figure 2). The initial presence of the K-Ras mutation, which guides the choice of first-line treatment for CRCs, loses consistency on the molecular cross-linked front of the advanced tumour. We hypothesize that in the case of advanced CRCs the therapeutic action of cancer containment should be concentrated by working on several molecular fronts. Without disregarding the target therapy but supporting it to favour the persistence and duration of a positive response, through direct inhibition of RAGE-ligand axis and PDL-1expression, previously verifying their sharing of the activity. From the data emerging in this review, patients with chemoresistant CRCs cannot be retrained only through the presence or absence of K-ras mutation, evaluated by i.e., cfDNA analysis [100, 101], but it is necessary, through the analysis of markers such as RAGE or PDL-1 expression, through immunocytochemical approach [102–104] on i.e., circulating tumour cells [105–107], to verify how to support the possible choice of a targeted therapy or in the absence of this, to identify an optional and personalized therapeutic intervention (Figure 3).
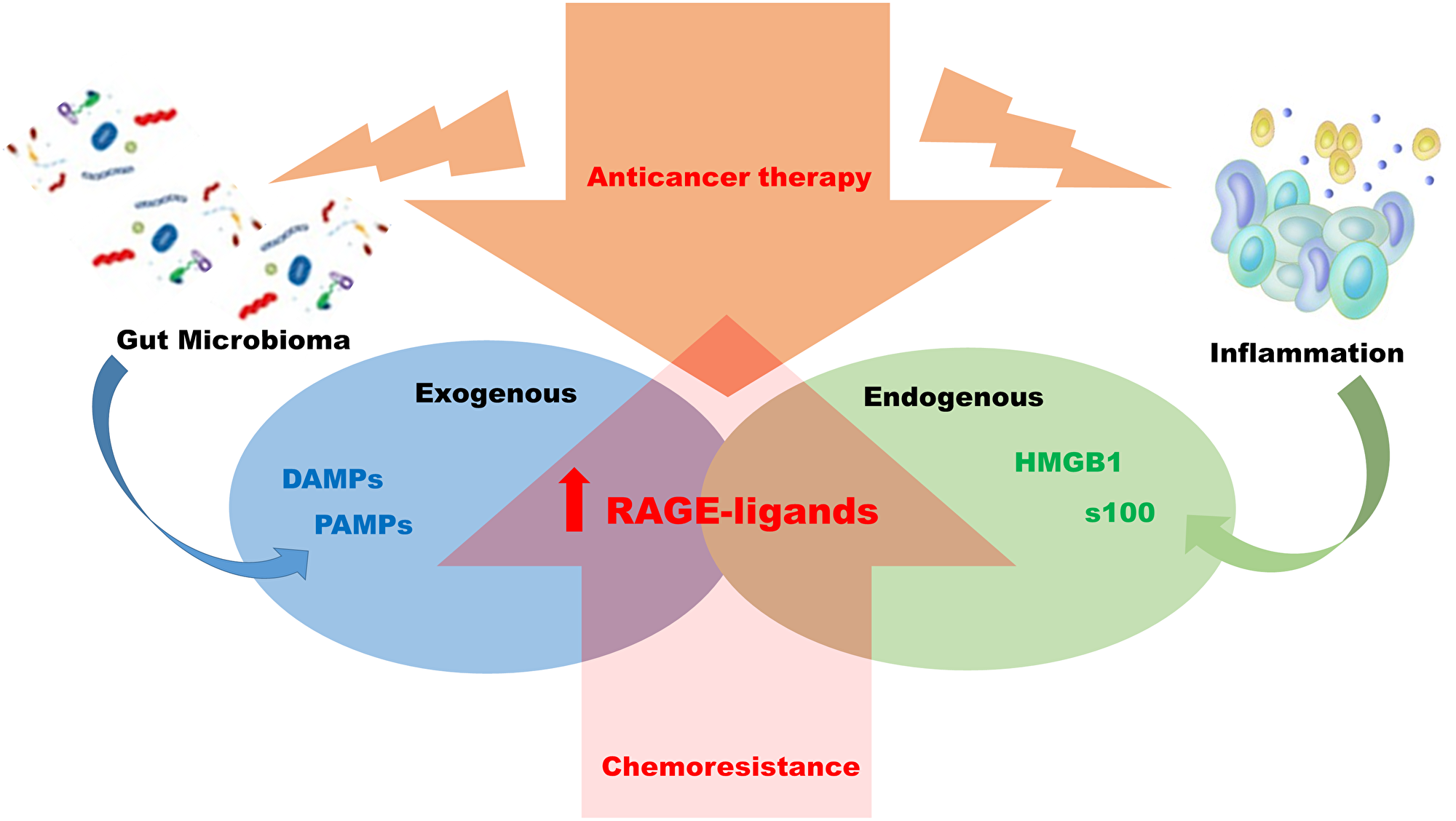
Figure 2: Mechanism of chemo resistance RAGE-ligands dependent. Anticancer treatments in colon rectal carcinoma, chemotherapy and radiotherapy have as a side effect the induction of local inflammation and alteration of the intestinal flora. The two biological phenomena can be associated with the induction of RAGE-ligand axis with consequent activation downstream of pro- inflammatory pathways favoring chemo resistance establishment.
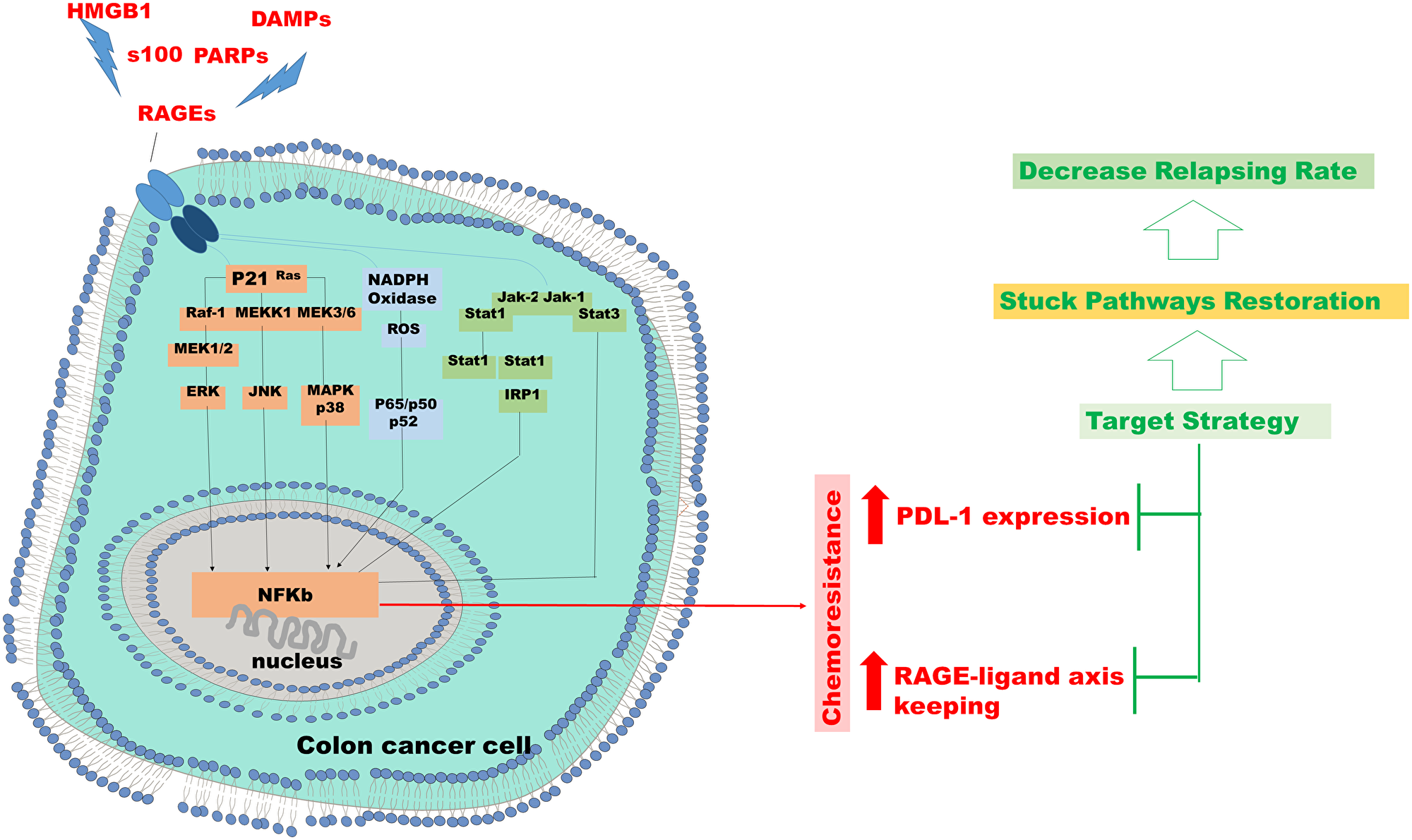
Figure 3: Chemo resistance in colon cancer cell. Hypothesis of targeted therapeutic intervention in resistant cases in which the RAGE or PDL-1, or both, overexpression is proven in colon cancer cells.
Abbreviations
CRC: Colorectal cancer; AGEs: Advanced glycation end products; RAGE: Receptor for advanced glycation end products; PRR: Pattern recognition receptor; CIN: Chromosomal instability; MSI: Microsatellite instability; APC: Adenomatous Polyposis Coli; MMR: Mismatch repair genes; CIMP: CpG island methylator phenotype; MAPK: mitogen-activating protein kinase; CMS: Consensus Molecular Subtypes; VEGF: Vascular endothelial growth factor; EGFR: Epidermal growth factor receptor; SAPK/JNK: Stress-activated protein kinases/c-Jun Nh(2)-terminal kinase; TAMs: Tumour Associated macrophages; TILs: Tumour-infiltrating lymphocytes; WE: Warburg effect; ADP: Adenosine diphosphate; G-6-P: Glucose 6-phosphate; LDH: Lactate dehydrogenase; PDK: Pyruvate dehydrogenase kinase; DAG: Diacylglycerol; MG: Methylglyoxal; KG: α-ketoglutarate; GLXI: Glyoxalase I; GLXII: Glyoxalase II; GS-H: Glutathione; DHAP: Dihydroxyacetone phosphate; GA-3P: Glycerol-3-phosphate dehydrogenase; ROS: Reactive oxygen species; NF-kB: Nuclear factor kappa B; JAK: Janus kinase; JNK: c-Jun N-terminal kinase; MAPK: Mitogen-activated protein kinase; MEK: Mitogen-activated protein kinase; ERK: Extracellular signal-regulated kinase; STAT: Signal transducer and activator of transcription; DAMPs: Damage-associated molecular pattern molecules; S100B: S100 calcium-binding protein B; HMGB1: High mobility group box 1 protein; PAMPs: Pathogen-associated molecular patterns; ChREBP: Carbohydrate response element binding protein; DCs: Dendritic cells; NK: Natural killer; HCT 116: Human, colon, carcinoma; MMPs: Matrix metallopeptidases; TLR: Toll-like receptors; PDL-1: Programmed death-ligand 1.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Sargent DJ, Wieand HS, Haller DG, Gray R, Benedetti JK, Buyse M, Labianca R, Seitz JF, O’Callaghan CJ, Francini G, Grothey A, O’Connell M, Catalano PJ, et al. Disease-free survival versus overall survival as a primary end point for adjuvant colon cancer studies: individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2005; 23:8664–70. https://doi.org/10.1200/JCO.2005.01.6071. [PubMed].
2. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990; 61:759–67. https://doi.org/10.1016/0092-8674(90)90186-i. [PubMed].
3. Harada S, Morlote D. Molecular Pathology of Colorectal Cancer. Adv Anat Pathol. 2020; 27:20–26. https://doi.org/10.1097/PAP.0000000000000247. [PubMed].
4. Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014; 120:3446–56. https://doi.org/10.1002/cncr.28864. [PubMed].
5. Papa S, Choy PM, Bubici C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene. 2019; 38:2223–40. https://doi.org/10.1038/s41388-018-0582-8. [PubMed].
6. Biswas SK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity. 2015; 43:435–49. https://doi.org/10.1016/j.immuni.2015.09.001. [PubMed].
7. Kuo CC, Ling HH, Chiang MC, Chung CH, Lee WY, Chu CY, Wu YC, Chen CH, Lai YW, Tsai IL, Cheng CH, Lin CW. Metastatic Colorectal Cancer Rewrites Metabolic Program Through a Glut3-YAP-dependent Signaling Circuit. Theranostics. 2019; 9:2526–40. https://doi.org/10.7150/thno.32915. [PubMed].
8. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014; 513:559–63. https://doi.org/10.1038/nature13490. [PubMed].
9. Väyrynen JP, Haruki K, Lau MC, Väyrynen SA, Zhong R, Dias Costa A, Borowsky J, Zhao M, Fujiyoshi K, Arima K, Twombly TS, Kishikawa J, Gu S, et al. The Prognostic Role of Macrophage Polarization in the Colorectal Cancer Microenvironment. Cancer Immunol Res. 2021; 9:8–19. https://doi.org/10.1158/2326-6066.CIR-20-0527. [PubMed].
10. Warburg O. [Metabolism of a cancer cell]. [Article in German]. Nat Sci. 1924; 12:1131–37. https://doi.org/10.1007/BF01504608.
11. Warburg O. On the origin of cancer cells. Science. 1956; 123:309–14. https://doi.org/10.1126/science.123.3191.309. [PubMed].
12. Brown RE, Short SP, Williams CS. Colorectal Cancer and Metabolism. Curr Colorectal Cancer Rep. 2018; 14:226–41. https://doi.org/10.1007/s11888-018-0420-y. [PubMed].
13. Pokorný J, Pokorný J, Kobilková J, Jandová A, Holaj R. Cancer development and damped electromagnetic activity. Appl Sci (Basel). 2020; 10:1826. https://doi.org/10.3390/app10051826.
14. Coluccio ML, Presta I, Greco M, Gervasi R, La Torre D, Renne M, Voci CP, Lunelli L, Donato G, Malara N. Microenvironment Molecular Profile Combining Glycation Adducts and Cytokines Patterns on Secretome of Short-term Blood-derived Cultures during Tumour Progression. Int J Mol Sci. 2020; 21:4711. https://doi.org/10.3390/ijms21134711. [PubMed].
15. Jandová A, Pokorný J, Pokorný J, Kobilková J, Nedbalová M, Čoček A, Jelínek F, Vrba J, Vrba J Jr, Dohnalová A, Kytnarová J, Tuszyński JA, Foletti A. Diseases caused by defects of energy level and loss of coherence in living cells. Electromagn Biol Med. 2015; 34:151–55. https://doi.org/10.3109/15368378.2015.1036076. [PubMed].
16. Kim MS, Gernapudi R, Cedeño YC, Polster BM, Martinez R, Shapiro P, Kesari S, Nurmemmedov E, Passaniti A. Targeting breast cancer metabolism with a novel inhibitor of mitochondrial ATP synthesis. Oncotarget. 2020; 11:3863–85. https://doi.org/10.18632/oncotarget.27743. [PubMed].
17. Mirzaei H, Hamblin MR. Regulation of Glycolysis by Non-coding RNAs in Cancer: Switching on the Warburg Effect. Mol Ther Oncolytics. 2020; 19:218–39. https://doi.org/10.1016/j.omto.2020.10.003. [PubMed].
18. Ripani P, Delp J, Bode K, Delgado ME, Dietrich L, Betzler VM, Yan N, von Scheven G, Mayer TU, Leist M, Brunner T. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain. Oncogene. 2020; 39:2345–57. https://doi.org/10.1038/s41388-019-1142-6. [PubMed].
19. Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett. 2012; 4:1151–57. https://doi.org/10.3892/ol.2012.928. [PubMed].
20. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8:3984–4001. https://doi.org/10.4161/cc.8.23.10238. [PubMed].
21. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, Witkiewicz AK, Vander Heiden MG, Migneco G, Chiavarina B, Frank PG, Capozza F, Flomenberg N, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010; 9:1960–71. https://doi.org/10.4161/cc.9.10.11601. [PubMed].
22. Ha TK, Chi SG. CAV1/caveolin 1 enhances aerobic glycolysis in colon cancer cells via activation of SLC2A3/GLUT3 transcription. Autophagy. 2012; 8:1684–85. https://doi.org/10.4161/auto.21487. [PubMed].
23. Donato G, Martinez Hoyos J, Amorosi A, Maltese L, Lavano A, Volpentesta G, Signorelli F, Pentimalli F, Pallante P, Ferraro G, Tucci L, Signorelli CD, Viglietto G, Fusco A. High mobility group A1 expression correlates with the histological grade of human glial tumors. Oncol Rep. 2004; 11:1209–13. https://doi.org/10.3892/or.11.6.1209. [PubMed].
24. Wang Y, Hu L, Zheng Y, Guo L. HMGA1 in cancer: Cancer classification by location. J Cell Mol Med. 2019; 23:2293–302. https://doi.org/10.1111/jcmm.14082. [PubMed].
25. El-Far AH, Sroga G, Jaouni SKA, Mousa SA. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int J Mol Sci. 2020; 21:3613. https://doi.org/10.3390/ijms21103613. [PubMed].
26. Kim J, DeBerardinis RJ. Mechanisms and Implications of Metabolic Heterogeneity in Cancer. Cell Metab. 2019; 30:434–46. https://doi.org/10.1016/j.cmet.2019.08.013. [PubMed].
27. Gomes-Osman J, Cabral DF, Morris TP, McInerney K, Cahalin LP, Rundek T, Oliveira A, Pascual-Leone A. Exercise for cognitive brain health in aging: A systematic review for an evaluation of dose. Neurol Clin Pract. 2018; 8:257–65. https://doi.org/10.1212/CPJ.0000000000000460. [PubMed].
28. Chen JH, Lin X, Bu C, Zhang X. Role of advanced glycation end products in mobility and considerations in possible dietary and nutritional intervention strategies. Nutr Metab (Lond). 2018; 15:72. https://doi.org/10.1186/s12986-018-0306-7. [PubMed].
29. Wautier MP, Guillausseau PJ, Wautier JL. Activation of the receptor for advanced glycation end products and consequences on health. Diabetes Metab Syndr. 2017; 11:305–09. https://doi.org/10.1016/j.dsx.2016.09.009. [PubMed].
30. Kuniyasu H, Chihara Y, Kondo H. Differential effects between amphoterin and advanced glycation end products on colon cancer cells. Int J Cancer. 2003; 104:722–27. https://doi.org/10.1002/ijc.11016. [PubMed].
31. Waghela BN, Vaidya FU, Ranjan K, Chhipa AS, Tiwari BS, Pathak C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol Cell Biochem. 2021; 476:585–98. https://doi.org/10.1007/s11010-020-03928-y. [PubMed].
32. Panduru NM, Forsblom C, Saraheimo M, Thorn LM, Gordin D, Elonen N, Harjusalo V, Bierhaus A, Humpert PM, Groop PH, and FinnDiane Study Group. Urinary liver-type fatty acid binding protein is an independent predictor of stroke and mortality in individuals with type 1 diabetes. Diabetologia. 2017; 60:1782–90. https://doi.org/10.1007/s00125-017-4328-x. [PubMed].
33. Presta I, Vismara M, Novellino F, Donato A, Zaffino P, Scali E, Pirrone KC, Spadea MF, Malara N, Donato G. Innate Immunity Cells and the Neurovascular Unit. Int J Mol Sci. 2018; 19:3856. https://doi.org/10.3390/ijms19123856. [PubMed].
34. Peppa M, Uribarri J, Vlassara H. Aging and glycoxidant stress. Hormones (Athens). 2008; 7:123–32. https://doi.org/10.1007/BF03401503. [PubMed].
35. Thelin EP, Tajsic T, Zeiler FA, Menon DK, Hutchinson PJA, Carpenter KLH, Morganti-Kossmann MC, Helmy A. Monitoring the Neuroinflammatory Response Following Acute Brain Injury. Front Neurol. 2017; 8:351. https://doi.org/10.3389/fneur.2017.00351. [PubMed].
36. Sparvero LJ, Asafu-Adjei D, Kang R, Tang D, Amin N, Im J, Rutledge R, Lin B, Amoscato AA, Zeh HJ, Lotze MT. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med. 2009; 7:17. https://doi.org/10.1186/1479-5876-7-17. [PubMed].
37. Onyeagucha BC, Mercado-Pimentel ME, Hutchison J, Flemington EK, Nelson MA. S100P/RAGE signaling regulates microRNA-155 expression via AP-1 activation in colon cancer. Exp Cell Res. 2013; 319:2081–90. https://doi.org/10.1016/j.yexcr.2013.05.009. [PubMed].
38. López-Díez R, Rastrojo A, Villate O, Aguado B. Complex tissue-specific patterns and distribution of multiple RAGE splice variants in different mammals. Genome Biol Evol. 2013; 5:2420–35. https://doi.org/10.1093/gbe/evt188. [PubMed].
39. Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004; 113:1641–50. https://doi.org/10.1172/JCI18704. [PubMed].
40. Gefter JV, Shaufl AL, Fink MP, Delude RL. Comparison of distinct protein isoforms of the receptor for advanced glycation end-products expressed in murine tissues and cell lines. Cell Tissue Res. 2009; 337:79–89. https://doi.org/10.1007/s00441-009-0791-0. [PubMed].
41. Jaskiewicz M, Conrath U, Peterhänsel C. Chromatin modification acts as a memory for systemic acquired resistance in the plant stress response. EMBO Rep. 2011; 12:50–55. https://doi.org/10.1038/embor.2010.186. [PubMed].
42. El-Far AH, Munesue S, Harashima A, Sato A, Shindo M, Nakajima S, Inada M, Tanaka M, Takeuchi A, Tsuchiya H, Yamamoto H, Shaheen HM, El-Sayed YS, et al. In vitro anticancer effects of a RAGE inhibitor discovered using a structure-based drug design system. Oncol Lett. 2018; 15:4627–34. https://doi.org/10.3892/ol.2018.7902. [PubMed].
43. Chen H, Wu L, Li Y, Meng J, Lin N, Yang D, Zhu Y, Li X, Li M, Xu Y, Wu Y, Tong X, Su Q. Advanced glycation end products increase carbohydrate responsive element binding protein expression and promote cancer cell proliferation. Mol Cell Endocrinol. 2014; 395:69–78. https://doi.org/10.1016/j.mce.2014.07.021. [PubMed].
44. Tong D, Rozas NS, Oakley TH, Mitchell J, Colley NJ, McFall-Ngai MJ. Evidence for light perception in a bioluminescent organ. Proc Natl Acad Sci U S A. 2009; 106:9836–41. https://doi.org/10.1073/pnas.0904571106. [PubMed].
45. Zill H, Günther R, Erbersdobler HF, Fölsch UR, Faist V. RAGE expression and AGE-induced MAP kinase activation in Caco-2 cells. Biochem Biophys Res Commun. 2001; 288:1108–11. https://doi.org/10.1006/bbrc.2001.5901. [PubMed].
46. Malara N, Gentile F, Coppedè N, Coluccio ML, Candeloro P, Perozziello G, Ferrara L, Giannetto M, Careri M, Castellini A, Mignogna C, Presta I, Pirrone CK, et al. Superhydrophobic lab-on-chip measures secretome protonation state and provides a personalized risk assessment of sporadic tumour. NPJ Precis Oncol. 2018; 2:26. https://doi.org/10.1038/s41698-018-0069-7. [PubMed].
47. Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 proteins. Curr Mol Med. 2013; 13:24–57. https://doi.org/10.2174/156652413804486214. [PubMed].
48. Bresnick AR, Weber DJ, Zimmer DB. S100 proteins in cancer. Nat Rev Cancer. 2015; 15:96–109. https://doi.org/10.1038/nrc3893. [PubMed].
49. Farokhzadian J, Mangolian Shahrbabaki P, Bagheri V. S100A12-CD36 axis: A novel player in the pathogenesis of atherosclerosis? Cytokine. 2019; 122:154104. https://doi.org/10.1016/j.cyto.2017.07.010. [PubMed].
50. Donato R. RAGE: a single receptor for several ligands and different cellular responses: the case of certain S100 proteins. Curr Mol Med. 2007; 7:711–24. https://doi.org/10.2174/156652407783220688. [PubMed].
51. Ichikawa M, Williams R, Wang L, Vogl T, Srikrishna G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol Cancer Res. 2011; 9:133–48. https://doi.org/10.1158/1541-7786.MCR-10-0394. [PubMed].
52. Fuentes MK, Nigavekar SS, Arumugam T, Logsdon CD, Schmidt AM, Park JC, Huang EH. RAGE activation by S100P in colon cancer stimulates growth, migration, and cell signaling pathways. Dis Colon Rectum. 2007; 50:1230–40. https://doi.org/10.1007/s10350-006-0850-5. [PubMed].
53. Sun X, Wang T, Zhang C, Ning K, Guan ZR, Chen SX, Hong TT, Hua D. S100A16 is a prognostic marker for colorectal cancer. J Surg Oncol. 2018; 117:275–83. https://doi.org/10.1002/jso.24822. [PubMed].
54. Sapkota D, Costea DE, Ibrahim SO, Johannessen AC, Bruland O. S100A14 interacts with S100A16 and regulates its expression in human cancer cells. PLoS One. 2013; 8:e76058. https://doi.org/10.1371/journal.pone.0076058. [PubMed].
55. Wang HY, Zhang JY, Cui JT, Tan XH, Li WM, Gu J, Lu YY. Expression status of S100A14 and S100A4 correlates with metastatic potential and clinical outcome in colorectal cancer after surgery. Oncol Rep. 2010; 23:45–52. [PubMed].
56. Sakic A, Chaabane C, Ambartsumian N, Klingelhöfer J, Lemeille S, Kwak BR, Grigorian M, Bochaton-Piallat ML. Neutralization of S100A4 induces stabilization of atherosclerotic plaques: role of smooth muscle cells. Cardiovasc Res. 2020 Nov 2. https://doi.org/10.1093/cvr/cvaa311. [Epub ahead of print]. [PubMed]
57. Zhang J, Jiao Y, Hou S, Tian T, Yuan Q, Hao H, Wu Z, Bao X. S100A4 contributes to colitis development by increasing the adherence of Citrobacter rodentium in intestinal epithelial cells. Sci Rep. 2017; 7:12099. https://doi.org/10.1038/s41598-017-12256-z. [PubMed].
58. Iseki Y, Shibutani M, Maeda K, Nagahara H, Fukuoka T, Matsutani S, Kashiwagi S, Tanaka H, Hirakawa K, Ohira M. A new method for evaluating tumor-infiltrating lymphocytes (TILs) in colorectal cancer using hematoxylin and eosin (H-E)-stained tumor sections. PLoS One. 2018; 13:e0192744. https://doi.org/10.1371/journal.pone.0192744. [PubMed].
59. Coluccio ML, Gentile F, Presta I, Donato G, Coppedè N, Valprapuram I, Mignogna C, Lavecchia A, Figuccia F, Garo VM, Fabrizio ED, Candeloro P, Viglietto G, Malara N. Tailoring Chemometric Models on Blood-Derived Cultures Secretome to Assess Personalized Cancer Risk Score. Cancers (Basel). 2020; 12:1362. https://doi.org/10.3390/cancers12061362. [PubMed].
60. VanPatten S, Al-Abed Y. High Mobility Group Box-1 (HMGb1): Current Wisdom and Advancement as a Potential Drug Target. J Med Chem. 2018; 61:5093–107. https://doi.org/10.1021/acs.jmedchem.7b01136. [PubMed].
61. Lee HJ, Kim A, Song IH, Park IA, Yu JH, Ahn JH, Gong G. Cytoplasmic expression of high mobility group B1 (HMGB1) is associated with tumor-infiltrating lymphocytes (TILs) in breast cancer. Pathol Int. 2016; 66:202–09. https://doi.org/10.1111/pin.12393. [PubMed].
62. Mignogna C, Staropoli N, Botta C, De Marco C, Rizzuto A, Morelli M, Di Cello A, Franco R, Camastra C, Presta I, Malara N, Salvino A, Tassone P, et al. Aurora Kinase A expression predicts platinum-resistance and adverse outcome in high-grade serous ovarian carcinoma patients. J Ovarian Res. 2016; 9:31. https://doi.org/10.1186/s13048-016-0238-7. [PubMed].
63. Völp K, Brezniceanu ML, Bösser S, Brabletz T, Kirchner T, Göttel D, Joos S, Zörnig M. Increased expression of high mobility group box 1 (HMGB1) is associated with an elevated level of the antiapoptotic c-IAP2 protein in human colon carcinomas. Gut. 2006; 55:234–42. https://doi.org/10.1136/gut.2004.062729. [PubMed].
64. Zhu L, Li X, Chen Y, Fang J, Ge Z. High-mobility group box 1: a novel inducer of the epithelial-mesenchymal transition in colorectal carcinoma. Cancer Lett. 2015; 357:527–34. https://doi.org/10.1016/j.canlet.2014.12.012. [PubMed].
65. Huang CY, Chiang SF, Ke TW, Chen TW, Lan YC, You YS, Shiau AC, Chen WT, Chao KSC. Cytosolic high-mobility group box protein 1 (HMGB1) and/or PD-1+ TILs in the tumor microenvironment may be contributing prognostic biomarkers for patients with locally advanced rectal cancer who have undergone neoadjuvant chemoradiotherapy. Cancer Immunol Immunother. 2018; 67:551–62. https://doi.org/10.1007/s00262-017-2109-5. [PubMed].
66. Kang HJ, Lee H, Choi HJ, Youn JH, Shin JS, Ahn YH, Yoo JS, Paik YK, Kim H. Non-histone nuclear factor HMGB1 is phosphorylated and secreted in colon cancers. Lab Invest. 2009; 89:948–59. https://doi.org/10.1038/labinvest.2009.47. [PubMed].
67. Malara NM, Leotta A, Sidoti A, Lio S, D’Angelo R, Caparello B, Munao F, Pino F, Amato A. Ageing, hormonal behaviour and cyclin D1 in ductal breast carcinomas. Breast. 2006; 15:81–89. https://doi.org/10.1016/j.breast.2004.12.008. [PubMed].
68. Patlolla JM, Swamy MV, Raju J, Rao CV. Overexpression of caveolin-1 in experimental colon adenocarcinomas and human colon cancer cell lines. Oncol Rep. 2004; 11:957–63. https://doi.org/10.3892/or.11.5.957. [PubMed].
69. Zhu C, Zhang L, Zhao S, Dai W, Xu Y, Zhang Y, Zheng H, Sheng W, Xu Y. UPF1 promotes chemoresistance to oxaliplatin through regulation of TOP2A activity and maintenance of stemness in colorectal cancer. Cell Death Dis. 2021; 12:519. https://doi.org/10.1038/s41419-021-03798-2. [PubMed].
70. Jiao Y, Wang HC, Fan SJ. Growth suppression and radiosensitivity increase by HMGB1 in breast cancer. Acta Pharmacol Sin. 2007; 28:1957–67. https://doi.org/10.1111/j.1745-7254.2007.00669.x. [PubMed].
71. Yao X, Zhao G, Yang H, Hong X, Bie L, Liu G. Overexpression of high-mobility group box 1 correlates with tumor progression and poor prognosis in human colorectal carcinoma. J Cancer Res Clin Oncol. 2010; 136:677–84. https://doi.org/10.1007/s00432-009-0706-1. [PubMed].
72. Fahmueller YN, Nagel D, Hoffmann RT, Tatsch K, Jakobs T, Stieber P, Holdenrieder S. CA 15-3 is a predictive and prognostic biomarker in patients with metastasized breast cancer undergoing Selective Internal Radiation Therapy. Int J Clin Pharmacol Ther. 2013; 51:63–66. https://doi.org/10.5414/cpp51063. [PubMed].
73. Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, Geuking MB, Beutler B, Tedder TF, Hardt WD, Bercik P, Verdu EF, McCoy KD, Macpherson AJ. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science. 2009; 325:617–20. https://doi.org/10.1126/science.1172747. [PubMed].
74. Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur J Immunol. 2015; 45:17–31. https://doi.org/10.1002/eji.201444972. [PubMed].
75. Escamilla-Tilch M, Filio-Rodríguez G, García-Rocha R, Mancilla-Herrera I, Mitchison NA, Ruiz-Pacheco JA, Sánchez-García FJ, Sandoval-Borrego D, Vázquez-Sánchez EA. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol. 2013; 91:601–10. https://doi.org/10.1038/icb.2013.58. [PubMed].
76. Stephens M, Liao S, von der Weid PY. Mesenteric Lymphatic Alterations Observed During DSS Induced Intestinal Inflammation Are Driven in a TLR4-PAMP/DAMP Discriminative Manner. Front Immunol. 2019; 10:557. https://doi.org/10.3389/fimmu.2019.00557. [PubMed].
77. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity. 2016; 45:374–88. https://doi.org/10.1016/j.immuni.2016.07.009. [PubMed].
78. Medeiros MC, Frasnelli SC, Bastos ADS, Orrico SR, Rossa C Jr. Modulation of cell proliferation, survival and gene expression by RAGE and TLR signaling in cells of the innate and adaptive immune response: role of p38 MAPK and NF-KB. J Appl Oral Sci. 2014; 22:185–93. https://doi.org/10.1590/1678-775720130593. [PubMed].
79. Bongers G, Pacer ME, Geraldino TH, Chen L, He Z, Hashimoto D, Furtado GC, Ochando J, Kelley KA, Clemente JC, Merad M, van Bakel H, Lira SA. Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. J Exp Med. 2014; 211:457–72. https://doi.org/10.1084/jem.20131587. [PubMed].
80. Perozziello G, La Rocca R, Cojoc G, Liberale C, Malara N, Simone G, Candeloro P, Anichini A, Tirinato L, Gentile F, Coluccio ML, Carbone E, Di Fabrizio E. Microfluidic devices modulate tumor cell line susceptibility to NK cell recognition. Small. 2012; 8:2886–94. https://doi.org/10.1002/smll.201200160. [PubMed].
81. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014; 39:91–100. https://doi.org/10.1016/j.tibs.2013.12.004. [PubMed].
82. Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013; 144:1199–209.e4. https://doi.org/10.1053/j.gastro.2013.02.007. [PubMed].
83. Bierhaus A, Nawroth PP. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia. 2009; 52:2251–63. https://doi.org/10.1007/s00125-009-1458-9. [PubMed].
84. Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, Hess J, Angel P. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008; 205:275–85. https://doi.org/10.1084/jem.20070679. [PubMed].
85. Perozziello G, Simone G, Candeloro P, Gentile F, Malara N, Larocca R, Coluccio M, Pullano SA, Tirinato L, Geschke O, di Fabrizio E. A fluidic motherboard for multiplexed simultaneous and modular detection in microfluidic systems for biological application. Micro Nanosyst. 2010; 2:227–38. https://doi.org/10.2174/1876402911002040227.
86. Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol. 2013; 56:739–44. https://doi.org/10.1016/j.molimm.2013.07.008. [PubMed].
87. Anunobi R, Boone BA, Cheh N, Tang D, Kang R, Loux T, Lotze MT, Zeh HJ. Extracellular DNA promotes colorectal tumor cell survival after cytotoxic chemotherapy. J Surg Res. 2018; 226:181–91. https://doi.org/10.1016/j.jss.2018.02.042. [PubMed].
88. Vernon PJ, Loux TJ, Schapiro NE, Kang R, Muthuswamy R, Kalinski P, Tang D, Lotze MT, Zeh HJ 3rd. The receptor for advanced glycation end products promotes pancreatic carcinogenesis and accumulation of myeloid-derived suppressor cells. J Immunol. 2013; 190:1372–79. https://doi.org/10.4049/jimmunol.1201151. [PubMed].
89. Lee H, Song M, Shin N, Shin CH, Min BS, Kim HS, Yoo JS, Kim H. Diagnostic significance of serum HMGB1 in colorectal carcinomas. PLoS One. 2012; 7:e34318. https://doi.org/10.1371/journal.pone.0034318. [PubMed].
90. Feng M, Zhao Z, Yang M, Ji J, Zhu D. T-cell-based immunotherapy in colorectal cancer. Cancer Lett. 2021; 498:201–09. https://doi.org/10.1016/j.canlet.2020.10.040. [PubMed].
91. Liu W, Zhang Z, Zhang Y, Chen X, Guo S, Lei Y, Xu Y, Ji C, Bi Z, Wang K. HMGB1-mediated autophagy modulates sensitivity of colorectal cancer cells to oxaliplatin via MEK/ERK signaling pathway. Cancer Biol Ther. 2015; 16:511–17. https://doi.org/10.1080/15384047.2015.1017691. [PubMed].
92. Huang CY, Chiang SF, Chen WT, Ke TW, Chen TW, You YS, Lin CY, Chao KSC, Huang CY. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018; 9:1004. https://doi.org/10.1038/s41419-018-1019-6. [PubMed].
93. Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, Taguchi A, Yan SF, Yan SD, Stern DM, Schmidt AM. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci. 2002; 59:1117–28. https://doi.org/10.1007/s00018-002-8491-x. [PubMed].
94. Klein JC, Moses K, Zelinskyy G, Sody S, Buer J, Lang S, Helfrich I, Dittmer U, Kirschning CJ, Brandau S. Combined toll-like receptor 3/7/9 deficiency on host cells results in T-cell-dependent control of tumour growth. Nat Commun. 2017; 8:14600. https://doi.org/10.1038/ncomms14600. [PubMed].
95. Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000; 405:354–60. https://doi.org/10.1038/35012626. [PubMed].
96. Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med. 2016; 375:1767–78. https://doi.org/10.1056/NEJMra1514296. [PubMed].
97. Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, Yu Z, Yang J, Wang B, Sun H, Xia H, Man Q, Zhong W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018; 560:382–86. https://doi.org/10.1038/s41586-018-0392-8. [PubMed].
98. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003; 100:5336–41. https://doi.org/10.1073/pnas.0931259100. [PubMed].
99. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016; 27:409–16. https://doi.org/10.1093/annonc/mdv615. [PubMed].
100. Shan T, Chen S, Wu T, Yang Y, Li S, Chen X. PD-L1 expression in colon cancer and its relationship with clinical prognosis. Int J Clin Exp Pathol. 2019; 12:1764–69. [PubMed].
101. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014; 40:1–116. https://doi.org/10.1016/j.mam.2014.05.001. [PubMed].
102. Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, Kim T, Chang WC, Hsu JL, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016; 30:925–39. https://doi.org/10.1016/j.ccell.2016.10.010. [PubMed].
103. Onesto V, Cancedda L, Coluccio ML, Nanni M, Pesce M, Malara N, Cesarelli M, Di Fabrizio E, Amato F, Gentile F. Nano-topography Enhances Communication in Neural Cells Networks. Sci Rep. 2017; 7:9841. https://doi.org/10.1038/s41598-017-09741-w. [PubMed].
104. Zhou J, Li XL, Chen ZR, Chng WJ. Tumor-derived exosomes in colorectal cancer progression and their clinical applications. Oncotarget. 2017; 8:100781–90. https://doi.org/10.18632/oncotarget.20117. [PubMed].
105. Xie W, Xie L, Song X. The diagnostic accuracy of circulating free DNA for the detection of KRAS mutation status in colorectal cancer: A meta-analysis. Cancer Med. 2019; 8:1218–31. https://doi.org/10.1002/cam4.1989. [PubMed].
106. Malara N, Guzzi G, Mignogna C, Trunzo V, Camastra C, Della Torre A, Di Vito A, Lavecchia AM, Gliozzi M, Ceccotti C, Volpentesta G, Lavano A, Donato G, Mollace V. Non-invasive real-time biopsy of intracranial lesions using short time expanded circulating tumor cells on glass slide: report of two cases. BMC Neurol. 2016; 16:127. https://doi.org/10.1186/s12883-016-0652-x. [PubMed].
107. Malara N, Trunzo V, Foresta U, Amodio N, De Vitis S, Roveda L, Fava M, Coluccio M, Macrì R, Di Vito A, Costa N, Mignogna C, Britti D, et al. Ex-vivo characterization of circulating colon cancer cells distinguished in stem and differentiated subset provides useful biomarker for personalized metastatic risk assessment. J Transl Med. 2016; 14:133. https://doi.org/10.1186/s12967-016-0876-y. [PubMed].