
The AP-1 transcription factor family – tumor suppressors and promoters
AP-1 (Activator Protein-1) transcription factors are basic leucine-zipper (bZIP) proteins [2-5]. They act as dimeric transcription factors composed of members of the JUN family (c-JUN, JUNB, JUND), forming homo- or heterodimers with members of the FOS, ATF (activating transcription factor) and MAF (musculoaponeurotic fibrosarcoma) protein family [6-7]. The expression and physiological role of the diverse AP-1 complexes is cell type- and even differentiation state-dependent [8-10]. AP-1 complexes modulate transcription of target genes by binding to their TRE or CRE consensus elements [3],[11-13],[8, 12, 14]. Consequently AP-1 dimers are involved in the regulation of various biological processes such as proliferation, differentiation and apoptosis [3, 15]. It is therefore not surprising that AP-1 factors are involved in cellular transformation and tumorigenesis [3, 16-17].
c-JUN was first discovered as the homologue of the viral oncoprotein v-JUN, which induces avian sarcoma [18-19]. Early evidence revealed a cooperation of c-JUN with oncogenic RAS in cellular transformation [4, 20-21]. On the other hand, some AP-1 family members suppress and block tumor development [4, 22-23]. This task has been previously been attributed to JUNB, inactivation of which provokes the development of myeloproliferative disorders [24].
Despite the similarity in their primary structure and DNA binding specificity, c-JUN and JUNB differ in transcriptional capacity. Both may act either as a transcriptional activator or as a repressor depending on the promoter context and the heterodimerization partner [4, 25-31]. In some tissues c-JUN and JUNB even exert antagonistic functions in biological processes such as cell proliferation [4, 26-29, 31]. Thus AP-1 is able to modulate opposing functions, such as promoting or suppressing tumor development.
c-JUN - accelerating tumorigenesis by driving transcription
Since the initial discovery of v-JUN, solid evidence has accumulated linking AP-1 members and in particular c-JUN to tumor development. Using mouse models, c-JUN has been implicated in tumor formation in both skin and liver [32-36]. Inhibiting c-JUN activity in basal keratinocytes blocks chemically induced papilloma formation because of the lack of expression of the AP-1 target genes [37]. In hepatocellular carcinoma, gene deletion of c-Jun after tumor onset leads to a significant reduction in tumor size. This effect has been explained as resulting from increased apoptosis induced by c-JUN dependent suppression of the pro-apoptotic gene p53 [36, 38]. Upon loss of c-JUN, p53 mediated apoptosis kicks in and leads to a significant reduction of tumor burden [36]. Similarly, murine embryonic fibroblasts (MEFs) lacking c-JUN show severe cell cycle abnormalities with a block in the G1-phase of the cell cycle [39]. These fibroblasts demonstrate increased expression of the pro-apoptotic gene p53 and its target gene, the cell cycle inhibitor p21CIP [38]. As in hepatocytes, c-JUN negatively regulates the transcription of p53 in MEFs by direct binding to the promoter region of p53. The concomitant deletion of p53 rescues the apoptotic and proliferative disadvantage [38, 40].
Besides c-JUN’s “survival” function in interfering with p53 induced apoptosis, c-JUN has been shown to drive cell proliferation. One defined “hallmark” of tumor formation is a deregulated cell cycle and c-JUN’s ability to effect this represents a further way in which the protein contributes to tumorigenesis. A direct pro-proliferative function of c-JUN is mediated through the regulation of CyclinD1 [38-39, 41]. c-JUN is able to bind to the CyclinD1 promoter and induces transcription in a phosphorylation-dependent manner, as the exchanges of critical serine residues to alanine (JunAA/AA) decrease CyclinD1 transcription [30]. Both CyclinD1 and c-JUN are overexpressed in a broad range of human cancer and both molecules are considered proto-oncogenes [10, 42-45]. Another cell-cycle regulator under the control of AP-1 members is the cell cycle inhibitor p16INK4a. Several AP-1 binding sites within the p16INK4a promoter enable AP-1 to control expression of the gene. JUNB has been defined as a positive regulator and induces p16INK4a expression [27, 46].
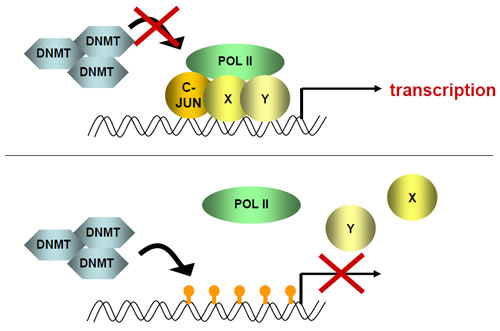
Scheme 1: A transcriptional complex, including c-JUN, binds to DNA and methylation is inhibited. In the absence of c-JUN, DNMTs can bind and methylate gene promoter region
A novel mechanism for c-JUN
An additional cell cycle component regulated by c-JUN is the G1-cell cycle kinase CDK6. CDK6 is most closely related to CDK4 and is required to allow cells to progress through the G1-phase of the cell cycle after binding to D-type Cyclins [47-50]. CDK6 and CDK4 generally have overlapping and redundant functions but CDK6, unlike CDK4, has been recently recognized to be a regulator of differentiation [51-54].
When we analyzed pro B-cells transformed by the oncoprotein BCR-ABL we found a peculiar regulation of CDK6 downstream of c-JUN [1]. Over time BCR-ABL-transformed cells lacking c-JUN had lower levels of CDK6 protein as well as mRNA. The decline in protein expression was independent of c-JUN phosphorylation as CDK6 was stably expressed in BCR-ABL+ JunAA/AA cells, in which the mutant JUN protein can no longer be phosphorylated. Interestingly, the reduction of CDK6 was accompanied by methylation of the 5’region of Cdk6 and could be reversed by treatment of the cells with the DNA methyltransferase (DNMT) inhibitor 5-aza-2’-deoxycytidine (Aza-dC). Two AP-1 binding sites are found in the vicinity of the methylated region. This led us to suggest that c-JUN exerts a “bodyguard” function, being required to prevent the Cdk6 promoter from being silenced. Only in the absence of c-JUN can methylation of the 5’region of Cdk6 take place and CDK6 expression be silenced. This effect is restricted to transformed cells and induced by the oncogenic event, as primary non-transformed lymphoid cells display unaltered CDK6 expression levels. It is attractive to speculate that c-Jun may also shape age-related DNA methylation by acting as a “bodyguard” to protect distinct DNA regions during aging [55-57].
Altering CDK6 expression has major consequences for leukemia and lymphoma development. Using Cdk6-/- animals we detected a significant delay in tumor formation. Thus, it is safe to conclude that by preventing the silencing of CDK6 c-JUN accelerates and promotes leukemogenesis. In the same murine model of BCR-ABL+ leukemia, JUNB exerts an antagonistic role. In accordance with its role as tumor suppressor, the enforced expression of JUNB suppresses tumor formation and the lack of JUNB lead to a highly aggressive and rapidly progressing disease. Again we found a role for CDK6; the lack of JUNB was accompanied by high CDK6 expression levels. How precisely JUNB interferes with the regulation of CDK6 remains to be determined [46].
CDK6 is not the only cell cycle regulator under the control of c-JUN. We found a consistent downregulation of the tumor suppressor and cell cycle inhibitor p16INK4a in c-JUN deficient BCR-ABL+ cells [1]. The levels of both the protein and the mRNA (Figure 1A) of p16INK4a decline with time after BCR-ABL-transformation, as verified by micro-array experiments [1].
As shown for Cdk6, we found promoter methylation of p16INK4a in all stable BCR-ABL+ c-JunΔ/Δ cell lines. In the case of wild type, only half the cell lines displayed methylated CpG islands (Figure 1B). Treatment of the cells with Aza-dC reverted the reduction of p16INK4a in cells lacking c-JUN (Figure 1C). Interestingly, no changes could be induced by treating wild type cells with this agent, indicating that additional regulatory mechanisms had shut off p16INK4a in the tumor cell lines. As is the case for Cdk6, the promoter region of the p16INK4a gene contains a number of AP1-binding sites, which is consistent with our model, that c-JUN is required to protect the p16INK4a promoter from being silenced by methylation.
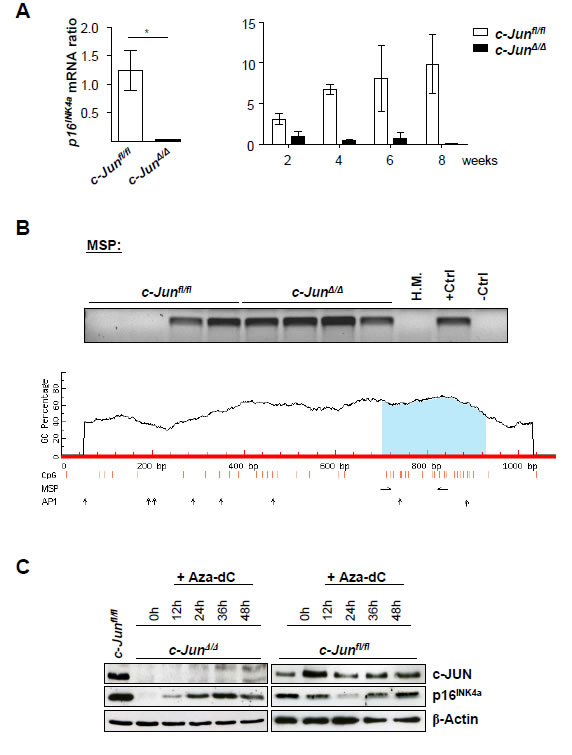
Figure 1: p16INK4a mRNA levels are down-regulated in c-JunΔ/Δ; p185BCR-ABL-transformed cell lines. A) p16INK4a mRNA levels of c-JunΔ/Δ and c-Junfl/fl cells 2, 4, 6 and 8 weeks after p185BCR-ABL transformation were analyzed by q-PCR. The fold change compared to c-JunΔ/Δ 2 weeks p16INK4a mRNA level is shown. Results were normalized by comparison to their Gapdh mRNA expression. B) upper panel: Methylation-specific PCR analysis of p16INK4a in stable c-Junfl/fl and c-JunΔ/Δ cell lines as detected by MSP analysis. A visible PCR product indicates the presence of methylated alleles. Abbreviations: H.M., bone marrow of a healthy mouse; +Ctrl (control for methylated samples); -Ctrl (control for unmethylated samples). Lower panel: Graphical overview of the CpG island associated with p16INK4a (Cdkn2a) (ENSMUSG00000044303). The following genomic region is shown: NCBIM37:4:88927717:88928797:-1. Vertical bars (orange) indicate the location of CpG dinucleotides, horizontal arrows indicate MSP primer binding sites and vertical arrows indicate AP1 transcription factor binding sites predicted using the transcription factor binding profile database JASPAR (http://jaspar.genereg.net/). C) Immunoblot for c-JUN and p16INK4a of c-JunΔ/Δ and c-Junfl/fl cells after 12, 24, 36 and 48 hours of Aza-dC treatment. β-Actin served as loading control. One representative set of data is depicted.
Conclusion
c-JUN is a common regulator of cell cycle components that has been described to have a direct role in regulating the transcription of p53 and CyclinD1. Based on our studies we postulate a novel mechanism for how c-JUN accelerates leukemogenesis and regulates genes required for cell cycle progression in tumor cells. We propose that binding of c-JUN to the promoter region exerts a protective function and prevents methylation and silencing of the genes. This function of c-JUN has recently been demonstrated for the cell cycle kinase CDK6. In the absence of c-JUN, CDK6 is downregulated accompanied by methylation of the 5’region. We now show that the same mechanism occurs at the promoter region of the cell cycle inhibitor and tumor suppressor p16INK4a. Our results indicate a novel mechanism by which AP-1 factors modulate tumor formation.
Acknowledgments
This work was supported by the Vienna Science and Technology Fund (WWTF, project number LS07-019) and by the Austrian Science Foundation (grants P19723).
References
1. Kollmann K, Heller G, Ott RG, Scheicher R, Zebedin-Brandl E, Schneckenleithner C, Simma O, Warsch W, Eckelhart E, Hoelbl A, Bilban M, Zochbauer-Muller S, Malumbres M, Sexl V. c-JUN promotes BCR-ABL-induced lymphoid leukemia by inhibiting methylation of the 5’ region of Cdk6. Blood. 2011; 117:4065-4075.
2. Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001; 20:2413-2423.
3. Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991; 1072:129-157.
4. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3:859-868.
5. Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001; 20:2401-2412.
6. Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001; 20:2438-2452.
7. Hai T, Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc Natl Acad Sci U S A. 1991; 88:3720-3724.
8. Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997; 9:240-246.
9. Schorpp-Kistner M, Wang ZQ, Angel P, Wagner EF. JunB is essential for mammalian placentation. EMBO J. 1999; 18:934-948.
10. Eferl R, Sibilia M, Hilberg F, Fuchsbichler A, Kufferath I, Guertl B, Zenz R, Wagner EF, Zatloukal K. Functions of c-Jun in liver and heart development. J Cell Biol. 1999; 145:1049-1061.
11. Angel P, Imagawa M, Chiu R, Stein B, Imbra RJ, Rahmsdorf HJ, Jonat C, Herrlich P, Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell. 1987; 49:729-739.
12. Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell. 1988; 55:917-924.
13. Turner R, Tjian R. Leucine repeats and an adjacent DNA binding domain mediate the formation of functional cFos-cJun heterodimers. Science. 1989; 243:1689-1694.
14. Lee W, Mitchell P, Tjian R. Purified transcription factor AP-1 interacts with TPA-inducible enhancer elements. Cell. 1987; 49:741-752.
15. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002; 4:E131-136.
16. Suzuki T, Hashimoto Y, Okuno H, Sato H, Nishina H, Iba H. High-level expression of human c-jun gene causes cellular transformation of chicken embryo fibroblasts. Jpn J Cancer Res. 1991; 82:58-64.
17. Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci U S A. 1995; 92:11746-11750.
18. Bos TJ, Bohmann D, Tsuchie H, Tjian R, Vogt PK. v-jun encodes a nuclear protein with enhancer binding properties of AP-1. Cell. 1988; 52:705-712.
19. Maki Y, Bos TJ, Davis C, Starbuck M, Vogt PK. Avian sarcoma virus 17 carries the jun oncogene. Proc Natl Acad Sci U S A. 1987; 84:2848-2852.
20. Johnson R, Spiegelman B, Hanahan D, Wisdom R. Cellular transformation and malignancy induced by ras require c-jun. Mol Cell Biol. 1996; 16:4504-4511.
21. Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991; 354:494-496.
22. Yang MY, Liu TC, Chang JG, Lin PM, Lin SF. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood. 2003; 101:3205-3211.
23. Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001; 20:2390-2400.
24. Passegue E, Wagner EF, Weissman IL. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell. 2004; 119:431-443.
25. Szabowski A, Maas-Szabowski N, Andrecht S, Kolbus A, Schorpp-Kistner M, Fusenig NE, Angel P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell. 2000; 103:745-755.
26. Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999; 21:326-329.
27. Passegue E, Wagner EF. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000; 19:2969-2979.
28. Szremska AP, Kenner L, Weisz E, Ott RG, Passegue E, Artwohl M, Freissmuth M, Stoxreiter R, Theussl HC, Parzer SB, Moriggl R, Wagner EF, Sexl V. JunB inhibits proliferation and transformation in B-lymphoid cells. Blood. 2003; 102:4159-4165.
29. Andrecht S, Kolbus A, Hartenstein B, Angel P, Schorpp-Kistner M. Cell cycle promoting activity of JunB through cyclin A activation. J Biol Chem. 2002; 277:35961-35968.
30. Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000; 19:2056-2068.
31. Chiu R, Angel P, Karin M. Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell. 1989; 59:979-986.
32. Young MR, Farrell L, Lambert P, Awasthi P, Colburn NH. Protection against human papillomavirus type 16-E7 oncogene-induced tumorigenesis by in vivo expression of dominant-negative c-jun. Mol Carcinog. 2002; 34:72-77.
33. Dhar A, Hu J, Reeves R, Resar LM, Colburn NH. Dominant-negative c-Jun (TAM67) target genes: HMGA1 is required for tumor promoter-induced transformation. Oncogene. 2004; 23:4466-4476.
34. Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol Cancer Res. 2003; 1:848-854.
35. Thompson EJ, MacGowan J, Young MR, Colburn N, Bowden GT. A dominant negative c-jun specifically blocks okadaic acid-induced skin tumor promotion. Cancer Res. 2002; 62:3044-3047.
36. Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003; 112:181-192.
37. Young MR, Li JJ, Rincon M, Flavell RA, Sathyanarayana BK, Hunziker R, Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A. 1999; 96:9827-9832.
38. Schreiber M, Kolbus A, Piu F, Szabowski A, Mohle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999; 13:607-619.
39. Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999; 18:188-197.
40. Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000; 103:897-907.
41. Jiang W, Kahn SM, Zhou P, Zhang YJ, Cacace AM, Infante AS, Doi S, Santella RM, Weinstein IB. Overexpression of cyclin D1 in rat fibroblasts causes abnormalities in growth control, cell cycle progression and gene expression. Oncogene. 1993; 8:3447-3457.
42. Maeno K, Masuda A, Yanagisawa K, Konishi H, Osada H, Saito T, Ueda R, Takahashi T. Altered regulation of c-jun and its involvement in anchorage-independent growth of human lung cancers. Oncogene. 2006; 25:271-277.
43. Liu JH, Yen CC, Lin YC, Gau JP, Yang MH, Chao TC, Hsiao LT, Wang WS, Tsai YC, Chen PM. Overexpression of cyclin D1 in accelerated-phase chronic myeloid leukemia. Leuk Lymphoma. 2004; 45:2419-2425.
44. Vleugel MM, Greijer AE, Bos R, van der Wall E, van Diest PJ. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum Pathol. 2006; 37:668-674.
45. Arykok AT, Onal BU, Han U. Expressions of cyclin D1, p53, bcl-2, and bax in infiltrative ductal carcinoma of the breast: correlations with clinicopathologic characteristics. Breast J. 2006; 12:391-392.
46. Ott RG, Simma O, Kollmann K, Weisz E, Zebedin EM, Schorpp-Kistner M, Heller G, Zochbauer S, Wagner EF, Freissmuth M, Sexl V. JunB is a gatekeeper for B-lymphoid leukemia. Oncogene. 2007; 26:4863-4871.
47. Sherr CJ. Mammalian G1 cyclins and cell cycle progression. Proc Assoc Am Physicians. 1995; 107:181-186.
48. Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005; 30:630-641.
49. Lundberg AS, Weinberg RA. Control of the cell cycle and apoptosis. Eur J Cancer. 1999; 35:1886-1894.
50. Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004; 118:493-504.
51. Matushansky I, Radparvar F, Skoultchi AI. CDK6 blocks differentiation: coupling cell proliferation to the block to differentiation in leukemic cells. Oncogene. 2003; 22:4143-4149.
52. Ericson KK, Krull D, Slomiany P, Grossel MJ. Expression of cyclin-dependent kinase 6, but not cyclin-dependent kinase 4, alters morphology of cultured mouse astrocytes. Mol Cancer Res. 2003; 1:654-664.
53. Ogasawara T, Kawaguchi H, Jinno S, Hoshi K, Itaka K, Takato T, Nakamura K, Okayama H. Bone morphogenetic protein 2-induced osteoblast differentiation requires Smad-mediated down-regulation of Cdk6. Mol Cell Biol. 2004; 24:6560-6568.
54. Ogasawara T, Katagiri M, Yamamoto A, Hoshi K, Takato T, Nakamura K, Tanaka S, Okayama H, Kawaguchi H. Osteoclast differentiation by RANKL requires NF-kappaB-mediated downregulation of cyclin-dependent kinase 6 (Cdk6). J Bone Miner Res. 2004; 19:1128-1136.
55. Murgatroyd C, Wu Y, Bockmuhl Y, Spengler D. The Janus face of DNA methylation in aging. Aging (Albany NY). 2010; 2:107-110.
56. Agrawal A, Tay J, Yang GE, Agrawal S, Gupta S. Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY). 2010; 2:93-100.
57. Pegoraro G, Misteli T. The central role of chromatin maintenance in aging. Aging (Albany NY). 2009; 1:1017-1022.