
Introduction
Iron is vital for normal cell growth and survival. Cancer is an evolutionary maverick, which exploits its trademark genomic instability to drain environmental resources. As an enzyme cofactor, iron is responsible for many cellular processes including mitochondrial metabolism and DNA synthesis [1–3]. As iron can drive cellular proliferation, cancer cells have an adapted iron metabolism allowing increased iron accumulation. Studies have demonstrated cancer cells have an aberrant expression of iron metabolism genes, as well as an overexpression of iron import proteins and underexpression of iron export proteins [4–7]. This has led to iron accumulation being considered as a target for cancer therapies. Among the potential therapies which target iron metabolism, iron chelators are one of the most well studied. Iron chelators selectively deplete cancer cells of iron, exploiting cancer’s iron addiction – a trait displayed by a range of different cancers.
Deferoxamine (DFO) was the first iron chelator taken forward for clinical trials in 1987. Initially DFO was designed as a treatment for iron overload, but promising research conducted in cell models prompted clinical testing [8–11]. Of note, in a Phase II trial neuroblastoma patients treated with DFO displayed reduced bone marrow infiltration and one patient had a significant decrease in tumour mass [12]. Overall patients’ response to DFO has been variable with some patients showing complete or partial response and some patients showing no response (See Supplementary Table 2) [12–16]. This has been attributed to the poor lipophilicity of DFO, as well as its rapid clearance by the kidneys and poor absorption in the small intestine [17, 18]. Moreover, DFO is administered through continuous infusions, which is inconvenient, time-consuming and painful for patients [19]. This mixed bag of results has fuelled further functional and structural studies aimed at designing improved iron chelators as potential anticancer therapies.
The thiosemicarbazone class is a later stage of iron chelator evolution which manifested in 1992 [20]. Unlike their predecessor DFO, thiosemicarbazone chelators are capable of inducing reactive oxygen species (ROS). Triapine (3-AP) is a thiosemicarbazone; its primary mode of action is thought to be ribonucleotide reductase inhibition with a higher potency than commonly used ribonucleotide reductase inhibitor, hydroxyurea [21, 22]. Interestingly, tumours resistant to hydroxyurea retain sensitivity to triapine [23]. Triapine was taken forward to clinical trials in 2002 where it was successful with blood cancers, but not solid tumours (See Supplementary Table 2) [24–27]. Triapine acted as a radiosensitiser during cervical, vaginal and ovarian cancer clinical trials (See Supplementary Table 2) [28, 29]. Another issue that manifested was a short-lived patient response, suggesting patients develop resistance [24, 30]. Triapine is rapidly metabolised and its metabolite is inactive, rendering it ineffective against solid tumours [31, 32].
More structural and functional studies lead to the emergence of Dp44mT – a terminally demethylated triapine derivative. ROS induction is pivotal for Dp44mT function as antioxidants inhibit Dp44mT cytotoxicity [33]. Dp44mT is almost 50-fold more potent than its predecessor triapine. Dp44mT is effective in vivo; treated mice experience no changes in body or organ weight and little change in hematologic indices [34, 35]. New innovative ways of drug delivery have been explored in an effort to lower the risk of Dp44mT side effects such as methemoglobin in clinic. Nanoparticles have been used to counteract Dp44mT high cytotoxicity and low bioavailability. This approach is predicted to protect healthy tissues from the cytotoxic effects as the timing and place of the drug release can be controlled [36]. Encapsulating Dp44mT in PLGA nanoparticles enhanced its ability to induce apoptosis and improved its selectivity towards cancer cells [37].
At present many more classes of iron chelators are being taken into consideration as potential cancer therapy candidates. VLX600 is a novel iron chelator capable of targeting both senescent and proliferative cells [38]. VLX600 has been taken forward to a phase I clinical trial where it showed limited adverse effects (See Supplementary Table 2) [39]. Promising preclinical and clinical work may prompt further VLX600 studies. Furthermore, there are many natural compounds, which chelate iron. Natural iron chelators have shown a similar impact on oncogenic signalling pathways as well-characterized iron chelators DFO and Dp44mT (See Supplementary Table 1). Silibinin (isolated from milk thistle), quercetin (plant flavonoid), epigallocatechin gallate (the most abundant component of green tea) have been suggested to have chemo-preventative properties in a range of cancers suggesting potential benefit to their dietary intake, however not without limitations [40–42]. Epigallocatechin gallate has poor bioavailability as it is degraded by the gut microbiota [43]. There are some potential risks associated with quercetin; many animal studies indicated nephrotoxicity, limiting the use of quercetin in patients with pre-existing kidney damage [44]. Although silibinin has been proven safe at high doses its use in clinic still limited by its poor bioavailability, poor solubility in water, and poor absorption in the small intestine [45, 46].
This review aims to explore the underlying mechanisms of action behind iron chelator driven cytotoxicity in the context of the hallmarks of cancer established by Hanahan and Weinberg [47, 48] (see Figure 1, Supplementary Table 1). This will in turn support further research into iron chelators as a potential effective anti-cancer therapy.
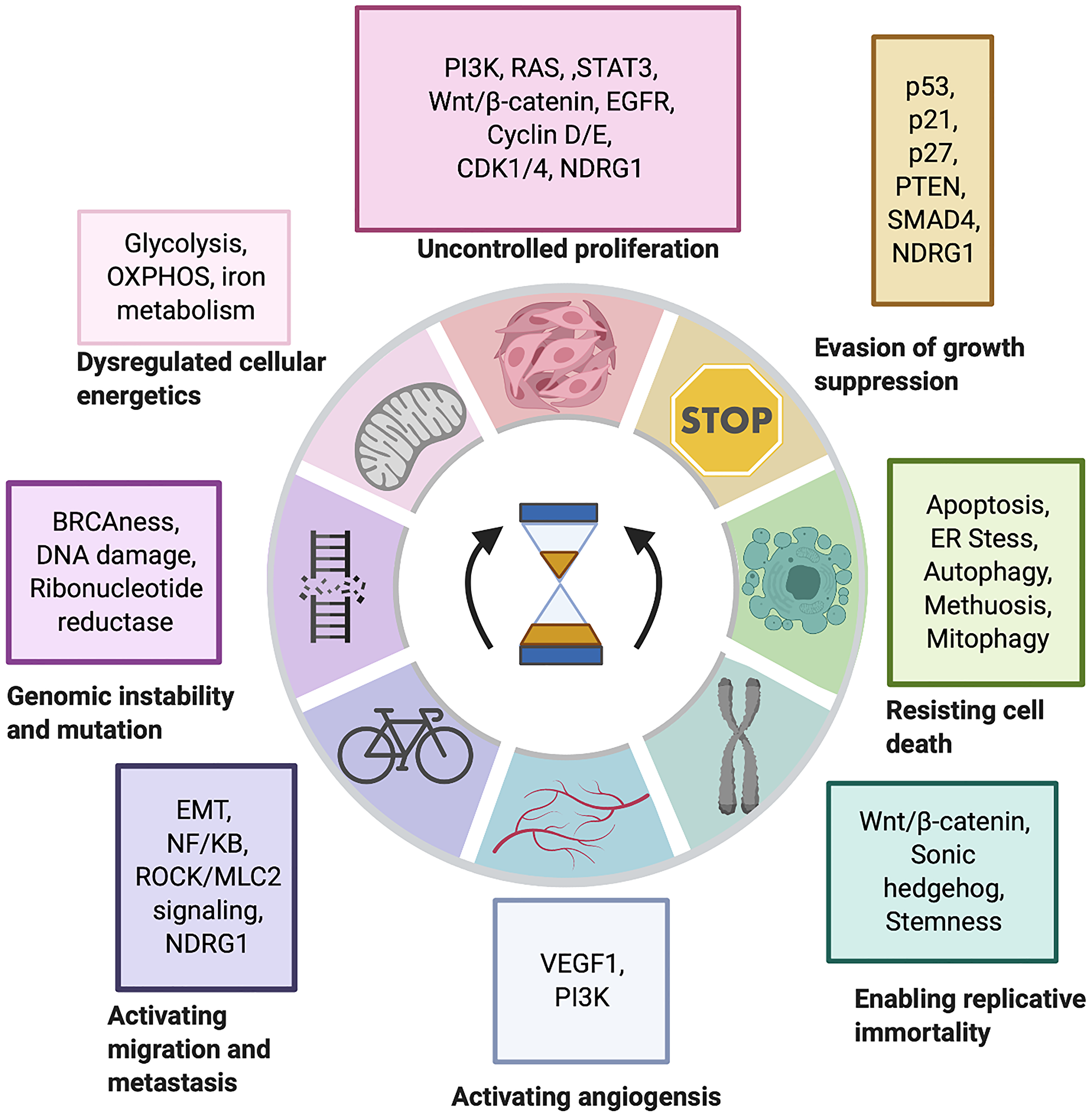
Figure 1: The impact of iron chelators on the hallmarks of cancer. Iron chelators have been shown to reverse many oncogenic signalling pathways associated with each hallmark of cancer with NDRG1 being a common thread. Generated through BioRender.com [47, 48].
NDRG1: The proposed target of iron chelators
N-MYC downregulated gene 1 (NDRG1) plays a critical role in inducing iron chelator mediated cytotoxicity [49]. The NDRG family is composed of 4 members: NDRG1, NDRG2, NDRG3, and NDRG4, sharing 53–65% sequence similarity [50] The C-terminus of NDRG proteins contains residues, which are potential targets for kinases [50, 51]. Numerous studies have implicated NDRG1 as the key target of iron chelation with few implicating NDRG2 and NDRG3. NDRG1 expression has been linked to cell differentiation, adhesion, development and p53 dependent apoptosis [52–55]. Despite a strong relation between iron chelators, a link between NDRG1 and natural iron chelators has not been explored in the literature.
There is conflicting evidence on whether NDRG1 should be classified as a tumour suppressor or an oncogene. NDRG1 can display pleiotropy - performing different roles in different cell types and conditions [56]. Low levels of NDRG1 have been associated positive patient prognosis in oesophageal squamous cell carcinoma and neuroblastoma, yet poor patient prognosis in gastric cancer, breast cancer and hepatocellular carcinoma [57–60]. Pre-metastatic tumours are more likely to be hypoxic; this potentially explains higher levels of NDRG1 expression potentially through HIF-1α. Post translational modifications (PTM) could be another driver of pleiotropy. PTMs have been shown to influence NDRG1 cellular localisation [61]. Phospo-NDRG1 (Serine 330) localises within the nucleus, while Phospho-NDRG1 (Threonine 346) localises within the cytoplasm. PTEN gene silencing has the ability to increase Thr346 phosphorylation in prostate cancer cells, but not in hepatocellular carcinoma [61]. This further supports a pleiotropic mode of action. Treatment with DFO and Dp44mT both increase Thr346 and Ser330 phosphorylation [62]. Western blot analysis of DU145 cell lysates demonstrated two bands corresponding to NDRG1 one at 41kDa and one at 46kDa from DU145. The 41kDa band represented the truncated isoform of NDRG1, which is expressed in prostate cancer cell lines; PC3, DU145, LNCaP but not normal prostate cells PrEC [63]. Truncated NDRG1 has also been observed in PANC-1 and HT-29 cells. The truncated isoform demonstrates lowered nuclear localisation than the full-length isoform [61]. Localisation of proteins can have a major impact on classification as an oncogene of a tumour suppressor. Taken together, this suggests the truncated isoform may serve a role during carcinogenesis. Future studies should be sure to classify and distinguish between truncated and wild type NDRG1 isoforms.
Uncontrolled cell growth and proliferation: Iron chelation attenuates cancer cell proliferation, enhancing signalling pathways
Uncontrolled proliferation is the most well recognised hallmark of cancer. During malignant transformation, cancer cells acquire changes in signalling pathways which amplify their proliferative potential. A plethora of evidence has linked iron chelation mediated NDRG1 upregulation to the inhibition of many oncogenic signalling pathways including STAT3, Wnt/ β-catenin, RAS and AKT/PI3K [62, 64–66]. Overexpression of NDRG1 - driven by iron chelators Dp44mT and DFO - in MIAPaCa-2 pancreatic cancer cell line lead to a downregulation of the oncogenic TGF-β and RAS pathways through an upregulation of SMAD4 (the pathway’s negative regulator). As a result, RAS downstream signalling molecules pERK, pSMAD2L are inhibited [64]. STAT3 is a transcription factor, which is constitutively active in tumours [67]. DFO, Dp44mT and DpC inhibit upstream kinases c-BCL and SRC, which promote STAT3 phosphorylation and subsequent dimerisation. As a result, STAT3 cannot promote the expression of its target genes: Bcl-2, cyclin D1, and c-myc [65].
NDRG1 can also suppress AKT and ERK signalling pathways. NDRG1 upregulation mediated through iron chelation has been shown to elevate PTEN expression levels in DU145 (prostate cancer cells), PrEC (normal prostate epithelial cells), but not PC3 (prostate cancer cells), which harbour a homozygous deletion in PTEN [62, 68]. While iron chelation did not increase the levels of AKT total protein it did increase the levels of pAKT (Ser473) in prostate cancer cells, whereas in Caco-2 cells there was a decrease in pAKT (Thr308) and no change in (Ser473) [62, 66]. The downstream effectors of pAKT – p-mTOR, S6K1, S6, 4E-BP1 and cyclin D1 - were not activated, despite the significant rise in pAKT [62, 66]. Both phosphorylation sites must be phosphorylated for pAKT to fulfil its role as a kinase [69]. Moreover, DFO stimulates REDD1 gene expression, which inhibits mTORC1 action [66]. Despite PC3 cells harbouring a deletion in PTEN, iron chelation still had an antiproliferative effect and still blocked AKT signalling, so hypothetically tumours with mutations in PTEN could still be treated with iron chelators. DFO and Dp44mT altered the ratio of p-SMAD2C and p-SMAD2L through decreasing the levels of oncogenic p-SMAD2L and p-SMAD2C levels remaining the same [62]. Levels of pERK1/2 dropped following iron chelation, suggesting pERK1/2 maybe responsible for the altered p-SMAD2L/p-SMAD2C ratio. NDRG1 overexpression mimicked the impact of iron chelation on AKT signalling, while NDRG1 silencing had the reverse effect, suggesting NDRG1 is responsible for the attenuation of AKT signalling. However, NDRG1 overexpression only slightly decreased the levels of pERK1/2 and p-SMAD2L so there may be another mechanism of signalling inhibition [62]. In contrast, treating triple negative breast cancer cells with DFO induces IL-6/PI3K/AKT signalling leading to an upregulation of iron uptake proteins Transferrin 1 and DMT1thus increasing iron uptake and supporting the pleiotropic function of NDRG1 [70]. It is possible that despite the capacity of NDRG1 to act as an oncogene, treating cells with iron chelators could return its tumour suppressive functions in certain conditions.
Wnt/β-catenin signalling represents another oncogenic signalling pathway attenuated by iron chelators. Deferasirox (DFX) inhibits Pyk2 phosphorylation without impacting total levels of Pyk2 protein, increasing β-catenin degradation through GSK-3β activation, thus impairing Wnt/β-catenin signalling [71]. Moreover, iron chelation by acyl hydrazones targeted β-catenin for ubiquitin mediated degradation even in cells with an abrogated destruction complex [72]. NDRG1 overexpression has been shown to increase β-catenin expression at the cell membrane and decrease β-catenin nuclear expression leading to lowered TCF/LEF signalling [73]. NDRG1 can interact with the Wnt receptor LRP6 in prostate cancer cells, inhibiting Wnt signalling cascade [74]. Cyclin D1 expression is a target gene of Wnt/ β-catenin signalling [75]. NDRG1 knockout lead to an increase in cyclin D1 levels [76].
It has been hypothesised that the ability of NDRG1 to block a range of oncogenic signalling pathways can be attributed to EGFR signalling as many of the pathways described previously are influenced by EGFR signalling. NDRG1 overexpression driven by Dp44mT and DpC in PANC-1 cells increases the half-life MI6 which facilitates EGFR degradation through lysosomal processing. This was dependent on PTEN expression [77]. Dp44mT can also prevent HER2 and HER3 activation leading to lower levels of EGFR/HER2 and EGF/HER3 heterodimer formation. Strikingly, Dp44mT has higher antiproliferative activity than the EGFR inhibitor erlotinib. The IC50 of erlotinib exceeds 80 μM in PANC-1, HT-29 and MIAPaCa-2 cells, whereas Dp44mT has an IC50 0.02–0.04 μM [78]. When proliferative signalling exceeds a certain threshold, it can induce senescence [47]. VLX600 is an iron chelator which can target senescent and actively proliferating proving that cytotoxicity driven by iron chelation is not always dependent on cell cycle progression [38].
Uncontrolled cell growth and proliferation: Iron chelation induces cell cycle arrest
The inhibition of cell proliferation pathways will have a domino effect across many cellular functions including cell cycle progression. Cells treated with iron chelators exhibit cell cycle arrest at two phases of the cell cycle: mid G1 phase and late G1/ early S phase [79, 80]. It has been predicted that an iron dependent checkpoint exists between G1 and S phase as cells depleted from iron will not enter S phase unless they had already reached S phase [79–81]. The mode of action of CDK1 is reliant on iron, which activates its kinase activities stimulating JAK/STAT3 signalling. Treatment with DFO inhibits CDK1 and its downstream signalling pathways [82]. Iron chelation can also elevate levels of cyclin E - a cyclin associated with mid G1 phase [83]. MDA MB 453 Breast cancer cells treated with mimosine have lowered levels of CDK4 and cyclin D – another CDK/cyclin complex which manifests during G1 phase [80].
Ribonucleotide reductase (RR) is an enzyme which provides cells with dNTPs - the building blocks of DNA. RR protein expression peaks during late G1 phase and early S phase to initiate the rate limiting step of DNA synthesis [84]. Iron is an enzyme cofactor for RR. When the binuclear iron reacts with oxygen a tyrosyl radical forms which is essential for RR enzyme activity [85]. The iron centre is labile and therefore requires constant replacing and as a result RR activity is dependent on the labile iron supply of the cell [85]. Iron chelators DFO, triapine and Dp44mT are capable of inhibiting RR, but through different mechanisms. DFO chelates the labile iron pool causing the disappearance of the tyrosyl radical and loss of RR enzyme activity [86]. Dp44mT inhibits RR activity through its impact on thiol antioxidant systems: thioredoxin, glutaredoxin and glutathione, which play a key role in maintaining RR protein double bonds [87]. Triapine has been suggested to bind directly to a binding pocket on the surface of the R2 subunit of RR freeing the diferric centre in mouse models [22]. Nevertheless, the net result is RR activity inhibition and cell cycle arrest.
Evading growth suppression: Iron chelation reactivates tumour suppressor genes
Tumour suppressor genes safeguard normal cells from uncontrolled growth and proliferation. During malignant transformation many tumour suppressor genes are mutated or switched off, therefore it is useful for new cancer therapies to be able to return of tumour suppressive signalling. NDRG1 is a potential tumour suppressor gene that has been implicated in iron chelator mediated cytotoxicity. NDRG1 gene expression is upregulated following treatment with DFO, 311 and Dp44mT and is attenuated when cells were supplemented with iron salts [88]. This suggests NDRG1 expression is stimulated by iron chelation rather than ROS induction. When NDRG1 is knocked out, cells become less sensitive to iron chelation, suggesting a crucial role for NDRG1 in the mode of iron chelator action. Interestingly, overexpression of NDRG1 combined with iron chelation treatment does not further sensitise cells to iron chelators [89]. There are several ways iron chelation could be inducing NDRG1 gene expression. Hypoxia-inducible factor 1α (HIF-1α) can promote NDRG1 gene expression through a Hif response element (HRE) sequence in the promoter region of NDRG1, however HIF-1α is not essential for NDRG1 gene expression [90]. In HIF-1α knockout (HIF-1α-/-) murine embryo fibroblasts (MEFs), which lack wild type HIF-1α protein expression iron chelation could still induce NDRG1 expression under normoxic conditions but not hypoxic conditions [88]. SP1, CEBPα, YB-1, Smad7 and p53 have also been proposed as transcriptional factors which upregulate NDRG1 gene expression [53, 88]. NDRG1 expression is repressed by oncogenic C-MYC and N-MYC [91]. NDRG1 was upregulated in p53 wild type MCF7 cells and p53 deficient H1299 to same extent in response to DFO and 311, suggesting this is independent of p53 expression [88].
Cell cycle progression is driven by components which are perceived as oncogenic (cyclins and cyclin dependent kinases) and inhibited by tumour suppressors (cyclin dependent kinase inhibitors). Iron chelation can inhibit oncogenic cell cycle drivers (e.g. cyclin D, CDK4), and simultaneously activate the tumour suppressive CDK inhibitors driving cell cycle arrest. Interestingly, iron chelation can modulate p21 levels and nuclear localisation independently of p53 status. Dp44mT can downregulate p21 in MCF7 (Breast cancer) cells, upregulate p21 in SK-MEL-28 (Melanoma) and CFPAC-1 (Pancreatic ductal adenocarcinoma) cells and has no effect on p21 in LNCaP (Prostate cancer) and SK-N-MC (Neuroepithelioma cell line derived from a supra orbital brain tumor) cells [92]. This is thought to be through MDM2. Iron chelation by DFO increases MDM2 gene expression, this is reversed by iron overload, as MDM2 expression is regulated by Iron Regulatory Protein 2 (IRP2) [93–95]. DFO can elevate the expression of p27 in serum-stimulated 3T3 cells inhibiting CDK2/cyclin E activity and preventing S phase entry [81]. DFO inhibits src kinase mediated phosphorylation and degradation of p27 [96]. Iron chelation increases levels of tumour suppressor gene PTEN and SMAD4 (detailed above) [64]. In addition to this, PTEN can upregulate NDRG1 gene expression suggesting a positive feedback loop [97].
Resisting programmed cell death: Iron chelation induces apoptotic signalling and the unfolded protein response
Normal cells have built-in mechanisms to prevent replication across damage thus passing on mutation. Among these mechanisms is apoptosis, which plays a vital role in growth and development of multicellular organisms and provides protection against carcinogenesis. Cancer cells develop mutations which prevent normal apoptotic signalling. Iron chelation has been shown to induce apoptosis in vitro and in vivo. This is accompanied by an increase in the levels of pro-apoptotic proteins such as BAX, caspase 8 caspase 9 and caspase 3 and a drop in anti-apoptotic proteins such as Bcl-2 as well as a release of cytochrome C [34, 98–103]. The dual involvement of caspase 8 and 9 suggests a death receptor and mitochondrial apoptotic cascade. Iron chelation by DFO induces an increase in p53 protein levels but not the mRNA levels indicating post-transcriptional regulation. The iron chelators 311 and Dp44mT have also been observed to increase p53 protein expression [99, 104, 105]. It has been established that an excess of iron leads to a downregulation of p53. Iron polyporphyrin heme triggers p53 degradation and blocks p53-DNA interactions [106]. However, iron chelator Dp44mT can induce p53 independent cell death in cell lines with non-functional p53 mutations such as PC-3 cells [33]. The effectiveness of iron chelation is therefore not dependent on p53.
Iron chelators DFO, DFX and Dp44mT induce ER stress through the four main modules of the unfolded protein response: protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1), activating transcription factor 6 (ATF6) and CaMKII. As a result of iron chelation mediated ER stress, JNK signalling is activated; a crucial signalling pathway for ROS induced cell death, and this initiates apoptotic signalling, therefore iron chelation mediated apoptosis is induced through ER stress [103, 107]. Interestingly, non-ROS inducing iron chelator DFO can stimulate JNK signalling [108].
The MAPK subfamilies p38, and JNK have been implicated in iron chelation induced death, however studies have not been consistent in assigning which signalling module is primarily responsible. The p38 MAPK was shown to be the primary inducer of apoptosis in HL-60 cells (human promyeloid leukemic cells), as p38 activation was observed prior to the early apoptotic traits and blocking p38 activity prevented the apoptotic cascade. Only low levels of JNK activation at the late stages of apoptosis was observed in HL-60 cells, whereas in gastric cancer cells AGS and SNU638 there was a rise in JNK phosphorylation and inhibiting JNK lead to a halt in iron chelation mediated cell death [101, 103].
In addition to this, iron chelation has been shown to induce autophagy. Interestingly, DFO and Dp44mT can drive LC3-I to LC3-II transformation - a marker of the autophagosome - through the PERK/eIF2α axis but NDRG1 overexpression represses pro-survival autophagy [109].
DFO and Dp44mT induced severe phenotypic changes in MDA MB 231 and MDA MB 157 breast cancer cell lines that closely resembled non-apoptotic non-autophagic cell death methuosis [110]. This was thought to be a survival adaptation allowing cells to accumulate nutrients extracellularly. The cells displayed lipid droplet accumulation, mitochondrial defects, a halt in protein translation and eventual cell death. These phenomena were undetected in any other cell lines [110]. Potentially different signalling pathways could trigger cell death in different cell lines. Another intriguing cellular response was silibinin induced mitophagy. Breast cancer cells MDA MB 231, MCF7 cells treated with silibinin suffered from mitochondrial fission, which lead up to mitophagy and apoptosis [111]. Ferroptosis is a form of cell death driven by iron accumulation, and ROS induction. Events observed during ferroptosis include lipid peroxidation, GPX4 depletion and cell rounding. As expected DFO can inhibit the onset of ferroptosis, but cannot reverse ongoing ferroptosis [112].
Enabling replicative immortality: Iron chelation inhibits stemness and Wnt/β-catenin signalling
In a normal cell population, there is a limit to the number of achievable cell doublings before cells reach their natural fate – senescence. Cancer cells lose this attribute to enable indefinite replication often showcasing stem cell like features. Enhanced telomerase activity is a driver of cancer’s immortality. Wnt/β-catenin signalling has been shown to directly induce telomerase transcription [113]. As mentioned previously iron chelation downregulates Wnt/β-catenin signalling and prevents nuclear translocation of β-catenin abrogating any changes in target gene expression. Iron chelation could potentially block replicative immortality, but this has not been directly investigated. Iron depletion by DFX hindered the expression of stemness markers Nanog, Oct3/4, Sox2, Klf4, and c-Myc and inhibited spherogenicity in colorectal and lung cancer stem cell models in vivo and in vitro. In addition to this, iron chelators DFO and DFX are cytotoxic to cancer stem cells which are resistant to classical chemotherapeutic drugs [114, 115]. Epigallocatechin gallate is also capable of inhibiting the expression of stem cell markers (CD)44, CD133, Oct4, ALDH1A1 and Nanog, through inhibiting sonic hedgehog signalling [116].
Sustained angiogenesis: Iron chelators demonstrate pro and anti-angiogenic capabilities
Angiogenesis is the creation of new blood vessels which sprout from pre-existing vessels improving access to nutrients and oxygen as well as increasing the likelihood of metastasis [47, 117]. Studies have been unclear and inconsistent regarding whether iron chelation can inhibit or stimulate angiogenesis. Iron chelation can inhibit PI3K signalling which is a proangiogenic pathway [64, 118]. NDRG1 upregulation has also been shown to downregulate MMP2 and MMP9 - key mediators of angiogenesis [119, 120]. On the other hand, DFO and Dp44mT upregulate VEGF expression through HIF- α, as hypoxia is a driver of angiogenesis [35, 117]. This is concerning as iron chelation has been proven to induce hypoxia [121]. Interestingly, VEGF gene expression is driven by HIF-α and STAT3 binding to the promoter region simultaneously [122]. As mentioned previously iron chelators prevent STAT3 activation and blocking STAT3 activity has been shown to inhibit VEGF gene expression [65, 123]. Iron chelation has been shown to induce macular edema in vivo [124]. This suggests iron chelation may induce angiogenesis. The reverse has been observed with natural iron chelators with silibinin, quercetin and epigallocatechin gallate decreasing the levels of angiogenesis associated proteins VEGF, MMP2, MMP9, and HIF-1α [125–128]. In fact, quercetin has been shown to inhibit angiogenic protein expression through STAT3 signalling inhibition [127]. The impact of iron chelation on angiogenesis in vivo has not been investigated. Iron chelators could potentially be combined with an angiogenesis inhibitor to prevent angiogenesis promotion induced by iron chelation.
Activating migration and metastasis Iron chelation prevents the initiation of metastasis through EMT and ROCK/MLC2 and NF-kB inhibition
Epithelial-mesenchymal transition (EMT) is a morphological change occurring during embryogenesis and wound healing. Cancer cells exploit the EMT to drive their migration and invasion. During the EMT cells undergo morphological and biochemical changes transforming them from an epithelial phenotype to a mesenchymal phenotype. TGF-β signalling promotes EMT and migration during the later stages of carcinogenesis [129]. When HT29 (colorectal cancer) or DU145 (prostate cancer) cells are treated with TGF-β, they undergo EMT which is characterised by a more spindle-like cell morphology, the presence of mesenchymal marker vimentin and a decrease in epithelial markers β-catenin and E-cadherin at the cell surface membrane. Combining TGF-β with iron chelators prevents TGF-β mediated EMT as displayed by a retained epithelial morphology and markers [76]. NDGR1 has been observed near the adhernes junctions and desomosomes in the cytoplasm so it possible that NDRG1 plays a role in cell adhesion through the formation of the E-cadherin/ β-catenin complex [56]. Another way the EMT is blocked is through the attenuation of NF-kB signalling. During tumorigenic conditions, the EMT is driven through TNF-α mediated LYRIC expression, which upregulates NF-kB signalling and in turn vimentin gene expression is induced. Treating cells with Dp44mT and DpC prevents this signalling cascade from occurring through NDRG1 upregulation. NDRG1 overexpression inhibits NF-ĸB signalling through reducing NEMO expression, preventing activation of Iĸĸα/β and blocking nuclear localisation of p65 [130]. During the EMT E-cadherin is downregulated by the transcriptional factors SLUG, SNAIL, TWIST and ZEB2. Iron chelation driven NDRG1 expression downregulates SLUG and SNAIL, whereas NDRG1 knockout has the reverse effect [76]. SMAD molecules are modulators of TGF-β signalling. Knockout of NDRG1 increased SMAD2, pSMAD3, and SMAD4 and overexpression lead to decreased SMAD2 and pSMAD3. These could mean NDRG1 regulation of TGF-β signalling could be through SMAD molecules [62, 76]. On the other hand, treating aggressive breast cancer cells, MDA MB 231 with DFO caused them to accumulate iron and acquire mesenchymal markers through activating TNF-alpha, NF-ĸB and TGF-β signalling. DFO was still effective in blocking EMT in non-aggressive MCF7 breast cancer cells [131].
Dp44mT and DFO have been shown to inhibit cell migration through NDRG1 mediated suppression of the Rho associated coiled-coil-forming protein kinase 1/myosin light chain 2 (ROCK1/pMLC2) pathway. ROCK1 phosphorylates MLC2 and this induces cell migration through actomyosin contractility [132, 133]. MLC2 drives cell motility through linking anti-parallel actin filaments causing sliding and forming stress fibres [132, 134]. Metastasis signalling is a trigger of stress fibre formation, therefore inhibiting MLC2 activity may inhibit metastasis [135]. Iron chelation treatment in HT29, HCT116 and DU145 cells decreases the levels of ROCK1 and phosphorylated MLC2 with no change in basal MLC2. Moreover, the levels of F-actin were also decreased making stress fibres less likely to form and attenuating cell motility and metastasis [136].
Genomic instability and mutation: Iron chelation exploits and mimics genomic instability
Many cancers have defective DNA damage repair, giving rise to mutants that will survive new selection pressures. As a result, many current cancer therapies exploit the genomic instability of cancer cells by causing irreparable DNA damage or inhibiting DNA damage repair eventually triggering cell death. ROS inducing iron chelators such as Dp44mT can induce double strand breaks [137–139]. Interestingly, iron chelators can not only induce DNA damage, but they can also inhibit homologous recombination repair DNA damage repair. Dp44mT and Triapine have been shown to inhibit ribonucleotide reductase preventing the production of the dNTP precursors of DNA damage repair [137]. Moreover, triapine can sensitise BRCA wildtype and PARP resistant epithelial ovarian cancer cells and xenograft mice to PARP inhibitor olaparib [140]. This is beneficial as reversal of BRCA mutations and a return of homologous recombination repair functioning have been observed in ovarian cancer patients [140–143]. Triapine combined with olaparib prevents homologous recombination repair of double strand breaks in wild type BRCA ovarian cancer cells through preventing RAD51 and BRCA1 foci formation as well as preventing BRCA1 from associating with the MRN complex and attenuation of CtIP phosphorylation [144]. In summary triapine blocks all olaparib driven homologous recombination repair in BRCA wildtype ovarian cancer cells, mimicking BRCAness [144, 145]. Cyclin D1 plays a role in mediating homologous recombination repair. Ionising radiation induces a cyclin D1/RAD51 interaction and inhibition of cyclin D1 expression sensitises cancer cells to ionising radiation [146]. Triapine could potentially be inhibiting HR through blocking cyclin D1 gene expression.
Metabolic reprogramming: Novel iron chelator VLX600 targets oxidative phosphorylation
Cancer cells undergo a metabolic transformation known as the Warburg effect, which shifts their source of energy from oxidative phosphorylation to glycolysis. This is another trait which is exploited by iron chelators. VLX600 diminishes the ability of MCF7 and HCT116 cells to undergo oxidative phosphorylation [38]. Triapine, DFO and CPX can inhibit oxidative phosphorylation but to a lesser extent. VLX600 reduces hypoxia which suggests it has lowered cell oxygen consumption. VLX600 also reduces cytochrome oxidase (complex IV) activity - the rate limiting step of oxidative phosphorylation [38]. The cells in the deep layers of a tumour are vulnerable to even the slightest decrease in oxidative phosphorylation, so theoretically they would be highly sensitive to VLX600 [38, 147, 148]. DFO can inhibit aerobic glycolysis and oxidative phosphorylation through ERK1/2 inhibition and reduce the gene expression of the 13 mitochondrial complex components [149].
Iron chelators can also impact iron metabolism. DFO targets ferritin for degradation through autophagy, whereas DFX and deferiprone target ferritin for proteasomal degradation [150]. Quercetin not only potently forms complexes with iron but can also induce cellular iron deficient behaviour such as induction of transferrin receptor-1 and iron regulatory protein-2 expression and lowered ferritin expression. Additionally, quercetin can modulate iron metabolism gene expression in rats decreasing expression of DMT1, Dcytb, FPN, and hepcidin. This decreased the level of iron absorption [151]. Dp44mT induced transferrin expression in DMS-53 xenograft mice, which could potentially lead to iron accumulation [35]. Although counterintuitive, iron chelation of MDA MB 231 cells by DFO lead to iron accumulation through an increase in TfR1 and DMT1 expression. This is thought to be a survival adaptation [152].
Conclusions and future work
Iron chelation has proven to be successful on a range of different tumour types in vitro and in vivo as iron addiction is a universal cancer trait. Even cancers which are notoriously hard to treat due to resistance have been subdued by iron chelation such as androgen insensitive prostate cancer and cancer stem cells or cells with loss of p53 and PTEN function.
Based on the data presented in this review iron chelators could potentially reverse many of the key hallmarks of cancer. Stripping the cells of iron impacts many cellular targets with some targets still undiscovered. NDRG1 has been proven to be the common link between the ability of iron chelators to reverse many of the hallmarks of cancer as overexpression of NDRG1 mimics the impact of iron chelation on several signalling pathways. Problems may arise from treating aggressive breast cancer cells with iron chelators. Aggressive breast cancer cells have demonstrated a unique reaction to iron chelators, including the accumulation of iron, activation of oncogenic signalling pathways and a methuosis-like death. Another potential issue is the ability of iron chelators to induce autophagy, which can function as a pro-survival response and a tumour suppressor response in cancer cells. There are still many unanswered questions about the mechanism of action of iron chelators. A consensus must be reached on the impact of iron chelation on angiogenesis through in vivo studies. As STAT3 is essential for VEGF gene expression and iron chelation attenuates STAT3 dimerisation and nuclear localisation. Studies must confirm if STAT3 is still capable of inducing VEGF gene expression in cells treated with iron chelators such as Dp44mT, as this has been confirmed with epigallocatechin gallate. If more evidence is found linking iron chelation and angiogenesis, iron chelation could potentially be combined with an angiogenesis inhibitor to prevent angiogenesis promotion. Any future studies on the impact of NDRG1 on patient prognosis or tumorigenesis must differentiate between the cleaved and full isoforms as well as the phosphorylation isoforms. The cleaved isoform is only present in cancer cells and could potentially be oncogenic. Although many mechanistic studies have been undertaken iron chelators, the complexity of cell signalling remains a hurdle preventing the discovery of all cellular targets of iron chelation. A potential way of discovering new targets is combining iron chelators with well-characterised cancer therapeutics. Triapine was discovered to inhibit homologous recombination repair as a result of combinatorial studies. We propose a combinatorial study of iron chelators with immune checkpoint inhibitors as they have shown success in clinic and could uncover more mechanisms of action. The full impact of iron chelators on the two remaining hallmarks of cancer inflammation and immune evasion must be established. Moreover, iron chelation has multiple targets within a cancer cell, so the question lies whether the ideal cancer therapy is overarching or specific.
ACKNOWLEDGMENTS
We would like to thank Biorender for the provision of their website for construction of our figure.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
References
1. Singer TP, Johnson MK. The prosthetic groups of succinate dehydrogenase: 30 years from discovery to identification. FEBS Lett. 1985; 190:189–98. https://doi.org/10.1016/0014-5793(85)81282-5. [PubMed].
2. Kolberg M, Strand KR, Graff P, Andersson KK. Structure, function, and mechanism of ribonucleotide reductases. Biochim Biophys Acta. 2004; 1699:1–34. https://doi.org/10.1016/j.bbapap.2004.02.007. [PubMed].
3. Puig S, Ramos-Alonso L, Romero AM, Martinez-Pastor MT. The elemental role of iron in DNA synthesis and repair. Metallomics. 2017; 9:1483–500. https://doi.org/10.1039/c7mt00116a. [PubMed].
4. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, Xian W, McKeon F, Lynch M, Crum CP, Hegde P, Brewer M, Wang X, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017; 36:4089–99. https://doi.org/10.1038/onc.2017.11. [PubMed].
5. Zhang S, Chang W, Wu H, Wang YH, Gong YW, Zhao YL, Liu SH, Wang HZ, Svatek RS, Rodriguez R, Wang ZP. Pan-cancer analysis of iron metabolic landscape across the Cancer Genome Atlas. J Cell Physiol. 2020; 235:1013–24. https://doi.org/10.1002/jcp.29017. [PubMed].
6. Xue D, Zhou CX, Shi YB, Lu H, He XZ. Decreased expression of ferroportin in prostate cancer. Oncol Lett. 2015; 10:913–6. https://doi.org/10.3892/ol.2015.3363. [PubMed].
7. Pinnix ZK, Miller LD, Wang W, D'Agostino R Jr, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010; 2:43ra56. https://doi.org/10.1126/scisignal.3001127. [PubMed].
8. Estrov Z, Tawa A, Wang XH, Dube ID, Sulh H, Cohen A, Gelfand EW, Freedman MH. In vitro and in vivo effects of deferoxamine in neonatal acute leukemia. Blood. 1987; 69:757–61. [PubMed].
9. Valle P, Timeus F, Piglione M, Rosso P, di Montezemolo LC, Crescenzio N, Marranca D, Ramenghi U. Effect of different exposures to desferrioxamine on neuroblastoma cell lines. Pediatr Hematol Oncol. 1995; 12:439–46. https://doi.org/10.3109/08880019509009473. [PubMed].
10. Helson L. In vivo effects of repeated exposure of human neuroblastoma cell lines to deferoxamine. Anticancer Res. 1991; 11:409–10. [PubMed].
11. Brodie C, Siriwardana G, Lucas J, Schleicher R, Terada N, Szepesi A, Gelfand E, Seligman P. Neuroblastoma sensitivity to growth inhibition by deferrioxamine: evidence for a block in G1 phase of the cell cycle. Cancer Res. 1993; 53:3968–75. [PubMed].
12. Donfrancesco A, De Bernardi B, Carli M, Mancini A, Nigro M, De Sio L, Casale F, Bagnulo S, Helson L, Deb G. Deferoxamine followed by cyclophosphamide, etoposide, carboplatin, thiotepa, induction regimen in advanced neuroblastoma: preliminary results. Italian Neuroblastoma Cooperative Group. Eur J Cancer. 1995; 31A:612–5. https://doi.org/10.1016/0959-8049(95)00068-t. [PubMed].
13. Frantz CN, Bernstein M, Castelberry R, Vietti T. Dose escalation of deferrioxamine (DFO) in children with refractory neuroblastoma: A Pediatric Oncology Group Study. Proc Am Soc Clin Oncol. 1994; 416.
14. Blatt J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994; 2109–12. [PubMed].
15. Donfrancesco A, Deb G, Dominici C, Angioni A, Caniglia M, De Sio L, Fidani P, Amici A, Helson L. Deferoxamine, cyclophosphamide, etoposide, carboplatin, and thiotepa (D-CECaT): a new cytoreductive chelation-chemotherapy regimen in patients with advanced neuroblastoma. Am J Clin Oncol. 1992; 15:319–22. https://doi.org/10.1097/00000421-199208000-00009. [PubMed].
16. Dayani PN, Bishop MC, Black K, Zeltzer PM. Desferoxamine (DFO)--mediated iron chelation: rationale for a novel approach to therapy for brain cancer. J Neurooncol. 2004; 67:367–77. https://doi.org/10.1023/b:neon.0000024238.21349.37. [PubMed].
17. Keberle H. The Biochemistry of Desferrioxamine and Its Relation to Iron Metabolism. Ann N Y Acad Sci. 1964; 119:758–68. https://doi.org/10.1111/j.1749-6632.1965.tb54077.x. [PubMed].
18. Donfrancesco A, Deb G, De Sio L, Cozza R, Castellano A. Role of deferoxamine in tumor therapy. Acta Haematol. 1996; 95:66–9. https://doi.org/10.1159/000203951. [PubMed].
19. Porter JB, Evangeli M, El-Beshlawy A. Challenges of adherence and persistence with iron chelation therapy. Int J Hematol. 2011; 94:453–60. https://doi.org/10.1007/s12185-011-0927-3. [PubMed].
20. Liu MC, Lin TS, Sartorelli AC. Synthesis and antitumor activity of amino derivatives of pyridine-2-carboxaldehyde thiosemicarbazone. J Med Chem. 1992; 35:3672–7. https://doi.org/10.1021/jm00098a012. [PubMed].
21. Zou Y, Zhou J, Xu B, Li W, Wang Z. Ribonucleotide reductase subunit M2 as a novel target for clear-cell renal cell carcinoma. Onco Targets Ther. 2019; 12:3267–75. https://doi.org/10.2147/OTT.S196347. [PubMed].
22. Popovic-Bijelic A, Kowol CR, Lind ME, Luo J, Himo F, Enyedy EA, Arion VB, Graslund A. Ribonucleotide reductase inhibition by metal complexes of Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone): a combined experimental and theoretical study. J Inorg Biochem. 2011; 105:1422–31. https://doi.org/10.1016/j.jinorgbio.2011.07.003. [PubMed].
23. Finch RA, Liu M, Grill SP, Rose WC, Loomis R, Vasquez KM, Cheng Y, Sartorelli AC. Triapine (3-aminopyridine-2-carboxaldehyde- thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem Pharmacol. 2000; 59:983–91. https://doi.org/10.1016/s0006-2952(99)00419-0. [PubMed].
24. Zeidner JF, Karp JE, Blackford AL, Smith BD, Gojo I, Gore SD, Levis MJ, Carraway HE, Greer JM, Ivy SP, Pratz KW, McDevitt MA. A phase II trial of sequential ribonucleotide reductase inhibition in aggressive myeloproliferative neoplasms. Haematologica. 2014; 99:672–8. https://doi.org/10.3324/haematol.2013.097246. [PubMed].
25. Attia S, Kolesar J, Mahoney MR, Pitot HC, Laheru D, Heun J, Huang W, Eickhoff J, Erlichman C, Holen KD. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Invest New Drugs. 2008; 26:369–79. https://doi.org/10.1007/s10637-008-9123-6. [PubMed].
26. Traynor AM, Lee JW, Bayer GK, Tate JM, Thomas SP, Mazurczak M, Graham DL, Kolesar JM, Schiller JH. A phase II trial of triapine (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Invest New Drugs. 2010; 28:91–7. https://doi.org/10.1007/s10637-009-9230-z. [PubMed].
27. Feun L, Modiano M, Lee K, Mao J, Marini A, Savaraj N, Plezia P, Almassian B, Colacino E, Fischer J, MacDonald S. Phase I and pharmacokinetic study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) using a single intravenous dose schedule. Cancer Chemother Pharmacol. 2002; 50:223–9. https://doi.org/10.1007/s00280-002-0480-0. [PubMed].
28. Kunos CA, Andrews SJ, Moore KN, Chon HS, Ivy SP. Randomized Phase II Trial of Triapine-Cisplatin-Radiotherapy for Locally Advanced Stage Uterine Cervix or Vaginal Cancers. Front Oncol. 2019; 9:1067. https://doi.org/10.3389/fonc.2019.01067. [PubMed].
29. Kunos CA, Sherertz TM. Long-Term Disease Control with Triapine-Based Radiochemotherapy for Patients with Stage IB2-IIIB Cervical Cancer. Front Oncol. 2014; 4:184. https://doi.org/10.3389/fonc.2014.00184. [PubMed].
30. Kunos CA, Chu E, Beumer JH, Sznol M, Ivy SP. Phase I trial of daily triapine in combination with cisplatin chemotherapy for advanced-stage malignancies. Cancer Chemother Pharmacol. 2017; 79:201–7. https://doi.org/10.1007/s00280-016-3200-x. [PubMed].
31. Pelivan K, Frensemeier LM, Karst U, Koellensperger G, Heffeter P, Keppler BK, Kowol CR. Comparison of metabolic pathways of different alpha-N-heterocyclic thiosemicarbazones. Anal Bioanal Chem. 2018; 410:2343–61. https://doi.org/10.1007/s00216-018-0889-x. [PubMed].
32. Pelivan K, Frensemeier L, Karst U, Koellensperger G, Bielec B, Hager S, Heffeter P, Keppler BK, Kowol CR. Understanding the metabolism of the anticancer drug Triapine: electrochemical oxidation, microsomal incubation and in vivo analysis using LC-HRMS. Analyst. 2017; 142:3165–76. https://doi.org/10.1039/c7an00902j. [PubMed].
33. Tian J, Peehl DM, Zheng W, Knox SJ. Anti-tumor and radiosensitization activities of the iron chelator HDp44mT are mediated by effects on intracellular redox status. Cancer Lett. 2010; 298:231–7. https://doi.org/10.1016/j.canlet.2010.07.010. [PubMed].
34. Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. 2004; 104:1450–8. https://doi.org/10.1182/blood-2004-03-0868. [PubMed].
35. Whitnall M, Howard J, Ponka P, Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006; 103:14901–6. https://doi.org/10.1073/pnas.0604979103. [PubMed].
36. Holley CK, Alkhalifah S, Majd S. Fabrication and Optimization of Dp44mT-Loaded Nanoparticles. Conf Proc IEEE Eng Med Biol Soc. 2018; 2018:5733–6. https://doi.org/10.1109/EMBC.2018.8513598. [PubMed].
37. Holley CK, Kang YJ, Kuo CF, Abidian MR, Majd S. Development and in vitro assessment of an anti-tumor nano-formulation. Colloids Surf B Biointerfaces. 2019; 184:110481. https://doi.org/10.1016/j.colsurfb.2019.110481. [PubMed].
38. Fryknas M, Zhang X, Bremberg U, Senkowski W, Olofsson MH, Brandt P, Persson I, D'Arcy P, Gullbo J, Nygren P, Schughart LK, Linder S, Larsson R. Iron chelators target both proliferating and quiescent cancer cells. Sci Rep. 2016; 6:38343. https://doi.org/10.1038/srep38343. [PubMed].
39. Mody K, Mansfield AS, Vemireddy L, Nygren P, Gulbo J, Borad M. A phase I study of the safety and tolerability of VLX600, an Iron Chelator, in patients with refractory advanced solid tumors. Invest New Drugs. 2019; 37:684–92. https://doi.org/10.1007/s10637-018-0703-9. [PubMed].
40. Ting H, Deep G, Agarwal R. Molecular mechanisms of silibinin-mediated cancer chemoprevention with major emphasis on prostate cancer. AAPS J. 2013; 15:707–16. https://doi.org/10.1208/s12248-013-9486-2. [PubMed].
41. Rauf A, Imran M, Khan IA, Ur-Rehman M, Gilani SA, Mehmood Z, Mubarak MS. Anticancer potential of quercetin: A comprehensive review. Phytother Res. 2018; 32:2109–30. https://doi.org/10.1002/ptr.6155. [PubMed].
42. Gan RY, Li HB, Sui ZQ, Corke H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit Rev Food Sci Nutr. 2018; 58:924–41. https://doi.org/10.1080/10408398.2016.1231168. [PubMed].
43. van't Slot G, Humpf HU. Degradation and metabolism of catechin, epigallocatechin-3-gallate (EGCG), and related compounds by the intestinal microbiota in the pig cecum model. J Agric Food Chem. 2009; 57:8041–8. https://doi.org/10.1021/jf900458e. [PubMed].
44. Andres S, Pevny S, Ziegenhagen R, Bakhiya N, Schafer B, Hirsch-Ernst KI, Lampen A. Safety Aspects of the Use of Quercetin as a Dietary Supplement. Mol Nutr Food Res. 2018; 62. https://doi.org/10.1002/mnfr.201700447. [PubMed].
45. Bijak M. Silybin, a Major Bioactive Component of Milk Thistle (Silybum marianum L. Gaernt.)-Chemistry, Bioavailability, and Metabolism. Molecules. 2017; 22:1942. https://doi.org/10.3390/molecules22111942. [PubMed].
46. Abenavoli L, Izzo AA, Milic N, Cicala C, Santini A, Capasso R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother Res. 2018; 32:2202–13. https://doi.org/10.1002/ptr.6171. [PubMed].
47. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013. [PubMed].
48. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70. https://doi.org/10.1016/s0092-8674(00)81683-9. [PubMed].
49. Lui GY, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS, Richardson DR. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget. 2015; 6:18748–79. https://doi.org/10.18632/oncotarget.4349. [PubMed].
50. Hwang J, Kim Y, Kang HB, Jaroszewski L, Deacon AM, Lee H, Choi WC, Kim KJ, Kim CH, Kang BS, Lee JO, Oh TK, Kim JW, et al. Crystal Structure of the Human N-Myc Downstream-regulated Gene 2 Protein Provides Insight into Its Role as a Tumor Suppressor. J Biol Chem. 2011; 286:12450–60. https://doi.org/10.1074/jbc.M110.170803. [PubMed].
51. Burchfield JG, Lennard AJ, Narasimhan S, Hughes WE, Wasinger VC, Corthals GL, Okuda T, Kondoh H, Biden TJ, Schmitz-Peiffer C. Akt mediates insulin-stimulated phosphorylation of Ndrg2: evidence for cross-talk with protein kinase C theta. J Biol Chem. 2004; 279:18623–32. https://doi.org/10.1074/jbc.M401504200. [PubMed].
52. Wakisaka Y, Furuta A, Masuda K, Morikawa W, Kuwano M, Iwaki T. Cellular distribution of NDRG1 protein in the rat kidney and brain during normal postnatal development. J Histochem Cytochem. 2003; 51:1515–25. https://doi.org/10.1177/002215540305101111. [PubMed].
53. Stein S, Thomas EK, Herzog B, Westfall MD, Rocheleau JV, Jackson RS 2nd, Wang M, Liang P. NDRG1 is necessary for p53-dependent apoptosis. J Biol Chem. 2004; 279:48930–40. https://doi.org/10.1074/jbc.M400386200. [PubMed].
54. Kachhap SK, Faith D, Qian DZ, Shabbeer S, Galloway NL, Pili R, Denmeade SR, DeMarzo AM, Carducci MA. The N-Myc down regulated Gene1 (NDRG1) Is a Rab4a effector involved in vesicular recycling of E-cadherin. PLoS One. 2007; 2:e844. https://doi.org/10.1371/journal.pone.0000844. [PubMed].
55. Okuda T, Higashi Y, Kokame K, Tanaka C, Kondoh H, Miyata T. Ndrg1-deficient mice exhibit a progressive demyelinating disorder of peripheral nerves. Mol Cell Biol. 2004; 24:3949–56. https://doi.org/10.1128/mcb.24.9.3949-3956.2004. [PubMed].
56. Lachat P, Shaw P, Gebhard S, van Belzen N, Chaubert P, Bosman FT. Expression of NDRG1, a differentiation-related gene, in human tissues. Histochem Cell Biol. 2002; 118:399–408. https://doi.org/10.1007/s00418-002-0460-9. [PubMed].
57. Mao XY, Fan CF, Wei J, Liu C, Zheng HC, Yao F, Jin F. Increased N-myc downstream-regulated gene 1 expression is associated with breast atypia-to-carcinoma progression. Tumour Biol. 2011; 32:1271–6. https://doi.org/10.1007/s13277-011-0232-z. [PubMed].
58. Ando T, Ishiguro H, Kimura M, Mitsui A, Kurehara H, Sugito N, Tomoda K, Mori R, Takashima N, Ogawa R, Fujii Y, Kuwabara Y. Decreased expression of NDRG1 is correlated with tumor progression and poor prognosis in patients with esophageal squamous cell carcinoma. Dis Esophagus. 2006; 19:454–8. https://doi.org/10.1111/j.1442-2050.2006.00618.x. [PubMed].
59. Yu CR, Hao XH, Zhang S, Hu WJ, Li JW, Sun J, Zheng MH. Characterization of the prognostic values of the NDRG family in gastric cancer. Therap Adv Gastroenterol. 2019; 12:1756284819858507. https://doi.org/10.1177/1756284819858507. [PubMed].
60. Matsushita K, Uchida K, Saigusa S, Ide S, Hashimoto K, Koike Y, Otake K, Inoue M, Tanaka K, Kusunoki M. Low NDRG1 mRNA expression predicts a poor prognosis in neuroblastoma patients. Pediatr Surg Int. 2013; 29:363–8. https://doi.org/10.1007/s00383-012-3248-6. [PubMed].
61. Park KC, Menezes SV, Kalinowski DS, Sahni S, Jansson PJ, Kovacevic Z, Richardson DR. Identification of differential phosphorylation and sub-cellular localization of the metastasis suppressor, NDRG1. Biochim Biophys Acta Mol Basis Dis. 2018; 1864:2644–63. https://doi.org/10.1016/j.bbadis.2018.04.011. [PubMed].
62. Dixon KM, Lui GY, Kovacevic Z, Zhang D, Yao M, Chen Z, Dong Q, Assinder SJ, Richardson DR. Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br J Cancer. 2013; 108:409–19. https://doi.org/10.1038/bjc.2012.582. [PubMed].
63. Ghalayini MK, Dong Q, Richardson DR, Assinder SJ. Proteolytic cleavage and truncation of NDRG1 in human prostate cancer cells, but not normal prostate epithelial cells. Biosci Rep. 2013; 33:e00042. https://doi.org/10.1042/BSR20130042. [PubMed].
64. Kovacevic Z, Chikhani S, Lui GYL, Sivagurunathan S, Richardson DR. The Iron-Regulated Metastasis Suppressor NDRG1 Targets NEDD4L, PTEN, and SMAD4 and Inhibits the PI3K and Ras Signaling Pathways. Antioxid Redox Signal. 2013; 18:874–87. https://doi.org/10.1089/ars.2011.4273. [PubMed].
65. Lui GYL, Kovacevic Z, Menezes SV, Kalinowski DS, Merlot AM, Sahni S, Richardson DR. Novel Thiosemicarbazones Regulate the Signal Transducer and Activator of Transcription 3 (STAT3) Pathway: Inhibition of Constitutive and Interleukin 6-Induced Activation by Iron Depletion. Mol Pharmacol. 2015; 87:543–60. https://doi.org/10.1124/mol.114.096529. [PubMed].
66. Watson A, Lipina C, McArdle HJ, Taylor PM, Hundal HS. Iron depletion suppresses mTORC1-directed signalling in intestinal Caco-2 cells via induction of REDD1. Cell Signal. 2016; 28:412–24. https://doi.org/10.1016/j.cellsig.2016.01.014. [PubMed].
67. Chen XM, Vinkemeier U, Zhao YX, Jeruzalmi D, Darnell JE, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998; 93:827–39. https://doi.org/10.1016/S0092-8674(00)81443-9. [PubMed].
68. Vlietstra RJ, van Alewijk DC, Hermans KG, van Steenbrugge GJ, Trapman J. Frequent inactivation of PTEN in prostate cancer cell lines and xenografts. Cancer Res. 1998; 58:2720–3. [PubMed].
69. Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997; 272:31515–24. https://doi.org/10.1074/jbc.272.50.31515. [PubMed].
70. Chen CL, Liu P, Duan XY, Cheng M, Xu LX. Deferoxamine-induced high expression of TfR1 and DMT1 enhanced iron uptake in triple-negative breast cancer cells by activating IL-6/PI3K/AKT pathway. Onco Targets Ther. 2019; 12:4359–77. https://doi.org/10.2147/Ott.S193507. [PubMed].
71. Kamihara Y, Takada K, Sato T, Kawano Y, Murase K, Arihara Y, Kikuchi S, Hayasaka N, Usami M, Iyama S, Miyanishi K, Sato Y, Kobune M, et al. The iron chelator deferasirox induces apoptosis by targeting oncogenic Pyk2/beta-catenin signaling in human multiple myeloma. Oncotarget. 2016; 7:64330–41. https://doi.org/10.18632/oncotarget.11830. [PubMed].
72. Song S, Christova T, Perusini S, Alizadeh S, Bao RY, Miller BW, Hurren R, Jitkova Y, Gronda M, Isaac M, Joseph B, Subramaniam R, Aman A, et al. Wnt inhibitor screen reveals iron dependence of beta-catenin signaling in cancers. Cancer Res. 2011; 71:7628–39. https://doi.org/10.1158/0008-5472.CAN-11-2745. [PubMed].
73. Wangpu X, Yang X, Zhao J, Lu J, Guan S, Lu J, Kovacevic Z, Liu W, Mi L, Jin R, Sun J, Yue F, Ma J, et al. The metastasis suppressor, NDRG1, inhibits "stemness" of colorectal cancer via down-regulation of nuclear beta-catenin and CD44. Oncotarget. 2015; 6:33893–911. https://doi.org/10.18632/oncotarget.5294. [PubMed].
74. Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo YY, Fukuda K, Li Y, Watabe K. N-myc downstream regulated gene 1 modulates Wnt-beta-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol Med. 2012; 4:93–108. https://doi.org/10.1002/emmm.201100190. [PubMed].
75. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999; 96:5522–7. https://doi.org/10.1073/pnas.96.10.5522. [PubMed].
76. Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z, Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. 2012; 287:17016–28. https://doi.org/10.1074/jbc.M112.350470. [PubMed].
77. Menezes SV, Kovacevic Z, Richardson DR. The metastasis suppressor NDRG1 down-regulates the epidermal growth factor receptor via a lysosomal mechanism by up-regulating mitogen-inducible gene 6. J Biol Chem. 2019; 294:4045–64. https://doi.org/10.1074/jbc.RA118.006279. [PubMed].
78. Kovacevic Z, Menezes SV, Sahni S, Kalinowski DS, Bae DH, Lane DJ, Richardson DR. The Metastasis Suppressor, N-MYC Downstream-regulated Gene-1 (NDRG1), Down-regulates the ErbB Family of Receptors to Inhibit Downstream Oncogenic Signaling Pathways. J Biol Chem. 2016; 291:1029–52. https://doi.org/10.1074/jbc.M115.689653. [PubMed].
79. Hoyes KP, Hider RC, Porter JB. Cell cycle synchronization and growth inhibition by 3-hydroxypyridin-4-one iron chelators in leukemia cell lines. Cancer Res. 1992; 52:4591–9. [PubMed].
80. Kulp KS, Green SL, Vulliet PR. Iron deprivation inhibits cyclin-dependent kinase activity and decreases cyclin D/CDK4 protein levels in asynchronous MDA-MB-453 human breast cancer cells. Exp Cell Res. 1996; 229:60–8. https://doi.org/10.1006/excr.1996.0343. [PubMed].
81. Wang G, Miskimins R, Miskimins WK. Regulation of p27(Kip1) by intracellular iron levels. Biometals. 2004; 17:15–24. https://doi.org/10.1023/A:1024417309370. [PubMed].
82. Kuang Y, Guo W, Ling J, Xu D, Liao Y, Zhao H, Du X, Wang H, Xu M, Song H, Wang T, Jing B, Li K, et al. Iron-dependent CDK1 activity promotes lung carcinogenesis via activation of the GP130/STAT3 signaling pathway. Cell Death Dis. 2019; 10:297. https://doi.org/10.1038/s41419-019-1528-y. [PubMed].
83. Siriwardana G, Seligman PA. Two cell cycle blocks caused by iron chelation of neuroblastoma cells: separating cell cycle events associated with each block. Physiol Rep. 2013; 1:e00176. https://doi.org/10.1002/phy2.176. [PubMed].
84. Elledge SJ, Zhou Z, Allen JB. Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem Sci. 1992; 17:119–23. https://doi.org/10.1016/0968-0004(92)90249-9. [PubMed].
85. Cooper CE, Lynagh GR, Hoyes KP, Hider RC, Cammack R, Porter JB. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J Biol Chem. 1996; 271:20291–9. https://doi.org/10.1074/jbc.271.34.20291. [PubMed].
86. Nyholm S, Mann GJ, Johansson AG, Bergeron RJ, Graslund A, Thelander L. Role of ribonucleotide reductase in inhibition of mammalian cell growth by potent iron chelators. J Biol Chem. 1993; 268:26200–5. [PubMed].
87. Yu Y, Suryo Rahmanto Y, Hawkins CL, Richardson DR. The potent and novel thiosemicarbazone chelators di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone and 2-benzoylpyridine-4,4-dimethyl-3-thiosemicarbazone affect crucial thiol systems required for ribonucleotide reductase activity. Mol Pharmacol. 2011; 79:921–31. https://doi.org/10.1124/mol.111.071324. [PubMed].
88. Le NTV, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood. 2004; 104:2967–75. https://doi.org/10.1182/blood-2004-05-1866. [PubMed].
89. Kovacevic Z, Fu D, Richardson DR. The iron-regulated metastasis suppressor, Ndrg-1: Identification of novel molecular targets. Biochim Biophys Acta. 2008; 1783:1981–92. https://doi.org/10.1016/j.bbamcr.2008.05.016. [PubMed].
90. Cangul H. Hypoxia upregulates the expression of the NDRG1 gene leading to its overexpression in various human cancers. BMC Genet. 2004; 5:27. https://doi.org/10.1186/1471-2156-5-27. [PubMed].
91. Okuda T, Kondoh H. Identification of new genes ndr2 and ndr3 which are related to Ndr1/RTP/Drg1 but show distinct tissue specificity and response to N-myc. Biochem Biophys Res Commun. 1999; 266:208–15. https://doi.org/10.1006/bbrc.1999.1780. [PubMed].
92. Moussa RS, Kovacevic Z, Richardson DR. Differential targeting of the cyclin-dependent kinase inhibitor, p21CIP1/WAF1, by chelators with anti-proliferative activity in a range of tumor cell-types. Oncotarget. 2015; 6:29694–711. https://doi.org/10.18632/oncotarget.5088. [PubMed].
93. Dongiovanni P, Fracanzani AL, Cairo G, Megazzini CP, Gatti S, Rametta R, Fargion S, Valenti L. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol. 2010; 176:1006–17. https://doi.org/10.2353/ajpath.2010.090249. [PubMed].
94. Kovacevic Z, Sivagurunathan S, Mangs H, Chikhani S, Zhang D, Richardson DR. The metastasis suppressor, N-myc downstream regulated gene 1 (NDRG1), upregulates p21 via p53-independent mechanisms. Carcinogenesis. 2011; 32:732–40. https://doi.org/10.1093/carcin/bgr046. [PubMed].
95. Zhang J, Kong X, Zhang Y, Sun W, Xu E, Chen X. Mdm2 is a target and mediator of IRP2 in cell growth control. FASEB J. 2020; 34:2301–2311. https://doi.org/10.1096/fj.201902278RR. [PubMed].
96. Siriwardana G, Seligman PA. Iron depletion results in Src kinase inhibition with associated cell cycle arrest in neuroblastoma cells. Physiol Rep. 2015; 3:e12341. https://doi.org/10.14814/phy2.12341. [PubMed].
97. Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, Takano Y, Saito K, Commes T, Piquemal D, Watabe M, Gross S, Wang Y, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004; 64:7655–60. https://doi.org/10.1158/0008-5472.Can-04-1623. [PubMed].
98. Li P, Zheng X, Shou K, Niu Y, Jian C, Zhao Y, Yi W, Hu X, Yu A. The iron chelator Dp44mT suppresses osteosarcoma's proliferation, invasion and migration: in vitro and in vivo. Am J Transl Res. 2016; 8:5370–85. [PubMed].
99. Lee SK, Lee JJ, Lee HJ, Lee J, Jeon BH, Jun CD, Lee SK, Kim EC. Iron chelator-induced growth arrest and cytochrome c-dependent apoptosis in immortalized and malignant oral keratinocytes. J Oral Pathol Med. 2006; 35:218–26. https://doi.org/10.1111/j.1600-0714.2006.00415.x. [PubMed].
100. Yang Y, Xu Y, Su A, Yang D, Zhang X. Effects of Deferoxamine on Leukemia In Vitro and Its Related Mechanism. Med Sci Monit. 2018; 24:6735–41. https://doi.org/10.12659/MSM.910325. [PubMed].
101. Kim BS, Yoon KH, Oh HM, Choi EY, Kim SW, Han WC, Kim EA, Choi SC, Kim TH, Yun KJ, Kim EC, Lyou JH, Nah YH, et al. Involvement of p38 MAP kinase during iron chelator-mediated apoptotic cell death. Cell Immunol. 2002; 220:96–106. https://doi.org/10.1016/s0008-8749(03)00031-5. [PubMed].
102. Harima H, Kaino S, Takami T, Shinoda S, Matsumoto T, Fujisawa K, Yamamoto N, Yamasaki T, Sakaida I. Deferasirox, a novel oral iron chelator, shows antiproliferative activity against pancreatic cancer in vitro and in vivo. BMC Cancer. 2016; 16:702. https://doi.org/10.1186/s12885-016-2744-9. [PubMed].
103. Kim JL, Lee DH, Na YJ, Kim BR, Jeong YA, Lee SI, Kang S, Joung SY, Lee SY, Oh SC, Min BW. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumour Biol. 2016; 37:9709–19. https://doi.org/10.1007/s13277-016-4878-4. [PubMed].
104. Fukuchi K, Tomoyasu S, Watanabe H, Kaetsu S, Tsuruoka N, Gomi K. Iron deprivation results in an increase in p53 expression. Biol Chem Hoppe Seyler. 1995; 376:627–30. https://doi.org/10.1515/bchm3.1995.376.10.627. [PubMed].
105. Le NT, Richardson DR. Potent iron chelators increase the mRNA levels of the universal cyclin-dependent kinase inhibitor p21(CIP1/WAF1), but paradoxically inhibit its translation: a potential mechanism of cell cycle dysregulation. Carcinogenesis. 2003; 24:1045–58. https://doi.org/10.1093/carcin/bgg042. [PubMed].
106. Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014; 7:180–93. https://doi.org/10.1016/j.celrep.2014.02.042. [PubMed].
107. Merlot AM, Shafie NH, Yu Y, Richardson V, Jansson PJ, Sahni S, Lane DJR, Kovacevic Z, Kalinowski DS, Richardson DR. Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): Activation of PERK/eIF2alpha, IRE1alpha, ATF6 and calmodulin kinase. Biochem Pharmacol. 2016; 109:27–47. https://doi.org/10.1016/j.bcp.2016.04.001. [PubMed].
108. Yu Y, Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. 2011; 286:15413–27. https://doi.org/10.1074/jbc.M111.225946. [PubMed].
109. Sahni S, Bae DH, Lane DJ, Kovacevic Z, Kalinowski DS, Jansson PJ, Richardson DR. The metastasis suppressor, N-myc downstream-regulated gene 1 (NDRG1), inhibits stress-induced autophagy in cancer cells. J Biol Chem. 2014; 289:9692–709. https://doi.org/10.1074/jbc.M113.529511. [PubMed].
110. De Bortoli M, Taverna E, Maffioli E, Casalini P, Crisafi F, Kumar V, Caccia C, Polli D, Tedeschi G, Bongarzone I. Lipid accumulation in human breast cancer cells injured by iron depletors. J Exp Clin Cancer Res. 2018; 37:75. https://doi.org/10.1186/s13046-018-0737-z. [PubMed].
111. Si L, Fu J, Liu W, Hayashi T, Mizuno K, Hattori S, Fujisaki H, Onodera S, Ikejima T. Silibinin-induced mitochondria fission leads to mitophagy, which attenuates silibinin-induced apoptosis in MCF-7 and MDA-MB-231 cells. Arch Biochem Biophys. 2020; 685:108284. https://doi.org/10.1016/j.abb.2020.108284. [PubMed].
112. Tang HM, Tang HL. Cell recovery by reversal of ferroptosis. Biol Open. 2019; 8:bio043182. https://doi.org/10.1242/bio.043182. [PubMed].
113. Zhang Y, Toh L, Lau P, Wang X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J Biol Chem. 2012; 287:32494–511. https://doi.org/10.1074/jbc.M112.368282. [PubMed].
114. Katsura Y, Ohara T, Noma K, Ninomiya T, Kashima H, Kato T, Sato H, Komoto S, Narusaka T, Tomono Y, Xing B, Chen Y, Tazawa H, et al. A Novel Combination Cancer Therapy with Iron Chelator Targeting Cancer Stem Cells via Suppressing Stemness. Cancers (Basel). 2019; 11:177. https://doi.org/10.3390/cancers11020177. [PubMed].
115. Ninomiya T, Ohara T, Noma K, Katsura Y, Katsube R, Kashima H, Kato T, Tomono Y, Tazawa H, Kagawa S, Shirakawa Y, Kimura F, Chen L, et al. Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget. 2017; 8:98405–16. https://doi.org/10.18632/oncotarget.21846. [PubMed].
116. Sun X, Song J, Li E, Geng H, Li Y, Yu D, Zhong C. ()Epigallocatechin3gallate inhibits bladder cancer stem cells via suppression of sonic hedgehog pathway. Oncol Rep. 2019; 42:425–35. https://doi.org/10.3892/or.2019.7170. [PubMed].
117. Loizzi V, Del Vecchio V, Gargano G, De Liso M, Kardashi A, Naglieri E, Resta L, Cicinelli E, Cormio G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. Int J Mol Sci. 2017; 18:1967. https://doi.org/10.3390/ijms18091967. [PubMed].
118. Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011; 4:51. https://doi.org/10.3389/fnmol.2011.00051. [PubMed].
119. Fang BA, Kovacevic Z, Park KC, Kalinowski DS, Jansson PJ, Lane DJ, Sahni S, Richardson DR. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim Biophys Acta. 2014; 1845:1–19. https://doi.org/10.1016/j.bbcan.2013.11.002. [PubMed].
120. Lee JC, Chung LC, Chen YJ, Feng TH, Juang HH. N-myc downstream-regulated gene 1 downregulates cell proliferation, invasiveness, and tumorigenesis in human oral squamous cell carcinoma. Cancer Lett. 2014; 355:242–52. https://doi.org/10.1016/j.canlet.2014.08.035. [PubMed].
121. Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, Richardson DR. Iron chelators for the treatment of cancer. Curr Med Chem. 2012; 19:2689–702. https://doi.org/10.2174/092986712800609706. [PubMed].
122. Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, Gallick GE. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene. 2005; 24:3110–20. https://doi.org/10.1038/sj.onc.1208513. [PubMed].
123. Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, Cheng JQ, Jove R, Yu H. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005; 24:5552–60. https://doi.org/10.1038/sj.onc.1208719. [PubMed].
124. Beerepoot LV, Shima DT, Kuroki M, Yeo KT, Voest EE. Up-regulation of vascular endothelial growth factor production by iron chelators. Cancer Res. 1996; 56:3747–51. [PubMed].
125. Li H, Li Z, Xu YM, Wu Y, Yu KK, Zhang C, Ji YH, Ding G, Chen FX. Epigallocatechin-3-gallate induces apoptosis, inhibits proliferation and decreases invasion of glioma cell. Neurosci Bull. 2014; 30:67–73. https://doi.org/10.1007/s12264-013-1394-z. [PubMed].
126. Zhu BH, Chen HY, Zhan WH, Wang CY, Cai SR, Wang Z, Zhang CH, He YL. (-)-Epigallocatechin-3-gallate inhibits VEGF expression induced by IL-6 via Stat3 in gastric cancer. World J Gastroenterol. 2011; 17:2315–25. https://doi.org/10.3748/wjg.v17.i18.2315. [PubMed].
127. Lan H, Hong W, Fan P, Qian D, Zhu J, Bai B. Quercetin Inhibits Cell Migration and Invasion in Human Osteosarcoma Cells. Cell Physiol Biochem. 2017; 43:553–67. https://doi.org/10.1159/000480528. [PubMed].
128. Byun HJ, Darvin P, Kang DY, Sp N, Joung YH, Park JH, Kim SJ, Yang YM. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol Rep. 2017; 37:3270–8. https://doi.org/10.3892/or.2017.5588. [PubMed].
129. Syed V. TGF-beta Signaling in Cancer. J Cell Biochem. 2016; 117:1279–87. https://doi.org/10.1002/jcb.25496. [PubMed].
130. Menezes SV, Fouani L, Huang MLH, Geleta B, Maleki S, Richardson A, Richardson DR, Kovacevic Z. The metastasis suppressor, NDRG1, attenuates oncogenic TGF-beta and NF-kappaB signaling to enhance membrane E-cadherin expression in pancreatic cancer cells. Carcinogenesis. 2019; 40:805–18. https://doi.org/10.1093/carcin/bgy178. [PubMed].
131. Liu P, He K, Song H, Ma Z, Yin W, Xu LX. Deferoxamine-induced increase in the intracellular iron levels in highly aggressive breast cancer cells leads to increased cell migration by enhancing TNF-alpha-dependent NF-kappaB signaling and TGF-beta signaling. J Inorg Biochem. 2016; 160:40–8. https://doi.org/10.1016/j.jinorgbio.2016.04.014. [PubMed].
132. Murrell M, Oakes PW, Lenz M, Gardel ML. Forcing cells into shape: the mechanics of actomyosin contractility. Nat Rev Mol Cell Biol. 2015; 16:486–98. https://doi.org/10.1038/nrm4012. [PubMed].
133. Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996; 392:189–93. https://doi.org/10.1016/0014-5793(96)00811-3. [PubMed].
134. Tanner K, Boudreau A, Bissell MJ, Kumar S. Dissecting Regional Variations in Stress Fiber Mechanics in Living Cells with Laser Nanosurgery. Biophys J. 2010; 99:2775–83. https://doi.org/10.1016/j.bpj.2010.08.071. [PubMed].
135. Wilkinson S, Paterson HF, Marshall CJ. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol. 2005; 7:255–U45. https://doi.org/10.1038/ncb1230. [PubMed].
136. Sun J, Zhang DH, Zheng Y, Zhao Q, Zheng MH, Kovacevic Z, Richardson DR. Targeting the Metastasis Suppressor, NDRG1, Using Novel Iron Chelators: Regulation of Stress Fiber-Mediated Tumor Cell Migration via Modulation of the ROCK1/pMLC2 Signaling Pathway. Mol Pharmacol. 2013; 83:454–69. https://doi.org/10.1124/mol.112.083097. [PubMed].
137. Ishiguro K, Lin ZP, Penketh PG, Shyam K, Zhu R, Baumann RP, Zhu YL, Sartorelli AC, Rutherford TJ, Ratner ES. Distinct mechanisms of cell-kill by triapine and its terminally dimethylated derivative Dp44mT due to a loss or gain of activity of their copper(II) complexes. Biochem Pharmacol. 2014; 91:312–22. https://doi.org/10.1016/j.bcp.2014.08.006. [PubMed].
138. Xu YX, Zeng ML, Yu D, Ren J, Li F, Zheng A, Wang YP, Chen C, Tao ZZ. In vitro assessment of the role of DpC in the treatment of head and neck squamous cell carcinoma. Oncol Lett. 2018; 15:7999–8004. https://doi.org/10.3892/ol.2018.8279. [PubMed].
139. Rao VA, Klein SR, Agama KK, Toyoda E, Adachi N, Pommier Y, Shacter EB. The iron chelator Dp44mT causes DNA damage and selective inhibition of topoisomerase IIalpha in breast cancer cells. Cancer Res. 2009; 69:948–57. https://doi.org/10.1158/0008-5472.CAN-08-1437. [PubMed].
140. Lin ZP, Zhu YL, Lo YC, Moscarelli J, Xiong A, Korayem Y, Huang PH, Giri S, LoRusso P, Ratner ES. Combination of triapine, olaparib, and cediranib suppresses progression of BRCA-wild type and PARP inhibitor-resistant epithelial ovarian cancer. PLoS One. 2018; 13:e0207399. https://doi.org/10.1371/journal.pone.0207399. [PubMed].
141. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, Urban N, Taniguchi T. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008; 451:1116–20. https://doi.org/10.1038/nature06633. [PubMed].
142. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008; 68:2581–6. https://doi.org/10.1158/0008-5472.CAN-08-0088. [PubMed].
143. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011; 29:3008–15. https://doi.org/10.1200/JCO.2010.34.2980. [PubMed].
144. Ratner ES, Zhu YL, Penketh PG, Berenblum J, Whicker ME, Huang PH, Lee Y, Ishiguro K, Zhu R, Sartorelli AC, Lin ZP. Triapine potentiates platinum-based combination therapy by disruption of homologous recombination repair. Br J Cancer. 2016; 114:777–86. https://doi.org/10.1038/bjc.2016.54. [PubMed].
145. Lin ZP, Ratner ES, Whicker ME, Lee Y, Sartorelli AC. Triapine disrupts CtIP-mediated homologous recombination repair and sensitizes ovarian cancer cells to PARP and topoisomerase inhibitors. Mol Cancer Res. 2014; 12:381–93. https://doi.org/10.1158/1541-7786.MCR-13-0480. [PubMed].
146. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 2011; 474:230–4. https://doi.org/10.1038/nature10155. [PubMed].
147. Zhang X, de Milito A, Olofsson MH, Gullbo J, D'Arcy P, Linder S. Targeting Mitochondrial Function to Treat Quiescent Tumor Cells in Solid Tumors. Int J Mol Sci. 2015; 16:27313–26. https://doi.org/10.3390/ijms161126020. [PubMed].
148. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015; 11:9–15. https://doi.org/10.1038/nchembio.1712. [PubMed].
149. Lan L, Wei W, Zheng Y, Niu L, Chen X, Huang D, Gao Y, Mo S, Lu J, Guo M, Liu Y, Lu B. Deferoxamine suppresses esophageal squamous cell carcinoma cell growth via ERK1/2 mediated mitochondrial dysfunction. Cancer Lett. 2018; 432:132–43. https://doi.org/10.1016/j.canlet.2018.06.012. [PubMed].
150. De Domenico I, Ward DM, Kaplan J. Specific iron chelators determine the route of ferritin degradation. Blood. 2009; 114:4546–51. https://doi.org/10.1182/blood-2009-05-224188. [PubMed].
151. Lesjak M, Balesaria S, Skinner V, Debnam ES, Srai SKS. Quercetin inhibits intestinal non-haem iron absorption by regulating iron metabolism genes in the tissues. Eur J Nutr. 2019; 58:743–53. https://doi.org/10.1007/s00394-018-1680-7. [PubMed].
152. Chen C, Wang S, Liu P. Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells Via a ROS-Dependent Mechanism. Int J Mol Sci. 2019; 20:4952. https://doi.org/10.3390/ijms20194952. [PubMed].
153. Chaston TB, Lovejoy DB, Watts RN, Richardson DR. Examination of the antiproliferative activity of iron chelators: multiple cellular targets and the different mechanism of action of triapine compared with desferrioxamine and the potent pyridoxal isonicotinoyl hydrazone analogue 311. Clin Cancer Res. 2003; 9:402–14. [PubMed].
154. Lee DH, Jang PS, Chung NG, Cho B, Jeong DC, Kim HK. Deferasirox shows in vitro and in vivo antileukemic effects on murine leukemic cell lines regardless of iron status. Exp Hematol. 2013; 41:539–46. https://doi.org/10.1016/j.exphem.2013.02.004. [PubMed].
155. Chang YC, Lo WJ, Huang YT, Lin CL, Feng CC, Lin HT, Cheng HC, Yeh SP. Deferasirox has strong anti-leukemia activity but may antagonize theanti-leukemia effect of doxorubicin. Leuk Lymphoma. 2017; 58:1–12. https://doi.org/10.1080/10428194.2017.1280604. [PubMed].
156. Oblender M, Carpentieri U. Effects of iron, copper and zinc on the activity of ribonucleotide reductase in normal and leukemic human lymphocytes. Anticancer Res. 1990; 10:123–7. [PubMed].
157. Kim BM, Choi JY, Kim YJ, Woo HD, Chung HW. Desferrioxamine (DFX) has genotoxic effects on cultured human lymphocytes and induces the p53-mediated damage response. Toxicology. 2007; 229:226–35. https://doi.org/10.1016/j.tox.2006.10.022. [PubMed].
158. Nakagawa H, Matsumiya T, Sakaki H, Imaizumi T, Kubota K, Kusumi A, Kobayashi W, Kimura H. Expression of vascular endothelial growth factor by photodynamic therapy with mono-L-aspartyl chlorin e6 (NPe6) in oral squamous cell carcinoma. Oral Oncol. 2007; 43:544–50. https://doi.org/10.1016/j.oraloncology.2006.03.020. [PubMed].
159. Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993; 82:3610–5. [PubMed].
160. Xu Y, Cai L. Lmimosine induces caspase9mediated apoptosis in human osteosarcoma cells. Mol Med Rep. 2018; 17:4695–701. https://doi.org/10.3892/mmr.2018.8403. [PubMed].
161. Alpan RS, Pardee AB. p21WAF1/CIP1/SDI1 is elevated through a p53-independent pathway by mimosine. Cell Growth Differ. 1996; 7:893–901. [PubMed].
162. Kyriakou S, Mitsiogianni M, Mantso T, Cheung W, Todryk S, Veuger S, Pappa A, Tetard D, Panayiotidis MI. Anticancer activity of a novel methylated analogue of L-mimosine against an in vitro model of human malignant melanoma. Invest New Drugs. 2020; 38:621–33. https://doi.org/10.1007/s10637-019-00809-0. [PubMed].
163. Alvero AB, Chen W, Sartorelli AC, Schwartz P, Rutherford T, Mor G. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone) induces apoptosis in ovarian cancer cells. J Soc Gynecol Investig. 2006; 13:145–52. https://doi.org/10.1016/j.jsgi.2005.11.004. [PubMed].
164. Guo ZL, Richardson DR, Kalinowski DS, Kovacevic Z, Tan-Un KC, Chan GC. The novel thiosemicarbazone, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), inhibits neuroblastoma growth in vitro and in vivo via multiple mechanisms. J Hematol Oncol. 2016; 9:98. https://doi.org/10.1186/s13045-016-0330-x. [PubMed].
165. Green DA, Antholine WE, Wong SJ, Richardson DR, Chitambar CR. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: effect on the R2 subunit of ribonucleotide reductase. Clin Cancer Res. 2001; 7:3574–9. [PubMed].
166. Liang SX, Richardson DR. The effect of potent iron chelators on the regulation of p53: examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis. 2003; 24:1601–14. https://doi.org/10.1093/carcin/bgg116. [PubMed].
167. Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, Rickardson L, D'Arcy P, Gullbo J, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014; 5:3295. https://doi.org/10.1038/ncomms4295. [PubMed].
168. Karlsson H, Senkowski W, Fryknas M, Mansoori S, Linder S, Gullbo J, Larsson R, Nygren P. A novel tumor spheroid model identifies selective enhancement of radiation by an inhibitor of oxidative phosphorylation. Oncotarget. 2019; 10:5372–82. https://doi.org/10.18632/oncotarget.27166. [PubMed].
169. Zhou J, Zhang H, Gu P, Margolick JB, Yin D, Zhang Y. Cancer stem/progenitor cell active compound 8-quinolinol in combination with paclitaxel achieves an improved cure of breast cancer in the mouse model. Breast Cancer Res Treat. 2009; 115:269–77. https://doi.org/10.1007/s10549-008-0072-8. [PubMed].
170. Leanderson P, Tagesson C. Iron bound to the lipophilic iron chelator, 8-hydroxyquinoline, causes DNA strand breakage in cultured lung cells. Carcinogenesis. 1996; 17:545–50. https://doi.org/10.1093/carcin/17.3.545. [PubMed].
171. Abeysinghe RD, Greene BT, Haynes R, Willingham MC, Turner J, Planalp RP, Brechbiel MW, Torti FM, Torti SV. p53-independent apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Carcinogenesis. 2001; 22:1607–14. https://doi.org/10.1093/carcin/22.10.1607. [PubMed].
172. Cho HJ, Suh DS, Moon SH, Song YJ, Yoon MS, Park DY, Choi KU, Kim YK, Kim KH. Silibinin inhibits tumor growth through downregulation of extracellular signal-regulated kinase and Akt in vitro and in vivo in human ovarian cancer cells. J Agric Food Chem. 2013; 61:4089–96. https://doi.org/10.1021/jf400192v. [PubMed].
173. Bai ZL, Tay V, Guo SZ, Ren J, Shu MG. Silibinin Induced Human Glioblastoma Cell Apoptosis Concomitant with Autophagy through Simultaneous Inhibition of mTOR and YAP. Biomed Res Int. 2018; 2018:6165192. https://doi.org/10.1155/2018/6165192. [PubMed].
174. Mateen S, Tyagi A, Agarwal C, Singh RP, Agarwal R. Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators. Mol Carcinog. 2010; 49:247–58. https://doi.org/10.1002/mc.20595. [PubMed].
175. Kauntz H, Bousserouel S, Gosse F, Raul F. The flavonolignan silibinin potentiates TRAIL-induced apoptosis in human colon adenocarcinoma and in derived TRAIL-resistant metastatic cells. Apoptosis. 2012; 17:797–809. https://doi.org/10.1007/s10495-012-0731-4. [PubMed].
176. Jiang K, Wang W, Jin X, Wang Z, Ji Z, Meng G. Silibinin, a natural flavonoid, induces autophagy via ROS-dependent mitochondrial dysfunction and loss of ATP involving BNIP3 in human MCF7 breast cancer cells. Oncol Rep. 2015; 33:2711–8. https://doi.org/10.3892/or.2015.3915. [PubMed].
177. Kumar S, Raina K, Agarwal C, Agarwal R. Silibinin strongly inhibits the growth kinetics of colon cancer stem cell-enriched spheroids by modulating interleukin 4/6-mediated survival signals. Oncotarget. 2014; 5:4972–89. https://doi.org/10.18632/oncotarget.2068. [PubMed].
178. Deep G, Gangar SC, Agarwal C, Agarwal R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev Res (Phila). 2011; 4:1222–32. https://doi.org/10.1158/1940-6207.CAPR-10-0370. [PubMed].
179. Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget. 2014; 5:10017–33. https://doi.org/10.18632/oncotarget.2488. [PubMed].
180. Rivera Rivera A, Castillo-Pichardo L, Gerena Y, Dharmawardhane S. Anti-Breast Cancer Potential of Quercetin via the Akt/AMPK/Mammalian Target of Rapamycin (mTOR) Signaling Cascade. PLoS One. 2016; 11:e0157251. https://doi.org/10.1371/journal.pone.0157251. [PubMed].
181. Srivastava NS, Srivastava RAK. Curcumin and quercetin synergistically inhibit cancer cell proliferation in multiple cancer cells and modulate Wnt/beta-catenin signaling and apoptotic pathways in A375 cells. Phytomedicine. 2019; 52:117–28. https://doi.org/10.1016/j.phymed.2018.09.224. [PubMed].
182. Mu C, Jia P, Yan Z, Liu X, Li X, Liu H. Quercetin induces cell cycle G1 arrest through elevating Cdk inhibitors p21 and p27 in human hepatoma cell line (HepG2). Methods Find Exp Clin Pharmacol. 2007; 29:179–83. https://doi.org/10.1358/mf.2007.29.3.1092095. [PubMed].
183. Srivastava S, Somasagara RR, Hegde M, Nishana M, Tadi SK, Srivastava M, Choudhary B, Raghavan SC. Quercetin, a Natural Flavonoid Interacts with DNA, Arrests Cell Cycle and Causes Tumor Regression by Activating Mitochondrial Pathway of Apoptosis. Sci Rep. 2016; 6:24049. https://doi.org/10.1038/srep24049. [PubMed].
184. Wang R, Yang L, Li S, Ye D, Yang L, Liu Q, Zhao Z, Cai Q, Tan J, Li X. Quercetin Inhibits Breast Cancer Stem Cells via Downregulation of Aldehyde Dehydrogenase 1A1 (ALDH1A1), Chemokine Receptor Type 4 (CXCR4), Mucin 1 (MUC1), and Epithelial Cell Adhesion Molecule (EpCAM). Med Sci Monit. 2018; 24:412–20. https://doi.org/10.12659/msm.908022. [PubMed].
185. Hoca M, Becer E, Kabadayi H, Yucecan S, Vatansever HS. The Effect of Resveratrol and Quercetin on Epithelial-Mesenchymal Transition in Pancreatic Cancer Stem Cell. Nutr Cancer. 2020; 72:1231–42. https://doi.org/10.1080/01635581.2019.1670853. [PubMed].
186. Horniblow RD, Henesy D, Iqbal TH, Tselepis C. Modulation of iron transport, metabolism and reactive oxygen status by quercetin-iron complexes in vitro. Mol Nutr Food Res. 2017; 61. https://doi.org/10.1002/mnfr.201600692. [PubMed].
187. Kurbitz C, Heise D, Redmer T, Goumas F, Arlt A, Lemke J, Rimbach G, Kalthoff H, Trauzold A. Epicatechin gallate and catechin gallate are superior to epigallocatechin gallate in growth suppression and anti-inflammatory activities in pancreatic tumor cells. Cancer Sci. 2011; 102:728–34. https://doi.org/10.1111/j.1349-7006.2011.01870.x. [PubMed].
188. Ganguly C, Saha P, Panda CK, Das S. Inhibition of growth, induction of apoptosis and alteration of gene expression by tea polyphenols in the highly metastatic human lung cancer cell line NCI-H460. Asian Pac J Cancer Prev. 2005; 6:326–31. [PubMed].
189. Yang JH, Hsia TC, Kuo HM, Chao PD, Chou CC, Wei YH, Chung JG. Inhibition of lung cancer cell growth by quercetin glucuronides via G2/M arrest and induction of apoptosis. Drug Metab Dispos. 2006; 34:296–304. https://doi.org/10.1124/dmd.105.005280. [PubMed].
190. Liu S, Xu ZL, Sun L, Liu Y, Li CC, Li HM, Zhang W, Li CJ, Qin W. Epigallocatechin3gallate induces apoptosis in human pancreatic cancer cells via PTEN. Mol Med Rep. 2016; 14:599–605. https://doi.org/10.3892/mmr.2016.5277. [PubMed].
191. Chen Y, Wang XQ, Zhang Q, Zhu JY, Li Y, Xie CF, Li XT, Wu JS, Geng SS, Zhong CY, Han HY. (-)-Epigallocatechin-3-Gallate Inhibits Colorectal Cancer Stem Cells by Suppressing Wnt/beta-Catenin Pathway. Nutrients. 2017; 9:572. https://doi.org/10.3390/nu9060572. [PubMed].
192. Li T, Zhao N, Lu J, Zhu Q, Liu X, Hao F, Jiao X. Epigallocatechin gallate (EGCG) suppresses epithelial-Mesenchymal transition (EMT) and invasion in anaplastic thyroid carcinoma cells through blocking of TGF-beta1/Smad signaling pathways. Bioengineered. 2019; 10:282–91. https://doi.org/10.1080/21655979.2019.1632669. [PubMed].
193. Gupta S, Hussain T, Mukhtar H. Molecular pathway for (-)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch Biochem Biophys. 2003; 410:177–85. https://doi.org/10.1016/s0003-9861(02)00668-9. [PubMed].
194. Masuda M, Suzui M, Lim JT, Weinstein IB. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clin Cancer Res. 2003; 9:3486–91. [PubMed].
195. Mayr C, Wagner A, Neureiter D, Pichler M, Jakab M, Illig R, Berr F, Kiesslich T. The green tea catechin epigallocatechin gallate induces cell cycle arrest and shows potential synergism with cisplatin in biliary tract cancer cells. BMC Complement Altern Med. 2015; 15:194. https://doi.org/10.1186/s12906-015-0721-5. [PubMed].
196. Jin L, Li C, Xu Y, Wang L, Liu J, Wang D, Hong C, Jiang Z, Ma Y, Chen Q, Yu F. Epigallocatechin gallate promotes p53 accumulation and activity via the inhibition of MDM2-mediated p53 ubiquitination in human lung cancer cells. Oncol Rep. 2013; 29:1983–90. https://doi.org/10.3892/or.2013.2343. [PubMed].
197. Donfrancesco A, Deb G, Dominici C, Pileggi D, Castello MA, Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990; 50:4929–30. [PubMed].
198. Mackenzie MJ, Saltman D, Hirte H, Low J, Johnson C, Pond G, Moore MJ. A Phase II study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) and gemcitabine in advanced pancreatic carcinoma. A trial of the Princess Margaret hospital Phase II consortium. Invest New Drugs. 2007; 25:553–8. https://doi.org/10.1007/s10637-007-9066-3. [PubMed].
199. Chao J, Synold TW, Morgan RJ Jr, Kunos C, Longmate J, Lenz HJ, Lim D, Shibata S, Chung V, Stoller RG, Belani CP, Gandara DR, McNamara M, et al. A phase I and pharmacokinetic study of oral 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) in the treatment of advanced-stage solid cancers: a California Cancer Consortium Study. Cancer Chemother Pharmacol. 2012; 69:835–43. https://doi.org/10.1007/s00280-011-1779-5. [PubMed].
200. Ocean AJ, Christos P, Sparano JA, Matulich D, Kaubish A, Siegel A, Sung M, Ward MM, Hamel N, Espinoza-Delgado I, Yen Y, Lane ME. Phase II trial of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehydethiosemicarbazone plus gemcitabine in patients with advanced biliary tract cancer. Cancer Chemother Pharmacol. 2011; 68:379–88. https://doi.org/10.1007/s00280-010-1481-z. [PubMed].
201. Odenike OM, Larson RA, Gajria D, Dolan ME, Delaney SM, Karrison TG, Ratain MJ, Stock W. Phase I study of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde-thiosemicarbazone (3-AP) in combination with high dose cytarabine in patients with advanced myeloid leukemia. Invest New Drugs. 2008; 26:233–9. https://doi.org/10.1007/s10637-008-9115-6. [PubMed].
202. Yen Y, Margolin K, Doroshow J, Fishman M, Johnson B, Clairmont C, Sullivan D, Sznol M. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother Pharmacol. 2004; 54:331–42. https://doi.org/10.1007/s00280-004-0821-2. [PubMed].
203. Wadler S, Makower D, Clairmont C, Lambert P, Fehn K, Sznol M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J Clin Oncol. 2004; 22:1553–63. https://doi.org/10.1200/JCO.2004.07.158. [PubMed].
204. Kunos CA, Waggoner S, von Gruenigen V, Eldermire E, Pink J, Dowlati A, Kinsella TJ. Phase I trial of pelvic radiation, weekly cisplatin, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) for locally advanced cervical cancer. Clin Cancer Res. 2010; 16:1298–306. https://doi.org/10.1158/1078-0432.CCR-09-2469. [PubMed].
205. Nutting CM, van Herpen CM, Miah AB, Bhide SA, Machiels JP, Buter J, Kelly C, de Raucourt D, Harrington KJ. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2009; 20:1275–9. https://doi.org/10.1093/annonc/mdn775. [PubMed].
206. Giles FJ, Fracasso PM, Kantarjian HM, Cortes JE, Brown RA, Verstovsek S, Alvarado Y, Thomas DA, Faderl S, Garcia-Manero G, Wright LP, Samson T, Cahill A, et al. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk Res. 2003; 27:1077–83. https://doi.org/10.1016/s0145-2126(03)00118-8. [PubMed].
207. Karp JE, Giles FJ, Gojo I, Morris L, Greer J, Johnson B, Thein M, Sznol M, Low J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk Res. 2008; 32:71–7. https://doi.org/10.1016/j.leukres.2007.05.003. [PubMed].
208. Ma B, Goh BC, Tan EH, Lam KC, Soo R, Leong SS, Wang LZ, Mo F, Chan AT, Zee B, Mok T. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest New Drugs. 2008; 26:169–73. https://doi.org/10.1007/s10637-007-9085-0. [PubMed].
209. Yee KW, Cortes J, Ferrajoli A, Garcia-Manero G, Verstovsek S, Wierda W, Thomas D, Faderl S, King I, O'Brien SM, Jeha S, Andreeff M, Cahill A, et al. Triapine and cytarabine is an active combination in patients with acute leukemia or myelodysplastic syndrome. Leuk Res. 2006; 30:813–22. https://doi.org/10.1016/j.leukres.2005.12.013. [PubMed].
210. Gojo I, Tidwell ML, Greer J, Takebe N, Seiter K, Pochron MF, Johnson B, Sznol M, Karp JE. Phase I and pharmacokinetic study of Triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk Res. 2007; 31:1165–73. https://doi.org/10.1016/j.leukres.2007.01.004. [PubMed].
211. Mortazavi A, Ling Y, Martin LK, Wei L, Phelps MA, Liu Z, Harper EJ, Ivy SP, Wu X, Zhou BS, Liu X, Deam D, Monk JP, et al. A phase I study of prolonged infusion of triapine in combination with fixed dose rate gemcitabine in patients with advanced solid tumors. Invest New Drugs. 2013; 31:685–95. https://doi.org/10.1007/s10637-012-9863-1. [PubMed].
212. Schelman WR, Morgan-Meadows S, Marnocha R, Lee F, Eickhoff J, Huang W, Pomplun M, Jiang Z, Alberti D, Kolesar JM, Ivy P, Wilding G, Traynor AM. A phase I study of Triapine in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2009; 63:1147–56. https://doi.org/10.1007/s00280-008-0890-8. [PubMed].
213. Murren J, Modiano M, Clairmont C, Lambert P, Savaraj N, Doyle T, Sznol M. Phase I and pharmacokinetic study of triapine, a potent ribonucleotide reductase inhibitor, administered daily for five days in patients with advanced solid tumors. Clin Cancer Res. 2003; 9:4092–100. [PubMed].
214. Knox JJ, Hotte SJ, Kollmannsberger C, Winquist E, Fisher B, Eisenhauer EA. Phase II study of Triapine in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161). Invest New Drugs. 2007; 25:471–7. https://doi.org/10.1007/s10637-007-9044-9. [PubMed].
215. Flaig TW, Glode M, Gustafson D, van Bokhoven A, Tao Y, Wilson S, Su LJ, Li Y, Harrison G, Agarwal R, Crawford ED, Lucia MS, Pollak M. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate. 2010; 70:848–55. https://doi.org/10.1002/pros.21118. [PubMed].
216. Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, Pierson AS, Agarwal R, Glode LM. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007; 25:139–46. https://doi.org/10.1007/s10637-006-9019-2. [PubMed].
217. Siegel AB, Narayan R, Rodriguez R, Goyal A, Jacobson JS, Kelly K, Ladas E, Lunghofer PJ, Hansen RJ, Gustafson DL, Flaig TW, Tsai WY, Wu DP, et al. A phase I dose-finding study of silybin phosphatidylcholine (milk thistle) in patients with advanced hepatocellular carcinoma. Integr Cancer Ther. 2014; 13:46–53. https://doi.org/10.1177/1534735413490798. [PubMed].
218. McLarty J, Bigelow RL, Smith M, Elmajian D, Ankem M, Cardelli JA. Tea polyphenols decrease serum levels of prostate-specific antigen, hepatocyte growth factor, and vascular endothelial growth factor in prostate cancer patients and inhibit production of hepatocyte growth factor and vascular endothelial growth factor in vitro. Cancer Prev Res (Phila). 2009; 2:673–82. https://doi.org/10.1158/1940-6207.CAPR-08-0167. [PubMed].