
Introduction
Acute myeloid leukemia (AML) is the second most common form of leukemia in adults, and has a very poor overall survival rate [1, 2]. For many years, chemotherapy based on the administration of anthracycline/cytarabine combinations has been the mainstay of AML treatment [3, 4]. More recently, advancements in next-generation sequencing technologies and understanding of genomic alterations involved in leukemogenesis have allowed the development of several novel targeted therapy approaches that complement the action of the currently available chemotherapeutic treatments. Among them are targeted drugs for specific mutations found in AML, such as the FLT3 inhibitors, IDH1/IDH2 inhibitors, pro-apoptotic agents, Hedgehog pathway inhibitors and others [5–7]. Even though these therapies have increased at variable degrees the response rates and survival benefit of subgroups of AML patients [8–11]; the development of resistance towards these novel drugs and subsequent relapse remains one of the major challenges for the treatment of this disease [12]. Therefore, there continues to be a need for new therapeutic modalities, including approaches targeting negative-feedback signaling pathways that may be activated in response to antileukemic treatments, leading to resistance.
The mitogen-activated protein kinase (MAPK) signaling pathway is a major regulatory cascade, activated in 70–80% of AML patients [13, 14]. MAPK-interacting kinases 1 and 2 (MNK1/2) are downstream effectors of this pathway, which control the activation of the eukaryotic translation initiation factor 4E (eIF4E) [15–17]. eIF4E has been demonstrated to be overexpressed in AML [18, 19] and has been linked to malignant cell transformation and proliferation [19–23]. The pro-neoplastic activity of eIF4E is associated with its phosphorylation/activation by MNK1/2 on serine 209 [24–26] and correlates with enhanced mRNA translation, as well as nuclear export of mRNAs involved in tumorigenesis and cell cycle control [26–31]. Several studies have shown that pharmacological targeting of MNK1/2 results in inhibitory activity against AML cells in pre-clinical models [32–37]. However, the extent of these effects has been frequently demonstrated using MNK1/2 inhibitors that lack high selectivity against MNK1/2 [32–37]. As a result, the full therapeutic potential of MNK1/2 inhibition for the treatment of AML has not been fully assessed. It should be noted that in addition to targeting eIF4E, MNK1/2 has been reported to phosphorylate other mRNA binding proteins such as the heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) [38] and the polypyrimidine tract-binding protein-associated splicing factor (PSF) [39]. However, only a few additional substrates of MNK1/2 have been identified beyond proteins involved in mRNA translation regulation, including cytosolic phospholipase A2 (cPLA2) [40] involved in arachidonate release, and Sprouty2 (Spry2) [41], which modulates signaling downstream of receptor tyrosine kinases. Therefore, identification of new components of the MNK1/2 pathway that could serve as potential targets may provide valuable information with therapeutic potential.
Tomivosertib, also known as eFT-508, is a potent, highly selective and orally bioavailable MNK1 and MNK2 inhibitor that is currently under investigation in Phase 1/2 clinical trials for the treatment of patients with advanced solid tumors and lymphomas [42]. This drug inhibits the activity of MNK1 and MNK2 with half-maximal inhibitory concentration (IC50) values of 2.4 nM and 1 nM; and has demonstrated potent anti-neoplastic effects against several cell lines and tumor models including diffuse large B-cell lymphoma (DLBCL), breast cancer, and lung cancer [42]. However, its potential activity against AML has not been explored. In this study, we investigated the efficacy of Tomivosertib in pre-clinical models of AML. We demonstrate that Tomivosertib suppresses eIF4E phosphorylation in AML cells and decreases leukemic cell survival and proliferation. We also provide evidence for synergy of Tomivosertib with Venetoclax, in vitro.
Results
Tomivosertib inhibits eIF4E phosphorylation and decreases leukemic cell survival and proliferation
Initially, we determined whether Tomivosertib blocks phosphorylation of the MNK effector eIF4E in AML cells. U937, MV411 and MM6 cells were treated with increasing doses of Tomivosertib for a period of 1 and 4 hours and eIF4E phosphorylation was assessed. Treatment with Tomivosertib abrogated the phosphorylation of eIF4E on serine 209 at a concentration of 0.1 μM, supporting potent inhibition of MNK1/2 activity in AML cells (Figure 1A–1C). To determine the potential antileukemic effects of Tomivosertib, we employed cellular viability assays as well as clonogenic assays in methylcellulose, using a panel of leukemia cell lines. Tomivosertib treatment resulted in dose-dependent inhibition of cellular viability in the AML lines tested. The compound showed the greatest suppressive activity against MV411, MM6 and KG-1 cells (Figure 1D–1F). Conversely, higher concentrations of the drug were required to suppress the viability of the myelomonocytic THP-1 and U937 cell lines (Figure 1G and 1H). A similar pattern was observed when the effects of Tomivosertib on leukemic progenitor cells were assessed. Treatment with Tomivosertib significantly inhibited KG-1-, MV411-, and MM6-derived leukemic progenitor (CFU-L) colony formation, while the effects were less pronounced on THP1- and U937-derived colony formation (Figure 2A–2E). Inhibitory effects were also seen on primary leukemic precursors from AML patients (Figure 2F). Taken together, these studies demonstrate that Tomivosertib inhibits eIF4E phosphorylation in AML cells, resulting in decreased leukemic cell survival and proliferation.
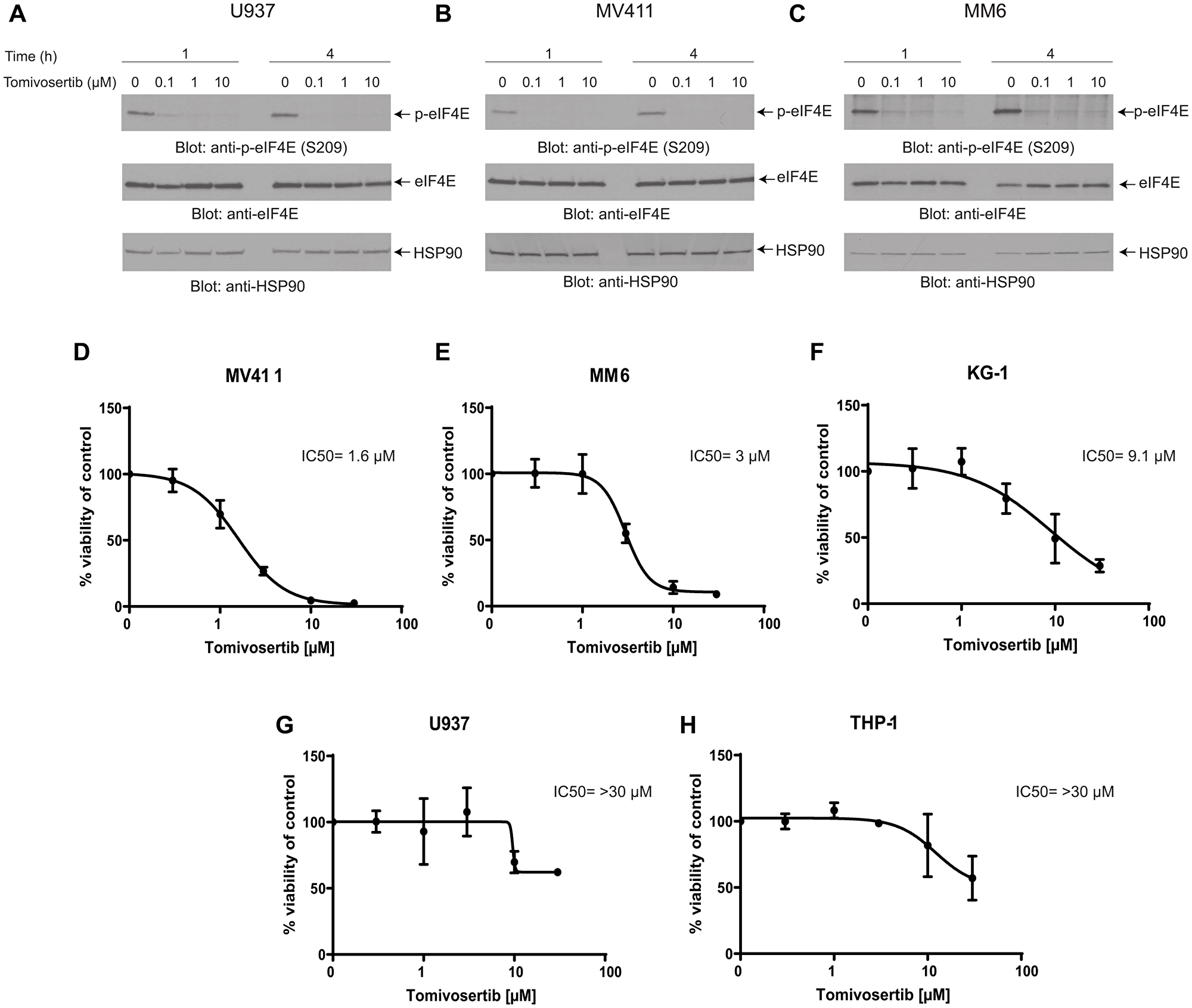
Figure 1: Tomivosertib blocks phosphorylation of eIF4E at Ser209 and inhibits cell viability in AML cells. (A) U937, (B) MV411, (C) MM6 cells were incubated with Tomivosertib for 1 hour and 4 hours at final concentrations of 0.1, 1 or 10 μM. Equal amounts of total cell lysates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. Membranes were probed with the p-eIF4E S209 antibody and then stripped and reprobed for total eIF4E. (D) MV411, (E) MM6, (F) KG-1, (G) U937 and (H) THP-1 cells were treated with increasing concentrations of Tomivosertib for 4 days. Viability was assessed using a WST-1 assay. Data are expressed as a percentage of control (DMSO-treated) cells. The means ± SE of three independent experiments, each done in triplicate, and IC50 values are shown for each cell line.
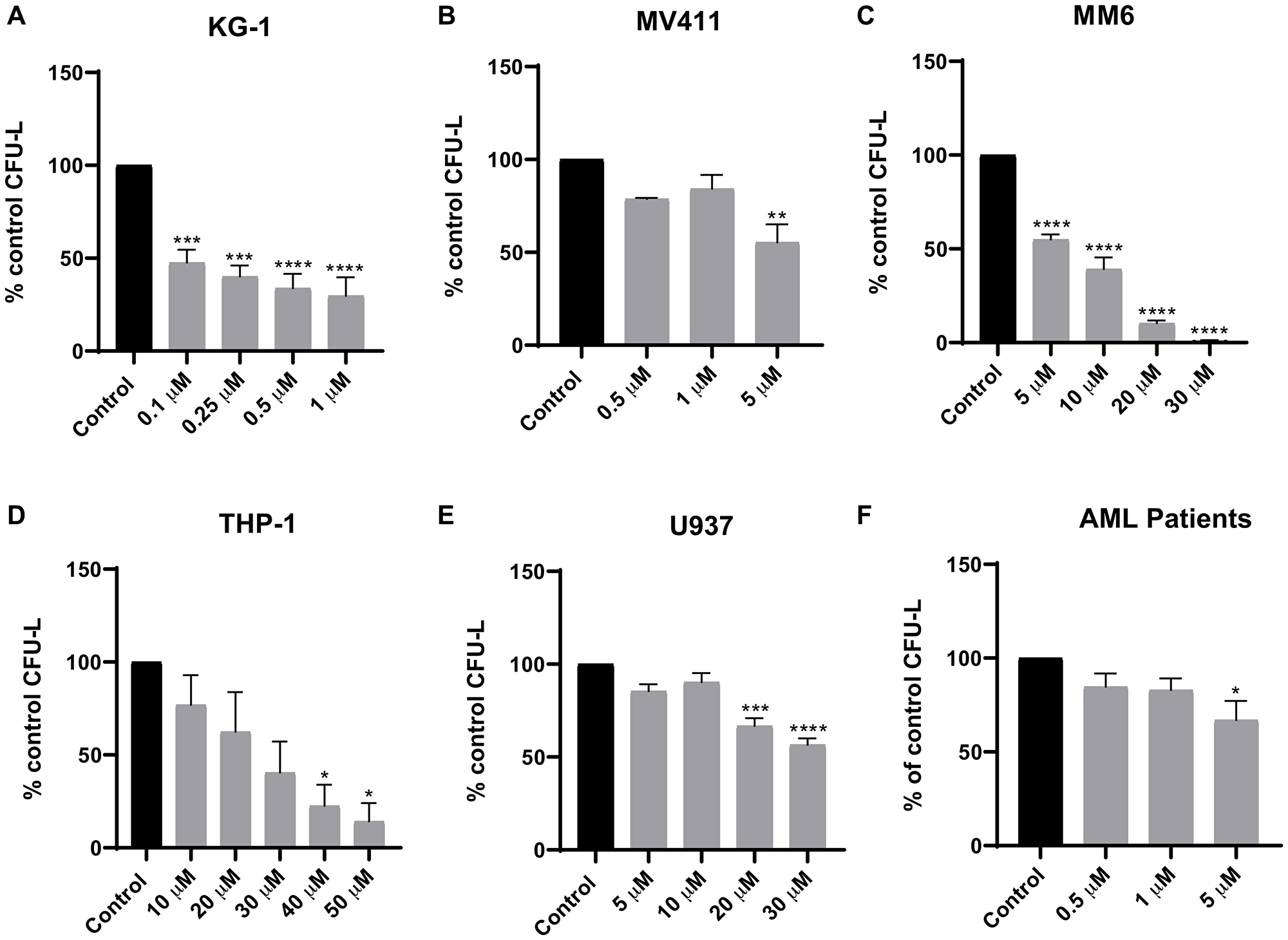
Figure 2: Tomivosertib suppresses growth of AML leukemic progenitors. The effects of Tomivosertib on leukemic-progenitors of (A) KG-1, (B) MV411, (C) MM6, (D) THP-1 and (E) U937 cells were assessed in clonogenic assays in methylcellulose. Data are expressed as percentage of CFU-L of control (DMSO-treated) cells and represent means ± SE of three independent experiments for U937, MV411, MM6 and THP-1 and four independent experiments for KG-1. (F) The inhibitory effects of Tomivosertib on primary leukemic progenitors from AML patients were assessed in clonogenic assays in methylcellulose. Data are expressed as percentage of CFU-L of control (DMSO-treated) and represent means ± SE from three independent experiments, using cells from three different patients with AML. One-way ANOVA analysis followed by Tukey’s test was used to evaluate statistically significant differences: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Tomivosertib synergistically enhances the anti-leukemic effects of Venetoclax against AML cells in vitro
In consequent studies, we investigated the antileukemic activity of a combined treatment of Tomivosertib with Venetoclax in AML cell lines. Venetoclax is a novel BCL-2 inhibitor that was recently approved by the FDA for the treatment of elderly patients with AML due to its significant clinical activity in combination with cytarabine or hypomethylating agents [43, 44]. However, the development of resistance to Venetoclax can occur through upregulation of other pro-survival proteins such as Myeloid cell leukemia 1 (MCL-1) [45, 46]. MNK1/2 was previously shown to be required for mRNA translation of MCL-1 [24, 33], and therefore, we hypothesized that concomitant treatment of cells with Venetoclax and a MNK1/2 inhibitor may help to overcome the resistance to this drug. We first investigated the effects of combination treatment with Tomivosertib and Venetoclax on cellular viability of different AML cell lines. The combination of Tomivosertib with Venetoclax resulted in synergistic inhibitory effects on cellular viability (Figure 3A and 3B). Similarly, the combination of Tomivosertib and Venetoclax resulted in potent inhibition of U937- and KG-1-derived CFU-L growth as compared to each agent alone (Figure 3C and 3D). Altogether, these studies provide evidence that the Tomivosertib/Venetoclax combination induces potent anti-leukemic responses.
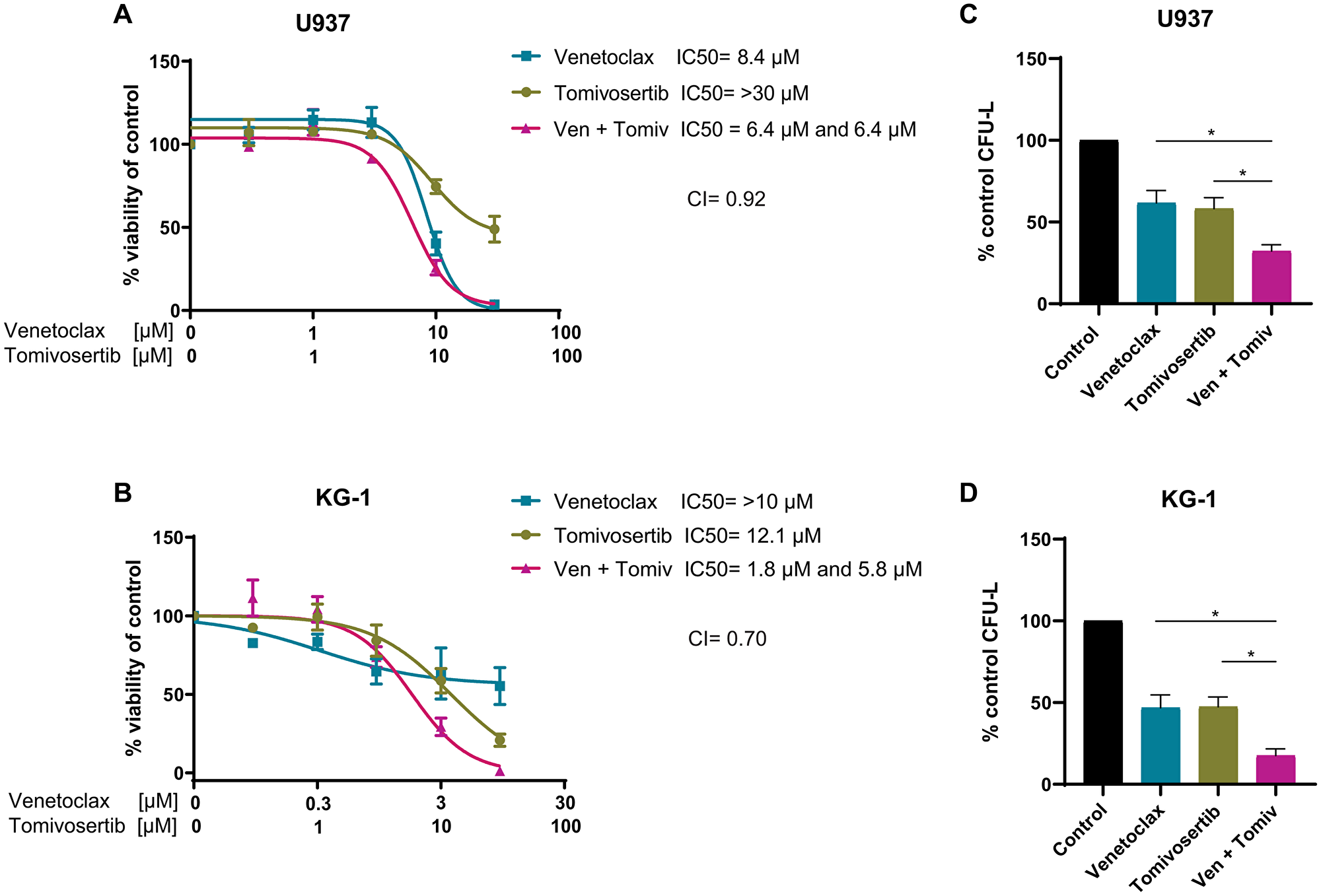
Figure 3: Tomivosertib synergizes with Venetoclax and enhances its anti-leukemic effects in vitro. (A) U937 and (B) KG-1 cells were treated with increasing concentrations of Tomivosertib and/or Venetoclax for 4 days. Viability was assessed using WST-1 assay. Data are expressed as percentage of control (DMSO-treated) cells. Shown are the means ± SE of three independent experiments. (C) U937 a were plated in methylcellulose with 30 μM Tomivosertib and 30 μM Venetoclax alone or in combination, as indicated. Data are expressed as percentage of colony formation of control (vehicle-treated) cells and represent means ± SE of four independent experiments. One-way ANOVA analysis followed by Tukey’s test was used to evaluate statistically significant differences: *p < 0.05. (D) KG-1 cells were plated in methylcellulose with 100 nM Tomivosertib and 100 nM Venetoclax alone or in combination, as indicated. Data are expressed as percentage of colony formation of control (vehicle-treated) cells and represent means ± SE of three independent experiments. One-way ANOVA analysis followed by Tukey’s test was used to evaluate statistically significant differences: *p < 0.05.
LC-MS/MS analysis identifies putative MNK1/2 targets and interactors
To identify novel binding partners of MNK1/2, liquid-chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed on protein-MNK1/2 complexes immunoprecipitated from 293T cells. We found that 16 proteins interacted with MNK1, 10 proteins interacted with MNK2 and 24 proteins interacted with both MNK1 and MNK2 (Figure 4A). Pathway and process enrichment analysis of the putative MNK1/2 interactors identified translation as one of the most significantly represented pathway activity, consistent with the role of MNK1/2 in this process (Figure 4B).
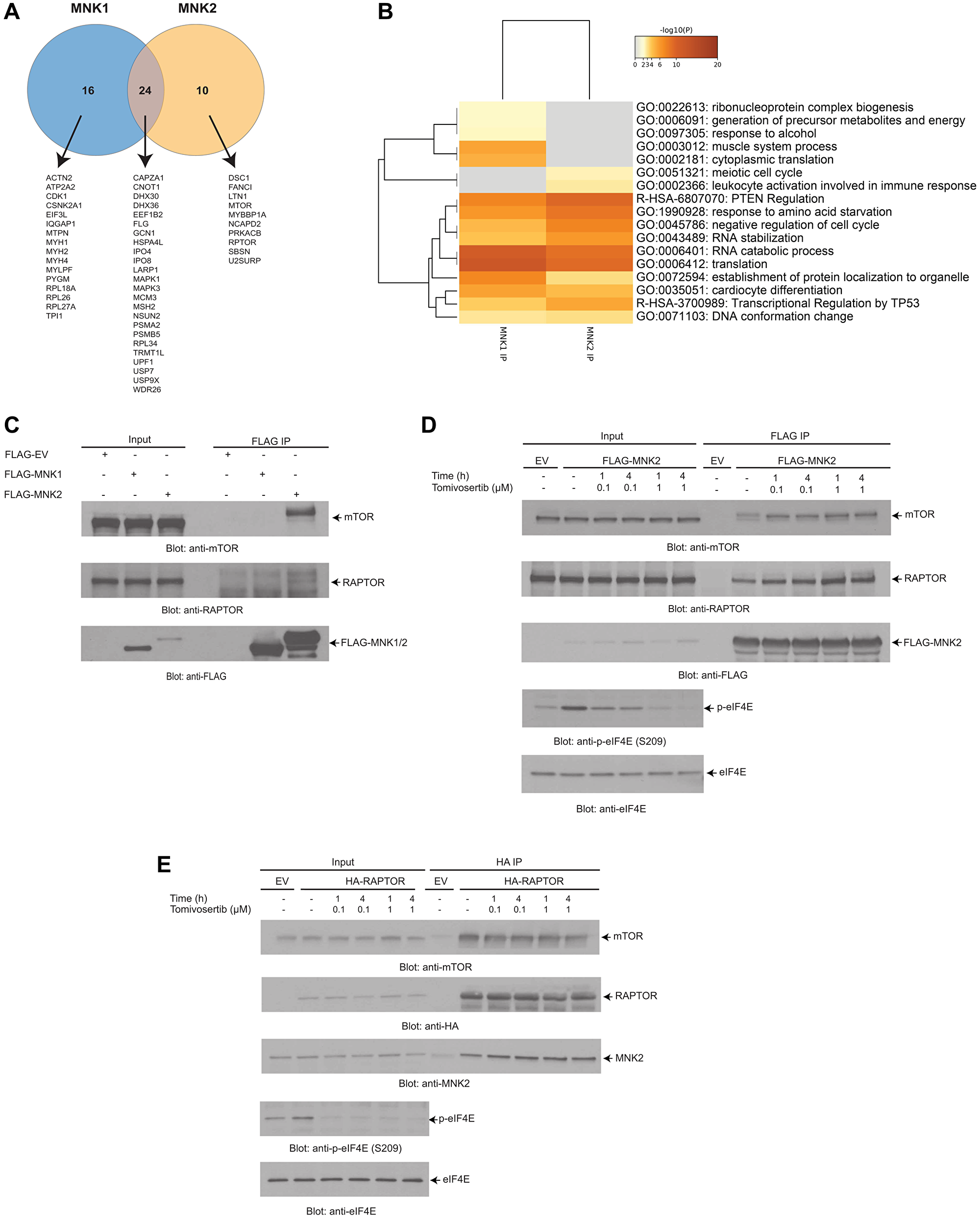
Figure 4: LC-MS/MS analysis identifies putative MNK1/2 targets and interactors. (A) FLAG-MNK2 or FLAG-MNK1 was overexpressed in 293T cells. MNK1/2 was immunoprecipitated from 293T cell lysates using anti-FLAG-M2 agarose conjugated beads. An empty vector was used as a negative control. Immunoprecipitated proteins were resolved by SDS-PAGE, and were prepared using standard techniques and then analyzed via LC-MS/MS. A Venn diagram was created depicting the number of proteins that interact with MNK1/2. (B) The results from (A) were annotated using Metascape. The heat map shows the most significant pathways and the overlap between the MNK1 IP and MNK2 IP. (C) FLAG-MNK2 or FLAG-MNK1 was immunoprecipitated as described in (A). Proteins were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (D) FLAG-MNK2 was overexpressed in 293T cells. Cells were treated with either DMSO (vehicle-control) or Tomivosertib at the indicated doses and time points, lysed and MNK2 was immunoprecipitated with anti-FLAG-M2 agarose conjugated beads. An empty vector (EV) was used as a negative control. Proteins were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (E) HA-RAPTOR was overexpressed in 293T cells. Cells were treated with either DMSO (vehicle-control) or Tomivosertib at the indicated doses and time points, lysed and RAPTOR was immunoprecipitated with anti-HA Sepharose conjugated beads. An empty vector (EV) was used as negative control. Proteins were resolved by SDS-PAGE and immunoblotted with the indicated antibodies. (C–E) Total cell lysates for each experimental condition were run in parallel with the immunoprecipitated proteins (Input).
Among the different proteins, RAPTOR and mTOR were identified as interacting partners of MNK2 in the LC-MS/MS studies (Figure 4A). We first confirmed the interaction between MNK2, mTOR and RAPTOR observed in the proteomic analysis. For this purpose, we overexpressed FLAG-MNK2 and FLAG-MNK1 in 293T cells and assessed the ability of MNK1 or MNK2 to interact with RAPTOR and mTOR by co-immunoprecipitation. Only MNK2 interacted with both RAPTOR and mTOR (Figure 4C). We then determined whether this interaction between MNK2 and RAPTOR was altered by treatment with Tomivosertib (Figure 4D and 4E). We also then further confirmed this interaction by overexpressing HA-RAPTOR and assessed the ability of RAPTOR to bind to MNK2 by co-immunoprecipitation (Figure 4E). Together, these findings indicate that MNK2 interacts with mTORC1 but that Tomivosertib does not affect the interaction.
DISCUSSION
Protein synthesis is commonly dysregulated in solid tumors and hematological malignancies [47, 48]. A key regulatory node in this process is the cap-binding complex that includes eIF4E, which controls translation initiation of many oncogenic mRNAs [26–31]. Both, overexpression of eIF4E or its phosphorylation on serine 209 by MNK1/2 have been implicated in cellular transformation [22, 24–28] and are associated with poor prognosis in a wide range of malignancies, among them AML [18, 19]. Due to the highly important role of eIF4E in tumorigenesis, several approaches to inhibit its function and/or target its expression have been developed, including anti-sense oligonucleotides, as well as drugs that mimic the 7-methylguanosine (m7G) cap or prevent the interaction of eIF4E with other proteins in the cap-binding complex [18, 49–55]. An alternative means to target oncogenic eIF4E activity, is by preventing its phosphorylation by MNK1/2 [24, 25, 56]. Given that the MNK1/2-induced eIF4E phosphorylation modulates the translation of mRNAs with tumorigenic potential rather than global protein synthesis [21, 25, 57–60]; and that MNK1/2 is dispensable for normal cell growth and development including normal hematopoiesis [61]; this strategy may constitute a safer and more specific option for cancer treatment due to a lack of toxicity on normal cells. Moreover, inhibition of the MNK1/2-eIF4E signaling axis results in robust anti-tumor responses, both in vitro and in vivo [21, 32–37].
In the current study, we assessed the anti-leukemic effects of Tomivosertib [42], a highly selective MNK1/2 inhibitor that has recently entered phase 1/2 clinical trials for the treatment of patients with several types of solid tumors and lymphomas. Our studies demonstrate that Tomivosertib induces potent suppressive effects on leukemic cell viability and clonogenicity through inhibititon of eIF4E phosphorylation on serine 209 in different AML cell lines. Similar to what was observed with other less potent or specific MNK inhibitors; Tomivosertib was most effective at suppressing the growth of M5 subtype AML cells that acquire FLT3 activating mutations, such as MV4-11 and MM6 [32–36]. This is consistent with the fact that the M5 subtype of AML is characterized by the expression of high levels of phosphorylated eIF4E, which correlates with poor prognosis in this cancer [18, 19]. Moreover, the MAPK signaling cascade can be activated downstream of the FLT3 receptor [62]. Therefore, the concomitant presence of mutant proteins upstream of MNK1/2 such as FLT3 and elevated eIF4E levels, may constitute the best model for exploring the potential clinical utility of Tomivosertib. In contrast, THP-1, U937 and KG-1 cells, which do not express FLT3 mutations, were less sensitive to the compound. These cell lines, however, all have p53 mutations which results in loss of function of p53. In our LC/MS/MS analysis of proteins bound to MNK1 and MNK2, we discovered transcription regulation of p53 as one of the pathways significantly represented. Therefore it may be relevant in future studies to focus on the role that MNKs play in the regulation of the p53 pathway and determine the effects of the p53 mutational status on responses to MNK inhibitors.
Our data also demonstrate that the combination of Tomivosertib with Venetoclax enhances the anti-leukemic responses in mutant p53-expressing AML cell lines. The combination of these two drugs resulted in synergistic suppression of cellular viability and CFU-L growth in U937 and KG-1 cells. It has recently been proposed that AML cells can develop resistance to BH3 mimetics, such as Venetoclax, through increased levels of the anti-apoptotic protein MCL-1 via regulation of MCL-1 expression and stability [45, 46, 63, 64]. Moreover, studies involving the use of Venetoclax and MEK inhibitors to simultaneously block BCL-2 and MAPK pathways have resulted in synergistic induction of apoptosis and suppression of cell proliferation in AML models [65]. The precise mechanism for the synergistic effect of Tomivosertib and Venetoclax remains to be elucidated in the future.
It is well-established that MNK1/2 drives mRNA translation through phosphorylation of its thoroughly-characterized substrate eIF4E [15–17]. Phosphorylation of eIF4E by MNK1/2 has been shown to promote cap-dependent translation and the nuclear export of mRNAs with oncogenic potential [26–31]. However, only a few other MNK1/2 substrates are known beyond eIF4E. We identified several potential novel interactors to both MNK1 and MNK2 in our LC/MS/MS analysis which require future validation studies to determine if they are bona fide substrates or regulatory partners. Notably, our LC-MS/MS studies identified mTOR and RAPTOR as interacting partners of MNK2. We demonstrated an interaction between MNK2, mTOR and RAPTOR through co-immunoprecipitation. A previous study has shown that MNK1/2 binds to mTORC1 and helps regulate mTORC1 substrate binding [66]. We only observed mTOR and RAPTOR binding to MNK2 in our LC/MS/MS analysis which supports previous published data that MNK2 binds with much more affinity than MNK1 [66]. This also supports previously published data that MNK2, not MNK1, is responsible for the activation of the feedback loop induced by the mTORC1 inhibitor rapamycin and that MNK2 plays a role in rapamycin insensitive mTORC1 complexes [67, 68]. We did not see an effect on binding of RAPTOR to MNK2 upon MNK kinase inhibition with Tomivosertib similarly to what was observed with another MNK inhibitor CGP 57380 [66]. Overall, our data further supports previously published data that MNK proteins may play a role in regulating mTORC1.
Viewed altogether, these studies indicate that MNK1/2 inhibition would most likely be a successful strategy in only a subset of AML patients. In future studies it will be crucial to ascertain what pathways are responsible for sensitivity to MNK inhibitors. These studies will help to identify potential regulatory programs through which MNK1/2 modulates cell signaling pathways critical for leukemic cell survival and may lead to the development of novel therapeutic interventions for AML.
Materials and Methods
Chemical reagents
Tomivosertib (eFT-508) and Venetoclax (ABT-199) were purchased from TargetMol. All compounds were dissolved in dimethyl sulfoxide (DMSO) and used at the indicated doses.
Cell lines
All cells were cultured as previously described [69–73, 77] and were authenticated through short tandem repeat (STR) profiling at least once per year.
Clonogenic leukemic progenitor assays in methylcellulose
Clonogenic assays in methylcellulose were conducted as in previous studies [69–73, 77]. For AML patient samples, informed consent was obtained prior to collection of peripheral blood or bone marrow cells as approved by the Institutional Review Board of Northwestern University.
Cell viability assays
Cell viability assays were performed and analyzed as previously reported using WST-1 reagent (Roche) [73–75, 77].
Cell lysis and immunoblotting
Cells were treated with either DMSO (vehicle-control) or Tomivosertib at the indicated doses and time points and processed for immunoblotting using an enhanced chemiluminescence method as described previously [69–75, 77]. Antibodies against phosphorylated-eIF4E at S209 (p-eIF4E (Ser209) (Cat. No. 9741) and mTOR (Cat. No. 4517) were purchased from Cell Signaling Technology. Antibodies against eIF4E (Cat. No. sc-9976) and HSP-90 (Cat. No. sc-7947) were purchased from Santa Cruz Biotechnology. Antibody against MNK2 (Cat. No. 17354-1-AP) was obtained from Proteintech. Antibody against RAPTOR (05-1470) was purchased from EMD Millipore. Anti-HA-tag HRP-conjugated (Cat. No. 14031) antibody was purchased from Cell Signaling Technology. Anti-FLAG-M2 HRP-conjugated (Cat. No. A8592) antibody was obtained from Sigma Aldrich.
Plasmids and transfections
MNK1 and MNK2a cDNAs were subcloned from pMX-puro (kindly provided by Dr. Rikiro Fukunaga, Osaka University) into pcDNA3.1-FLAG plasmid. pRK5-HA-RAPTOR [76] was purchased from Addgene. 293T cells were transfected using lipofectamine 2000, as stated by the manufacturer’s protocol.
Co-immunoprecipitation assays
Cells were treated with either DMSO (vehicle-control) or Tomivosertib at the indicated doses and time points. Samples were processed and immunoprecipitation was performed using anti-FLAG-M2 agarose conjugated beads (Sigma-Aldrich) or anti-HA Sepharose conjugated beads (Cell Signaling) as previously described [77].
Proteomics immunoprecipitation analysis using LC-MS/MS
Samples were prepared and processed by LC-MS/MS analysis as previously reported [77]. Immunoprecipitation was performed with anti-FLAG-M2 agarose conjugated beads.
Gene annotation and protein function enrichment analysis
This was performed as previously described using the Metascape database [77, 78].
Statistical analyses
All experiments were performed in triplicate and repeated at least twice; variations about mean were presented as standard error. One-way ANOVA was used to compare more than two groups followed by Tukey’s multiple comparison test. Student’s t-test was used to assess differences between two groups. Differences were considered statistically significant when p values were less than 0.05. All statistical analyses were performed using Prism GraphPad 6.0.
ACKNOWLEDGMENTS
We would like to thank Dr. Rikiro Fukunaga from Osaka University for kindly providing the cDNA for MNK1 and MNK2a. The authors also thank Northwestern University’s Proteomics Center of Excellence Core Facility for assistance.
CONFLICTS OF INTEREST
J.K.A. receives research funding to institution for clinical trials from AbbVie, ALX Oncology, Amgen, Aprea, Astellas, BioSight, BMS, Boehringer Ingelheim, Fujifilm, Immunogen, Kartos, and Kura. She serves on a data safety and monitoring committee for GlycoMimetics and has served on advisory boards for AbbVie, Astellas, Kura, and Syros. All other authors have no conflicts of interests to declare.
FUNDING
This work was supported by grant I01CX000916 from the Department of Veterans Affairs, NIH grants R01-CA77816, R01-CA121192, and funds from the Vassilatos foundation. Proteomics services were performed by the Northwestern Proteomics Center of Excellence Core Facility, supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70:7–30. https://doi.org/10.3322/caac.21590. [PubMed].
2. Surveillance, Epidemiology, and End Results Program Cancer Stat Facts: Leukemia–Acute Myeloid Leukemia (AML). https://seer.cancer.gov/statfacts/html/amyl.html. Accessed 1 April 2020.
3. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129:424–47. https://doi.org/10.1182/blood-2016-08-733196. [PubMed].
4. Dombret H, Gardin C. An update of current treatments for adult acute myeloid leukemia. Blood. 2016; 127:53–61. https://doi.org/10.1182/blood-2015-08-604520. [PubMed].
5. Bohl SR, Bullinger L, Rücker FG. New Targeted Agents in Acute Myeloid Leukemia: New Hope on the Rise. Int J Mol Sci. 2019; 20:1983. https://doi.org/10.3390/ijms20081983. [PubMed].
6. Wei AH, Tiong IS. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood. 2017; 130:2469–74. https://doi.org/10.1182/blood-2017-08-784066. [PubMed].
7. Fiorentini A, Capelli D, Saraceni F, Menotti D, Poloni A, Olivieri A. The Time Has Come for Targeted Therapies for AML: Lights and Shadows. Oncol Ther. 2020; 8:13–32. https://doi.org/10.1007/s40487-019-00108-x. [PubMed].
8. Castaigne S, Pautas C, Terré C, Raffoux E, Bordessoule D, Bastie JN, Legrand O, Thomas X, Turlure P, Reman O, de Revel T, Gastaud L, de Gunzburg N, et al, and Acute Leukemia French Association. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012; 379:1508–16. https://doi.org/10.1016/S0140-6736(12)60485-1. [PubMed].
9. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, Thiede C, Prior TW, Döhner K, Marcucci G, Lo-Coco F, Klisovic RB, Wei A, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017; 377:454–64. https://doi.org/10.1056/NEJMoa1614359. [PubMed].
10. Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart RK, Strickland SA, Hogge D, Solomon SR, Stone RM, Bixby DL, Kolitz JE, et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J Clin Oncol. 2018; 36:2684–92. https://doi.org/10.1200/JCO.2017.77.6112. [PubMed].
11. Pollyea DA, Pratz KW, Jonas BA, Letai A, Pullarkat VA, Wei A, Konopleva MY, Recher C, Frankfurt O, Rizzieri D, Xu T, Dail M, Chyla B, et al. Venetoclax in Combination with Hypomethylating Agents Induces Rapid, Deep, and Durable Responses in Patients with AML Ineligible for Intensive Therapy. Blood. 2018; 132:285.
12. Zhang J, Gu Y, Chen B. Mechanisms of drug resistance in acute myeloid leukemia. Onco Targets Ther. 2019; 12:1937–45. https://doi.org/10.2147/OTT.S191621. [PubMed].
13. Ricciardi MR, McQueen T, Chism D, Milella M, Estey E, Kaldjian E, Sebolt-Leopold J, Konopleva M, Andreeff M. Quantitative single cell determination of ERK phosphorylation and regulation in relapsed and refractory primary acute myeloid leukemia. Leukemia. 2005; 19:1543–49. https://doi.org/10.1038/sj.leu.2403859. [PubMed].
14. Milella M, Kornblau SM, Estrov Z, Carter BZ, Lapillonne H, Harris D, Konopleva M, Zhao S, Estey E, Andreeff M. Therapeutic targeting of the MEK/MAPK signal transduction module in acute myeloid leukemia. J Clin Invest. 2001; 108:851–59. https://doi.org/10.1172/JCI12807. [PubMed].
15. Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997; 16:1909–20. https://doi.org/10.1093/emboj/16.8.1909. [PubMed].
16. Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999; 19:1871–80. https://doi.org/10.1128/mcb.19.3.1871. [PubMed].
17. Flynn A, Proud CG. Serine 209, not serine 53, is the major site of phosphorylation in initiation factor eIF-4E in serum-treated Chinese hamster ovary cells. J Biol Chem. 1995; 270:21684–88. https://doi.org/10.1074/jbc.270.37.21684. [PubMed].
18. Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH Jr, Borden KL. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): a proof-of-principle clinical trial with ribavirin. Blood. 2009; 114:257–60. https://doi.org/10.1182/blood-2009-02-205153.
19. Topisirovic I, Guzman ML, McConnell MJ, Licht JD, Culjkovic B, Neering SJ, Jordan CT, Borden KL. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol. 2003; 23:8992–9002. https://doi.org/10.1128/mcb.23.24.8992-9002.2003. [PubMed].
20. Landon AL, Muniandy PA, Shetty AC, Lehrmann E, Volpon L, Houng S, Zhang Y, Dai B, Peroutka R, Mazan-Mamczarz K, Steinhardt J, Mahurkar A, Becker KG, et al. MNKs act as a regulatory switch for eIF4E1 and eIF4E3 driven mRNA translation in DLBCL. Nat Commun. 2014; 5:5413. https://doi.org/10.1038/ncomms6413. [PubMed].
21. Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, Lim AQ, Chang CT, Fruman DA, Rizzieri DA, Tan SY, Fan H, Chuah CT, Ong ST. Targeting of the MNK-eIF4E axis in blast crisis chronic myeloid leukemia inhibits leukemia stem cell function. Proc Natl Acad Sci U S A. 2013; 110:E2298–307. https://doi.org/10.1073/pnas.1301838110. [PubMed].
22. Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004; 10:484–86. https://doi.org/10.1038/nm1042. [PubMed].
23. Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004; 428:332–37. https://doi.org/10.1038/nature02369. [PubMed].
24. Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Teruya-Feldstein J, Pelletier J, Lowe SW. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007; 21:3232–37. https://doi.org/10.1101/gad.1604407. [PubMed].
25. Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, Pandolfi PP, Saad F, Sonenberg N. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A. 2010; 107:14134–39. https://doi.org/10.1073/pnas.1005320107. [PubMed].
26. Topisirovic I, Ruiz-Gutierrez M, Borden KL. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res. 2004; 64:8639–42. https://doi.org/10.1158/0008-5472.CAN-04-2677. [PubMed].
27. Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell. 2015; 162:59–71. https://doi.org/10.1016/j.cell.2015.05.049. [PubMed].
28. De Benedetti A, Harris AL. eIF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol. 1999; 31:59–72. https://doi.org/10.1016/s1357-2725(98)00132-0. [PubMed].
29. Phillips A, Blaydes JP. MNK1 and EIF4E are downstream effectors of MEKs in the regulation of the nuclear export of HDM2 mRNA. Oncogene. 2008; 27:1645–49. https://doi.org/10.1038/sj.onc.1210785. [PubMed].
30. Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KL. eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3′UTR. J Cell Biol. 2005; 169:245–56. https://doi.org/10.1083/jcb.200501019. [PubMed].
31. Culjkovic-Kraljacic B, Fernando TM, Marullo R, Calvo-Vidal N, Verma A, Yang S, Tabbò F, Gaudiano M, Zahreddine H, Goldstein RL, Patel J, Taldone T, Chiosis G, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood. 2016; 127:858–68. https://doi.org/10.1182/blood-2015-05-645069. [PubMed].
32. Altman JK, Szilard A, Konicek BW, Iversen PW, Kroczynska B, Glaser H, Sassano A, Vakana E, Graff JR, Platanias LC. Inhibition of Mnk kinase activity by cercosporamide and suppressive effects on acute myeloid leukemia precursors. Blood. 2013; 121:3675–81. https://doi.org/10.1182/blood-2013-01-477216. [PubMed].
33. Kosciuczuk EM, Saleiro D, Kroczynska B, Beauchamp EM, Eckerdt F, Blyth GT, Abedin SM, Giles FJ, Altman JK, Platanias LC. Merestinib blocks Mnk kinase activity in acute myeloid leukemia progenitors and exhibits antileukemic effects in vitro and in vivo. Blood. 2016; 128:410–14. https://doi.org/10.1182/blood-2016-02-698704. [PubMed].
34. Abdelaziz AM, Diab S, Islam S, Basnet SKC, Noll B, Li P, Mekonnen LB, Lu J, Albrecht H, Milne RW, Gerber C, Yu M, Wang S. Discovery of N-Phenyl-4-(1H-pyrrol-3-yl)pyrimidin-2-amine Derivatives as Potent Mnk2 Inhibitors: Design, Synthesis, SAR Analysis, and Evaluation of in vitro Anti-leukaemic Activity. Med Chem. 2019; 15:602–23. https://doi.org/10.2174/1573406415666181219111511. [PubMed].
35. Kosciuczuk EM, Kar AK, Blyth GT, Fischietti M, Abedin S, Mina AA, Siliezar R, Rzymski T, Brzozka K, Eklund EA, Beauchamp EM, Eckerdt F, Saleiro D, Platanias LC. Inhibitory effects of SEL201 in acute myeloid leukemia. Oncotarget. 2019; 10:7112–21. https://doi.org/10.18632/oncotarget.27388. [PubMed].
36. Teo T, Lam F, Yu M, Yang Y, Basnet SK, Albrecht H, Sykes MJ, Wang S. Pharmacologic Inhibition of MNKs in Acute Myeloid Leukemia. Mol Pharmacol. 2015; 88:380–89. https://doi.org/10.1124/mol.115.098012. [PubMed].
37. Santag S, Siegel F, Wengner AM, Lange C, Bömer U, Eis K, Pühler F, Lienau P, Bergemann L, Michels M, von Nussbaum F, Mumberg D, Petersen K. BAY 1143269, a novel MNK1 inhibitor, targets oncogenic protein expression and shows potent anti-tumor activity. Cancer Lett. 2017; 390:21–29. https://doi.org/10.1016/j.canlet.2016.12.029. [PubMed].
38. Buxadé M, Parra JL, Rousseau S, Shpiro N, Marquez R, Morrice N, Bain J, Espel E, Proud CG. The Mnks are novel components in the control of TNF alpha biosynthesis and phosphorylate and regulate hnRNP A1. Immunity. 2005; 23:177–89. https://doi.org/10.1016/j.immuni.2005.06.009. [PubMed].
39. Buxadé M, Morrice N, Krebs DL, Proud CG. The PSF.p54nrb complex is a novel Mnk substrate that binds the mRNA for tumor necrosis factor alpha. J Biol Chem. 2008; 283:57–65. https://doi.org/10.1074/jbc.M705286200. [PubMed].
40. Hefner Y, Borsch-Haubold AG, Murakami M, Wilde JI, Pasquet S, Schieltz D, Ghomashchi F, Yates JR 3rd, Armstrong CG, Paterson A, Cohen P, Fukunaga R, Hunter T, et al. Serine 727 phosphorylation and activation of cytosolic phospholipase A2 by MNK1-related protein kinases. J Biol Chem. 2000; 275:37542–51. https://doi.org/10.1074/jbc.M003395200. [PubMed].
41. DaSilva J, Xu L, Kim HJ, Miller WT, Bar-Sagi D. Regulation of sprouty stability by Mnk1-dependent phosphorylation. Mol Cell Biol. 2006; 26:1898–907. https://doi.org/10.1128/MCB.26.5.1898-1907.2006. [PubMed].
42. Reich SH, Sprengeler PA, Chiang GG, Appleman JR, Chen J, Clarine J, Eam B, Ernst JT, Han Q, Goel VK, Han EZR, Huang V, Hung INJ, et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J Med Chem. 2018; 61:3516–40. https://doi.org/10.1021/acs.jmedchem.7b01795. [PubMed].
43. DiNardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, Arellano M, Frattini MG, Kantarjian H, Popovic R, Chyla B, Xu T, Dunbar M, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018; 19:216–28. https://doi.org/10.1016/S1470-2045(18)30010-X. [PubMed].
44. Wei AH, Strickland SA Jr, Hou JZ, Fiedler W, Lin TL, Walter RB, Enjeti A, Tiong IS, Savona M, Lee S, Chyla B, Popovic R, Salem AH, et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol. 2019; 37:1277–84. https://doi.org/10.1200/JCO.18.01600. [PubMed].
45. Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley JP, Wood KC. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep. 2016; 6:27696. https://doi.org/10.1038/srep27696. [PubMed].
46. Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, Blum W, DiNardo CD, Kadia T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016; 6:1106–17. https://doi.org/10.1158/2159-8290.CD-16-0313. [PubMed].
47. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015; 14:261–78. https://doi.org/10.1038/nrd4505. [PubMed].
48. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013. [PubMed].
49. Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007; 117:2638–48. https://doi.org/10.1172/JCI32044. [PubMed].
50. Hong DS, Kurzrock R, Oh Y, Wheler J, Naing A, Brail L, Callies S, André V, Kadam SK, Nasir A, Holzer TR, Meric-Bernstam F, Fishman M, Simon G. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin Cancer Res. 2011; 17:6582–91. https://doi.org/10.1158/1078-0432.CCR-11-0430. [PubMed].
51. Jacobson BA, Thumma SC, Jay-Dixon J, Patel MR, Dubear Kroening K, Kratzke MG, Etchison RG, Konicek BW, Graff JR, Kratzke RA. Targeting eukaryotic translation in mesothelioma cells with an eIF4E-specific antisense oligonucleotide. PLoS One. 2013; 8:e81669. https://doi.org/10.1371/journal.pone.0081669. [PubMed].
52. Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez-Mias RA, Wagner G, Léger-Abraham M. Molecular mechanism of the dual activity of 4EGI-1: Dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E-BP1. Proc Natl Acad Sci U S A. 2015; 112:E4036–45. https://doi.org/10.1073/pnas.1512118112. [PubMed].
53. Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011; 108:1046–51. https://doi.org/10.1073/pnas.1011477108. [PubMed].
54. Cao J, He L, Lin G, Hu C, Dong R, Zhang J, Zhu H, Hu Y, Wagner CR, He Q, Yang B. Cap-dependent translation initiation factor, eIF4E, is the target for Ouabain-mediated inhibition of HIF-1α. Biochem Pharmacol. 2014; 89:20–30. https://doi.org/10.1016/j.bcp.2013.12.002. [PubMed].
55. Peffley DM, Sharma C, Hentosh P, Buechler RD. Perillyl alcohol and genistein differentially regulate PKB/Akt and 4E-BP1 phosphorylation as well as eIF4E/eIF4G interactions in human tumor cells. Arch Biochem Biophys. 2007; 465:266–73. https://doi.org/10.1016/j.abb.2007.05.022. [PubMed].
56. Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, Dowless MS, Iversen PW, Parsons S, Ellis KE, McCann DJ, Pelletier J, Furic L, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011; 71:1849–57. https://doi.org/10.1158/0008-5472.CAN-10-3298. [PubMed].
57. Geter PA, Ernlund AW, Bakogianni S, Alard A, Arju R, Giashuddin S, Gadi A, Bromberg J, Schneider RJ. Hyperactive mTOR and MNK1 phosphorylation of eIF4E confer tamoxifen resistance and estrogen independence through selective mRNA translation reprogramming. Genes Dev. 2017; 31:2235–49. https://doi.org/10.1101/gad.305631.117. [PubMed].
58. Beggs JE, Tian S, Jones GG, Xie J, Iadevaia V, Jenei V, Thomas G, Proud CG. The MAP kinase-interacting kinases regulate cell migration, vimentin expression and eIF4E/CYFIP1 binding. Biochem J. 2015; 467:63–76. https://doi.org/10.1042/BJ20141066. [PubMed].
59. Robichaud N, del Rincon SV, Huor B, Alain T, Petruccelli LA, Hearnden J, Goncalves C, Grotegut S, Spruck CH, Furic L, Larsson O, Muller WJ, Miller WH, Sonenberg N. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene. 2015; 34:2032–42. https://doi.org/10.1038/onc.2014.146. [PubMed].
60. Grzmil M, Morin P Jr, Lino MM, Merlo A, Frank S, Wang Y, Moncayo G, Hemmings BA. MAP kinase-interacting kinase 1 regulates SMAD2-dependent TGF-β signaling pathway in human glioblastoma. Cancer Res. 2011; 71:2392–402. https://doi.org/10.1158/0008-5472.CAN-10-3112. [PubMed].
61. Ueda T, Watanabe-Fukunaga R, Fukuyama H, Nagata S, Fukunaga R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol Cell Biol. 2004; 24:6539–49. https://doi.org/10.1128/MCB.24.15.6539-6549.2004. [PubMed].
62. Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, Naoe T. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000; 19:624–31. https://doi.org/10.1038/sj.onc.1203354. [PubMed].
63. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017; 58:1–17. https://doi.org/10.1080/10428194.2017.1283032. [PubMed].
64. Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, Riemondy KA, Gillen AE, Sheridan RM, Kim J, Costello JC, Amaya ML, Inguva A, et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020; 10:536–51. https://doi.org/10.1158/2159-8290.CD-19-0710. [PubMed].
65. Han L, Zhang Q, Dail M, Shi C, Cavazos A, Ruvolo VR, Zhao Y, Kim E, Rahmani M, Mak DH, Jin SS, Chen J, Phillips DC, et al. Concomitant targeting of BCL2 with venetoclax and MAPK signaling with cobimetinib in acute myeloid leukemia models. Haematologica. 2020; 105:697–707. https://doi.org/10.3324/haematol.2018.205534. [PubMed].
66. Brown MC, Gromeier M. MNK Controls mTORC1:Substrate Association through Regulation of TELO2 Binding with mTORC1. Cell Rep. 2017; 18:1444–57. https://doi.org/10.1016/j.celrep.2017.01.023. [PubMed].
67. Eckerdt F, Beauchamp E, Bell J, Iqbal A, Su B, Fukunaga R, Lulla RR, Goldman S, Platanias LC. Regulatory effects of a Mnk2-eIF4E feedback loop during mTORC1 targeting of human medulloblastoma cells. Oncotarget. 2014; 5:8442–51. https://doi.org/10.18632/oncotarget.2319. [PubMed].
68. Stead RL, Proud CG. Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a). FEBS Lett. 2013; 587:2623–28. https://doi.org/10.1016/j.febslet.2013.06.045. [PubMed].
69. Altman JK, Glaser H, Sassano A, Joshi S, Ueda T, Watanabe-Fukunaga R, Fukunaga R, Tallman MS, Platanias LC. Negative regulatory effects of Mnk kinases in the generation of chemotherapy-induced antileukemic responses. Mol Pharmacol. 2010; 78:778–84. https://doi.org/10.1124/mol.110.064642. [PubMed].
70. Altman JK, Szilard A, Goussetis DJ, Sassano A, Colamonici M, Gounaris E, Frankfurt O, Giles FJ, Eklund EA, Beauchamp EM, Platanias LC. Autophagy is a survival mechanism of acute myelogenous leukemia precursors during dual mTORC2/mTORC1 targeting. Clin Cancer Res. 2014; 20:2400–09. https://doi.org/10.1158/1078-0432.CCR-13-3218. [PubMed].
71. Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, Russo S, Barr S, Platanias LC. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res. 2011; 17:4378–88. https://doi.org/10.1158/1078-0432.CCR-10-2285. [PubMed].
72. Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, Platanias LC. Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci U S A. 2010; 107:12469–74. https://doi.org/10.1073/pnas.1005114107. [PubMed].
73. Colamonici M, Blyth G, Saleiro D, Szilard A, Bliss-Moreau M, Giles FJ, Altman JK, Beauchamp EM, Platanias LC. Dual targeting of acute myeloid leukemia progenitors by catalytic mTOR inhibition and blockade of the p110α subunit of PI3 kinase. Oncotarget. 2015; 6:8062–70. https://doi.org/10.18632/oncotarget.3509. [PubMed].
74. Beauchamp EM, Kosciuczuk EM, Serrano R, Nanavati D, Swindell EP, Viollet B, O’Halloran TV, Altman JK, Platanias LC. Direct binding of arsenic trioxide to AMPK and generation of inhibitory effects on acute myeloid leukemia precursors. Mol Cancer Ther. 2015; 14:202–12. https://doi.org/10.1158/1535-7163.MCT-14-0665-T. [PubMed].
75. Curi DA, Beauchamp EM, Blyth GT, Arslan AD, Donato NJ, Giles FJ, Altman JK, Platanias LC. Pre-clinical evidence of PIM kinase inhibitor activity in BCR-ABL1 unmutated and mutated Philadelphia chromosome-positive (Ph+) leukemias. Oncotarget. 2015; 6:33206–16. https://doi.org/10.18632/oncotarget.5091. [PubMed].
76. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002; 110:163–75. https://doi.org/10.1016/s0092-8674(02)00808-5. [PubMed].
77. Beauchamp EM, Abedin SM, Radecki SG, Fischietti M, Arslan AD, Blyth GT, Yang A, Lantz C, Nelson A, Goo YA, Akpan I, Eklund EA, Frankfurt O, et al. Identification and targeting of novel CDK9 complexes in acute myeloid leukemia. Blood. 2019; 133:1171–85. https://doi.org/10.1182/blood-2018-08-870089. [PubMed].
78. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019; 10:1523. https://doi.org/10.1038/s41467-019-09234-6. [PubMed].