
Introduction
The increasing incidence of gastrointestinal (GI) malignancies may be linked to a Westernization of lifestyle and risk behavior [1–3]. According to the World Health Organization estimate for the last few years, cancers of different parts of GI tract are considered among 15 most common malignancies with the highest rates of mortality [4–6]. Cytochrome P450 (CYP) epoxygenases include a superfamily of enzymes generally expressed in the liver, kidney, and the cardiovascular system [7]. Up to 18 mammalian CYP gene families and 44 subfamilies that include 57 human CYP genes have been identified [8]. CYP2, CYP3, and CYP4 are the largest human gene families [9]. These enzymes catalyze reactions involved in the metabolism of xenobiotics and lipids synthesis such as of cholesterol, bile acids, fatty acids and steroids [10]. Particularly, endogenous arachidonic acid (AA) as a substrate [11] is converted by CYP ω-hydroxylases to hydroxyeicosatetraenoic acids (HETEs) and by CYP epoxygenases to 5,6-, 8,9-, 11,12-, and 14,15 epoxyeicosatrienoic acids (EETs) [12]. EETs are potent endogenous vasodilators [13, 14], inhibitors of vascular inflammation [15], and contribute to blood pressure homeostasis through the regulation of Na+ transport in kidney epithelium [16, 17]. The role of CYPs and their metabolites in oncogenesis, angiogenesis, and metastasis is a popular field of research in eicosanoid biology [18].
CYPs highly expressed in the liver [19] play an important role in oxidative metabolism of xenobiotics and clearance of toxic drugs [20]. However, extrahepatic CYP enzymes and their biologically active lipid products might exhibit different tissue-specific functions. For instance, CYP-mediated metabolism in the gut wall affects the bioavailability of oral drugs [21, 22]. Orally administered xenobiotics undergo the bioactivation processes in target tissues following further detoxification through the digestive system associated CYPs [7]. While metabolism of xenobiotics commonly leads to detoxification of exogenous compounds, the CYP-mediated reactions can also produce toxic metabolites that increase risks of pathologies, including development of cancers and birth defects [10]. Therefore, alteration in CYPs function within alimentary canal could contribute to GI carcinogenesis and affect the response of tumors to chemotherapy.
Expression of CYP epoxygenases in human GI tract
The differential expression of CYPs in different parts of alimentary canal suggests different biological functions of these enzymes and their products outside of the liver [23]. In fact, studies of CYPs expression, localization, and function in human esophagus highlight the capacity of these enzymes to activate DNA-reactive carcinogens. For instance, the protein expression of CYP1A, CYP2A, CYP2E1, CYP3A, and CYP4A enzymes has been shown in 25 normal tissue specimens of human esophageal mucosa [24]. Early immunohistochemical evidence of CYPs expression in intestinal mucosa suggested that human intestinal wall, in addition to its absorptive function, might metabolize exogenous substances [25]. Therefore, members of the CYP1A, CYP2C, CYP2D, CYP2E, and CYP3A subfamilies have been shown to be constitutively expressed in the human intestinal mucosa [26, 27]. The highest CYPs expression was detected in the duodenum and it progressively decreased distally to the ileum [28]. Interestingly, most highly expressed CYPs were detected from mid-villus to villous tip, with little expression in the crypts of Lieberkühn [26]. However, crypt-specific expression of CYP2W1 was demonstrated by immunohistochemical analysis of human fetal colon [29]. Notably, in the small intestine, CYP3A has been found as the most abundant isoform with 50–82% expression of the total intestinal CYPs [30].
Studies of RNA preparations from the human and rat intestine revealed the mRNA expression of CYP2J2 and its corresponding rat homologue CYP2J3 mostly in the small intestine and colon, while CYP2J proteins were expressed throughout the entire GI tract [31]. Particularly, high levels of CYP2J2 protein were detected in nerve cells of autonomic ganglia, epithelial cells, intestinal smooth muscle cells, and vascular endothelium [31]. Furthermore, NADPH-dependent AA conversion to EETs was demonstrated in microsomal fractions from human jejunum, and gas chromatography/mass spectrometry analysis confirmed the presence of EETs. The authors suggested that CYP2J associated products may be involved in the release of intestinal neuropeptides, regulation of intestinal motility, and transport of intestinal electrolytes [31].
Rylander and co-authors characterized CYP2S1 isoform, which is selectively expressed in the intestine, demonstrated its importance for extrahepatic xenobiotic metabolism [32]. Notably, using subcellular fractionation and immunostaining methods, CYP2S1 protein expression was localized in the endoplasmic reticulum [32]. Expression of several CYPs is frequently induced by accumulation of a specific substrate [33]. Tissue specific CYPs produce different ratios of EET regioisomers [34], thereby alterations in CYPs activity could reduce the availability of AA for other metabolic pathways. This affects the synthesis of other bioactive metabolites such as proinflammatory prostaglandins. Particularly, the changes in AA metabolism through cyclooxygenase (COX) and lipoxygenase (LOX) pathways is a common feature of numerous malignancies and has been shown to play key roles in cancer progression [35]. The synthesis of AA-derived biologically active lipid metabolites can take place in the tumor and inflamed stromal tissues [36]. A summary of CYP enzymes expression in normal GI tissues is shown in Table 1.
Table 1: Overview of CYP protein expression in normal GI tract tissues
| GI organ/tissue localization | CYP isoforms | References |
|---|---|---|
| Normal tissues | ||
| Esophagus | Mucosa: CYP1A, CYP2A, CYP2E1, CYP3A, CYP4A | [24] |
| CYP2J2 | ||
| C1A1, 1A2, 2A, 2E1, 2J2, 3A5 | [31] | |
| Stomach | CYP2J2 | [31] |
| CYP2S1 | [32] | |
| Intestine | Mucosa: CYP1A, CYP2C, CYP2D, CYP2E, CYP3A | [26, 49] [30] |
| Enterocytes: CYP 3A4. CYP3A5, CYP2J2, CYP 2C9, CYP2C19, CYP2D6 | ||
| Autonomic ganglion cells, epithelial cells, smooth muscle cells, and vascular endothelium: CYP2J2 | [31] [29, 32] | |
| Fetal colon: CYP2W1, CYP2S1 | ||
Role of CYP epoxygenases in GI cancers
Multiple factors, including diet, infectious agents, environmental toxins and oral drugs have been associated with GI carcinogenesis [37]. Because CYP pathways mediate the effect of exogenous factors, studying the expression patterns and the activity changes of these enzymes in GI cancer tissues could lead to the development of new clinical and therapeutic approaches based on extrahepatic CYPs expression and activity in various GI cancers.
Esophageal cancer
Esophageal cancer, which consists of two histological subtypes, esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC), represents the eighth most frequently occurring malignancy and the sixth most common cause of cancer-related death worldwide [6]. The known risk factors of ESCC include cigarette smoking, alcohol beverage consumption, and low intake of fruits and vegetables [38]. Chronic gastroesophageal reflux disease (GERD) and precancerous Barrett’s esophagus are major risk factors of EAC [39]. Disparity between phase I drug metabolism mediated by CYPs and phase II detoxification by other enzymes such as glutathione-S-transferases (GST) has been suggested as a contributing factor to pathogenesis of these cancers [40]. In fact, early immunohistochemical studies of esophageal cancer and non-neoplastic esophageal tissue samples revealed the expression of CYP1A, CYP2C, CYP3A and the functionally associated enzymes Epoxide Hydrolase (sEH) and GST in 60% of studied cancer samples [41]. Western blot analysis showed that CYP3A4, CYP3A5, and CYP2C8 protein levels in ESCC patients were significantly higher than in control group of healthy patients. Conversely, CYP2E1 protein level was significantly lower in ESCC patients than in healthy control group [42].
Surprisingly, the analysis of CYP1A, CYP2A, CYP2E1, CYP3A, and CYP4A protein levels in 25 non-neoplastic surgical tissue specimens of human esophageal mucosa did not reveal any significant associations with the patients’ medical history data and known esophageal cancer risk factors, such as tobacco smoke and GERD [24]. These unexpected results were explained by the small number of studied samples that impeded the authors from making any conclusions regarding the connection between CYPs expression and esophageal cancer. Studies of CYPs expression in esophagectomy specimens demonstrated the expression of CYP1A2, CYP3A4, CYP2E1, and CYP2C9/10 proteins in esophageal squamous mucosa and in the basal glandular actively proliferating areas of Barrett’s esophagus [43]. Another study showed that mRNA levels of CYP3A4 and CYP2C were significantly lower in malignant tissue than in normal tissue in ESCC patients [42].
CYP2J2, a well-characterized epoxygenase, is highly expressed in the cardiac tissues and vascular endothelium [15, 44]. In addition to its cardioprotective functions, the implication of CYP2J2 in carcinogenesis has been investigated intensively. Notably, abundant mRNA and protein expression of CYP2J subfamily has been demonstrated through the entire human and rat GI tract from esophagus to colon [31]. Moreover, high and selective CYP2J2 expression was demonstrated in various human tumor tissue samples and cancer cell lines [3, 45]. In fact, Jiang and co-authors [3] characterized CYP2J2 expression in tissue samples obtained from 130 patients with different types of cancer. In 77% of the patients, CYP2J2 mRNA and protein levels were markedly higher in tumors than surrounding noncancer tissues. The CYP2J2 expression was observed in most samples from all tumor types, including ESSC (20 of 31) and EAC (4 of 4) [3]. The data demonstrated that CYP2J2 plays an important role in the pathogenesis and progression of several types of human cancers [3]. Notably, CYP2J2 has been associated with inflammation and the pathogenesis of Crohn’s disease [46]. Interestingly, inhibition of CYP2J2 with terfenadine-related compounds decreased EET production and suppressed growth and proliferation in human tongue carcinoma cells and in murine xenograft models [45]. Together, these findings suggest that CYP2J2 might contribute to neoplastic pathogenesis of GI epithelium. Alterations of CYP expression in patients with ESCC support a potential role of these enzymes in the pathogenesis of esophageal malignancies. Additionally, the known ability of CYP system to activate carcinogens could lead to oncogenic transformation in metaplastic esophagus.
Gastric cancer
Gastric cancer is one of the most frequent malignancies in the world and the third leading cause of cancer mortality [47]. Helicobacter pylori infection in combination with genetic polymorphisms associated with a predisposition to cancer development are risk factors for gastric carcinogenesis [48]. Based on early histochemical data, protein expression of CYP1A and CYP3A was detected in 51% and 28% of studied gastric cancer cases, respectively, and undetected in normal stomach tissues [49]. Additionally, the increased expression of CYP2J2 relative to adjacent normal tissue was shown in 5 out of 5 studied human gastric cancer samples [3]. These data support the CYP2J2 expression trend observed in other cancerous tissue samples [3]. Interestingly, a new report showed that elevated expression of CYP3A4 could be associated with the progression of chronic atrophic gastritis to gastric cancer and might predict poor prognosis [50]. A recent study has shed some light on the role of CYP2E1 in the development and progression of gastric adenocarcinoma [51]. Overexpression of CYP2E1 in gastric cancer cells enhanced proliferation, invasion, and survival. Mechanistic investigations showed that CYP2E1 overexpression upregulated the oncogenic PI3K-AKT-mTOR signaling pathway in gastric cancer cells [51]. Notably, little research on CYPs in the stomach was done because CYPs are generally expressed less in the normal gastric mucosa than in the other parts of the GI tract. The fact that gastric epithelium has rather a secretory than absorptive function in addition to the mucous barriers suggests that the stomach may be protected from chemical agents [52]. Therefore, the role of CYPs in driving gastric carcinogenesis remains largely unknown. Additional studies will be required to gain more information on the potential implication of CYPs in gastric cancer.
Colon cancer
Colon cancer is the third occurring malignancy and the second leading cause of cancer mortality in the United States [53]. Hereditary syndromes, such as familial adenomatous polyposis and Lynch Syndrome, and inflammatory bowel disease are major predisposition risks for the development of colon cancer [54]. Tumor-specific expression of CYP2W1 was detected in approximately 30% of higher-grade colon cancers, while the expression of this enzyme was insignificant in normal colon tissues [55–57]. Interestingly, mouse and human developmental studies showed that CYP2W1 is expressed in the small intestine and colon tissues in the early stages of embryonic development and silenced shortly after birth [29]. Postnatal silencing of both murine and human CYP2W1 gene was associated with increased methylation of CpG-rich promoter regions [58]. CYP2W1 expression can be induced by the treatment with the antitumor agent imatinib, linoleic acid and its derivatives in the colon adenocarcinoma cell line HCC2998 [29]. Although activation of CYP2W1 by demethylation in colorectal cancer (CRC) has been confirmed [58], the precise mechanisms of epigenetic modifications of CYP2W1 gene remain unclear. The positive correlation of the increasing CYP2W1 expression with tumor progression and metastasis in CRC [59–61] could be used as a diagnostic tool. Larger scale clinical studies will be required to validate the potential application of CYP2W1 as a prognostic cancer biomarker.
A recent study based on a metabolomics approach demonstrated that epoxygenated fatty acids, which are eicosanoid metabolites produced by CYP epoxygenases, were elevated in the plasma and colon of azoxymethane (AOM)/dextran sodium sulfate (DSS)-induced colon cancer mouse model [62]. Genetic knockdown or pharmacologic inhibition of CYPs decreased AOM/DSS-induced colon tumorigenesis in mice. Unlike other eicosanoid metabolites, treatment with 12,13-epoxyoctadecenoic acid (EpOME) increased AOM/DSS-induced colon tumorigenesis in vivo. These findings demonstrate that the previously understudied CYP epoxygenases and their lipid metabolites contribute to colon tumorigenesis [62]. CYP enzymes expression in neoplastic GI tissues is summarized in Table 2.
Table 2: Overview of CYP protein expression in neoplastic GI tissues
| GI organ/tissue localization | CYP isoforms | References |
|---|---|---|
| Neoplastic tissues | ||
| Barrett’s esophagus/esophageal squamous mucosa | CYP1A2, CYP3A4, CYP2E1, CYP2C9/10 | [43] |
| ESCC | CYP1A↑, CYP2C↑, CYP3A↑ | [41] |
| CYP3A4, CYP3A5, CYP2C8, CYP2E1 | [42] | |
| CYP2J2↑ | [3] | |
| EAC | CYP2J2↑ | [3] |
| Gastric cancer | CYP1A, CYP3A | [49] |
| CYP2J2↑ | [3] | |
| Colon adenocarcinoma | CYP2J2↑ | [3] |
| CYP2W1↑ | [55, 59] | |
CYP epoxygenases in the activation of pro-carcinogens
The GI tract is exposed to various exogenous compounds, including pro-carcinogens and orally consumed drugs. CYPs expressed in GI tissues might be involved in the metabolic activation of potential carcinogens [63]. Extrahepatic tissues play a key role in the CYP-mediated metabolism of xenobiotic compounds affecting the susceptibility of certain organs to neoplastic transformation [64]. It is established that chronic exposure to cigarette smoke and chewing tobacco have been associated with the development of esophageal cancer [65, 66]. The expression of xenobiotic-metabolizing CYPs in the esophagus may determine the susceptibility of this organ to the carcinogenic effect of tobacco-derived nitrosamines [67]. A study of esophageal microsomal samples from patients in the United States and Henan Province (China), a high-risk area for esophageal cancer, demonstrated that CYP3A4 and CYP2E1 are involved in the activation of tobacco carcinogens N’-nitrosonornicotine and N-nitrosodimethylamine, respectively, in the human esophagus [66]. The activities of xenobiotic-metabolizing enzymes were decreased by 30–50% in the squamous cell carcinomas as compared to their corresponding non-cancerous mucosa [66]. Another potent esophageal carcinogen, N-methyl-N-pentyl-nitrosamine, was metabolized by CYPs in microsomal fractions of human and rat esophagus [68].
CYP epoxygenases as cancer drugs metabolizing systems
Xenobiotic-metabolizing CYPs in GI tissues are involved in the first-pass clearance and could contribute to the activation of anticancer drugs [7, 63]. The limitation to the current knowledge is that CYP-mediated metabolism has been investigated mostly in the liver and the drug-metabolizing function of GI CYPs remains incomplete. The anticancer drugs tamoxifen and cyclophosphamide have been shown to be metabolized by CYP2D6, CYP2C19 and CYP2B6 [63]. Notably, the CYP3A subfamily enzymes play a key role in the metabolism of approximately 30% of all clinically used drugs [69, 70]. Anticancer drugs metabolized by CYP3A include paclitaxel, ifosfamide, tamoxifen [69, 71, 72], and irinotecan [73]. A potential substrate-overlap between CYP3A4 and the multidrug resistance protein 1 (Mdr1) has been proposed [30, 74]. While selective expression of CYPs in GI tumors suggests a mechanism for drug resistance, both CYP- and Mdr1-mediated pathways may synergistically contribute to the metabolism and detoxification of oral drugs.
Gene-Directed Enzyme Prodrug Therapy (GDEPT) utilizing drug metabolizing CYPs that activate bio-reductive cytotoxins is a novel approach in increasing the efficacy of targeted therapy of drug-resistant hypoxic tumors. Hypoxia is a restricting factor in the clinical outcome of conventional cancer therapies, and promotes the malignant tumor progression [75]. The anti-cancer prodrug AQ4N [1,4-bis [2-(dimethylamino-N-oxide) ethyl] amino 5, 8-di-hydroxyanthracene-9, 10-dione] (banoxantrone) is converted by CYPs to the cytotoxin AQ4 in the hypoxic tumor microenvironment. AQ4N does not bind to DNA, while its derivative AQ4 has a high DNA affinity and is a potent topoisomerase II inhibitor [76], preventing tumor cells from proliferating [77, 78]. Targeting topoisomerase II, a key player in cell cycle regulation, may also sensitize tumors to radiotherapy. AQ4N, as a potential hypoxia-activated cancer chemotherapy drug, underwent clinical trials (NCT00394628) [79]. It has been shown that intra-tumoral injection of CYP3A4, CYP2B6 [77] and CYP1A1 [80] gene constructs in combination with AQ4N and radiation suppresses the growth of tumors in RIF-1 sarcoma mouse model. To our knowledge, there are no known GDEPT studies on GI cancer models. Indeed, because of their bio-metabolic characteristics, GI tumors may be a good candidate for radio-sensitization by exogenous CYPs-mediated drugs activation in a hypoxic tumor microenvironment.
Tumor-specific expression of CYP2W1 and its ability to activate multiple prodrugs to cytotoxic metabolites in mouse xenograft models of colorectal carcinoma (CRC) suggest that this enzyme may be an important target for CRC treatment [57, 60]. An approach was proposed to use CYP2W1 as a new tumor-associated antigen for cancer immunotherapy [57]. Notably, the imatinib-induced expression of tumor CYP2W1 followed by activation of duocarmycin prodrugs was suggested as an adjuvant therapy of CRC [57]. Additionally, CYP2W1 was reported to metabolize high affinity exogenous indolines, especially chloromethylindolines, into cytotoxic metabolites that inhibit growth of human colon tumors in a mouse xenograft model [60, 61]. The CRC specific expression of CYP2W1 and its effective activation of prodrugs makes it a valuable target for novel cancer therapeutics. Although CYP2W1 substrates comprise various endogenous compounds, including arachidonic acid, retinoic acid, and lysophospholipids [60, 61], additional substrates for this enzyme need to be investigated and further studies are required to verify if CYP2W1 is a specific drug target in CRC treatment.
An increasing body of evidence indicates that many medications, including those used in cancer treatment, are substrates for CYP2J2 [81]. CYP2J2 and CYP3A4 play a key role in the metabolism of cancer drugs known to cause cardiotoxicity [82]. The most studied case is the chronic and irreversible dose-dependent toxicity induced by Doxorubicin (DOX), an anthracycline used for the treatment of solid tumors and hematologic carcinomas. Frequent adverse side effects related to DOX include acute cardiomyopathy, chronic heart failure, and arrhythmias [82, 83]. Production of reactive oxygen species has been shown as the underlying mechanism of DOX cardiotoxicity [84]. Furthermore, DOX and its metabolite 7-deoxydoxorubicin aglycone (7-de-aDOX) have been shown to inhibit CYP2J2-mediated synthesis of EETs through binding to the active site of the enzyme [85]. It is highly possible that the metabolic activity of GI tract associated CYPs could also contribute to the cardiotoxicity of anti-cancer drugs. More studies on how the expression and activity of CYP2J and CYP4A in the digestive system affect drug concentration, production and elimination of toxic metabolites can help in the development of new effective cancer drugs. A summary of GI associated CYP enzymes that are important for the cancer drug metabolism is shown in Table 3.
Table 3: CYPs of clinical importance for cancer drug metabolism and clearance expressed in the GI tract
| CYP isoforms | Substrates | References |
|---|---|---|
| CYP3A, CYP3A4 | Taxol, Ifosfamide, Tamoxifen | [71] |
| Irinotecan | [72] | |
| Docetaxel | [73] | |
| Cyclophosphamide | [100] | |
| CYP2B6 | Tamoxifen | [71, 100, 101] |
| Cyclophosphamide | ||
| CYP2C19 | Tamoxifen | [63, 100] |
| Cyclophosphamide | ||
| CYP2C9 | Cyclophosphamide | [100] |
| CYP2D6 | Tamoxifen | [70, 72, 100] |
| Cyclophosphamide | ||
| CYP2J2 | Tamoxifen | [102] |
| Doxorubicin | [84, 85] | |
| CYP2S1 | 1,4-bis [2-(dimethylamino-N-oxide) ethyl] amino 5, 8-di-hydroxyanthracene-9, 10-dione (AQ4N) | [103–105] |
| N-hydroxylamine drug 2-(4-amino-3-methylphenyl)-5-fluorobenzothiazole | ||
| CYP2W1 | Chloromethylindolines | [59, 60] |
CYP polymorphisms and gastric cancer risk
The drug-metabolizing function of CYPs can be affected by genetic polymorphism of these enzymes. Adverse side effects and cancer therapy failure are associated with individual CYPs variability in drug pharmacokinetics and response [69, 86]. Most of human CYPs implicated in xenobiotics turnover belong to CYP1, CYP2, CYP3, and CYP4 families [10]. Multiple allelic variants within each of these gene families generate a high level of pharmacogenetic heterogeneity. Although several studies have established a link between genetic polymorphisms of CYPs and various pathologies, it remains unclear whether genetic polymorphisms of CYPs are associated with increased risks of GI cancers.
While multiple studies have shown that CYP1A1 genetic polymorphisms (CYP1A1 Msp I and CYP1A1 Ile/Val) could be risk factors for esophageal, gastric, and colorectal cancers [87], the current data remain controversial. Notably, a meta-analysis of the published data of CYP1A1 and CYP1A2 polymorphisms in different ethnicities revealed possible associations between CYP1A1 MspI and CYP1A2*1F polymorphisms and gastric cancer, and no significant associations between CYP1A1 Ile462Val polymorphism and gastric cancer [88]. Conversely, a meta-analysis of available clinical data in the Chinese population performed by Liu and colleagues demonstrated that CYP1A1 Ile/Val genetic polymorphisms, but not CYP1A1 MspI polymorphisms, are associated with an increased GI cancers risk [87]. Particularly, the study of single nucleotide polymorphisms (SNPs) of CYP1A1 (rs4646421, rs4646422, and rs1048943), GSTM1, and GSTT1, the key enzymes in the carcinogen metabolizing pathway, revealed that CYP1A1 (rs4646422) polymorphism could be implicated in gastric carcinogenesis in the Japanese population [89]. Interindividual variation in CYP2J2 expression has been assessed in relation to genetic polymorphism. Notably, 10 distinct star alleles have been identified [69]. The most common CYP2J2 allele variant with functional relevance is CYP2J2*7, which arises at frequencies of 2–17% in various populations. The key SNP, rs890293, is in the proximal promoter at (−76 G>T) and disrupts one of the SP1 binding sites, which results in 50% decrease of promoter activity as compared to the wild-type promoter [90].
Pharmacogenetic studies in patients with gastric ulcers under H. pylori eradication therapy demonstrated the effect of CYP2C19 polymorphism on pharmacokinetics of proton pump inhibitors (PPI), omeprazole, lansoprazole, pantoprazole, and to a lesser extent, rabeprazole [91, 92]. Therefore, genotypes of CYP2C19 were classified as rapid metabolizers (RM: *1/*1), intermediate metabolizers (IM: *1/*X), and poor metabolizers (PM: *X/*X). *1 and *X represent the wild-type and mutant allele, respectively [93]. The pharmacokinetics and pharmacodynamics of PPIs varied among these three CYP2C19 genotype groups. The lowest plasma PPI levels and intragastric pH following PPI treatment were the lowest in the RM group, intermediate in the IM group, and the highest in the PM group [93]. Several pharmacogenomic studies that include patients of different ethnicities demonstrated that PPI induce the increase of intragastric pH, promote ulcer healing, and improve efficacy of the antibiotics and overall treatment outcome according to the CYP2C19 polymorphism [93–96]. Since PPIs are commonly used in the treatment of reflux esophagitis, gastroesophageal reflux disease (GERD), Zollinger–Ellison syndrome, non-ulcer dyspepsia, and NSAID-related damage, the healing process in the therapy of these diseases was predictably affected by CYP2C19 genotype [91, 93, 96, 97]. Surprisingly, esomeprazole-induced healing of GERD was not associated with the CYP2C19 polymorphism and was explained by the CYP3A4 metabolic activity [98]. Therefore, personalized H. pylori eradication protocols that include inhibition of excessive acid secretion and antimicrobials can achieve higher eradication rates, improve healing process, and prevent neoplastic transformation of gastric epithelium.
Knowledge of the intrinsic and extrinsic factors that regulate expression and function of the CYP enzymes is a requirement for predicting variable pharmacokinetics and drug treatment response. While monogenic polymorphisms explain the variability for only few enzymes, most enzymes are controlled by several factors that include additional polymorphisms in regulatory genes and factors such as sex, age, disease, and hormones [69].
Conclusions and perspectives
There is a growing body of evidence that changes in the CYPs expression and enzymatic activity may play a major role in the GI cancer pathogenesis and progression. The expression of CYPs varies throughout the different parts of alimentary canal and this pattern is altered in cancerous tissues. Xenobiotics following absorption through intestinal wall and their metabolism by intestinal CYPs pass to liver through the portal vein [55, 99]. Hepatic CYPs in a coordinated fashion perform the final stages of drug metabolism to maintain the capacity of the digestive organs for first-pass clearance of orally administered drugs [99]. Some studies have recently implicated the CYP metabolites in inflammation and tumorigenesis [62]. The published data strongly suggest that exposure of GI tract-associated tissues to endogenous and exogenous substrates alters expression of CYPs, leading to production of proinflammatory metabolites that play a key role in tumorigenesis (Figure 1).
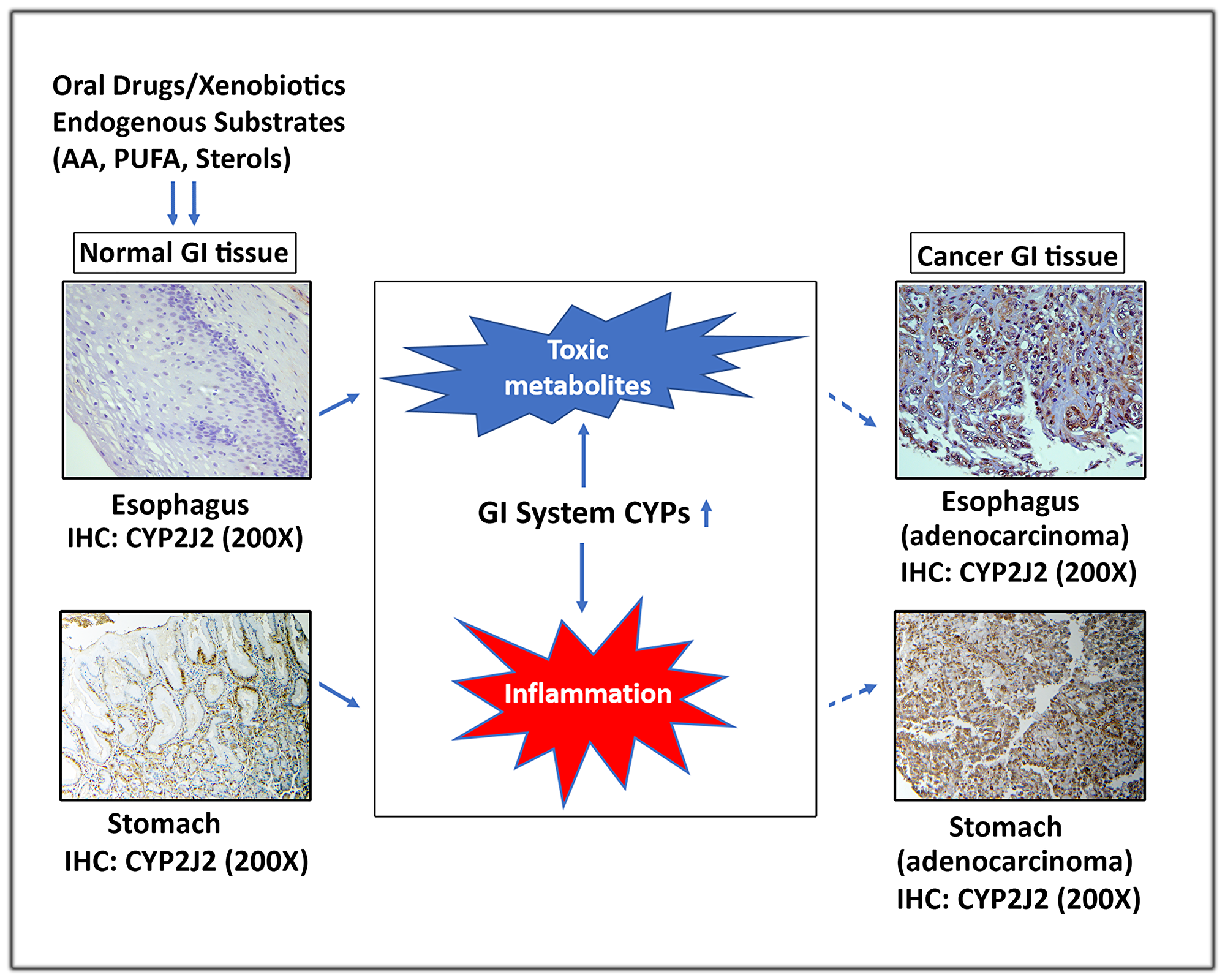
Figure 1: A schematic representation depicting the role of extrahepatic GI system CYPs in carcinogenesis. Exposure of normal GI tissues to xenobiotics and endogenous substrates (AA, PUFA & sterols) alters expression of CYPs, leading to production of toxic and proinflammatory metabolites that contribute to development of GI cancers. For illustration, protein of CYP2J2 was evaluated by IHC in normal and cancer GI tissues.
The available clinical data point to an important role for GI tract associated extrahepatic CYPs in cancer pathogenesis. Notably, increased expression of individual CYPs in GI cancer cells provides an opportunity for development of compounds that would be specifically metabolized. In addition, selective expression of CYPs in GI tumors strongly suggests a mechanism for drug resistance. Targeting CYP-mediated mechanisms of cancer drugs breakdown could help to address the major challenges in current chemotherapies, especially drug toxicity and resistance. The CYP2C19 genetic polymorphism has been shown that it could predict the clinical outcome of patients with gastric ulcers or GERD treated with PPIs [91, 93, 96, 97]. This pharmacogenetic approach could be applied to individual GI cancer patients to predict sensitivity or resistance to anti-cancer drugs. The development of this precision medicine strategy requires extensive genotyping of highly expressed CYPs in GI tumors and assessing the pharmacokinetics of various drugs and response to treatments. It is plausible that metabolizing drugs by specific CYP genotypes could lead to drug neutralization or generation of more toxic metabolites for tumors. Additionally, activation of bio-reductive cytotoxins through CYP gene-directed prodrug therapy is another potentially highly effective approach in the targeted treatment and radio-sensitization of drug-resistant hypoxic GI tumors. More studies investigating the mechanisms of CYPs expression and activity in the digestive tract system in relation to the drug-metabolizing functions will be required for the development of new and effective targeted cancer therapeutic approaches.
Abbreviations
AA: Arachidonic acid; EAC: Esophageal adenocarcinoma; AOM: Azoxymethane; COX: Cyclooxygenase; CRC: Colorectal carcinoma; GI: Gastrointestinal; CYP: Cytochrome P450 epoxygenase; DOX: Doxorubicin; DSS: Dextran sodium sulfate; EET: Epoxyeicosatrienoic acid; ESCC: Esophageal squamous cell carcinoma; EpOME: Epoxyoctadecenoic acid; FAP: Familial adenomatous polyposis; GDEPT: Gene-directed enzyme prodrug therapy; GERD: Gastroesophageal reflux disease; GST: Glutathione S transferase; HETE: Hydroxyeicosatetraenoic acid; LOX: Lipoxygenase; PPI: Proton pump inhibitor; PUFA: Polyunsaturated fatty acid; ROS: Reactive oxygen species; sEH: Soluble epoxide hydrolase.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number RO1CA193219 and Vanderbilt-Ingram Cancer Center (VICC) Specialized Project of Research Excellence (SPORE) in GI cancer (P50CA95103). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health and Vanderbilt University Medical Center.
References
1. Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the Human Development Index (2008–2030): a population-based study. Lancet Oncol. 2012; 13:790–801. https://doi.org/10.1016/s1470-2045(12)70211-5. [PubMed].
2. Bray F, Soerjomataram I. The Changing Global Burden of Cancer: Transitions in Human Development and Implications for Cancer Prevention and Control. Disease Control Priorities, Third Edition (Volume 3): Cancer. 2015; 23–44. https://doi.org/10.1596/978-1-4648-0349-9_ch2.
3. Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, Ning YG, Xiao X, Zeldin DC, Wang DW. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005; 65:4707–4715. https://doi.org/10.1158/0008-5472.can-04-4173. [PubMed].
4. Global Health Estimates 2015: Disease burden by Cause, Age, Sex, by Country and by Region, 2000–2015. Geneva: World Health Organization. 2016.
5. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2016. Geneva: World Health Organization. 2018.
6. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–386. https://doi.org/10.1002/ijc.29210. [PubMed].
7. Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003; 43:149–173. https://doi.org/10.1146/annurev.pharmtox.43.100901.140251. [PubMed].
8. Nelson DR. The Cytochrome P450 Homepage. Human Genomics. 2009; 4:59–65. https://doi.org/10.1186/1479-7364-4-1-59. [PubMed].
9. Sim SC, Ingelman-Sundberg M. Update on allele nomenclature for human cytochromes P450 and the Human Cytochrome P450 Allele (CYP-allele) Nomenclature Database. Methods Mol Biol. 2013; 987:251–259. https://doi.org/10.1007/978-1-62703-321-3_21. [PubMed].
10. Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002; 360:1155–1162. https://doi.org/10.1016/s0140-6736(02)11203-7. [PubMed].
11. Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW. Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci U S A. 1981; 78:5362–5366. https://doi.org/10.1073/pnas.78.9.5362. [PubMed].
12. Capdevila J, Yadagiri P, Manna S, Falck JR. Absolute configuration of the hydroxyeicosatetraenoic acids (HETEs) formed during catalytic oxygenation of arachidonic acid by microsomal cytochrome P-450. Biochem Biophys Res Commun. 1986; 141:1007–1011. https://doi.org/10.1016/s0006-291x(86)80144-9. [PubMed].
13. Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ Res. 1999; 84:484–488. https://doi.org/10.1161/01.res.84.4.484. [PubMed].
14. Imig JD, Falck JR, Wei S, Capdevila JH. Epoxygenase metabolites contribute to nitric oxide-independent afferent arteriolar vasodilation in response to bradykinin. J Vasc Res. 2001; 38:247–255. https://doi.org/10.1159/000051053. [PubMed].
15. Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999; 285:1276–1279. https://doi.org/10.1126/science.285.5431.1276. [PubMed].
16. Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, Harris RC, Wang W. The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem. 2014; 289:4377–4386. https://doi.org/10.1074/jbc.m113.508416. [PubMed].
17. Pidkovka N, Rao R, Mei S, Gong Y, Harris RC, Wang WH, Capdevila JH. Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation. J Biol Chem. 2013; 288:5223–5231. https://doi.org/10.1074/jbc.m112.407981. [PubMed].
18. Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005; 314:522–532. https://doi.org/10.1124/jpet.105.083477. [PubMed].
19. Ince I, Knibbe CA, Danhof M, de Wildt SN. Developmental changes in the expression and function of cytochrome P450 3A isoforms: evidence from in vitro and in vivo investigations. Clin Pharmacokinet. 2013; 52:333–345. https://doi.org/10.1007/s40262-013-0041-1. [PubMed].
20. Sheweita SA. Drug-metabolizing enzymes: mechanisms and functions. Curr Drug Metab. 2000; 1:107–132. https://doi.org/10.2174/1389200003339117. [PubMed].
21. Darwich AS, Neuhoff S, Jamei M, Rostami-Hodjegan A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the "Advanced Dissolution, Absorption, Metabolism (ADAM)" model. Curr Drug Metab. 2010; 11:716–729. https://doi.org/10.2174/138920010794328913. [PubMed].
22. Zhang QY, Kaminsky LS, Dunbar D, Zhang J, Ding X. Role of small intestinal cytochromes p450 in the bioavailability of oral nifedipine. Drug Metab Dispos. 2007; 35:1617–1623. https://doi.org/10.1124/dmd.107.016543. [PubMed].
23. Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004; 52:447–454. https://doi.org/10.1177/002215540405200403. [PubMed].
24. Lechevrel M, Casson AG, Wolf CR, Hardie LJ, Flinterman MB, Montesano R, Wild CP. Characterization of cytochrome P450 expression in human oesophageal mucosa. Carcinogenesis. 1999; 20:243–248. https://doi.org/10.1093/carcin/20.2.243. [PubMed].
25. Watkins PB, Wrighton SA, Schuetz EG, Molowa DT, Guzelian PS. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest. 1987; 80:1029–1036. https://doi.org/10.1172/jci113156. [PubMed].
26. Kaminsky LS, Fasco MJ. Small intestinal cytochromes P450. Crit Rev Toxicol. 1991; 21:407–422. https://doi.org/10.3109/10408449209089881. [PubMed].
27. Murray GI, Barnes TS, Sewell HF, Ewen SW, Melvin WT, Burke MD. The immunocytochemical localisation and distribution of cytochrome P-450 in normal human hepatic and extrahepatic tissues with a monoclonal antibody to human cytochrome P-450. Br J Clin Pharmacol. 1988; 25:465–475. https://doi.org/10.1111/j.1365-2125.1988.tb03331.x. [PubMed].
28. Peters WHM, Kremers PG. Cytochromes P-450 in the intestinal mucosa of man. Biochem Pharmacol. 1989; 38:1535–1538. https://doi.org/10.1016/0006-2952(89)90194-9. [PubMed].
29. Choong E, Guo J, Persson A, Virding S, Johansson I, Mkrtchian S, Ingelman-Sundberg M. Developmental regulation and induction of cytochrome P450 2W1, an enzyme expressed in colon tumors. PLoS One. 2015; 10:e0122820. https://doi.org/10.1371/journal.pone.0122820.
30. Karlgren M, Bergström CAS. CHAPTER 1 How Physicochemical Properties of Drugs Affect Their Metabolism and Clearance. New Horizons in Predictive Drug Metabolism and Pharmacokinetics. The Royal Society of Chemistry. 2016; 1–26. https://doi.org/10.1039/9781782622376-00001.
31. Zeldin DC, Foley J, Goldsworthy SM, Cook ME, Boyle JE, Ma J, Moomaw CR, Tomer KB, Steenbergen C, Wu S. CYP2J subfamily cytochrome P450s in the gastrointestinal tract: expression, localization, and potential functional significance. Mol Pharmacol. 1997; 51:931–943. https://doi.org/10.1124/mol.51.6.931. [PubMed].
32. Rylander T, Neve EP, Ingelman-Sundberg M, Oscarson M. Identification and tissue distribution of the novel human cytochrome P450 2S1 (CYP2S1). Biochem Biophys Res Commun. 2001; 281:529–535. https://doi.org/10.1006/bbrc.2001.4390. [PubMed].
33. Schuetz EG. Induction of cytochromes P450. Curr Drug Metab. 2001; 2:139–147. https://doi.org/10.2174/1389200013338595. [PubMed].
34. Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000; 41:163–181. https://doi.org/10.1016/S0022-2275(20)32049-6. [PubMed].
35. Rigas B, Goldman IS, Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med. 1993; 122:518–523. [PubMed].
36. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010; 10:181–193. https://doi.org/10.1038/nrc2809. [PubMed].
37. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–209. https://doi.org/10.1038/nature13480. [PubMed].
38. Abnet CC, Arnold M, Wei WQ. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterology. 2018; 154:360–373. https://doi.org/10.1053/j.gastro.2017.08.023. [PubMed].
39. Coleman HG, Xie SH, Lagergren J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology. 2018; 154:390–405. https://doi.org/10.1053/j.gastro.2017.07.046. [PubMed].
40. van Lieshout EM, Roelofs HM, Dekker S, Mulder CJ, Wobbes T, Jansen JB, Peters WH. Polymorphic expression of the glutathione S-transferase P1 gene and its susceptibility to Barrett's esophagus and esophageal carcinoma. Cancer Res. 1999; 59:586–589. [PubMed].
41. Murray GI, Shaw D, Weaver RJ, McKay JA, Ewen SW, Melvin WT, Burke MD. Cytochrome P450 expression in oesophageal cancer. Gut. 1994; 35:599–603. https://doi.org/10.1136/gut.35.5.599. [PubMed].
42. Bergheim I, Wolfgarten E, Bollschweiler E, Holscher AH, Bode C, Parlesak A. Cytochrome P450 levels are altered in patients with esophageal squamous-cell carcinoma. World J Gastroenterol. 2007; 13:997–1002. https://doi.org/10.3748/wjg.v13.i7.997. [PubMed].
43. Hughes SJ, Morse MA, Weghorst CM, Kim H, Watkins PB, Guengerich FP, Orringer MB, Beer DG. Cytochromes P450 are expressed in proliferating cells in Barrett's metaplasia. Neoplasia. 1999; 1:145–153. https://doi.org/10.1038/sj.neo.7900017. [PubMed].
44. Delozier TC, Kissling GE, Coulter SJ, Dai D, Foley JF, Bradbury JA, Murphy E, Steenbergen C, Zeldin DC, Goldstein JA. Detection of human CYP2C8, CYP2C9, and CYP2J2 in cardiovascular tissues. Drug Metab Dispos. 2007; 35:682–688. https://doi.org/10.1124/dmd.106.012823. [PubMed].
45. Chen C, Li G, Liao W, Wu J, Liu L, Ma D, Zhou J, Elbekai RH, Edin ML, Zeldin DC, Wang DW. Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. J Pharmacol Exp Ther. 2009; 329:908–918. https://doi.org/10.1124/jpet.109.152017. [PubMed].
46. Bystrom J, Thomson SJ, Johansson J, Edin ML, Zeldin DC, Gilroy DW, Smith AM, Bishop-Bailey D. Inducible CYP2J2 and its product 11,12-EET promotes bacterial phagocytosis: a role for CYP2J2 deficiency in the pathogenesis of Crohn's disease? PLoS One. 2013; 8:e75107. https://doi.org/10.1371/journal.pone.0075107. [PubMed].
47. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492. [PubMed].
48. Polk DB, Peek RM Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010; 10:403–414. https://doi.org/10.1038/nrc2857. [PubMed].
49. Murray GI, Taylor MC, Burke MD, Melvin WT. Enhanced expression of cytochrome P450 in stomach cancer. Br J Cancer. 1998; 77:1040–1044. https://doi.org/10.1038/bjc.1998.173. [PubMed].
50. Zhang F, Wang F, Chen C, Wang T, Hu J, Su R, Li X, Gu B, Tang S, Chen H, Li Y. Prediction of progression of chronic atrophic gastritis with Helicobacter pylori and poor prognosis of gastric cancer by CYP3A4. J Gastroenterol Hepatol. 2020; 35:425–432. https://doi.org/10.1111/jgh.14844. [PubMed].
51. Wang RY, Chen XW, Zhang WW, Jiang F, Liu MQ, Shen XB. CYP2E1 changes the biological function of gastric cancer cells via the PI3K/Akt/mTOR signaling pathway. Mol Med Rep. 2020; 21:842–850. https://doi.org/10.3892/mmr.2019.10890. [PubMed].
52. Sorbye H, Kvinnsland S, Svanes K. Effect of salt-induced mucosal damage and healing on penetration of N-methyl-N'-nitro-N-nitrosoguanidine to proliferative cells in the gastric mucosa of rats. Carcinogenesis. 1994; 15:673–679. https://doi.org/10.1093/carcin/15.4.673. [PubMed].
53. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5–29. https://doi.org/10.3322/caac.21254. [PubMed].
54. Healy MA, Thirumurthi S, You YN. Screening high-risk populations for colon and rectal cancers. J Surg Oncol. 2019; 120:858–63. https://doi.org/10.1002/jso.25648. [PubMed].
55. Karlgren M, Gomez A, Stark K, Svard J, Rodriguez-Antona C, Oliw E, Bernal ML, Ramon y Cajal S, Johansson I, Ingelman-Sundberg M. Tumor-specific expression of the novel cytochrome P450 enzyme, CYP2W1. Biochem Biophys Res Commun. 2006; 341:451–458. https://doi.org/10.1016/j.bbrc.2005.12.200. [PubMed].
56. Tan BS, Tiong KH, Muruhadas A, Randhawa N, Choo HL, Bradshaw TD, Stevens MF, Leong CO. CYP2S1 and CYP2W1 mediate 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (GW-610, NSC 721648) sensitivity in breast and colorectal cancer cells. Mol Cancer Ther. 2011; 10:1982–1992. https://doi.org/10.1158/1535-7163.mct-11-0391. [PubMed].
57. Travica S, Pors K, Loadman PM, Shnyder SD, Johansson I, Alandas MN, Sheldrake HM, Mkrtchian S, Patterson LH, Ingelman-Sundberg M. Colon cancer-specific cytochrome P450 2W1 converts duocarmycin analogues into potent tumor cytotoxins. Clin Cancer Res. 2013; 19:2952–2961. https://doi.org/10.1158/1078-0432.ccr-13-0238. [PubMed].
58. Gomez A, Karlgren M, Edler D, Bernal ML, Mkrtchian S, Ingelman-Sundberg M. Expression of CYP2W1 in colon tumors: regulation by gene methylation. Pharmacogenomics. 2007; 8:1315–1325. https://doi.org/10.2217/14622416.8.10.1315. [PubMed].
59. Stenstedt K, Hallstrom M, Ledel F, Ragnhammar P, Ingelman-Sundberg M, Johansson I, Edler D. The expression of CYP2W1 in colorectal primary tumors, corresponding lymph node metastases and liver metastases. Acta Oncol. 2014; 53:885–891. https://doi.org/10.3109/0284186x.2014.887224. [PubMed].
60. Guo J, Johansson I, Mkrtchian S, Ingelman-Sundberg M. The CYP2W1 enzyme: regulation, properties and activation of prodrugs. Drug Metab Rev. 2016; 48:369–378. https://doi.org/10.1080/03602532.2016.1188939. [PubMed].
61. Stenstedt K, Hallstrom M, Johansson I, Ingelman-Sundberg M, Ragnhammar P, Edler D. The expression of CYP2W1: a prognostic marker in colon cancer. Anticancer Res. 2012; 32:3869–3874. [PubMed].
62. Wang W, Yang J, Edin ML, Wang Y, Luo Y, Wan D, Yang H, Song CQ, Xue W, Sanidad KZ, Song M, Bisbee HA, Bradbury JA, et al. Targeted Metabolomics Identifies the Cytochrome P450 Monooxygenase Eicosanoid Pathway as a Novel Therapeutic Target of Colon Tumorigenesis. Cancer Res. 2019; 79:1822–1830. https://doi.org/10.1158/0008-5472.can-18-3221. [PubMed].
63. Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006; 25:1679–1691. https://doi.org/10.1038/sj.onc.1209377. [PubMed].
64. McFadyen MCE, Melvin WT, Murray GI. Cytochrome P 450 enzymes: Novel options for cancer therapeutics. Mol Cancer Ther. 2004; 3:363–371. [PubMed].
65. Dong J, Thrift AP. Alcohol, smoking and risk of oesophago-gastric cancer. Best Pract Res Clin Gastroenterol. 2017; 31:509–517. https://doi.org/10.1016/j.bpg.2017.09.002. [PubMed].
66. Smith TJ, Liao A, Wang LD, Yang GY, Starcic S, Philbert MA, Yang CS. Characterization of xenobiotic-metabolizing enzymes and nitrosamine metabolism in the human esophagus. Carcinogenesis. 1998; 19:667–672. https://doi.org/10.1093/carcin/19.4.667. [PubMed].
67. Chakradeo PP, Nair J, Bhide SV. Metabolism of N'-nitrosonornicotine by adult and fetal human oesophagal cultures. Cell Biol Int. 1995; 19:53–58. https://doi.org/10.1006/cbir.1995.1007. [PubMed].
68. Huang Q, Stoner G, Resau J, Nickols J, Mirvish SS. Metabolism of N-nitrosomethyl-n-amylamine by microsomes from human and rat esophagus. Cancer Res. 1992; 52:3547–3551. [PubMed].
69. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013; 138:103–141. https://doi.org/10.1016/j.pharmthera.2012.12.007. [PubMed].
70. Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008; 392:1093–1108. https://doi.org/10.1007/s00216-008-2291-6. [PubMed].
71. Chang TK, Weber GF, Crespi CL, Waxman DJ. Differential activation of cyclophosphamide and ifosphamide by cytochromes P-450 2B and 3A in human liver microsomes. Cancer Res. 1993; 53:5629–5637. [PubMed].
72. Desta Z, Ward BA, Soukhova NV, Flockhart DA. Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A, CYP2D6. J Pharmacol Exp Ther. 2004; 310:1062–1075. https://doi.org/10.1124/jpet.104.065607. [PubMed].
73. Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res. 2001; 7:2182–2194. [PubMed].
74. Abbott KL, Chaudhury CS, Chandran A, Vishveshwara S, Dvorak Z, Jiskrova E, Poulikova K, Vyhlidalova B, Mani S, Pondugula SR. Belinostat, at Its Clinically Relevant Concentrations, Inhibits Rifampicin-Induced CYP3A4 and MDR1 Gene Expression. Mol Pharmacol. 2019; 95:324–334. https://doi.org/10.1124/mol.118.114587. [PubMed].
75. Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004; 9:4–9. https://doi.org/10.1634/theoncologist.9-90005-4. [PubMed].
76. Smith PJ, Desnoyers R, Blunt N, Giles Y, Patterson LH, Watson JV. Flow cytometric analysis and confocal imaging of anticancer alkylaminoanthraquinones and their N-oxides in intact human cells using 647-nm krypton laser excitation. Cytometry. 1997; 27:43–53. [PubMed].
77. McCarthy HO, Yakkundi A, McErlane V, Hughes CM, Keilty G, Murray M, Patterson LH, Hirst DG, McKeown SR, Robson T. Bioreductive GDEPT using cytochrome P450 3A4 in combination with AQ4N. Cancer Gene Ther. 2003; 10:40–48. https://doi.org/10.1038/sj.cgt.7700522. [PubMed].
78. Patterson LH, McKeown SR. AQ4N: a new approach to hypoxia-activated cancer chemotherapy. Br J Cancer. 2000; 83:1589–1593. https://doi.org/10.1054/bjoc.2000.1564. [PubMed].
79. Albertella MR, Loadman PM, Jones PH, Phillips RM, Rampling R, Burnet N, Alcock C, Anthoney A, Vjaters E, Dunk CR, Harris PA, Wong A, Lalani AS, et al. Hypoxia-selective targeting by the bioreductive prodrug AQ4N in patients with solid tumors: results of a phase I study. Clin Cancer Res. 2008; 14:1096–1104. https://doi.org/10.1158/1078-0432.ccr-07-4020. [PubMed].
80. Yakkundi A, McErlane V, Murray M, McCarthy HO, Ward C, Hughes CM, Patterson LH, Hirst DG, McKeown SR, Robson T. Tumor-selective drug activation: a GDEPT approach utilizing cytochrome P450 1A1 and AQ4N. Cancer Gene Ther. 2006; 13:598–605. https://doi.org/10.1038/sj.cgt.7700933. [PubMed].
81. Aparicio-Gallego G, Blanco M, Figueroa A, Garcia-Campelo R, Valladares-Ayerbes M, Grande-Pulido E, Anton-Aparicio L. New insights into molecular mechanisms of sunitinib-associated side effects. Mol Cancer Ther. 2011; 10:2215–2223. https://doi.org/10.1158/1535-7163.mct-10-1124. [PubMed].
82. Solanki M, Pointon A, Jones B, Herbert K. Cytochrome P450 2J2: Potential Role in Drug Metabolism and Cardiotoxicity. Drug Metab Dispos. 2018; 46:1053–1065. https://doi.org/10.1124/dmd.117.078964. [PubMed].
83. Belham M, Kruger A, Mepham S, Faganello G, Pritchard C. Monitoring left ventricular function in adults receiving anthracycline-containing chemotherapy. Eur J Heart Fail. 2007; 9:409–414. https://doi.org/10.1016/j.ejheart.2006.09.007. [PubMed].
84. Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012; 18:1639–1642. https://doi.org/10.1038/nm.2919. [PubMed].
85. Arnold WR, Baylon JL, Tajkhorshid E, Das A. Arachidonic Acid Metabolism by Human Cardiovascular CYP2J2 Is Modulated by Doxorubicin. Biochemistry. 2017; 56:6700–6712. https://doi.org/10.1021/acs.biochem.7b01025. [PubMed].
86. Sim SC, Ingelman-Sundberg M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer-reviewed database of CYP variants and their associated effects. Hum Genomics. 2010; 4:278–281. https://doi.org/10.1186/1479-7364-4-4-278. [PubMed].
87. Liu C, Jiang Z, Deng QX, Zhao YN. Meta-analysis of association studies of CYP1A1 genetic polymorphisms with digestive tract cancer susceptibility in Chinese. Asian Pac J Cancer Prev. 2014; 15:4689–4695. https://doi.org/10.7314/apjcp.2014.15.11.4689. [PubMed].
88. Xue H, Lu Y, Xue Z, Lin B, Chen J, Tang F, Huang G. The effect of CYP1A1 and CYP1A2 polymorphisms on gastric cancer risk among different ethnicities: a systematic review and meta-analysis. Tumour Biol. 2014; 35:4741–4756. https://doi.org/10.1007/s13277-014-1620-y. [PubMed].
89. Hidaka A, Sasazuki S, Matsuo K, Ito H, Charvat H, Sawada N, Shimazu T, Yamaji T, Iwasaki M, Inoue M, Tsugane S, Group JS. CYP1A1, GSTM1 and GSTT1 genetic polymorphisms and gastric cancer risk among Japanese: A nested case-control study within a large-scale population-based prospective study. Int J Cancer. 2016; 139:759–768. https://doi.org/10.1002/ijc.30130. [PubMed].
90. Spiecker M, Darius H, Hankeln T, Soufi M, Sattler AM, Schaefer JR, Node K, Borgel J, Mugge A, Lindpaintner K, Huesing A, Maisch B, Zeldin DC, et al. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004; 110:2132–2136. https://doi.org/10.1161/01.cir.0000143832.91812.60. [PubMed].
91. Lay CS, Lin CJ. Correlation of CYP2C19 genetic polymorphisms with helicobacter pylori eradication in patients with cirrhosis and peptic ulcer. J Chin Med Assoc. 2010; 73:188–193. https://doi.org/10.1016/s1726-4901(10)70039-3. [PubMed].
92. Yang JC, Lin CJ. CYP2C19 genotypes in the pharmacokinetics/pharmacodynamics of proton pump inhibitor-based therapy of Helicobacter pylori infection. Expert Opin Drug Metab Toxicol. 2010; 6:29–41. https://doi.org/10.1517/17425250903386251. [PubMed].
93. Furuta T, Sugimoto M, Shirai N, Ishizaki T. CYP2C19 pharmacogenomics associated with therapy of Helicobacter pylori infection and gastro-esophageal reflux diseases with a proton pump inhibitor. Pharmacogenomics. 2007; 8:1199–1210. https://doi.org/10.2217/14622416.8.9.1199. [PubMed].
94. Furuta T, Ohashi K, Kosuge K, Zhao XJ, Takashima M, Kimura M, Nishimoto M, Hanai H, Kaneko E, Ishizaki T. CYP2C19 genotype status and effect of omeprazole on intragastric pH in humans. Clin Pharmacol Ther. 1999; 65:552–561. https://doi.org/10.1016/s0009-9236(99)70075-5. [PubMed].
95. Schwab M, Schaeffeler E, Klotz U, Treiber G. CYP2C19 polymorphism is a major predictor of treatment failure in white patients by use of lansoprazole-based quadruple therapy for eradication of Helicobacter pylori. Clin Pharmacol Ther. 2004; 76:201–209. https://doi.org/10.1016/j.clpt.2004.05.002. [PubMed].
96. Sugimoto M, Furuta T. Efficacy of tailored Helicobacter pylori eradication therapy based on antibiotic susceptibility and CYP2C19 genotype. World J Gastroenterol. 2014; 20:6400–6411. https://doi.org/10.3748/wjg.v20.i21.6400. [PubMed].
97. Sugimoto M, Shirai N, Nishino M, Kodaira C, Uotani T, Sahara S, Ichikawa H, Kagami T, Sugimoto K, Furuta T. Comparison of acid inhibition with standard dosages of proton pump inhibitors in relation to CYP2C19 genotype in Japanese. Eur J Clin Pharmacol. 2014; 70:1073–1078. https://doi.org/10.1007/s00228-014-1713-y. [PubMed].
98. Schwab M, Klotz U, Hofmann U, Schaeffeler E, Leodolter A, Malfertheiner P, Treiber G. Esomeprazole-induced healing of gastroesophageal reflux disease is unrelated to the genotype of CYP2C19: evidence from clinical and pharmacokinetic data. Clin Pharmacol Ther. 2005; 78:627–634. https://doi.org/10.1016/j.clpt.2005.08.017. [PubMed].
99. Xie F, Ding X, Zhang QY. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016; 6:374–383. https://doi.org/10.1016/j.apsb.2016.07.012. [PubMed].
100. Kishino Y, Hasegawa T, Kato A, Nishiya Y, Rozhnal V, Watanabe K, Takasaki W, Yamoto T, Mori K. Effect of inter-individual variability in human liver cytochrome P450 isozymes on cyclophosphamide-induced micronucleus formation. Mutat Res Genet Toxicol Environ Mutagen. 2019; 838:37–45. https://doi.org/10.1016/j.mrgentox.2018.11.016. [PubMed].
101. Xie HJ, Yasar U, Lundgren S, Griskevicius L, Terelius Y, Hassan M, Rane A. Role of polymorphic human CYP2B6 in cyclophosphamide bioactivation. Pharmacogenomics J. 2003; 3:53–61. https://doi.org/10.1038/sj.tpj.6500157. [PubMed].
102. Lee CA, Neul D, Clouser-Roche A, Dalvie D, Wester MR, Jiang Y, Jones JP 3rd, Freiwald S, Zientek M, Totah RA. Identification of novel substrates for human cytochrome P450 2J2. Drug Metab Dispos. 2010; 38:347–356. https://doi.org/10.1124/dmd.109.030270. [PubMed].
103. Nishida CR, Lee M, de Montellano PR. Efficient hypoxic activation of the anticancer agent AQ4N by CYP2S1 and CYP2W1. Mol Pharmacol. 2010; 78:497–502. https://doi.org/10.1124/mol.110.065045. [PubMed].
104. Xiao Y, Shinkyo R, Guengerich FP. Cytochrome P450 2S1 is reduced by NADPH-cytochrome P450 reductase. Drug Metab Dispos. 2011; 39:944–946. https://doi.org/10.1124/dmd.111.039321. [PubMed].
105. Wang K, Guengerich FP. Bioactivation of fluorinated 2-aryl-benzothiazole antitumor molecules by human cytochrome P450s 1A1 and 2W1 and deactivation by cytochrome P450 2S1. Chem Res Toxicol. 2012; 25:1740–1751. https://doi.org/10.1021/tx3001994. [PubMed].