
Introduction
Covalent modifications of histones sophisticatedly regulate chromatin architecture [1–4]. Of the histone modifications, histone lysine (K) methylation is crucial to the epigenetic and transcriptional regulation of gene expression. This modification is associated with either activation or silencing of gene expression, and its effect on gene expression is dependent on methylated lysine sites. For instance, methylations at histone H3 lysine 4 (H3K4), H3K36, and H3K79 are generally considered activation marks (or signals), whereas methylations at H3K9, H3K27, and H4K20 are known to be repressive marks. Lysine methylation can take place at three different levels (mono-, di- or trimethyl) at specific lysine residues within histones and is catalyzed by lysine methyltransferases (KMTs) [4, 5].
In particular, H3K4 methylation is critical for epigenetic and spatiotemporal activation of gene expression [6–10]. It can be catalyzed by the COMPASS (Complex of Proteins Associated with Set1) family of histone H3K4 methyltransferases, which is conserved from yeast to human and includes SET1A, SET1B, and KMT2A-D (MLL1-4) in mammals (e.g., mouse and human) [11] (Figure 1). Notably, KMT2D (also called MLL4, ALR, and MLL2) is the biggest H3K4 methyltransferase and is frequently mutated in many different types of cancer.
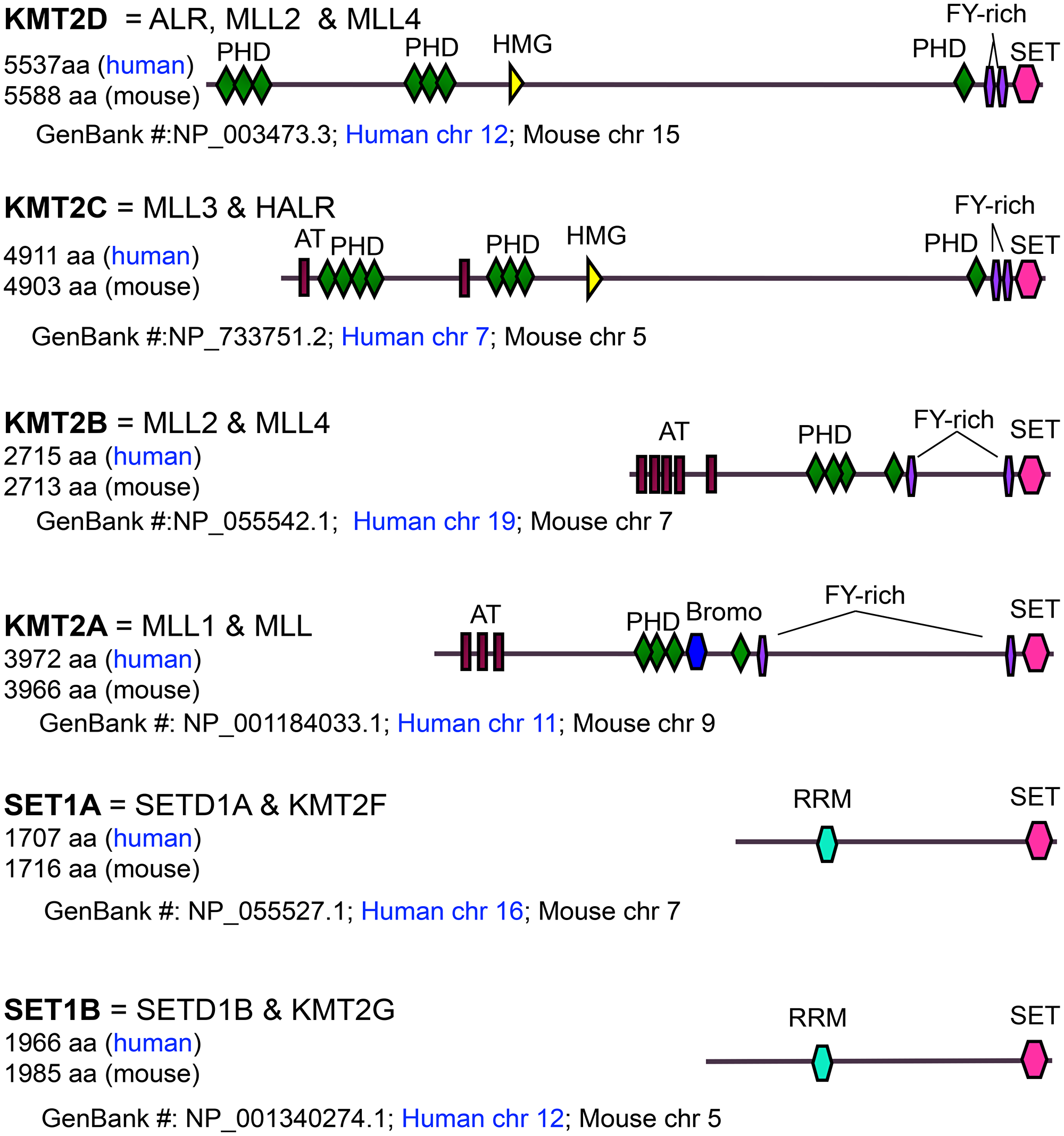
Figure 1: Domain organization, size, and chromosomal location of MLL/SET1 H3K4 methyltransferases. AT: AT-hook DNA binding domain; PHD: Plant Homeodomain; Bromo: Bromodomain; FYR: FY-rich domain; SET: Su(var)3-9, Enhancer of zeste, Trithorax domain; HMG: High Mobility Group domain; RRM: RNA Recognition Motif.
KMT2D’s nomenclature
KMT2D (human chromosome 12 and mouse chromosome 15) is different from KMT2B (also called MLL2 and MLL4) in both size and chromosomal location, although, confusingly, both of KMT2D and KMT2B have been called MLL2 (Figure 1).
KMT2D’s mutations in cancer
The KMT2D gene is mutated and deleted in many different types of cancer, including medulloblastoma, melanoma, lymphoma, leukemia, and lung, prostate, renal, bladder, ovarian, pancreatic, esophageal, and gastric cancers [12–28]. KMT2D has two frequently occurring genomic alterations: truncation and missense mutations (Figure 2). The frequency of these mutations varies according to cancer type. For example, according to The Cancer Genome Atlas database, melanoma contains much more missense mutations than truncations. In contrast, in medulloblastoma, most KMT2D mutations are truncations [29]. Most, if not all, of KMT2D truncations cause catalytic inactivity because the catalytic SET domain (5397aa‒5513aa) is located at the C-terminus of KMT2D (5537aa). Because KMT2D harbors truncating mutations in many types of cancer, KMT2D have been considered a tumor-suppressor in such instances. Of KMT2D’s missense mutations in lymphoma, several in the catalytic domain reduce KMT2D’s enzyme activity [30]. However, most missense mutations are located outside the catalytic SET domain in many different types of cancer, including lymphoma. It is unclear whether and how such KMT2D’s missense mutations affect its function, and more research in this area is needed
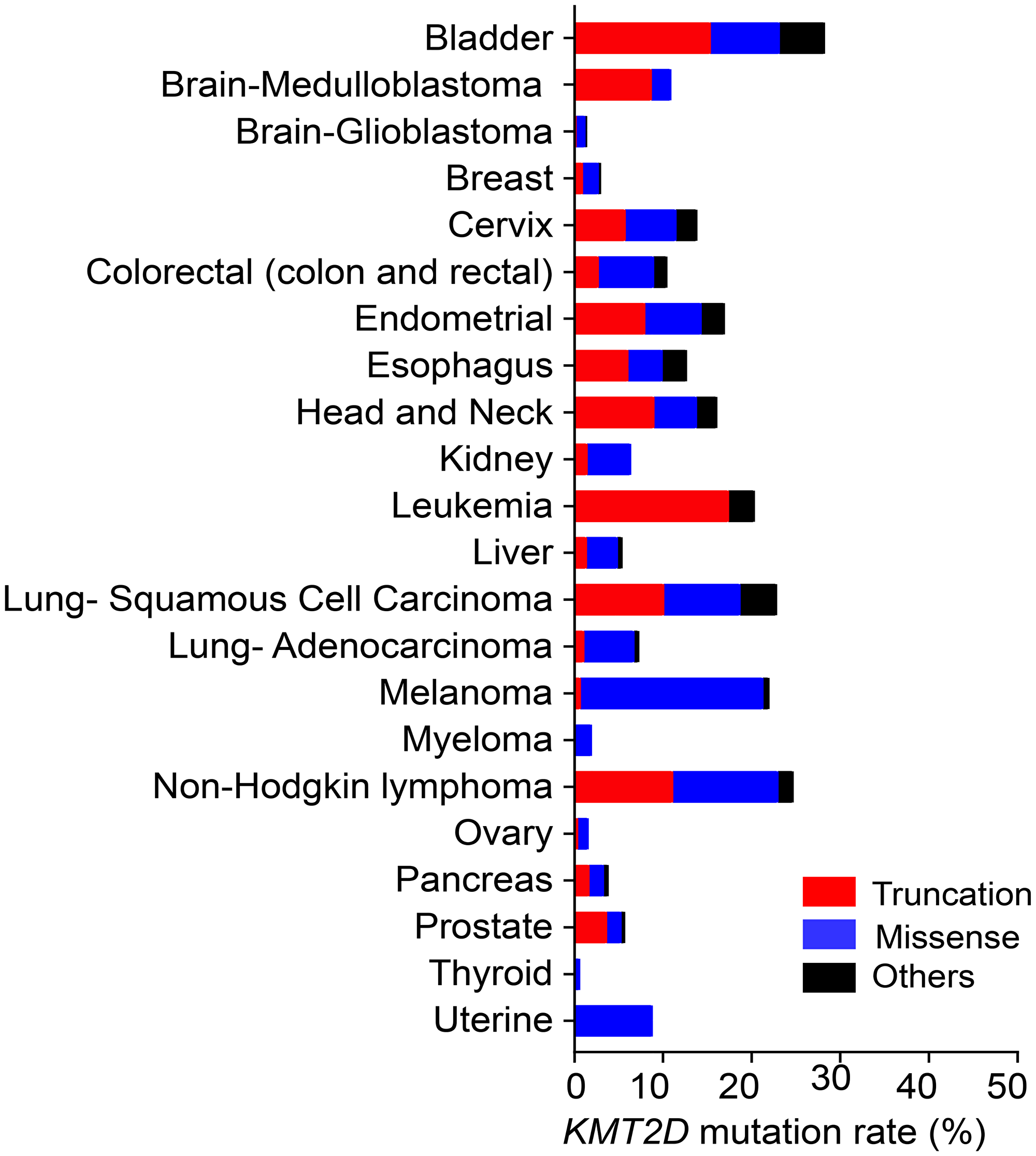
Figure 2: The frequency of truncating, missense, and other (inframe and splice-site) mutations in KMT2D in different types of cancer. The mutation frequencies for the cancer types listed, except medulloblastoma (Broad database), leukemia (St Jude database), and Non-Hodgkin lymphoma (Duke database), were obtained from data in TCGA PanCancer Atlas. All data were from the cBioportal database.
KMT2D as a tumor suppressor
Consistent with its loss-of-function mutations, KMT2D can act as a tumor suppressor (Table 1). We have shown that brain-specific loss of Kmt2d alone induces spontaneous medulloblastoma in 34.6% of brains in a genetically engineered mouse model (GEMM) [31]. Mechanistically, KMT2D upregulates expression of the tumor suppressor gene Dnmt3a whose protein, DNMT3A, downregulates expression of several oncogenic Ras activators to suppress medulloblastoma [31]. In addition, KMT2D activates expression of the tumor suppressor genes Bcl6 and Sirt1, whose proteins repress expression of several oncogenic Notch pathway genes to suppress medulloblastoma [31].
Table 1: The tumor-suppressive or pro-tumorigenic functions of KMT2D
| Cancer type | Experimental systems for tumorigenesis study | Anti-tumor or pro-tumor | KMT2D-regulated pathways | Ref |
|---|---|---|---|---|
| Bladder cancer | Cell lines & xenograft model | Anti-tumor | KMT2D → Tumor suppressor genes | [36] |
| Brain cancer (Medulloblastoma) | GEMM (Kmt2dfl/fl; Nestin-Cre), cerebellar neurospheres & mouse transplantation | Anti-tumor | KMT2D → Dnmt3a ┤ Ras activators KMT2D → Sirt1/Bcl6 ┤ Notch pathway | [31] |
| Cell lines | Pro-tumor | N/D | [47] | |
| Breast cancer | Cell lines | Pro-tumor | AKT-mediated phosphorylation of KMT2D ┤ KMT2D activity & ER function | [43] |
| Cell lines & xenograft model | Pro-tumor | KMT2D → Genes for tumor growth and cell invasion | [44] | |
| Esophageal cancer | Cell line | Pro-tumor | KMT2D → ? → Epithelial mesenchymal transition | [42] |
| Gastric cancer | Cell lines & xenograft model | Pro-tumor | KMT2D → ? → Phospho-AKT | [41] |
| Melanoma | GEMM (Kmt2dfl/fl ;Tyr-CreERT2, Rosa26-rtta; TetO-BRAFV600E; PTEN fl/fl; INK/ARF fl/fl), cell lines & xenograft model | Anti-tumor | KMT2D → IGFBP5 ┤ IGF1R signaling → Glycolysis genes | [33] |
| PDX models | Pro-tumor | KMT2D → Genes for tumor growth and cell migration | [45] | |
| Leukemia | GEMM (MLL-AF9–transduced Kmt2dfl/fl;Mx-Cre cells and transplantation) | Pro-tumor | KMT2D → Pro-tumorigenic HOXA9 target genes | [37] |
| GEMM (HOXA9/MEIS1–transduced Kmt2dfl/fl;ER-Cre cells and transplantation) | Pro-tumor | KMT2D → Antioxidant response genes | [38] | |
| Lung cancer | GEMM (Kmt2dfl/fl ;K-Ras LSL-G12D and intratracheal intubation of Ad5-CMV-Cre), cell lines & xenograft model | Anti-tumor | KMT2D → Circadian rhythm tumor suppressor gene Per2 ┤ Glycolysis genes | [32] |
| Lymphoma | GEMM (Kmt2dfl/fl;Cγ1-Cre; VavP-Bcl2) & B cells | Anti-tumor | KMT2D → ? ┤ Cell cycle and anti-apoptotic genes | [30] |
| GEMM (Kmt2dfl/fl;CD19-Cre and Kmt2dfl/fl;CD19-Cre; AID-Tg) & B cells | Anti-tumor | KMT2D → Tumor suppressor genes | [35] | |
| Pancreatic cancer | Cell lines & xenograft model | Anti-tumor | KMT2D → ? ┤ Glucose transporter SLC2A3 (alias GLUT3) | [34] |
| Cell lines | Pro-tumor | KMT2D → Cell cycle genes | [46] | |
| Prostate cancer | Cell lines & xenograft model | Pro-tumor | KMT2D → KLF4 and LIFR | [39] |
We also demonstrated that lung-specific loss of Kmt2d significantly promoted K-RasG12D–driven lung tumorigenesis by upregulating tumor-promoting programs [32]. Interestingly, KMT2D activates expression of the tumor suppressor Per2 and thereby downregulates expression of tumor-promoting glycolytic genes in K-RasG12D–driven lung tumors [32]. Similarly, Maitituoheti et al. [33] showed that Kmt2d loss promoted melanoma in a GEMM and that KMT2D downregulated expression of glycolytic genes by increasing expression of IGFBP5, a tumor suppressor and negative regulator of IGF1R signaling. In a study of pancreatic cancer, Koutsioumpa et al. [34] showed that KMT2D levels were downregulated in pancreatic tumors compared to normal pancreatic tissues and that KMT2D decreased glycolysis via downregulation of the glucose transporter SLC2A3 (alias GLUT3) to suppress tumorigenicity of pancreatic cancer cell lines.
Studies have shown that genetic ablation of Kmt2d or KMT2D knockdown accelerated the oncogene (Bcl2)-driven lymphomagenesis of B cells in mice [30, 35]. In addition, Kmt2d deletion in CD19+ early B cells induced spontaneous lymphoma in 58% of mice [35]. KMT2D suppressed lymphoma by activating pro-apoptotic genes while repressing genes associated with cell growth and survival pathways, such as cell cycle and anti-apoptotic genes [30, 35]. A study of bladder cancer showed that KMT2D was required to maintain tumor suppressor genes and that it impeded the tumorigenicity and invasiveness of bladder cancer cells [36].
KMT2D’s pro-tumorigenic function
In contrast to its tumor-suppressive function, KMT2D appears to activate tumor-promoting pathways in certain contexts or tissue-specific manners (Table 1). Santos et al. [37] have shown that KMT2D upregulates gene expression programs for antioxidant responses to protect against reactive oxygen species and DNA damage. Thus, it is indispensable for leukemogenesis induced by the fusion oncogene MLL–AF9 in vivo. Another leukemia study showed that KMT2D occupied and activated pro-tumorigenic HOXA9 target genes and that KMT2D was required for leukemogenesis mediated by co-expression of HOXA9 and MEIS1 in vivo [38].
In addition, Lv et al. [39] have shown that KMT2D knockdown reduced the PI3K/AKT pathway to decrease the proliferation and invasion of prostate cancer cell lines. This study also showed that KMT2D activated expression of KLF4 (a transcription factor promoting cell invasion and LIFR expression) and LIFR (a receptor activating the PI3K/AKT pathway). Similarly, studies using gastric cancer cell lines showed that KMT2D knockdown reduced phospho-AKT signals and cell proliferation [40, 41]. In esophageal squamous cell carcinoma cells, KMT2D knockout inhibited cell proliferation and migration and reduced epithelial mesenchymal transition [42].
A study using estrogen receptor (ER)-positive breast cancer cell lines showed that KMT2D knockdown reduced cell viability and increased the sensitivity of xenograft tumors to a PI3Kα inhibitor. In addition, KMT2D played a critical role in recruiting ER to ER target genes and activating such genes [43]. In a breast cancer study, we showed that KMT2D knockdown inhibited cell proliferation and invasion of triple–negative breast cancer cell lines and that KMT2D and KMT2D-interacting histone demethylase UTX co-activated gene expression programs for cell proliferation and invasion [44].
In patient-derived xenograft (PDX) melanoma models, KMT2D was required for the growth and invasion of N-RAS-mutated melanoma and positively regulated genes for tumor growth and cell migration [45]. This finding appears to be in disagreement with the above-described melanoma study by Maitituoheti et al. [33], which showed a tumor-suppressive role for KMT2D. Dawkins et al. [46] reported that high KMT2D levels correlated with worse prognosis in patients with pancreatic cancer and that KMT2D positively regulated cell cycle genes in several pancreatic cancer cell lines. This conclusion appears to be discordant with the aforementioned pancreatic study by Koutsioumpa et al. [34], although these two studies used different pancreatic cancer cell lines. Guo et al. [47] showed that KMT2D deficiency attenuated the proliferative and migratory ability of medulloblastoma cell lines and others. This is also in discrepancy with ours, which showed medulloblastoma-suppressive function of KMT2D using a GEMM [31]. In these studies, although the same types of cancer were studied, KMT2D could be anti-tumorigenic or pro-tumorigenic, depending on the model systems used (cell lines, PDX models, and GEMM). These discrepancies are further discussed in the following section.
KMT2D: tumor suppressor vs pro-tumorigenic functions
An important question is how KMT2D can have tumor-suppressive or pro-tumorigenic function for a specific tissue, depending on whether the model system is a GEMM, a human cancer cell line, or a PDX model? GEMM models can harbor tumors that result from a few defined genetic alterations, whereas human cancer cell lines and PDX models often harbor multiple mutations. Such mutations may render a cancer cell line or PDX tumor dependent on KMT2D for survival. In such cases, KMT2D deficiency can attenuate the proliferation of affected cell lines or the growth of PDX tumors, even if KMT2D is a tumor suppressor. This point can be supported by the notion that simultaneous inactivation of two tumor suppressor genes, such as BRCA1 and Poly(ADP-ribose) polymerase, leads to synthetic lethality (cell death) [48]. Another point is that human cell lines, xenograft tumors, and PDX tumors are usually grown in immunodeficient conditions. In contrast, the autochthonous growth of tumors in GEMMs occurs in native microenvironments. These microenvironments may involve intact immune systems that would better support the growth of KMT2D-deficient tumors than that of KMT2D-wild type (WT) tumors. Supporting this point, it was shown that KMT2D-deficient syngeneic tumors were more sensitive to immunotherapy than were KMT2D-WT tumors [49] and that immune response pathways were more enriched in KMT2D-deficient melanoma than in KMT2D-WT melanoma [33].
Another critical question is how KMT2D can act as a tumor suppressor or a pro-tumorigenic factor in a tissue-dependent manner. One possibility is that tissue-specific transcription factors direct the KMT2D complex to a unique set of tumor-promoting genes or tumor suppressor genes in a tissue-dependent manner. For example, KMT2D associates with the tumorigenic factor HOXA9 during leukemogenesis [38]. Relevant to this question, the KMT2D complex can associate with lineage-specific DNA-binding transcription factors. Studies have shown that p63 interacts with KMT2D, which occupies and establishes enhancers for p63 target genes in keratinocytes [50] and that MyoD associates with the KMT2D complex to induce myocyte differentiation [51]. In addition, PPARγ and C/EBP interact with the KMT2D complex to induce adipocyte differentiation [51]. Another possibility—not mutually exclusive with the above possibility—is that post-translational modifications may regulate KMT2D function in a tissue-dependent manner.
KMT2D-catalyzed H3K4 methylation in cancer cells
KMT2D was initially identified as ALR in a multi-protein complex containing activating signal cointegrator 2 (ASC2) and other proteins [52]. Later, we showed that the KMT2D complex included the H3K27 demethylase UTX, PTIP, and other subunits (e.g., RBBP5 and WDR5) [53]. In parallel, other groups found these subunits in the KMT2D complex [54, 55]. We demonstrated that immunoprecipitates containing full-length or minimal versions of KMT2D can catalyze H3K4me1, H3K4me2, and H3K4me3 in vitro [53, 56]. In addition, recombinant core subunits of KMT2D (WDR5, ASH2L, RBBP5 and DPY30) conferred H3K4 tri-methyltransferase activity on a recombinant KMT2D fragment containing the catalytic domain SET, which has a monomethyltransferase H3K4 activity along with a much weaker dimethyltransferase activity [57]. Therefore, KMT2D is capable of catalyzing H3K4m1, H3K4me2, and H3K4me3.
Interestingly, it has been shown that KMT2D acts as a H3K4 mono-methyltransferase or a H3K4 mono- and dimethyltransferase in mammalian cells [51, 58, 59]. In these studies, the effects of KMT2D loss on H3K4 methylation were examined using mammalian cells with MLL3-null states (e.g., human HCT116 colon cancer cells, mouse brown adipocytes, and mouse embryonic stem cells) because KMT2D and MLL3 (homologues of the Drosophila H3K4 methyltransferase Trr) are closely related to each other. Our study also showed that KMT2D loss in K-RasG12D–driven lung tumors reduced global H3K4me1 levels while not having an obvious effect on global H3K4me3 levels [32].
In addition to KMT2D-catalyzed mono- and dimethylation of H3K4, KMT2D-mediated H3K4 trimethylation has been reported in other cell lines and animal models, consistent with the in vitro activity of KMT2D. We have shown that loss of the Kmt2d gene in the mouse brain reduced H3K4me1 and H3K4me3 levels in many genes during medulloblastoma genesis. We have also reported that KMT2D activates gene expression by upregulating H3K4me3 at gene promoters in breast cancer cells and in the neuron-committed human embryonal carcinoma cell line NT2/D1 [44, 56]. Consistent with this, Kmt2d loss in mouse B cells during lymphomagenesis decreases global H3K4me1, H3K4me2, and H3K4me3 levels [30].
These studies indicate that KMT2D monomethylates H3K4 and can trimethylate H3K4 in a cell-type-specific manner. Although it is not clear how KMT2D–catalyzed H3K4 trimethylation is regulated in a cell-type-specific manner, one possibility is that the distinct composition or expression patterns of the core subunits WDR5, ASH2L, RBBP5, and DPY30 in different cell types play a role in KMT2D-mediated H3K4 trimethylation. Future studies may determine the detailed mechanism underlying KMT2D-mediated H3K4 trimethylation.
KMT2D-mediated regulation of enhancers and super-enhancers
Enhancers are a critical gene-activating signature that spatiotemporally enhances gene expression by interacting with and activating the promoters. Monomethyl H3K4 (H3K4me1) and acetyl H3K27 (H3K27ac) decorate the enhancers, which are co-occupied by transcriptional activators and co-activators (e.g., the histone acetyltransferases p300 and CBP) [60, 61]. H3K27ac signifies active states of the enhancers and can be generated by p300 and CBP [62]. Large clusters of enhancers, called super-enhancers, highly activate gene expression and determine cell fate and cell identity [63–65]. In addition, super-enhancers can activate expression of oncogenes [66] and be associated with specific cancer subtypes [67].
It has been reported that KMT2D activates pro-tumorigenic enhancers in different types of cancer (Figure 3A). For instance, in N-RAS-mutated melanoma, KMT2D activated enhancers for the cell migration-associated genes MFGE8 and RPL39L [45]. In ER-positive breast cancer cells, SGK1-mediated phosphorylation of KMT2D attenuated the activity and chromatin occupancy of KMT2D to downregulate the enhancer signal H3K4me1 at oncogenes (e.g., MYC and cFOS) [68].
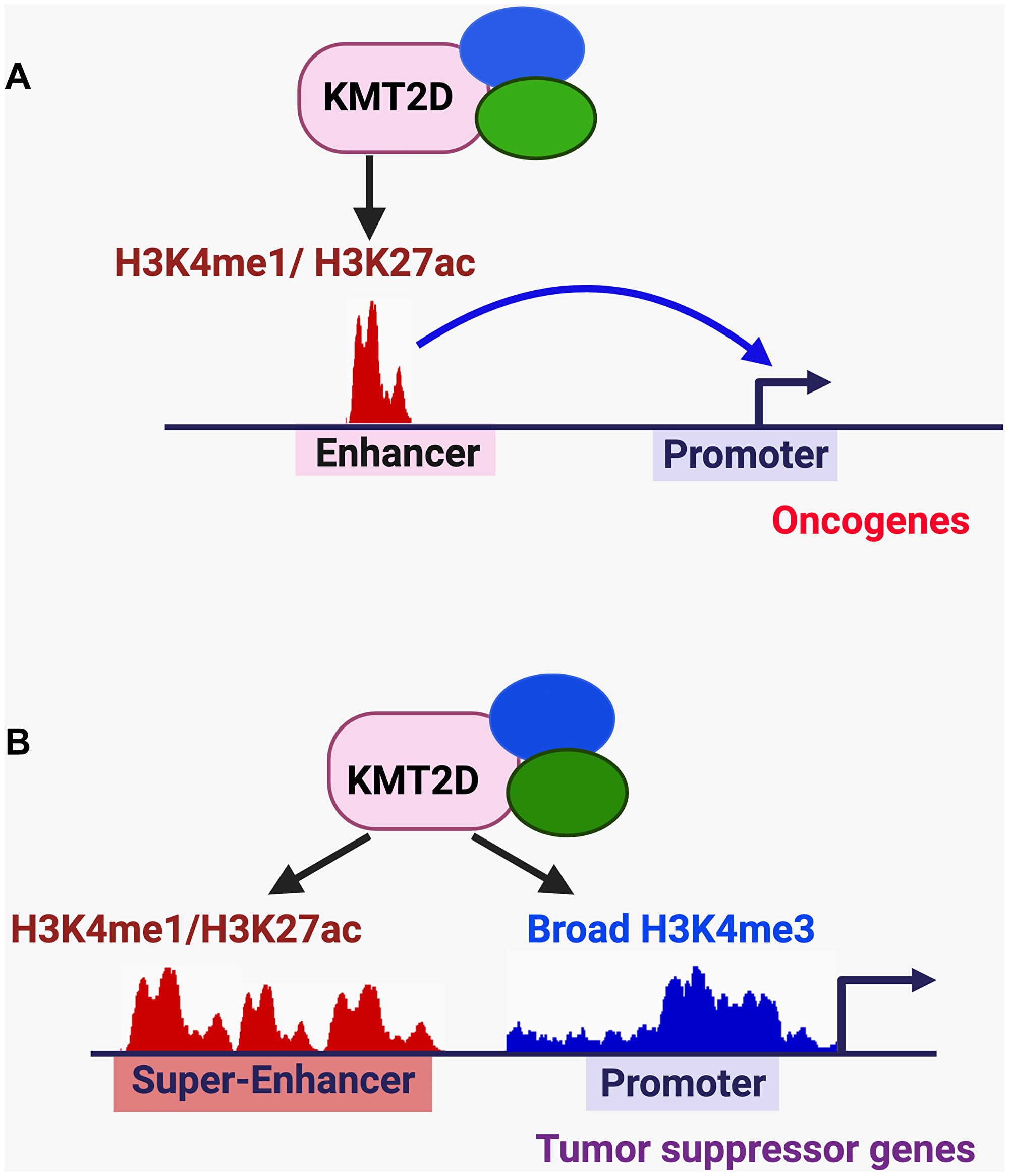
Figure 3: KMT2D-mediated regulation of enhancers, super-enhancers, and broad H3K4me3 signature in cancer. (A) KMT2D-mediated activation of pro-tumorigenic enhancers. (B) KMT2D-mediated activation of tumor-suppressive super-enhancers/enhancers and broad H3K4me3 peaks.
In contrast to oncogenic super-enhancers, some super-enhancers are associated with tumor suppressor genes (Figure 3B). We have shown that KMT2D-activated tumor-suppressive super-enhancers interact with the promoters of the DNA methyltransferase gene Dnmt3a and the medulloblastoma suppressor gene Bcl6 and upregulate these genes to suppress medulloblastoma genesis [31]. We have also shown that expression of the circadian transcription–repressive tumor suppressor Per2 is positively regulated by a KMT2D–activated, tumor-suppressive super-enhancer to attenuate lung tumorigenesis [32]. Moreover, KMT2D positively regulates the enhancer of the tumor suppressor gene IGFBP5 for melanoma suppression [33] and the enhancers of several tumor suppressor genes (e.g., Socs3, Asxl1 and Arid1A) for lymphoma suppression [35]. Therefore, KMT2D plays a critical role in activating tumor-suppressive enhancers and super-enhancers. Of note, KMT2D is also required for the positive regulation of enhancers and super-enhancers in other cellular processes, such as the exit from naive pluripotency [69], embryonic stem cell differentiation [59], and adipogenesis [70].
KMT2D-mediated regulation of the tumor-suppressive, broad H3K4me3 signature
H3K4me3 is generally located in gene-regulatory regions spanning the promoters and the transcription start sites [10, 71]. This mark is positively associated with the transcription frequency of active genes [7, 8], although the co-occupancy of H3K4me3 and the repressive mark H3K27me3 (called bivalent domains) represents poised (activatable but repressed) states [72, 73]. Remarkably, H3K4me3 occupies up to 79% of all protein-coding gene promoters in human cells, such as human embryonic stem cells [74–76]. Thus, it is one of most abundant and important histone marks.
Sharp and broad H3K4me3 peaks are two major types of H3K4me3 signatures. Sharp H3K4me3 peaks are a high and narrow signature that is enriched in the approximately 1-kb symmetric regions around the transcription start sites [77]. Broad H3K4me3 peaks (also called H3K4me3 breadth) [78] cover at least from -500 bp to +3,500 bp and signify highly active genes. Broad H3K4me3 peaks are associated with cell identity and tumor suppression, are reduced in cancer cells, and inversely correlate with DNA methylation [77, 78]. We have previously demonstrated that homozygous kmt2d loss in the brain downregulates broad H3K4me3 peaks to decrease expression of tumor suppressor genes (Figure 3B) [31]. In contrast, we showed that homozygous kmt2d loss in K-RasG12D–driven lung tumors did not obviously change the average size of H3K4me3 peaks [32]. These studies suggest that KMT2D positively regulates broad H3K4me3 peaks in a tissue-dependent manner.
KMT2D-regulating pathways
KMT2D function is regulated in several different mechanisms (Figure 4A–4D). The kinases AKT and SGK1 interact with and phosphorylate KMT2D to reduce its methyltransferase activity, which is important for ER’s tumor-promoting function in ER-positive breast cancer cell lines [43, 68]. It was reported that miR-217 promoted the proliferation and migration of bladder cancer cells by targeting KMT2D and that miR-217 levels were negatively correlated with KMT2D expression in bladder cancer samples [79]. KMT2D expression levels can be downregulated by DNA CpG methylation in pancreatic cancer cells [34]. Saffie et al. [80] have shown that the E3 ligase FBXW7 interacts with KMT2D and promotes KMT2D degradation to increase the proliferation of diffuse large B-cell lymphoma cells.
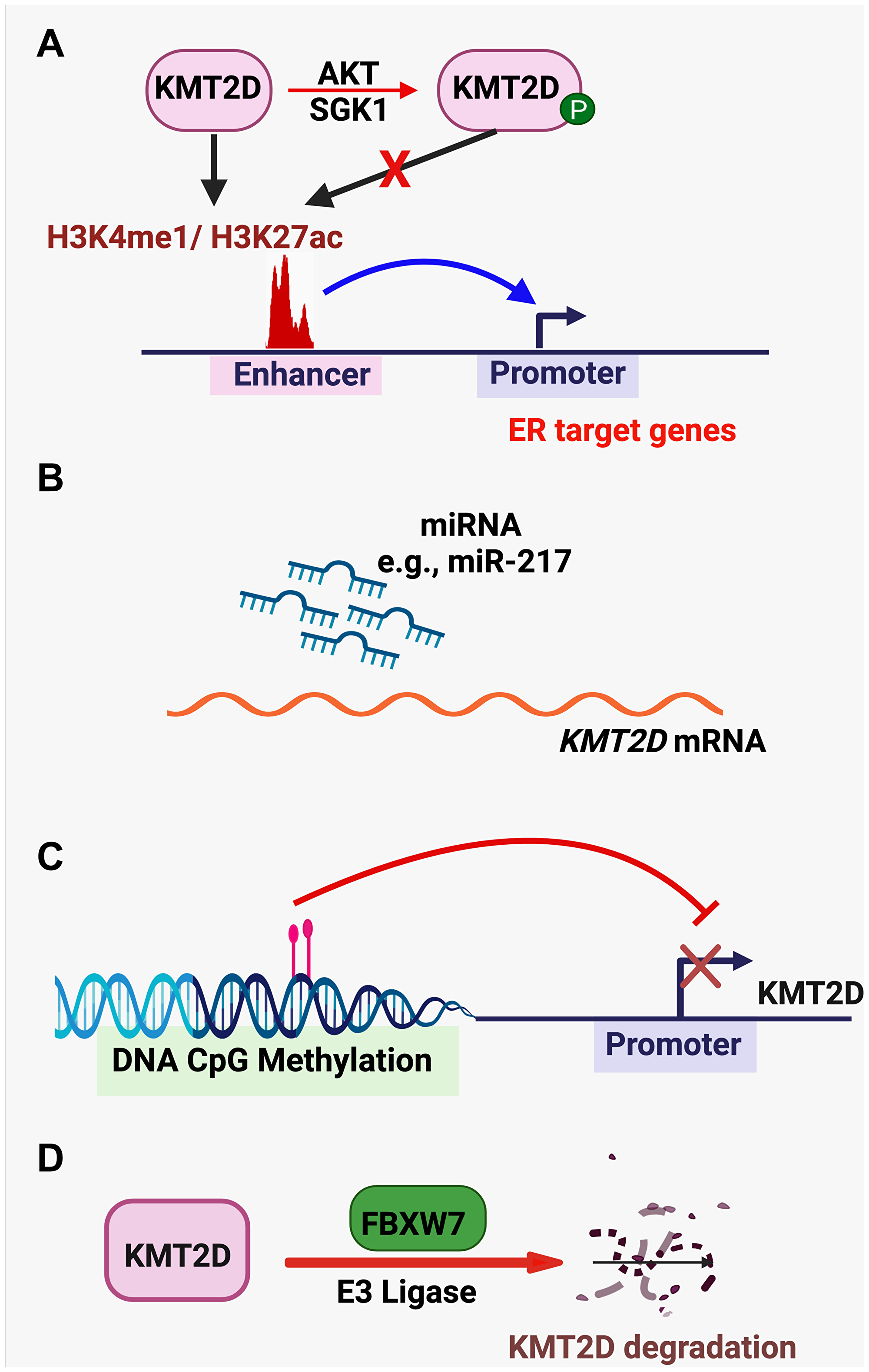
Figure 4: KMT2D-regulating pathways. (A) The kinases AKT and SGK1 phosphorylate KMT2D to reduce its methyltransferase activity. (B) miRNAs (e.g., miR-217) targets KMT2D mRNA to reduce KMT2D expression. (C) KMT2D expression is repressed by DNA CpG methylation. (D) The E3 ligase FBXW7 targets KMT2D for protein degradation.
The clonality and therapeutic implications of KMT2D’s mutations
If gene mutations occur as early events and are present in all cancer cells during tumorigenesis, such mutations are clonal (truncal). The clonality of mutations is important because subclonal (branched) alterations may exist in only a subset of cancer cells and thus limit treatment efficacy if tumors bearing such mutations are treated [81]. If mutations of a gene are clonal, the resultant gene products or their downstream effectors can be candidate actionable targets for cancer treatment. A study has shown that the majority of KMT2D mutations (e.g., 6 out of 9 mutations examined) in non-small cell lung cancer were clonal [82]. Moreover, all KMT2D mutations examined in esophageal tumors was clonal [83]. These studies would justify pharmacological targeting of oncogenic pathways altered by KMT2D deficiency (KMT2D truncation or loss) for the treatment of non-small cell lung tumors and esophageal tumors bearing KMT2D mutations. For other types of cancer, the clonality of KMT2D mutations remains to be investigated.
Therapeutic opportunities for the treatment of KMT2D-deficient tumors
Consistent with our finding that Kmt2d loss increases glycolysis in lung tumorigenesis, [32], we showed that pharmacological inhibition of glycolysis using 2-deoxy-D-glucose impeded the proliferation of human lung cancer cell lines bearing KMT2D truncations to a greater extent than that of KMT2D-WT lung cancer cell lines and selectively inhibited the tumorigenicity of KMT2D-deficient lung cancer cells in mice [32]. Similarly, pharmacological inhibition of the glycolysis pathway using 2-deoxy-D-glucose, POMHEX (an enolase1 inhibitor), and lonidamine (a hexokinase inhibitor) selectively impeded the proliferation of KMT2D-mutant melanoma cells compared with that of KMT2D WT melanoma cells [33]. However, the OXPHOS inhibitor IACS-010759 did not have a significant selective inhibitory effect on the proliferation of KMT2D-deficient cancer cells compared to that of KMT2D-WT cancer cells [32, 33].
Interestingly, pharmacological inhibition of the KDM5 family of H3K4 demethylases diminished tumorigenic growth of KMT2D-mutant lymphoma cells, as certain KDM5 family members can antagonize the function and methylation activity of KMT2D [84]. Other study has shown that KMT2D deficiency sensitizes different types of syngeneic tumors (including lung tumor, bladder tumor, breast tumor, and melanoma) to anti-PD1 [49], suggesting that anti-PD1 may have a stronger inhibitory effect on KMT2D-mutant tumors than on KMT2D-WT tumors. In addition, KMT2D deficiency augments the tumor-inhibitory effect of the curcumin analog L48H37 on pancreatic cancer cells [85] and increases the sensitivity of other cancer cells to chemotherapeutic drugs, such as doxorubicin, carboplatin, and the nucleotide analog Fluorouracil [46, 86]. Furthermore, KMT2D knockdown increases the sensitivity of head and neck squamous cell carcinoma cells to Aurora kinase inhibitors [87]. These studies indicate therapeutic opportunities for the treatment of KMT2D-deficient tumors.
Other aspects of KMT2D studies
As mentioned above, KMT2D has been described as either a tumorigenic factor or a tumor suppressor on the basis of results from the use of human cancer cell lines and their xenograft tumor models. Such results have also provided mechanistic insights into how KMT2D regulates tumorigenicity. However, because experiments using human cancer cell lines do not use native tumor microenvironments and human cancer cell lines often harbor multiple uncharacterized mutations, future studies using genetically defined mouse models (e.g., GEMMs) are desirable. This approach may allow researchers to assess whether results from cancer cell line experiments would be reproduced using GEMM experiments.
Histone modifiers often posttranslationally modify non-histone substrates relevant to tumorigenesis. For instance, the histone lysine methyltransferase SMYD3 methylates the kinase MAP3K2 to upregulate MAP kinase signaling and accelerate K-RasG12D-driven tumorigenesis [88]. It is possible that KMT2D-mediated methylation alters the functions of certain oncogenic factors or tumor suppressors to regulate tumorigenesis. Therefore, in future studies, it would be interesting to determine whether KMT2D regulates oncogenic factors or tumor suppressors via methylation.
The use of standard chemotherapy medicines, small molecular inhibitors, and immune checkpoint inhibitors has provided survival benefits for cancer patients. To improve cancer treatment, drug-combination therapies have frequently been used. Thus, it would be useful to study drug-combination approaches as therapeutic strategies of the treatment of KMT2D-deficient tumors. Recently, Tazemetostat, which inhibits the histone methyltransferase EZH2, was approved as the first epigenetic drug of solid tumors (epithelioid sarcomas and follicular lymphoma). Other epigenetic inhibitors are in clinical trials for the treatment of different cancers [89]. Thus, targeting altered epigenetic modifiers and their downstream effectors is a viable and promising option for cancer treatment.
ACKNOWLEDGMENTS
We thank Laura L. Russell, scientific editor, Research Medical Library in UT MD Anderson Cancer Center, for editing this article. This work was supported by grants to MG Lee from the National Institutes of Health (NIH): R01 CA207109 and R01 CA207098. Figures were created with https://biorender.com. We sincerely apologize for not citing and summarizing a number of relevant articles due to space limitation.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293:1074–80. https://doi.org/10.1126/science.1063127. [PubMed].
2. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000; 403:41–45. https://doi.org/10.1038/47412. [PubMed].
3. Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002; 12:142–48. https://doi.org/10.1016/s0959-437x(02)00279-4. [PubMed].
4. Sims RJ 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003; 19:629–39. https://doi.org/10.1016/j.tig.2003.09.007. [PubMed].
5. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005; 6:838–49. https://doi.org/10.1038/nrm1761. [PubMed].
6. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007; 25:15–30. https://doi.org/10.1016/j.molcel.2006.12.014. [PubMed].
7. Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005; 122:517–27. https://doi.org/10.1016/j.cell.2005.06.026. [PubMed].
8. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–37. https://doi.org/10.1016/j.cell.2007.05.009. [PubMed].
9. Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008; 40:897–903. https://doi.org/10.1038/ng.154. [PubMed].
10. Strahl BD, Ohba R, Cook RG, Allis CD. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci U S A. 1999; 96:14967–72. https://doi.org/10.1073/pnas.96.26.14967. [PubMed].
11. Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012; 81:65–95. https://doi.org/10.1146/annurev-biochem-051710-134100. [PubMed].
12. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MDM, Miller CA, Welch JS, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333–39. https://doi.org/10.1038/nature12634. [PubMed].
13. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, Imielinski M, Hu X, Ling S, et al, Cancer Genome Atlas Research Network. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016; 48:607–16. https://doi.org/10.1038/ng.3564. [PubMed].
14. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012; 487:239–43. https://doi.org/10.1038/nature11125. [PubMed].
15. Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stütz AM, Korshunov A, Reimand J, Schumacher SE, Beroukhim R, Ellison DW, Marshall CR, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012; 488:49–56. https://doi.org/10.1038/nature11327. [PubMed].
16. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, Gallia GL, Jallo GI, Binder ZA, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011; 331:435–39. https://doi.org/10.1126/science.1198056. [PubMed].
17. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012; 488:106–10. https://doi.org/10.1038/nature11329. [PubMed].
18. Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stütz AM, Rausch T, Warnatz HJ, Ryzhova M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012; 488:100–05. https://doi.org/10.1038/nature11284. [PubMed].
19. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012; 488:43–48. https://doi.org/10.1038/nature11213. [PubMed].
20. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, et al. A landscape of driver mutations in melanoma. Cell. 2012; 150:251–63. https://doi.org/10.1016/j.cell.2012.06.024. [PubMed].
21. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011; 476:298–303. https://doi.org/10.1038/nature10351. [PubMed].
22. Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Döhner K, Armstrong SA. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia. 2012; 26:1881–83. https://doi.org/10.1038/leu.2012.56. [PubMed].
23. Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010; 463:360–63. https://doi.org/10.1038/nature08672. [PubMed].
24. Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011; 43:875–78. https://doi.org/10.1038/ng.907. [PubMed].
25. Hillman RT, Celestino J, Terranova C, Beird HC, Gumbs C, Little L, Nguyen T, Thornton R, Tippen S, Zhang J, Lu KH, Gershenson DM, Rai K, et al. KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary. Nat Commun. 2018; 9:2496. https://doi.org/10.1038/s41467-018-04950-x. [PubMed].
26. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, et al, Australian Pancreatic Cancer Genome Initiative. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016; 531:47–52. https://doi.org/10.1038/nature16965. [PubMed].
27. Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017; 541:169–75. https://doi.org/10.1038/nature20805. [PubMed].
28. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–09. https://doi.org/10.1038/nature13480. [PubMed].
29. Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, Kool M, Jones DT, Unterberger A, Morrissy AS, Shih D, Peacock J, Ramaswamy V, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013; 125:373–84. https://doi.org/10.1007/s00401-012-1070-9. [PubMed].
30. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K, Ge K, Dalla-Favera R, Pasqualucci L. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015; 21:1190–98. https://doi.org/10.1038/nm.3940. [PubMed].
31. Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V, Flores ER, Northcott PA, Rajaram V, et al. MLL4 Is Required to Maintain Broad H3K4me3 Peaks and Super-Enhancers at Tumor Suppressor Genes. Mol Cell. 2018; 70:825–41.e6. https://doi.org/10.1016/j.molcel.2018.04.028. [PubMed].
32. Alam H, Tang M, Maitituoheti M, Dhar SS, Kumar M, Han CY, Ambati CR, Amin SB, Gu B, Chen TY, Lin YH, Chen J, Muller FL, et al. KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer. Cancer Cell. 2020; 37:599–617.e7. https://doi.org/10.1016/j.ccell.2020.03.005. [PubMed].
33. Maitituoheti M, Keung EZ, Tang M, Yan L, Alam H, Han G, Singh AK, Raman AT, Terranova C, Sarkar S, Orouji E, Amin SB, Sharma S, et al. Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2D Mutant Melanoma. Cell Rep. 2020; 33:108293. https://doi.org/10.1016/j.celrep.2020.108293. [PubMed].
34. Koutsioumpa M, Hatziapostolou M, Polytarchou C, Tolosa EJ, Almada LL, Mahurkar-Joshi S, Williams J, Tirado-Rodriguez AB, Huerta-Yepez S, Karavias D, Kourea H, Poultsides GA, Struhl K, et al. Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut. 2019; 68:1271–86. https://doi.org/10.1136/gutjnl-2017-315690. [PubMed].
35. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, Lee JE, Chen HT, Ennishi D, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015; 21:1199–208. https://doi.org/10.1038/nm.3943. [PubMed].
36. Sun P, Wu T, Sun X, Cui Z, Zhang H, Xia Q, Zhang D. KMT2D inhibits the growth and metastasis of bladder Cancer cells by maintaining the tumor suppressor genes. Biomed Pharmacother. 2019; 115:108924. https://doi.org/10.1016/j.biopha.2019.108924. [PubMed].
37. Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, Chen HT, Wong N, Finkel N, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014; 514:107–11. https://doi.org/10.1038/nature13483. [PubMed].
38. Sun Y, Zhou B, Mao F, Xu J, Miao H, Zou Z, Phuc Khoa LT, Jang Y, Cai S, Witkin M, Koche R, Ge K, Dressler GR, et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell. 2018; 34:643–58.e5. https://doi.org/10.1016/j.ccell.2018.08.018. [PubMed].
39. Lv S, Ji L, Chen B, Liu S, Lei C, Liu X, Qi X, Wang Y, Lai-Han Leung E, Wang H, Zhang L, Yu X, Liu Z, et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene. 2018; 37:1354–68. https://doi.org/10.1038/s41388-017-0026-x. [PubMed].
40. Li Q, Wu R, Wu F, Chen Q. KMT2D promotes proliferation of gastric cancer cells: evidence from ctDNA sequencing. J Clin Lab Anal. 2021; 35:e23721. https://doi.org/10.1002/jcla.23721. [PubMed].
41. Xiong W, Deng Z, Tang Y, Deng Z, Li M. Downregulation of KMT2D suppresses proliferation and induces apoptosis of gastric cancer. Biochem Biophys Res Commun. 2018; 504:129–36. https://doi.org/10.1016/j.bbrc.2018.08.143. [PubMed].
42. Abudureheman A, Ainiwaer J, Hou Z, Niyaz M, Turghun A, Hasim A, Zhang H, Lu X, Sheyhidin I. High MLL2 expression predicts poor prognosis and promotes tumor progression by inducing EMT in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2018; 144:1025–35. https://doi.org/10.1007/s00432-018-2625-5. [PubMed].
43. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, Baselga J. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017; 355:1324–30. https://doi.org/10.1126/science.aah6893. [PubMed].
44. Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, Lee MG. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014; 74:1705–17. https://doi.org/10.1158/0008-5472.CAN-13-1896. [PubMed].
45. Bossi D, Cicalese A, Dellino GI, Luzi L, Riva L, D'Alesio C, Diaferia GR, Carugo A, Cavallaro E, Piccioni R, Barberis M, Mazzarol G, Testori A, et al. In Vivo Genetic Screens of Patient-Derived Tumors Revealed Unexpected Frailty of the Transformed Phenotype. Cancer Discov. 2016; 6:650–63. https://doi.org/10.1158/2159-8290.CD-15-1200. [PubMed].
46. Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, Martin SA, Chelala C, Balkwill FR, Fitzgibbon J, Grose RP. Reduced Expression of Histone Methyltransferases KMT2C and KMT2D Correlates with Improved Outcome in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2016; 76:4861–71. https://doi.org/10.1158/0008-5472.CAN-16-0481. [PubMed].
47. Guo C, Chen LH, Huang Y, Chang CC, Wang P, Pirozzi CJ, Qin X, Bao X, Greer PK, McLendon RE, Yan H, Keir ST, Bigner DD, He Y. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget. 2013; 4:2144–53. https://doi.org/10.18632/oncotarget.1555. [PubMed].
48. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5:689–98. https://doi.org/10.1038/nrc1691. [PubMed].
49. Wang G, Chow RD, Zhu L, Bai Z, Ye L, Zhang F, Renauer PA, Dong MB, Dai X, Zhang X, Du Y, Cheng Y, Niu L, et al. CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 2020; 10:1912–33. https://doi.org/10.1158/2159-8290.CD-19-1448. [PubMed].
50. Lin-Shiao E, Lan Y, Coradin M, Anderson A, Donahue G, Simpson CL, Sen P, Saffie R, Busino L, Garcia BA, Berger SL, Capell BC. KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev. 2018; 32:181–93. https://doi.org/10.1101/gad.306241.117. [PubMed].
51. Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, Ge K. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife. 2013; 2:e01503. https://doi.org/10.7554/eLife.01503. [PubMed].
52. Goo YH, Sohn YC, Kim DH, Kim SW, Kang MJ, Jung DJ, Kwak E, Barlev NA, Berger SL, Chow VT, Roeder RG, Azorsa DO, Meltzer PS, et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol Cell Biol. 2003; 23:140–49. https://doi.org/10.1128/MCB.23.1.140-149.2003. [PubMed].
53. Lee MG, Villa R, Trojer P, Norman J, Yan KP, Reinberg D, Di Croce L, Shiekhattar R. Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science. 2007; 318:447–50. https://doi.org/10.1126/science.1149042. [PubMed].
54. Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007; 27:1889–903. https://doi.org/10.1128/MCB.01506-06. [PubMed].
55. Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem. 2007; 282:20395–406. https://doi.org/10.1074/jbc.M701574200. [PubMed].
56. Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012; 26:2749–62. https://doi.org/10.1101/gad.203356.112. [PubMed].
57. Zhang Y, Mittal A, Reid J, Reich S, Gamblin SJ, Wilson JR. Evolving Catalytic Properties of the MLL Family SET Domain. Structure. 2015; 23:1921–33. https://doi.org/10.1016/j.str.2015.07.018. [PubMed].
58. Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol. 2013; 33:4745–54. https://doi.org/10.1128/MCB.01181-13. [PubMed].
59. Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K. Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci U S A. 2016; 113:11871–76. https://doi.org/10.1073/pnas.1606857113. [PubMed].
60. Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM, Pennacchio LA. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009; 457:854–58. https://doi.org/10.1038/nature07730. [PubMed].
61. Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–12. https://doi.org/10.1038/nature07829. [PubMed].
62. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013; 49:825–37. https://doi.org/10.1016/j.molcel.2013.01.038. [PubMed].
63. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320–34. https://doi.org/10.1016/j.cell.2013.03.036. [PubMed].
64. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013; 153:307–19. https://doi.org/10.1016/j.cell.2013.03.035. [PubMed].
65. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013; 155:934–47. https://doi.org/10.1016/j.cell.2013.09.053. [PubMed].
66. Sur I, Taipale J. The role of enhancers in cancer. Nat Rev Cancer. 2016; 16:483–93. https://doi.org/10.1038/nrc.2016.62. [PubMed].
67. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz HJ, Worst BC, Ju B, Orr BA, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016; 530:57–62. https://doi.org/10.1038/nature16546. [PubMed].
68. Toska E, Castel P, Chhangawala S, Arruabarrena-Aristorena A, Chan C, Hristidis VC, Cocco E, Sallaku M, Xu G, Park J, Minuesa G, Shifman SG, Socci ND, et al. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep. 2019; 27:294–306.e5. https://doi.org/10.1016/j.celrep.2019.02.111. [PubMed].
69. Cao K, Collings CK, Morgan MA, Marshall SA, Rendleman EJ, Ozark PA, Smith ER, Shilatifard A. An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci Adv. 2018; 4:eaap8747. https://doi.org/10.1126/sciadv.aap8747. [PubMed].
70. Lai B, Lee JE, Jang Y, Wang L, Peng W, Ge K. MLL3/MLL4 are required for CBP/p300 binding on enhancers and super-enhancer formation in brown adipogenesis. Nucleic Acids Res. 2017; 45:6388–403. https://doi.org/10.1093/nar/gkx234. [PubMed].
71. Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc Natl Acad Sci U S A. 2002; 99:8695–700. https://doi.org/10.1073/pnas.082249499. [PubMed].
72. Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006; 125:315–26. https://doi.org/10.1016/j.cell.2006.02.041. [PubMed].
73. Dhar SS, Lee SH, Chen K, Zhu G, Oh W, Allton K, Gafni O, Kim YZ, Tomoiga AS, Barton MC, Hanna JH, Wang Z, Li W, Lee MG. An essential role for UTX in resolution and activation of bivalent promoters. Nucleic Acids Res. 2016; 44:3659–74. https://doi.org/10.1093/nar/gkv1516. [PubMed].
74. Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007; 130:77–88. https://doi.org/10.1016/j.cell.2007.05.042. [PubMed].
75. Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007; 1:299–312. https://doi.org/10.1016/j.stem.2007.08.003. [PubMed].
76. Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, Ruan Y, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007; 1:286–98. https://doi.org/10.1016/j.stem.2007.08.004. [PubMed].
77. Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, Shi X, Wang Q, Li W. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015; 47:1149–57. https://doi.org/10.1038/ng.3385. [PubMed].
78. Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, Hitz BC, Gupta R, Rando TA, et al. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014; 158:673–88. https://doi.org/10.1016/j.cell.2014.06.027. [PubMed].
79. Hou G, Xu W, Jin Y, Wu J, Pan Y, Zhou F. MiRNA-217 accelerates the proliferation and migration of bladder cancer via inhibiting KMT2D. Biochem Biophys Res Commun. 2019; 519:747–53. https://doi.org/10.1016/j.bbrc.2019.09.029. [PubMed].
80. Saffie R, Zhou N, Rolland D, Önder Ö, Basrur V, Campbell S, Wellen KE, Elenitoba-Johnson KSJ, Capell BC, Busino L. FBXW7 Triggers Degradation of KMT2D to Favor Growth of Diffuse Large B-cell Lymphoma Cells. Cancer Res. 2020; 80:2498–511. https://doi.org/10.1158/0008-5472.CAN-19-2247. [PubMed].
81. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, Cibulskis K, McKenna A, Chapman MA, et al, Multiple Myeloma Research Consortium. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014; 25:91–101. https://doi.org/10.1016/j.ccr.2013.12.015. [PubMed].
82. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, et al, and TRACERx Consortium. Tracking the Evolution of Non-Small-Cell Lung Cancer. N Engl J Med. 2017; 376:2109–21. https://doi.org/10.1056/NEJMoa1616288. [PubMed].
83. Hao JJ, Lin DC, Dinh HQ, Mayakonda A, Jiang YY, Chang C, Jiang Y, Lu CC, Shi ZZ, Xu X, Zhang Y, Cai Y, Wang JW, et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat Genet. 2016; 48:1500–07. https://doi.org/10.1038/ng.3683. [PubMed].
84. Heward JA, Koniali L, D'Avola A, Close K, Yeomans A, Philpott M, Dunford JE, Rahim T, Al Seraihi AF, Wang J, Korfi K, Araf S, Iqbal S, et al. KDM5 inhibition offers a novel therapeutic strategy for the treatment of KMT2D mutant lymphomas. Blood. 2021 Mar 30. https://doi.org/10.1182/blood.2020008743. [Epub ahead of print]. [PubMed]
85. Li SS, Jiang WL, Xiao WQ, Li K, Zhang YF, Guo XY, Dai YQ, Zhao QY, Jiang MJ, Lu ZJ, Wan R. KMT2D deficiency enhances the anti-cancer activity of L48H37 in pancreatic ductal adenocarcinoma. World J Gastrointest Oncol. 2019; 11:599–621. https://doi.org/10.4251/wjgo.v11.i8.599. [PubMed].
86. Lv S, Wen H, Shan X, Li J, Wu Y, Yu X, Huang W, Wei Q. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics. 2019; 14:1194–208. https://doi.org/10.1080/15592294.2019.1634985. [PubMed].
87. Kalu NN, Mazumdar T, Peng S, Tong P, Shen L, Wang J, Banerjee U, Myers JN, Pickering CR, Brunell D, Stephan CC, Johnson FM. Comprehensive pharmacogenomic profiling of human papillomavirus-positive and -negative squamous cell carcinoma identifies sensitivity to aurora kinase inhibition in KMT2D mutants. Cancer Lett. 2018; 431:64–72. https://doi.org/10.1016/j.canlet.2018.05.029. [PubMed].
88. Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, Tummino PJ, Kruger RG, Garcia BA, et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014; 510:283–87. https://doi.org/10.1038/nature13320. [PubMed].
89. Bennett RL, Licht JD. Targeting Epigenetics in Cancer. Annu Rev Pharmacol Toxicol. 2018; 58:187–207. https://doi.org/10.1146/annurev-pharmtox-010716-105106. [PubMed].