
Introduction
Sorafenib is an oral multitarget tyrosine kinase inhibitor directed to vascular endothelial growth factor receptor (VEGFR)-1, -2, -3, platelet-derived growth factor receptor (PDGFR)- b, c-KIT, RET, FLT-3 and BRAF. This agent demonstrated survival benefit in patients with hepatocellular carcinoma (HCC) [1, 2] and represents one of the first line treatments for HCC patients. Besides HCC, sorafenib is also indicated for advanced renal cell carcinoma [3], locally recurrent or metastatic radioactive iodine-refractory differentiated thyroid carcinoma [4] and refractory desmoid tumors [5].
Sorafenib-related dermatologic adverse events (DAE) affect approximately 40% of the patients and are notable for a wide spectrum of manifestations, including maculopapular rash, hand-foot skin reaction, alopecia, xerosis, generalized exanthema and erythema multiforme-Stevens-Johnson syndrome [3, 6]. The occurrence of DAE within the first 60 days, defined as ‘early DAE’ (eDAE), is associated with favorable outcomes in patients with HCC [7–9].
Since the approval of sorafenib, case reports and small series have described the onset of proliferative and inflammatory skin lesions (SL) in patients undergoing treatment, such as squamous cell carcinoma (SCC), keratoacanthoma (KA), actinic keratosis, cystic folliculitis and basal cell carcinoma (BCC) [10–17]. These lesions were predominantly reported in patients with renal cell carcinoma, while anecdotical cases have been described in HCC [11, 12].
It is hypothesized that proliferative keratinocytic SL, specifically SCC, are generated due to a paradoxical activation of the mitogen-activated protein kinase (MAPK) signaling in the presence of a pre-existing mutation in RAS [18]. This molecular background differs from the sporadic cases, in which the pathogenesis in highly influenced by UV-radiation-induced mutations in the tumor-suppressor gene TP53, that encodes the protein p53 [19].
In daily practice, the occurrence of SL can impair efficacy and quality of life because it often requires treatment interruptions and excisional procedures, potentially leading to functional and/or esthetical sequelae.
There is lack of detailed information regarding the incidence, spectrum of subtypes and molecular hallmarks of SL in HCC patients treated with sorafenib. In the present study, we aimed to describe clinicopathological and molecular features of SL requiring excisional procedures, in a prospective cohort of patients under sorafenib treatment for HCC.
Results
Clinical characteristics and treatment outcomes
Between October 2007 and January 2018, a total of 311 HCC patients started sorafenib treatment. The median age of the entire cohort was 63.7 years-old [IQR: 55.7–70.8], 269 (86.5%) patients were male, 278 (89.4%) had ECOG-PS of 0, 270 (86.8%) had preserved liver function (Child-Pugh A). Nine (2.9%) were previously submitted to liver transplantation. Median follow-up was 12 months [IQR: 6.1–22.5], and the median duration of sorafenib treatment was 6.2 months [IQR: 2.1–12.5]. Grade > II DAEs within the first 60 days of treatment (eDAE) were observed in 88 (28.1%) patients (Table 1).
Table 1: Baseline characteristics of all patients (n = 311) and those who developed SL (n = 23)
| Patients, n | All cohort | SL cohort |
|---|---|---|
| 311 | 23 | |
| Age (Years), median [IQR] | 63.69 [55.7–70.8 ] | 58 [52–66] |
| Gender (male), n (%) | 269 (86.5) | 19 (82.6) |
| ECOG-PS (0/1), n (%) | 278 (89.4)/33 (10.6) | 21 (91.3)/2 (8.7) |
| Child-Pugh score (A/B), n (%) | 270 (86.8)*/41 (13.2) | 21 (91.3)/2 (8.7) |
| Prior liver transplantation (Yes), n (%) | 9 (2.9) | 3 (13) |
| BCLC stage (A/B/C), n (%) | 2 (0.6)/139 (44.7)/170 (54.7) | 0/12 (52.2)/11 (47.8) |
| Etiology, n (%) | ||
| HCV | 126 (40.5) | 14 (60.9) |
| HBV | 21 (6.8) | 0 |
| Alcohol | 71 (22.8) | 2 (8.7) |
| HCV & Alcohol | 37 (11.9) | 5 (21.7) |
| NASH | 9 (2.9) | 1 (4.3) |
| HIV-HCV | 9 (2.9) | 1 (4.3) |
| co-infection HBV-HCV | 5 (1.6) | 0 |
| Combination of more than one of last categories | 5 (1.6) | 0 |
| Others | 19 (6.1) | 0 |
| Healthy liver | 9 (2.9) | 0 |
From the initial cohort, 23 (7.4%) patients developed 32 SL that were submitted to biopsy or excisional procedure, with pathologic assessment. Five patients developed more than 1 SL. The median age of the group who developed SL was 58 years-old [IQR: 52–66], 19 (82.6%) patients were male, 21 (91.3%) had ECOG-PS 0, 21 (91.3%) were Child-Pugh A and 3 (13%) had previous liver transplantation. Eleven (47.8%) were BCLC-C and 12 (52.2%) were BCLC-B. The main liver disease etiologies were chronic hepatitis C virus infection (n = 20; 86.9%), alcohol (n = 2; 8.7%) and non-alcoholic steatohepatitis (n = 1; 4.3%). Coexistence of alcohol and chronic hepatitis C was found in 5 (21.7%) patients (Table 1). The median time from sorafenib initiation to SL biopsy was 9.9 months [IQR: 4.7 to 20.2] and no SL was diagnosed within 60 days of treatment initiation. No differences were found in median time from treatment starting to SL biopsy in eDAE group 10.6 months [IQR: 4.1 to 22.8] and non-eDAE group 9.9 months [IQR: 4.8 to 18].
The incidence of SL was similar between the 88 patients who presented eDAE (n = 7; 8%) and the 223 patients who did not present eDAE, 16 (7.2%) (Figure 1).
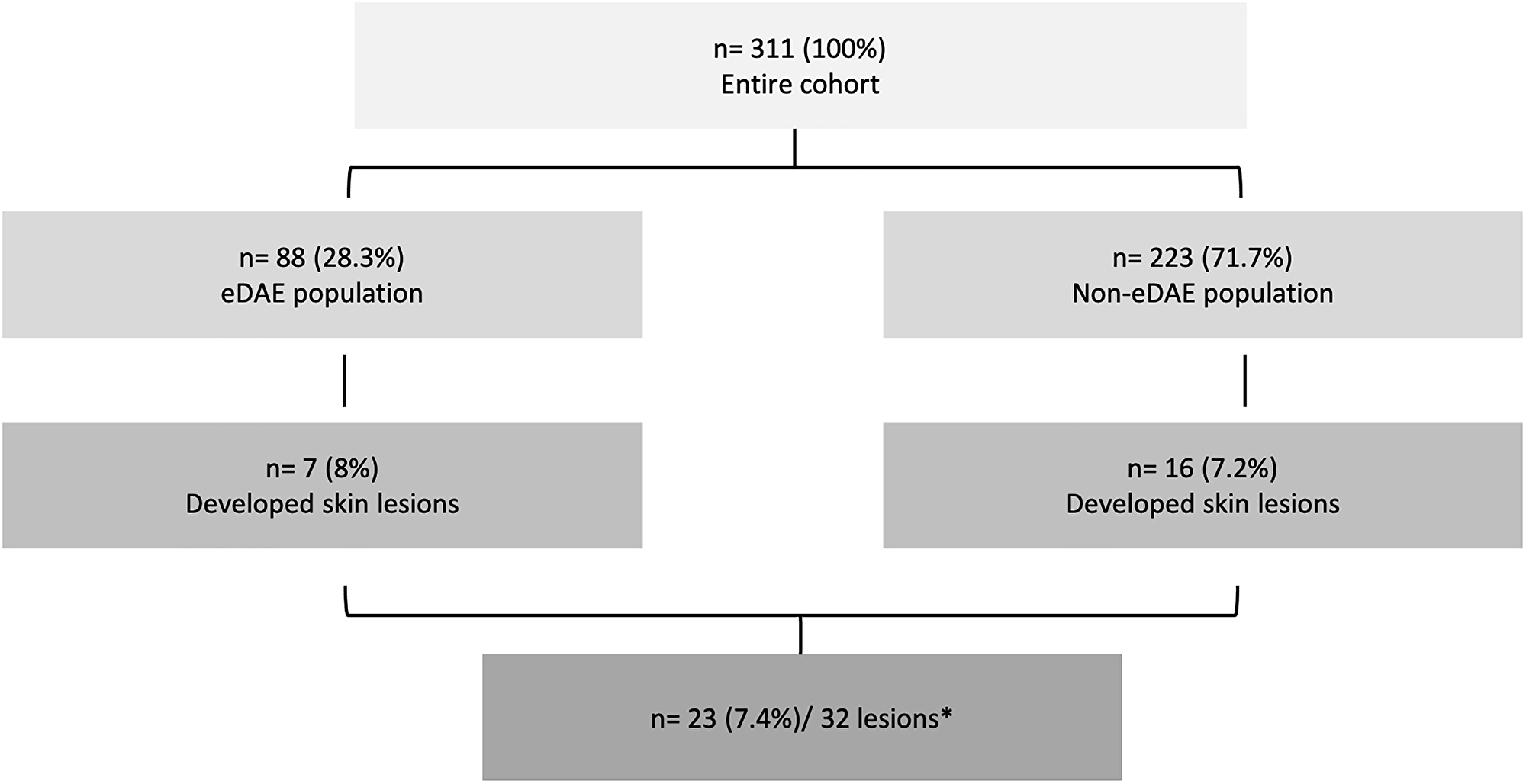
Figure 1: Consort diagram of the patients evaluated in the study. eDAE: dermatologic adverse event within the first 60 days of treatment. *5 patients presented more than 1 skin lesion.
Clinical outcomes and treatment course
None of the patients required definitive discontinuation of sorafenib due to SL management. The median overall survival of the patients with SL (n = 23) was 26.5 months (95% CI 21.6–44) with a median treatment duration of 15.5 months [IQR: 9.5–23]. Patients who develop eDAE had a median overall survival of 18.2 months (95% CI 13.9–23.6) with median treatment duration of 9 months [IQR: 4.5–14]. Finally, those patients with eDAE and SL (n = 7) had a median OS of 26.5 months (95% CI 22–51.6) with a median treatment duration of 16.7 months [IQR: 10–22.6]. Detailed clinical characteristics and management are shown in Supplementary Table 1.
Pathology characterization of skin lesions
For additional pathology and molecular characterization, a total of 28 samples of SL from 19 HCC patients were available. We found that 18 of the 28 SL (64.3%) could be classified as proliferative lesions while the rest were non-proliferative entities (n = 10/35.7%). The most frequent types of proliferative SL were KAs (n = 4/22.2%), SCCs (n = 5/27.7%) (Figure 2), BCCs (n = 3/6.6%) and seborrheic keratosis (n = 5/27.7%). The whole spectrum of SL is depicted in Supplementary Table 2. Marked immune cell infiltration was observed in 15 out of the 18 proliferative lesions (83.3%).
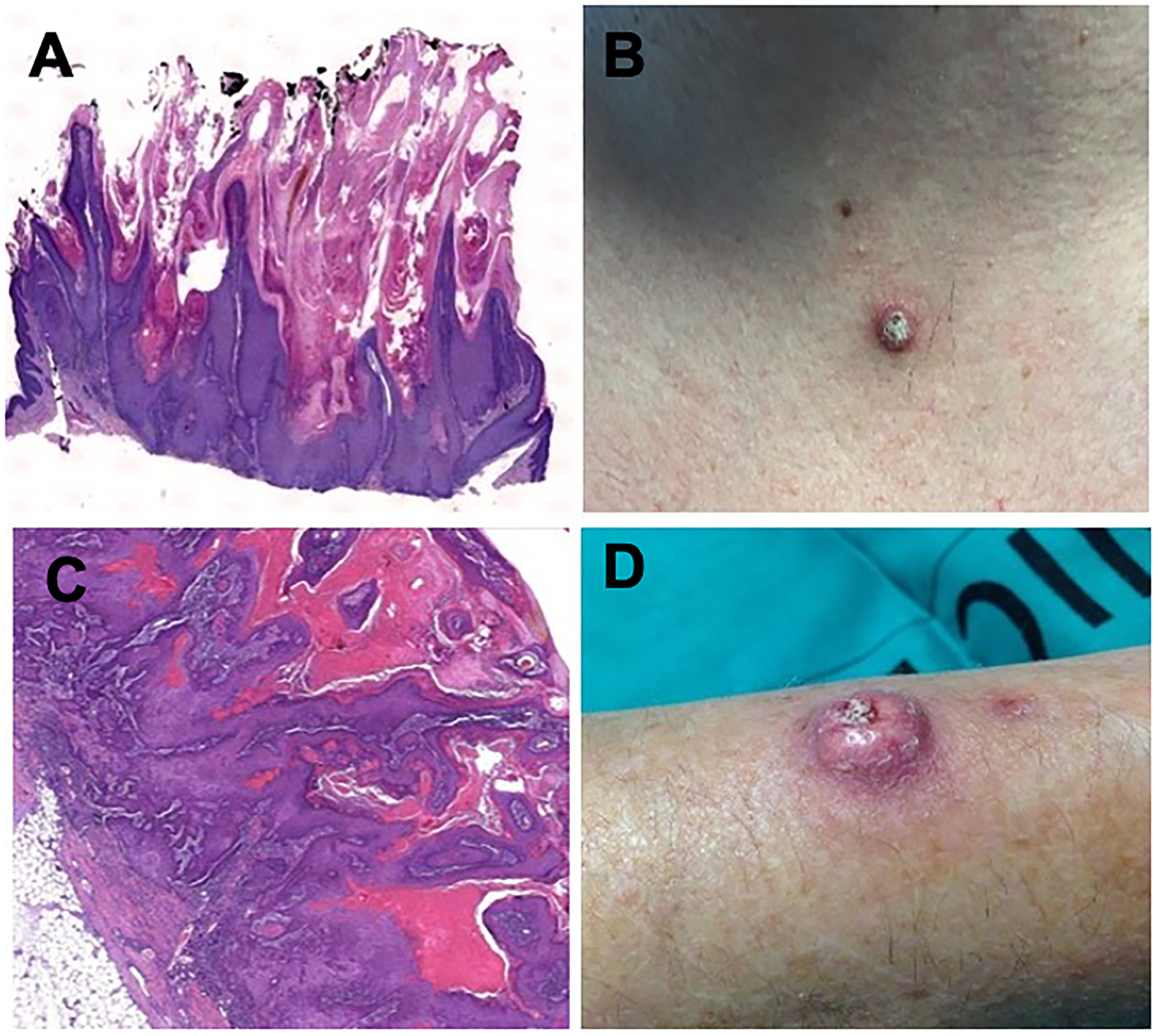
Figure 2: Histologic findings in hematoxylin and eosin staining 2x magnification (A) and clinical aspect (B) of a keratoacanthoma in a patient under sorafenib treatment. Histologic findings in hematoxylin and eosin staining 2x magnification (C) and clinical aspect (D) of a squamous-cell carcinoma in a patient under sorafenib treatment.
When we divided the 28 SL according to their development in eDAE group of patients (n = 11) or non-eDAE patients (n = 17) we found that the great majority of SL in eDAE group were proliferative lesions (n = 10/ 90.9% vs n = 8/47%) with higher immune infiltration (n = 8/72.7% vs n = 6/35.3%).
Mutations and immuhistochemistry profile
We further characterized those 28 SL for their HRAS, KRAS BRAF and TP53 mutational status, MAPK pathway activation. These cases were paired with 19 control samples by age, gender and type of SL. The whole description of the mutations found in cases and controls as well as the immunohistochemical findings are described in Supplementary Table 2 and Table 2.
Table 2: Clinical and molecular features of control samples
| IHC pERK1/2 | IHC P53 | HRAS G12D | HRAS Q61K | HRAS Q61L | BRAF V6OOE | KRAS G12D | |
|---|---|---|---|---|---|---|---|
| Squamous cell carcinoma | negative | < 5% | WT | WT | WT | NE | NE |
| negative | 10% | WT | WT | WT | WT | WT | |
| negative | 30% | WT | WT | WT | WT | WT | |
| Basal cell carcinoma | 10% | 50% | WT | WT | WT | WT | WT |
| negative | 20% | WT | WT | WT | WT | WT | |
| negative | < 5% | WT | WT | WT | WT | WT | |
| negative | < 5% | WT | WT | WT | WT | WT | |
| negative | negative | WT | WT | WT | WT | WT | |
| negative | 60% | WT | WT | WT | WT | WT | |
| Seborrheic queratosis | negative | 10% | WT | WT | WT | NE | NE |
| negative | < 5% | WT | WT | WT | WT | WT | |
| negative | < 5% | WT | WT | WT | WT | WT | |
| negative | < 5% | WT | WT | WT | WT | WT | |
| negative | < 5% | WT | WT | WT | WT | WT | |
| Keratoacanthoma | NE | NE | WT | WT | WT | WT | WT |
Briefly, we observed a HRAS G12D mutation in 1 out of 5 SCC, KRAS G12D mutation 1 (out of 3) BBC and in the sebaceous hyperplasia case, HRAS Q61L in 1 (out of 3) BCC and a HRAS Q61K mutation was found in 1 out of 5 seborrheic keratosis. The latter also presented strong immunostaining for nuclear pERK, suggesting activation of the MAPK pathway. None of the samples presented BRAF V600E mutation.
Five out of 24 cases that could be analyzed (20.8%) showed immunostaining for nuclear pERK and only 1 (5.3%) control presented a weak (10%) immunostaining for pERK, suggesting that activation of MAPK pathway was more frequent in HCC patients compared to the control group.
Additionally, we found 4 (16.6%) cases and 8 (42.1%) controls with immune expression of mutated p53 (in > 5% of the cells). This result evidence a higher p53 mutation in control patients compared to sorafenib-treated patients.
DISCUSSION
We reported the largest series dedicated exclusively to patients with HCC under sorafenib focusing on the onset of SL requiring excisional procedures and their management, mutational profile and clinical outcomes. A wide spectrum of lesions was observed ranging from inflammatory to proliferative profile, with a varying range of time between treatment initiation and biopsy (2.4 to 54.1 months). Despite SL may appear early after sorafenib start, we found no difference in time to biopsy between patients with eDAE and patients without eDAE. Sporadic skin tumors and other proliferative cutaneous lesions development generally occurs with gradual accumulation of genetic mutations along years or decades. The fact that patients receiving sorafenib treatment may develop SL within months after its start, suggest that this agent may hyperactivate the pathways for keratinocyte transformation. While the incidence of SL was similar between patients who presented eDAE (n = 7; 8%) and those who did not present eDAE, 16 (7.2%), our study shows that 90% of the SL from patients who developed eDAE, clusters into the proliferative pattern with associated immune cell infiltration in 73% of them. Our study also shows that a more exacerbated and early “skin response” evidenced a longer OS for those patients with eDAE and SL (median 26.5 months 95% CI 22–51.6) compared to that of all eDAE patients (median 18.2 months 95% CI 13.9–23.6) with a higher median treatment time also for eDAE plus SL group. Of note, the longer median sorafenib duration treatment in patients developing SL together with the late onset of SL raise the “chicken or egg paradox” hampering any robust inference in terms of survival.
In order to assess if sorafenib-treated group of patients were enduring a pro-proliferative stimulus for SL development due to a potential paradoxical increase in MAPK signaling pathway in skin cells, we checked mutational status for BRAF, KRAS, HRAS genes in our patients compared to mutational status for the same genes in a control group paired according to age, gender, type of skin lesions and demographics. Although none of our samples were positive for BRAF V600E mutation, the expression of pERK observed in a number of our samples are in line with the hypothetical paradoxical activation of the MAPK signaling in BRAF wild-type cells already described by other groups [15, 20, 21]. The concept that a targeted agent that blocks a pro-oncogenic mechanism in tumor cell has the potential to activate cell proliferation in a distinct tissue is well-known, not only in patients treated with sorafenib who develop skin cancer, but more frequently with BRAF inhibitors used in melanoma, such as vemurafenib, dabrafenib and encorafenib [22–24]. Once BRAF is located downstream from RAS, it is proposed that the inhibition of BRAF may lead to an upregulation of RAS, including H-RAS, that is often mutated with keratinocytes with ultraviolet radiation damage, resulting in a RAS-induced proliferation and activation of ERK-mediated transcription [20]. In accordance, we did not observe BRAF V600E mutations in our cohort. RAS mutations were present only in 2 cases of BCC, 1 SCC, 1 seborrheic keratosis and 1 sebaceous hyperplasia. Although the low prevalence of RAS mutations both in cases and controls do not fully support the hypothesis of paradoxical activation as it is reported in literature, the hyperactivation of MAPK pathway we have seen in sorafenib-treated group may be due to a compensatory mechanism resulting from its inhibition by sorafenib. Since skin toxicities are frequent in patients receiving multikinase inhibitors, these mechanisms deserves further evaluation in patients under similar agents with kinase inhibition activity.
The current knowledge of skin tumors pathogenesis has been expanding over the last 2 decades [25]. It is reported that 25% of middle-aged people’s skin contains a mutation in one of well-known genes reported to be skin cancer drivers and clones of cells with mutations in p53 gene are reported in the 4% of the epidermis [26]. SCC are molecularly guided by ultraviolet radiation-induced mutagenesis combined with an inability of the immune system to recognize and eliminate mutated cells [27, 28]
The mutation in the gene encoding the tumor-suppressor p53 is a well described alteration in SCC [29], corroborated by the IHC expression both in samples and in controls found in the present study.
Other gene mutations found in SCC include the NOTCH pathway genes, CDKN2A, HRAS, KNSTRN and the KMT2 family [19, 30]. On the other side, KAs origin remains controversial. There are histological similarities between SCC and KA, what could suggest that KA may be part of a spectrum of premalignant lesions associated with SCC. However, the gene expression profile of KA was revealed to be distinct of that of SCC, what suggests that they should be individualized into two distinct entities [31, 32]. The BCC molecular background was clarified by the identification of variants of the PTCH1, that is the causative mutation of the Gorlin´s syndrome (an autosomal disorder characterized by early occurrence of multicentric BCC), and by the mechanisms behind the activation of the hedgehog signaling pathways [33]. There are other mechanisms, not explored in the present study, that may also act concomitantly. For example, sorafenib may decrease the transforming growth factor (TGF)-beta by inhibiting the PDGFR. TGF-beta acts on downregulation of cellular proliferation and differentiation, what may turn the keratinocytes more susceptible to oncogenic pathways [24]. Other potential mechanism relies in evidence that sorafenib decrease primary T-cell immune response and impair cutaneous anti-tumor immunosurveillance [21]. Impairment in skin immune environment may be actively involved in the occurrence of inflammatory lesions in our cohort of patients because of sorafenib-treatment and/or immunosuppression regimes after liver transplantation. The association with prior development of DAEs further supports such immune dysregulation. In turn, vascular lesions such as livedo reticularis and thrombotic microangiopathy may derive from the direct impact of sorafenib on endothelial cell regulation and modulation of systemic coagulation in cancer patients [34, 35].
Our study is limited by the low number of patients with each type of SL and to the restricted set of mutations analyzed. This prevents us from drawing a complete causative chain between sorafenib and molecular alterations driving to SL. We expect our data may prime other groups to explore skin lesions under cancer therapy and increase the knowledge in this field. The underlying molecular events and the influence of the immune system should be further explored in order to provide useful information on the impact on these events in the clinical course and outcomes of patients with HCC under any other treatment besides sorafenib. From a clinical point of view, it is important to pay attention to the need to closely monitoring patients in order to detect newly developed SL during treatment.
Materials and Methods
Patients
A prospective database of consecutive patients treated at Hospital Clinic de Barcelona, from October 2007 to January 2018 was evaluated. Informed consent was obtained from all patients involved in the study that was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Hospital Clínic de Barcelona.
Included population consisted of patients with 1) HCC diagnosed based on radiologic and/or histologic features; 2) preserved liver function classified as Child-Pugh class A or B7 (without clinical ascites and/or encephalopathy); 3) Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 or 1; 4) absence of active cardiovascular disease, except for controlled arterial hypertension and/or stable peripheral vascular disease; 5) adequate renal and hematologic profile, 6) development of a SL detected during sorafenib treatment and 7) requirement of excisional or incisional biopsy for diagnostic and/or therapeutic purposes.
Relevant data were selected including age, gender, ECOG-PS, preexisting liver disease, Child-Pugh score, Barcelona Clinic Liver Cancer (BCLC) stage, prior treatment for HCC, sorafenib treatment duration, toxicities and overall survival. Outcome data were last updated on June 2020.
Treatment procedures
According to the local protocol, sorafenib was administered orally at starting dose of 400 mg twice daily, which could be modified upon development of adverse events according to its type and severity [36]. The management of DAE and SL was based on a multidisciplinary approach that combined dermatology and hepatology teams. Treatment was continued until symptomatic progression, radiologic progression with option to a second-line trial, treatment intolerance or death. Clinical and laboratory assessments were done monthly while radiology evaluation was done at the end of the first month and bimonthly afterwards. In each clinical evaluation, a complete physical examination including skin inspection was realized by the treating physician. Once a suspected SL was detected, the patient was referred to the dermatology consultant and, if indicated, a biopsy was performed.
Pathology evaluation and making patients’ study groups
SL samples from patients under sorafenib treatment were collected and sent to Pathology Department. Each skin sample was formalin-fixed, paraffin-embedded and stained with hematoxylin and eosin. SLs were clustered by two pathologists (AD and CF) according to the etiology as 1) proliferative lesions (BCC, KA, SCC and seborrheic keratosis) and 2) non proliferative.
For the control group, samples from patients with SL (6 BCC, 5 KA, 5 seborrheic keratosis and 3 SCC) without diagnosis of HCC nor use of sorafenib were selected. Control group was paired by age, gender and demographics to comparatively characterize molecular and immunohistochemical (IHC) findings.
Immunohistochemistry was used to analyze the nuclear positivity of phospho-ERK 1/2 (pERK) and mutant p53 proteins in SL cells.
Immunostainings were performed on formalin fixed and paraffined tissue sections submitted to antigen retrieval technique using pH 6.0 buffer. Incubation was done with specific antibodies for pERK (Cell Signaling ref 4370 at a 1:300 dilution) and mutant p53 (Abcam ref ab32049 at a dilution 1:400). The result was given as the percentage of SL cells with nuclear expression of pERK and p53 out of the total of SL counted. We considered that nuclear positivity of pERK stands for activation of MAPK pathway. No uniform cut-off has been established in literature for reduced or increased immunostaining of pERK. Based on available evidence, we arbitrarily set the cut-off of 20%, meaning that > 20% of SL cells with positive pERK means activation of MAPK pathway [37]. For p53, a cut-off of > 5% was considered as indicative of p53 mutation [38, 39].
Mutation detection assay – competitive allele-specificTaqMan-PCR
Mutation analysis was done in the same paraffined-embedded tissues used for IHC analysis. DNA was purified from 10 μm sections using PureLink Genomic DNA mini kit (Invitrogene, ref K1820-02). Detection and measurement of BRAF V600E (BRAF_476_mu: Hs00000111_mu; BRAF_rf: Hs00000172_rf), HRAS G12D (HRAS_484_mu: Hs00000778_mu; HRAS_ref: Hs00001018_rf), HRAS Q61K (HRAS_496_mu: Hs00000785_mu; HRAS_ref: Hs00001018_rf), HRAS Q61L (HRAS_498_mu: Hs00000786_mu; HRAS_ref: Hs00001018_rf) and KRAS G12D (KRAS_521_mu: Hs00000121_mu; KRAS_rf: Hs00000174_rf) somatic mutations was done by means of competitive allele-specific TaqMan™ PCR technology (castPCR™ Technology). The CAST-PCR technology can detect rare amounts of mutated DNA in a sample that contains large amounts of normal, wild-type DNA and works in formalin-fixed paraffin-embedded (FFPE) tissue samples. Real-Time PCR data obtained was analyzed using specific Applied Biosystems™ Mutation Detector™ Software.
Statistical analysis
Categorical variables were described as absolute frequency and percentages (%). Continuous or ordinal variables were described as median and interquartile ranges [IQR: percentile 25th–75th]. Time to-event data were estimated by Kaplan Meier with median and 95% Confidence Interval (95%CI). All the analysis were performed using SAS v. 9.4 (SAS Institute, Cary, NC).
ACKNOWLEDGMENTS
Investigators are indebted to CERCA Program from Generalitat de Catalunya and Fondo Europeo de Desarrollo Regional (FEDER) from European Commission. This work was developed at the building Centro Esther Koplowitz, Barcelona. Special thanks to patients and families participating in the study.
CONFLICTS OF INTEREST
Leonardo G. da Fonseca: None. -Carla Fuster-Anglada: None. -Cristina Carrera: None. Cristina Millán: None. - Esther Samper: None. Víctor Sapena: Travel funding from Bayer. Álvaro Díaz-González: Speaker fees and travel funding from Bayer. Travel funding from BTG and GILEAD. Marco Sanduzzi-Zamparelli: Speaker fees and travel funding from Bayer. Travel grant from BTG. Alejandro Forner: Consultancy fees from Bayer, Guerbert, AstraZeneca. Speaker fees from Bayer, MSD-Eisai and Gilead. Jordi Bruix: Consultancy fees from Arqule, Bayer, Novartis, BMS, BTGBiocompatibles, Eisai, Kowa, Terumo, Gilead, Bio-Alliance/Onxeo, Roche, AbbVie, Merck, Sirtex, Ipsen, Astra-Medimmune, Incyte, Quirem, Adaptimmune, Lilly, Basilea, Nerviano. Research grants from Bayer and BTG. Educational grants from Bayer and BTG. Lecture fees from Bayer, BTG- Biocompatibles, Eisai, Terumo, Sirtex, Ipsen. María Reig: Consultancy fees from Bayer, BMS, Roche, Ipsen, AstraZeneca and Lilly. Lecture fees from Bayer, BMS, Gilead, and Lilly. Research grants from Bayer and Ipsen. Loreto Boix: None. Alba Díaz: None.
FUNDING
This study was funded by Instituto de Salud Carlos III (PI18/00768 project) together with Fondo Europeo de Desarrollo Regional. “Una manera de hacer Europa”. AD-G: Received grant support from Instituto de Salud Carlos III, Río Hortega Grant (CM15/00050), Ayuda Clínico Junior 2018 (CLJUN18016DIAZ) from the Asociación Española Contra el Cáncer (AECC) and Beca Iniciació a la Recerca 2017 from the Societat Catalana de Digestologia. AF: received grant support from Instituto de Salud Carlos III (PI13/01229 and PI18/00542). MS-Z: received grants from “Ajuts per a la iniciació a la recerca 2019 from Societat Catalana de Digestologia (SCD)” and FI19/041958 from Instituto de Salud Carlos III. JB: received grant support from Instituto de Salud Carlos III (PI18/0768), AECC (PI044031), Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement (2014 SGR 605) and WCR (AICR) 16-0026. CIBERehd is funded by the Instituto de Salud Carlos III. MR: received grant support from Instituto de Salud Carlos III (PI15/00145 and PI18/00358). CF-A: received grant support from Contractes Clínic de Recerca “Emili Letang-Josep Font” 2020, granted by Hospital Clínic de Barcelona.
References
1. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, Cosme de Oliveira A, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–390. https://doi.org/10.1056/NEJMoa0708857. [PubMed].
2. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10:25–34. https://doi.org/10.1016/S1470-2045(08)70285-7. [PubMed].
3. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007; 356:125–34. https://doi.org/10.1056/NEJMoa060655. [PubMed].
4. Thomas L, Lai SY, Dong W, Feng L, Dadu R, Regone RM, Cabanillas ME. Sorafenib in metastatic thyroid cancer: a systematic review. The Oncologist. 2014; 19:251–258. https://doi.org/10.1634/theoncologist.2013-0362. [PubMed].
5. Gounder MM, Mahoney MR, Van Tine BA, Ravi V, Attia S, Deshpande HA, Gupta AA, Milhem MM, Conry RM, Movva S, Pishvaian MJ, Riedel RF, Sabagh T, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018; 379:2417–2428. https://doi.org/10.1056/NEJMoa1805052. [PubMed].
6. Ikeda M, Fujita T, Amoh Y, Mii S, Matsumoto K, Iwamura M. Stevens-Johnson syndrome induced by sorafenib for metastatic renal cell carcinoma. Urol Int. 2013; 91:482–483. https://doi.org/10.1159/000351918. [PubMed].
7. Díaz-González A, Sanduzzi-Zamparelli M, Sapena V, Torres F, Llarch N, Iserte G, Forner A, Fonseca LG, Rios J, Bruix J, Reig M. Systematic review with meta-analysis: the critical role of dermatological events in patients with hepatocellular carcinoma treated with sorafenib. Aliment Pharmacol Ther. 2019; 49:482–491. https://doi.org/10.1111/apt.15088. [PubMed].
8. Branco F, Alencar RS, Volt F, Sartori G, Dode A, Kikuchi L, Tani CM, Chagas AL, Pfiffer T, Hoff P, Carrilho FJ, Mattos AA. The Impact of Early Dermatologic Events in the Survival of Patients with Hepatocellular Carcinoma Treated with Sorafenib. Ann Hepatol. 2017; 16:263–68. https://doi.org/10.5604/16652681.1231585. [PubMed].
9. Reig M, Torres F, Rodríguez-Lope C, Forner A, LLarch N, Rimola J, Darnell A, Ríos J, Ayuso C, Bruix J. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J Hepatol. 2014; 61:318–24. https://doi.org/10.1016/j.hep.2014.03.030. [PubMed].
10. Kong HH, Cowen EW, Azad NS, Dahut W, Gutierrez M, Turner ML. Keratoacanthomas associated with sorafenib therapy. J Am Acad Dermatol. 2007; 56:171–172. https://doi.org/10.1016/j.jaad.2006.10.032. [PubMed].
11. Arnault JP, Wechsler J, Escudier B, Spatz A, Tomasic G, Sibaud V, Aractingi S, Grange JD, Poirier-Colame V, Malka D, Soria JC, Mateus C, Robert C. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009; 27:e59–61. https://doi.org/10.1200/JCO.2009.23.4823. [PubMed].
12. Smith KJ, Haley H, Hamza S, Skelton HG. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009; 35:1766–1770. https://doi.org/10.1111/j.1524-4725.2009.01289.x. [PubMed].
13. Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011; 23:177–182. https://doi.org/10.1097/CCO.0b013e3283436e8c. [PubMed].
14. Dubauskas Z, Kunishige J, Prieto VG, Jonasch E, Hwu P, Tannir NM. Cutaneous squamous cell carcinoma anf inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer. 2009; 7:20–23. https://doi.org/10.3816/CGC.2009.n.003. [PubMed].
15. Arnault JP, Mateus C, Escudier B, Tomasic G, Wechsler J, Hollville E, Soria JC, Malka D, Sarasin A, Larcher M, André J, Kamsu-Kom N, Boussemart L, et al. Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res. 2012; 18:263–272. https://doi.org/10.1158/1078-0432.CCR-11-1344. [PubMed].
16. Degen A, Satzger I, Voelker B, Kapp A, Hauschild A, Gutzmer R. Does basal cell carcinoma belong to the spectrum of sorafenib-induced epithelial skin cancers? Dermatology. 2010; 221:193–196. https://doi.org/10.1159/000317081. [PubMed].
17. Breaker K, Naam M, La Rosa FG, Flaig IP, Flaig TW. Skin cancer associated with the use of sorafenib and sunitinib for renal cell carcinoma. Dermatol Surg. 2013; 39:981–987. https://doi.org/10.1111/dsu.12184. [PubMed].
18. Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, Roden C, Chalk CJ, Ardlie K, Palescandolo E, Piris A, MacConaill LE, Robert C, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012; 30:316–21. https://doi.org/10.1200/JCO.2011.36.7680. [PubMed].
19. Yilmaz AS, Ozer HG, Gillespie JL, Allain DC, Bernhardt MN, Furlan KC, Castro LT, Peters SB, Nagarajan P, Kang SY, Iwenofu OH, Olencki T, Teknos TN, Toland AE. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumours. Cancer. 2017; 123:1184–1193. https://doi.org/10.1002/cncr.30459. [PubMed].
20. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012; 366:207–15. https://doi.org/10.1056/NEJMoa1105358. [PubMed].
21. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010; 464:431–435. https://doi.org/10.1038/nature08833. [PubMed].
22. Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KSM. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol. 2013; 10:390–399. https://doi.org/10.1038/nrclinonc.2013.83. [PubMed].
23. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014; 14:455–467. https://doi.org/10.1038/nrc3760. [PubMed].
24. Carlos G, Anforth R, Clements A, Menzies AM, Carlino MS, Chou S, Fernandez-Peñas P. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatology. 2015; 151:1103–1109. https://doi.org/10.1001/jamadermatol.2015.1745. [PubMed].
25. Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015; 348:880–886. https://doi.org/10.1126/science.aaa6806. [PubMed].
26. Jonason AS, Kunala S, Price GJ, Restifo RJ, Spinelli HM, Persing JA, Leffell DJ, Tarone RE, Brash DE. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci USA. 1996; 93:14025–14029. https://doi.org/10.1073/pnas.93.24.14025. [PubMed].
27. Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013; 5:a012559. https://doi.org/10.1101/cshperspect.a012559. [PubMed].
28. Xiang F, Lucas R, Hales S, Neale R. Incidence of nonmelanoma skin cancer in relation to ambient UV radiation in white populations, 1978–2012:empirical relationships. JAMA Dermatology. 2014; 150:1063–71. https://doi.org/10.1001/jamadermatol.2014.762. [PubMed].
29. Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, Tsai KY, Curry JL, Tetzlaff MT, Lai SY, Yu J, Muzny DM, Doddapaneni H, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014; 20:6582–92. https://doi.org/10.1158/1078-0432.CCR-14-1768. [PubMed].
30. South AP, Purdie KJ, Watt SA, Haldenby S, den Breems N, Dimon M, Arron ST, Kluk MJ, Aster JC, McHugh A, Xue DJ, Dayal JH, Robinson KS, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014; 134:2630–2636. https://doi.org/10.1038/jid.2014.154. [PubMed].
31. Gleich T, Chiticariu E, Huber M, Hohl D. Keratocanthoma:a distinct entity? Exp Dermatol. 2016; 25:85–91. https://doi.org/10.111/exd.12880. [PubMed].
32. Ra SH, Su A, Li X, Zhou J, Cochran AJ, Kulkarni RP, Binder SW. Keratoacanthoma and squamous cell carcinoma are distinct from a molecular perspective. Mod Pathol. 2015; 28:799–806. https://doi.org/10.1038/modpathol.2015.5. [PubMed].
33. Athar M, Li C, Kim AL, Spiegelman VS, Bickers DR. Sonic hedgehog signaling in Basal cell nevus syndrome. Cancer Res. 2014; 74:4967–75. https://doi.org/10.1158/0008-5472.CAN-14-1666. [PubMed].
34. Elice F, Rodeghiero F, Falanga A, Rickles FR. Thrombosis associated with angiogenesis inhibitors. Best Pract Res Clin Haematol. 2009; 22:115–28. https://doi.org/10.1016/j.beha.2009.01.001. [PubMed].
35. Patel TV, Morgan JA, Demetri GD, George S, Maki RG, Quigley M, Humphreys BD. A preeclampsia-like syndrome characterized by reversible hypertension and proteinuria induced by the multitargeted kinase inhibitors sunitinib and sorafenib. J Natl Cancer Inst. 2008; 100:282–284. https://doi.org/10.1093/jnci/djm311. [PubMed].
36. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.0. 2009.
37. Levidou G, Saetta AA, Gigelou F, Karlou M, Papanastasiou P, Stamatelli A, Kavantzas N, Michalopoulos NV, Agrogiannis G, Patsouris E, Korkolopoulou P. ERK/pERK expression and B-raf mutations in colon adenocarcinomas: correlation with clinicopathological characteristics. World J Surg Oncol. 2012; 10:47. https://doi.org/10.1186/1477-7819-10-47. [PubMed].
38. Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih IM, Kurman RJ. Immunohistochemical staining patterns pf p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011; 24:1248–53. https://doi.org/10.1038/modpathol.2011.85. [PubMed].
39. Khodaeiani E, Fakhrjou A, Amirnia M, Babaei-Nezhad S, Taghvamanesh F, Razzagh-Karimi E, Alikhah H. Immunohistochemical evaluation of p53 and KI67 expression in skin epithelial tumors. Indian J Dermatol. 2013; 58:181–187. https://doi.org/10.4103/0019-5154.110824.