
EDITORIAL COMMENTARY
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive, deadly soft tissue sarcomas that lack effective therapies [1–3]. These Schwann cell derived tumors arise spontaneously as well as in patients with the hereditary cancer predisposition syndrome, Neurofibromatosis Type I (NF1). The 5-year survival rate for patients with MPNSTs is only 20–35%, and MPNSTs are the leading cause of death in NF1 patients. In all contexts, loss of the Ras inhibitor, neurofibromin (encoded by the NF1 gene), is a defining event in MPNST genesis [4]. Current treatment with traditional chemotherapy and radiation is highly toxic and does not reduce patient mortality, necessitating the development of more targeted treatments [5–9].
In most MPNSTs, cyclin-dependent kinases (CDKs) 2 and 4/6 are hyperactivated due to loss of their endogenous inhibitors, p16 and p27, or amplification of the cyclin and CDK genes [3]. This results in functional loss of the retinoblastoma (RB1) tumor suppressor, one of the most important guardians against cellular transformation and cancer development [10]. Because RB1 remains genetically wild type in the majority of MPNSTs, its reactivation in tumors represents an exciting new approach for treating this disease.
Recent studies of a novel RB1 regulator, named RABL6A, strengthens the rationale for RB1 targeted therapy in MPNST [11]. RABL6A is an oncogenic, RAB-like GTPase previously shown to inhibit RB1 signaling in pancreatic neuroendocrine tumors via downregulation of p27 [12]. Because p27 protein loss is a frequent event associated with worse MPNST patient survival [13], we explored RABL6A expression and function in those tumors. RABL6A protein was found to be robustly upregulated in patient MPNSTs relative to benign precursor lesions, with p27 expression patterns inverse to RABL6A levels [11]. Moreover, RABL6A knockdown studies showed it is required for MPNST cell viability and cell cycle progression. At the molecular level, RABL6A promotes MPNST proliferation and survival by decreasing p27 expression, increasing CDK4/6 activity and thereby inactivating RB1 (Figure 1). In addition, RABL6A has been shown to activate MEK [14, 15] and upregulate Myc mRNA [11, 16]. This can contribute to CDK-RB1 dysregulation because MEK and Myc are both hyperactivated in Ras-driven MPNSTs, and Myc is a transcriptional activator of cyclin D, CDK4, and CDK6 genes [17–19]. Together, these observations reinforced the notion that MPNST growth would be strongly suppressed by reactivating RB1.
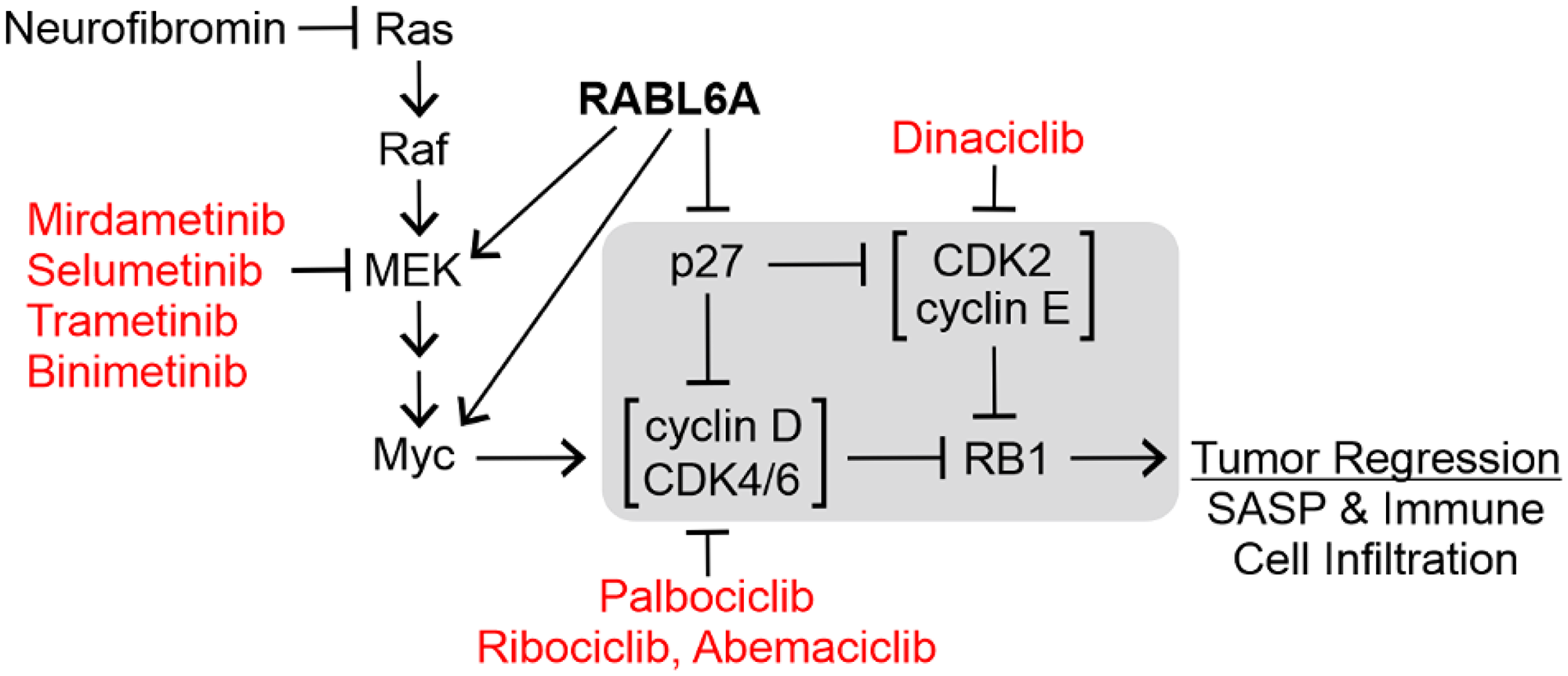
Figure 1: Pharmacologic targeting of the RABL6A-RB1 pathway in tumors. The central regulators of the RB1 tumor suppressor are highlighted in the gray box. Representative inhibitors of the indicated kinases are depicted in red. RABL6A inhibits RB1 by reducing the CDK inhibitor, p27, and activating MEK and Myc. Arrows, activating events; perpendicular bars, inhibitory events.
At present, the most effective way to reactivate RB1 in tumors is using drugs that selectively block CDK4/6 activity. CDK inhibitors were first introduced in the late 1990s as anti-cancer therapeutics; however, their lack of specificity for particular CDKs caused significant toxicity and undermined their usage in the clinic [3, 20–22]. Newer generation CDK inhibitors with greatly reduced toxicity include a group of related drugs with high specificity for CDK4/6, namely palbociclib, ribociclib, and abemaciclib (see Figure 1). These drugs are FDA-approved for the treatment of estrogen receptor-positive/HER2-negative metastatic breast cancer in combination with aromatase inhibitors [23, 24]. Excitingly, palbociclib and its relatives are showing great promise in clinical trials for other solid tumors, including another aggressive sarcoma, dedifferentiated liposarcoma (DDLPS) [25], which frequently harbors amplification of CDK4 (NCT01209598, NCT02897375) [26].
With the above considerations in mind, we examined the efficacy of CDK4/6 inhibition in pre-clinical models of MPNST [11]. Reactivation of RB1 with palbociclib halted MPNST growth both in vitro and in vivo in orthotopic (sciatic nerve) mouse tumor models. The response to palbociclib was significantly reduced by RABL6A depletion in the tumor cells, suggesting that patient MPNSTs bearing elevated RABL6A may be more responsive to CDK4/6 inhibitor therapy. However, tumors in immunodeficient mice invariably acquired resistance to the monotherapy. This was not surprising since tumors in people and mouse models have been shown to employ many mechanisms to override the effects of CDK4/6 inhibitor monotherapy [24, 27, 28].
Upregulation of CDK2 is one of the most common mechanisms through which tumors overcome sustained CDK4/6 inhibition [27–30]. As depicted in Figure 1, CDK2 can compensate for the loss of CDK4/6 by phosphorylating and inactivating RB1. This, along with the high levels of CDK2 observed in many MPNST patient samples [11, 13, 31], prompted us to test combination therapy targeting both CDK4/6 and CDK2. Treatment with palbociclib plus the non-specific CDK2 inhibitor, dinaciclib, displayed greater antitumor activity in MPNST-bearing mice [11]. Unfortunately, dinaciclib was not well tolerated by the animals, reflecting the toxic effects of dinaciclib in patients that have limited its clinical use. This has steered ongoing investigations toward other RABL6A-RB1 targeted combination therapies.
Hyperactivation of Ras-MEK-ERK signaling is another way tumor cells acquire resistance to CDK4/6 inhibitor therapy as the pathway converges on RB1 [24, 32, 33]. This is illustrated by the fact activated MEK-ERK stimulates Myc, a transcription factor that directly increases the mRNA expression of CDK4, CDK6 and their regulatory partners, the D cyclins (Figure 1). Since MPNSTs are initiated by neurofibromin loss and enhanced Ras activity, they may have a higher likelihood or increased rate of resistance to CDK4/6 inhibition. Therefore, it stands to reason that agents targeting CDK4/6 in combination with clinically proven inhibitors of MEK may negate a key mechanism of resistance and effectively treat MPNSTs.
Indeed, the combination of CDK4/6 and MEK inhibitors has shown remarkable promise in pre-clinical and clinical studies of Ras-driven cancers, such as non-small cell lung cancer (NSCLC) and pancreatic adenocarcinomas [34–36]. The pre-clinical studies revealed a crucial role of drug-induced tumor cell senescence and the immune system in the anti-tumor response. The combination induces a senescence-associated secretory phenotype, called SASP, in which secreted cytokines promote the infiltration of immune cells (natural killer or CD8+ T cells) that cause tumor regression. The use of immune competent mouse tumor models is critical to observe that response and accurately evaluate the efficacy of dual therapies targeting CDK4/6 and MEK, such as palbociclib plus mirdametinib (Figure 1). This is possible for studies of MPNST given the availability of genetically engineered and CRISPR-based mouse models that develop de novo MPNSTs in the context of fully active immune systems [37–41]. To fully appreciate the complete role of the microenvironment in these studies, it will be important to evaluate therapies using multiple preclinical platforms, as murine background strain has a strong impact on the immune landscape of MPNSTs [42].
Clinical studies targeting CDK4/6 and MEK in solid tumors are highly encouraging. In a Phase 1 dose escalation study evaluating the combination of palbociclib with mirdametinib in patients with RAS-mutant solid tumors, promising progression free survival was reported. Of 25 patients, 11 were noted to be progression free for > 3 months with 6 patients displaying no disease progression for > 6 months. Moreover, 72% of patients achieved stable disease as their best response and 1 patient achieved partial response. This combination at maximum administered doses was deemed to be well tolerated [25]. A Phase 1b/2 trial evaluating the combination of ribociclib with binimetinib in NRAS-mutant melanoma recently reported their results from 41 patients treated on a 28-day schedule Phase 2 dose expansion cohort. The median duration of response was 10.3 months with a median time to progression and median progression free survival of 3.7 months. Slightly more than half of the patients (51.2%) achieved stable disease and 8 patients (19.5%) achieved partial response. The overall survival of the Phase 2 cohort was reported to be 11.3 months (NCT01781572). These are compelling results for NRAS-mutant melanoma patients since there are no targeted therapies currently approved. Other clinical studies evaluating similar combinations in RAS-mutant colorectal cancer (NCT03981614), KRAS-mutant NSCLC (NCT03170206), and in children and young adults with brain tumors (NCT03434262) are ongoing.
While there is good reason to be excited about the anti-tumor potential of therapies inhibiting both CDK4/6 and MEK, combination therapies targeting other RABL6A effectors may also have clinical efficacy in MPNSTs. That is because RABL6A upregulation is so prominent in this disease [11]. Moreover, RABL6A is a potent oncoprotein that controls many druggable cancer targets besides CDK4/6 and MEK, including Myc (Figure 1), PP2A-Akt-mTOR, and receptor tyrosine kinase pathways like VEGFR and EGFR [11, 12, 16]. As advances in drug development yield more specific targeted therapeutics, the future looks bright for the number of novel combination therapies that could be evaluated for rare malignancies, like MPNSTs, which currently lack effective therapy.
ACKNOWLEDGMENTS AND FUNDING
We wish to acknowledge support from the Children’s Tumor Foundation Synodos Grant (DEQ, BWD and RDD) and Young Investigator Award (JLK), NCI Neuroendocrine Tumor SPORE P50 CA174521 (DEQ and BWD), as well as grants to DEQ, JLK and the team through the Holden Comprehensive Cancer Center (NCI P30 Grant CA086862, Sarcoma Multidisciplinary Oncology Group Award, and the Mezhir Translational Award).
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep. 2009; 11:322–8. https://doi.org/10.1007/s11912-009-0045-z. [PubMed].
2. Farid M, Demicco EG, Garcia R, Ahn L, Merola PR, Cioffi A, Maki RG. Malignant peripheral nerve sheath tumors. Oncologist. 2014; 19:193–201. https://doi.org/10.1634/theoncologist.2013-0328. [PubMed].
3. Kohlmeyer JL, Gordon DJ, Tanas MR, Monga V, Dodd RD, Quelle DE. CDKs in sarcoma: Mediators of disease and emerging therapeutic targets. Int J Mol Sci. 2020; 21:3018. https://doi.org/10.3390/ijms21083018. [PubMed].
4. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015; 15:290–301. https://doi.org/10.1038/nrc3911. [PubMed].
5. Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, Zhang W, McCutcheon IE, Slopis JM, Lazar AJ, Pollock RE, Lev D. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009; 249:1014–22. https://doi.org/10.1097/SLA.0b013e3181a77e9a. [PubMed].
6. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002; 39:311–4. https://doi.org/10.1136/jmg.39.5.311. [PubMed].
7. Reilly KM, Kim A, Blakely J, Ferner RE, Gutmann DH, Legius E, Miettinen MM, Randall RL, Ratner N, Jumbe NL, Bakker A, Viskochil D, Widemann BC, et al. Neurofibromatosis Type 1-associated MPNST state of the science: Outlining a research agenda for the future. J Natl Cancer Inst. 2017; 109:djx124. https://doi.org/10.1093/jnci/djx124. [PubMed].
8. Kaushal A, Citrin D. The role of radiation therapy in the management of sarcomas. Surg Clin North Am. 2008; 88:629–46. https://doi.org/10.1016/j.suc.2008.03.005. [PubMed].
9. Zehou O, Fabre E, Zelek L, Sbidian E, Ortonne N, Banu E, Wolkenstein P, Valeyrie-Allanore L. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis. 2013; 8:127. https://doi.org/10.1186/1750-1172-8-127. [PubMed].
10. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70. https://doi.org/10.1016/s0092-8674(00)81683-9. [PubMed].
11. Kohlmeyer JL, Kaemmer CA, Pulliam C, Maharjan CK, Samayoa AM, Major HJ, Cornick KE, Knepper-Adrian V, Khanna R, Sieren JC, Leidinger MR, Meyerholz DK, Zamba KD, et al. RABL6A is an essential driver of MPNSTs that negatively regulates the RB1 pathway and sensitizes tumor cells to CDK4/6 inhibitors. Clin Cancer Res. 2020; 26:2997–3011. https://doi.org/10.1158/1078-0432.CCR-19-2706. [PubMed].
12. Hagen J, Muniz VP, Falls KC, Reed SM, Taghiyev AF, Quelle FW, Gourronc FA, Klingelhutz AJ, Major HJ, Askeland RW, Sherman SK, O'Dorisio TM, Bellizzi AM, et al. RABL6A promotes G1-S phase progression and pancreatic neuroendocrine tumor cell proliferation in an Rb1-dependent manner. Cancer Res. 2014; 74:6661–70. https://doi.org/10.1158/0008-5472.CAN-13-3742. [PubMed].
13. Kourea HP, Cordon-Cardo C, Dudas M, Leung D, Woodruff JM. Expression of p27(kip) and other cell cycle regulators in malignant peripheral nerve sheath tumors and neurofibromas: the emerging role of p27(kip) in malignant transformation of neurofibromas. Am J Pathol. 1999; 155:1885–91. https://doi.org/10.1016/S0002-9440(10)65508-3. [PubMed].
14. Muniz VP, Askeland RW, Zhang X, Reed SM, Tompkins VS, Hagen J, McDowell BD, Button A, Smith BJ, Weydert JA, Mezhir JJ, Quelle DE. RABL6A promotes oxaliplatin resistance in tumor cells and is a new marker of survival for resected pancreatic ductal adenocarcinoma patients. Genes Cancer. 2013; 4:273–84. https://doi.org/10.1177/1947601913501074. [PubMed].
15. Montalbano J, Lui K, Sheikh MS, Huang Y. Identification and characterization of RBEL1 subfamily of GTPases in the Ras superfamily involved in cell growth regulation. J Biol Chem. 2009; 284:18129–42. https://doi.org/10.1074/jbc.M109.009597. [PubMed].
16. Umesalma S, Kaemmer CA, Kohlmeyer JL, Letney B, Schab AM, Reilly JA, Sheehy RM, Hagen J, Tiwari N, Zhan F, Leidinger MR, O'Dorisio TM, Dillon J, et al. RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth. J Clin Invest. 2019; 129:1641–1653. https://doi.org/10.1172/JCI123049. [PubMed].
17. Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta. 2015; 1849:506–16. https://doi.org/10.1016/j.bbagrm.2014.03.013. [PubMed].
18. Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006; 16:253–64. https://doi.org/10.1016/j.semcancer.2006.07.014. [PubMed].
19. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011; 10:57–67. https://doi.org/10.4161/cc.10.1.14449. [PubMed].
20. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015; 14:130–46. https://doi.org/10.1038/nrd4504. [PubMed].
21. Peyressatre M, Prevel C, Pellerano M, Morris MC. Targeting cyclin-dependent kinases in human cancers: from small molecules to Peptide inhibitors. Cancers (Basel). 2015; 7:179–237. https://doi.org/10.3390/cancers7010179. [PubMed].
22. Sanchez-Martinez C, Gelbert LM, Lallena MJ, de Dios A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg Med Chem Lett. 2015; 25:3420–35. https://doi.org/10.1016/j.bmcl.2015.05.100. [PubMed].
23. Laderian B, Fojo T. CDK4/6 Inhibition as a therapeutic strategy in breast cancer: palbociclib, ribociclib, and abemaciclib. Semin Oncol. 2017; 44:395–403. https://doi.org/10.1053/j.seminoncol.2018.03.006. [PubMed].
24. Alvarez-Fernandez M, Malumbres M. Mechanisms of sensitivity and resistance to CDK4/6 inhibition. Cancer Cell. 2020; 37:514–29. https://doi.org/10.1016/j.ccell.2020.03.010. [PubMed].
25. Dickson MA, Koff A, D'Angelo SP, Gounder MM, Keohan ML, Kelly CM, Chi P, Antonescu CR, Landa J, Qin LX, Crago AM, Singer S, Tap WD. Phase 2 study of the CDK4 inhibitor abemaciclib in dedifferentiated liposarcoma. Journal of Clinical Oncology. 2019; 37:11004. https://doi.org/10.1200/JCO.2019.37.15_suppl.11004.
26. Dickson MA, Schwartz GK, Keohan ML, D'Angelo SP, Gounder MM, Chi P, Antonescu CR, Landa J, Qin LX, Crago AM, Singer S, Koff A, Tap WD. Progression-free survival among patients with well-differentiated or dedifferentiated liposarcoma treated with CDK4 inhibitor palbociclib: A phase 2 clinical trial. JAMA Oncol. 2016; 2:937–40. https://doi.org/10.1001/jamaoncol.2016.0264. [PubMed].
27. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M, Cortes J, Elliott R, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016; 76:2301–13. https://doi.org/10.1158/0008-5472.CAN-15-0728. [PubMed].
28. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016; 6:353–67. https://doi.org/10.1158/2159-8290.CD-15-0894. [PubMed].
29. Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017; 173:83–105. https://doi.org/10.1016/j.pharmthera.2017.02.008. [PubMed].
30. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016; 13:417–30. https://doi.org/10.1038/nrclinonc.2016.26. [PubMed].
31. Agesen TH, Florenes VA, Molenaar WM, Lind GE, Berner JM, Plaat BE, Komdeur R, Myklebost O, van den Berg E, Lothe RA. Expression patterns of cell cycle components in sporadic and neurofibromatosis type 1-related malignant peripheral nerve sheath tumors. J Neuropathol Exp Neurol. 2005; 64:74–81. https://doi.org/10.1093/jnen/64.1.74. [PubMed].
32. Rodriguez-Puebla ML, Robles AI, Conti CJ. Ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol Carcinog. 1999; 24:1–6. [PubMed].
33. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011; 11:761–74. https://doi.org/10.1038/nrc3106. [PubMed].
34. Ruscetti M, Leibold J, Bott MJ, Fennell M, Kulick A, Salgado NR, Chen CC, Ho YJ, Sanchez-Rivera FJ, Fencht J, Baslan T, Tian S, Chen HA, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018; 362:1416. https://doi.org/10.1126/science.aas9090. [PubMed].
35. Ruscetti M, Morris JP 4th, Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, Li J, Norgard RJ, Wilkinson JE, et al. Senescence-induced vascular remodeling creates therapeutic vulnerabilities in pancreas cancer. Cell. 2020; 181:424–41.e21. https://doi.org/10.1016/j.cell.2020.03.008. [PubMed].
36. Shapiro GI, Hilton J, Gandi L, Chau N, Cleary J, Wolanski A, Anderson A, Beardslee B, Hassinger F, Bhushan K, Downey E, Gibson J, Pruitt-Thompson S, et al. Abstract CT046: Phase I dose escalation study of the CDK4/6 inhibitor palbociclib in combination with the MEK inhibitor PD-0325901 in patients with RAS mutant solid tumors. Cancer Res. 2017; 77:CT046–CT. https://doi.org/10.1158/1538-7445.Am2017-ct046.
37. Dodd RD, Mito JK, Eward WC, Chitalia R, Sachdeva M, Ma Y, Barretina J, Dodd L, Kirsch DG. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013; 12:1906–17. https://doi.org/10.1158/1535-7163.MCT-13-0189. [PubMed].
38. Huang J, Chen M, Whitley MJ, Kuo HC, Xu ES, Walens A, Mowery YM, Van Mater D, Eward WC, Cardona DM, Luo L, Ma Y, Lopez OM, et al. Generation and comparison of CRISPR-Cas9 and Cre-mediated genetically engineered mouse models of sarcoma. Nat Commun. 2017; 8:15999. https://doi.org/10.1038/ncomms15999. [PubMed].
39. Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999; 286:2172–6. https://doi.org/10.1126/science.286.5447.2172. [PubMed].
40. Keng VW, Rahrmann EP, Watson AL, Tschida BR, Moertel CL, Jessen WJ, Rizvi TA, Collins MH, Ratner N, Largaespada DA. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012; 72:3405–13. https://doi.org/10.1158/0008-5472.CAN-11-4092. [PubMed].
41. Dodd RD, Mito JK, Kirsch DG. Animal models of soft-tissue sarcoma. Dis Model Mech. 2010; 3:557–66. https://doi.org/10.1242/dmm.005223. [PubMed].
42. Scherer A, Stephens VR, McGivney GR, Gutierrez WR, Laverty EA, Knepper-Adrian V, Dodd RD. Distinct Tumor Microenvironments Are a Defining Feature of Strain-Specific CRISPR/Cas9-Induced MPNSTs. Genes (Basel). 2020; 11:583. https://doi.org/10.3390/genes11050583. [PubMed].