
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent human cancer and third-highest cause of cancer-related deaths worldwide, and its incidence is significantly increasing [1, 2]. Despite advances in surgical techniques, radiofrequency ablation, and trans-arterial therapy, the prognosis of HCC patients is unacceptable because of post-treatment relapse and distant metastasis. Therefore, great efforts have been made to identify novel prognostic factors for managing patients with HCC as well as molecular targets to prevent oncogenesis and tumor progression [3–6].
Several genome-wide studies of the molecular alterations in tumor and non-tumor liver tissues addressed DNA, mRNA, and microRNA (miRNA) expression as well as epigenetic alterations to predict recurrence and hepatocarcinogenesis after treating patients with curative intent according to distinct gene expression or alteration patterns [7–11]. Here we focused on the epigenetic alterations of the genomes of tumor and non-tumor cells of patients with HCC to identify the mechanisms of tumor progression and hepatocarcinogenesis., because global DNA hypomethylation or cancer specific DNA hypermethylation occurs in certain carcinomas [12]. And we firstly chose long interspersed nuclear element-1 (LINE-1) sequences provided a surrogate marker of global DNA methylation levels, and detected hypomethylation of LINE-1 in HCC tissues, which was significantly associated with poor prognosis of patients with HCC through the activation of MET [13]. Further, we investigated epigenetic characteristics with array-based analysis of DNA methylation using the Illumina Human Methylation 450 BeadChip, and identified specific DNA methylation profiling in nontumor liver tissue of patients without HCV and HBV detectable infection (NBNC-HCC), which possibly contributed to the development of HCC [14].
In this study, we analyzed DNA methylation profiles in tumor tissue of HCC using those case series of our recent study [14], and found the most hypermethylated gene in tumor compared with nontumor tissues, encodes T-box 15 (TBX15), implicating TBX15 as a candidate regulator of tumor progression. Hence, the aim of the present study was to identify novel biomarkers for early diagnosis, risk assessment, and chemoprevention of HCC. For this purpose, we further evaluated the clinical significance of TBX15 expression levels associated with tumor phenotype as well as with the prognosis of patients with HCC who underwent hepatectomy.
Results
Detection of differentially methylated genes
Figure 1 shows volcano plots of 3,139 differentially methylated CpG sites in tumor tissues compared with nontumor liver tissues (beta-value difference > 0.2, P < 0.05). Consistent with previous reports [15, 16], here we detected global differential DNA hypomethylation (3,108 CpG sites) in tumor tissues compared with nontumor tissues. Further, 31 CpG sites were hypermethylated, corresponding to 16 gene promoters that are commonly hypermethylated in tumor tissues compared with nontumor tissues (Table 1). Among them, TBX15 of tumor tissue was the most hypermethylated (mean difference of beta-value = 0.518765).
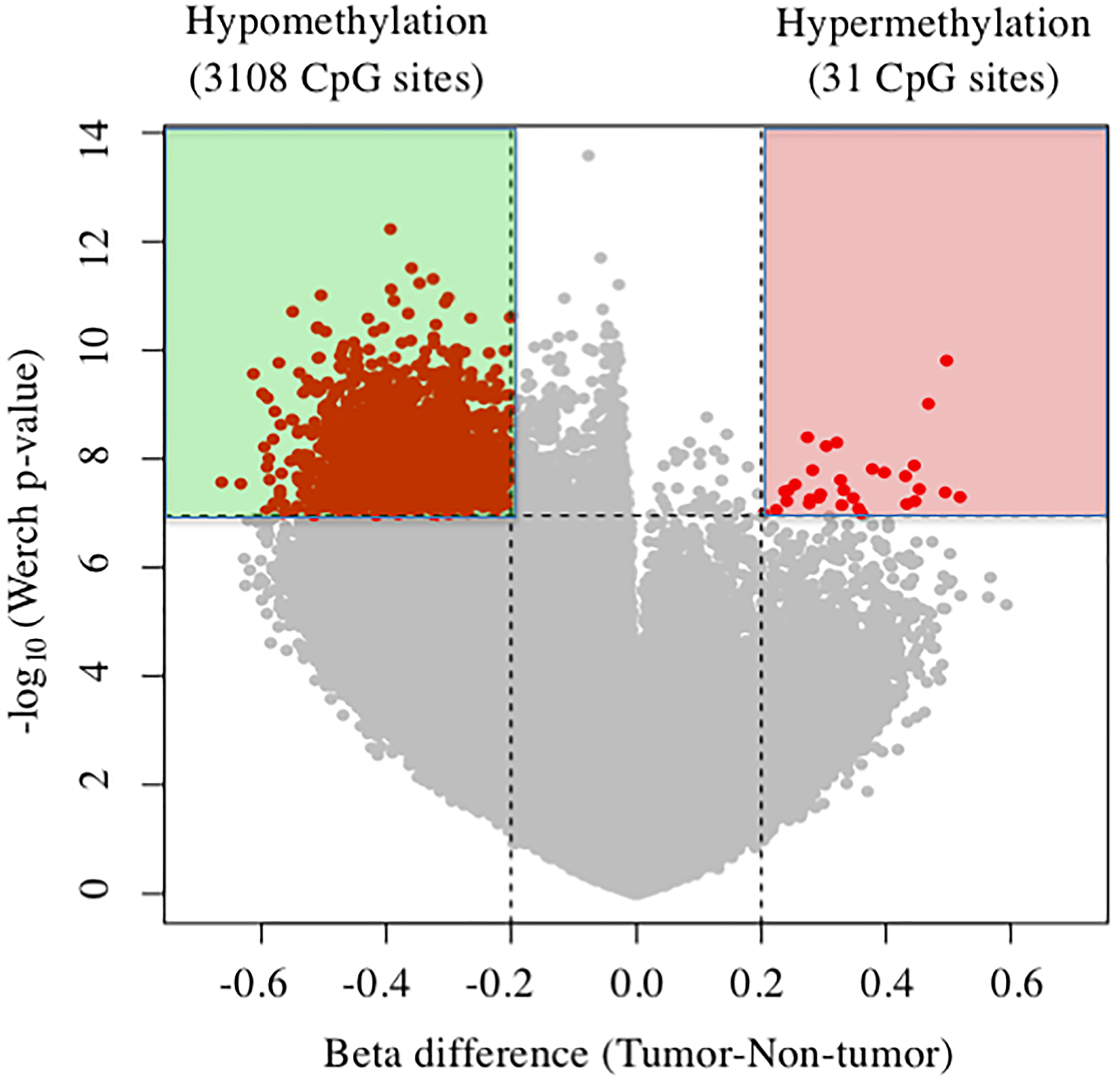
Figure 1: Volcano plots of DNA methylation in tumor tissues compared with nontumor tissue. Significant associations are indicated in red.
Table 1: Genes with hypermethylated CpG sites in HCC tissues
| Gene name | Target ID | Chromosome | Position | Mean difference |
|---|---|---|---|---|
| TBX15 | cg10703826 | 1 | 119333639 | 0.518765 |
| PITX1 | cg00396667 | 5 | 134392286 | 0.446505 |
| ILDR2 | cg25147376 | 1 | 165156917 | 0.360129286 |
| GRHL2 | cg23973429 | 8 | 102573922 | 0.347427857 |
| LOC146880 | cg01720033 | 17 | 60208206 | 0.327155 |
| RTN4R | cg12393104 | 22 | 18613758 | 0.320887143 |
| INF2 | cg07155724 | 14 | 104236775 | 0.304177857 |
| CYB5R2 | cg05919312 | 11 | 7650739 | 0.294705 |
| GAB1 | cg17453767 | 4 | 144497246 | 0.282047857 |
| PITPNM2 | cg01973483 | 12 | 122157076 | 0.278242143 |
| C1QTNF4 | cg18356785 | 11 | 47568356 | 0.277035714 |
| KIAA0664 | cg12229632 | 17 | 2554656 | 0.273881429 |
| PPFIA3 | cg08319905 | 19 | 54328082 | 0.254201429 |
| HOXA10 | cg10724867 | 7 | 27185392 | 0.242810714 |
| PKP4 | cg13168187 | 2 | 159231927 | 0.237381429 |
| ANKS1A | cg07254055 | 6 | 35119746 | 0.203343571 |
To explore the relationship between TBX15 methylation status and HCC tumor malignancy, we investigated the TBX15 methylation analysis of HCC patients from TCGA data. Among those data, 59 registered CpG sites of TBX15 were identified and could be classified into 3 clusters according to its beta-value (Supplementary Figure 1). Consequently, the high methylation beta-value of TBX15 was significantly associated with poor disease-free survival in HCC patients, regardless of cluster classification, while poor overall survival only in cluster 3 (Supplementary Figure 2). Next, we investigated the TBX15 mRNA expression in several cancers from TCGA. TBX15 mRNA expression in tumor tissue was varied according to type of carcinoma, while it was significantly lower in tumor tissue of HCC compared to nontumor tissue (Supplementary Figure 3). Also, we found that the same results of HCC were obtained in breast cancer and cholangiocarcinoma.
In our series, the levels of TBX15 mRNA in tumor tissues were significantly lower compared with those in nontumor tissues (p < 0.0001) (Figure 2A). And then, immunohistochemistry revealed that TBX15 protein expression in tumor tissue was weak compared to that of nontumor tissue (Figure 2B).
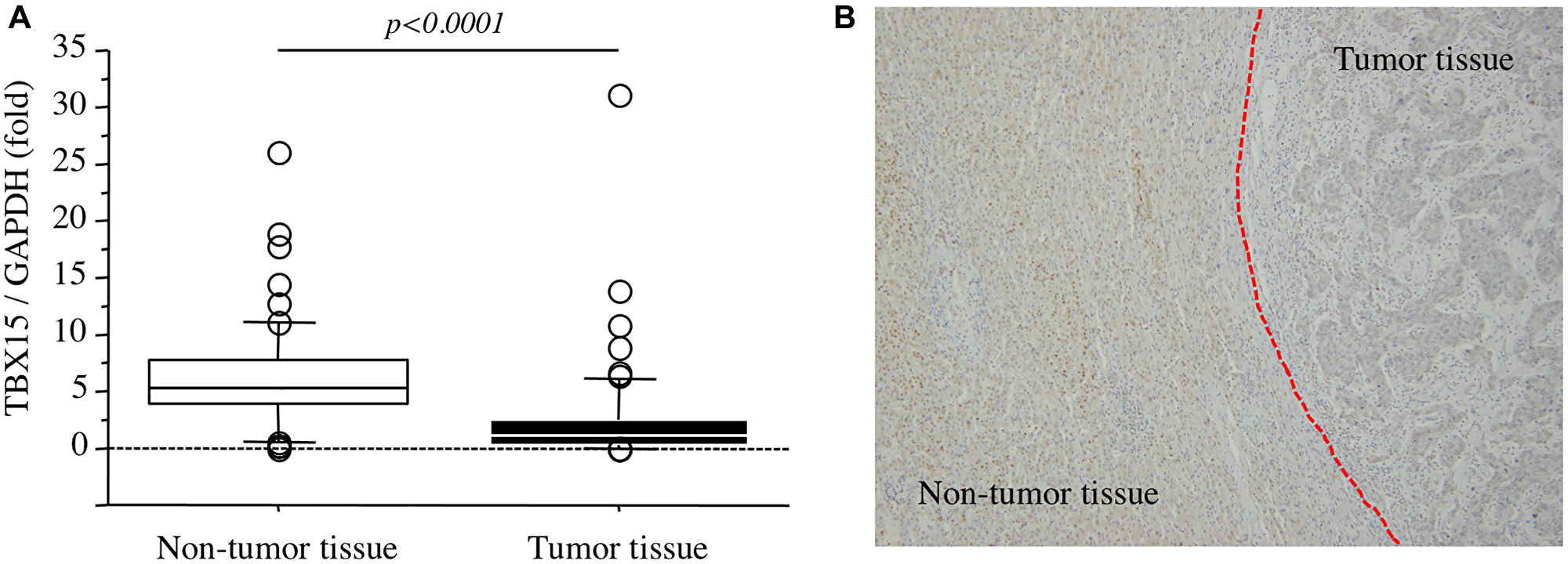
Figure 2: (A) TBX15 mRNA expression in tumor tissues and nontumor tissues. (B) TBX15 protein expression in tumor tissues and nontumor tissues.
Relationship between TBX15 mRNA levels and patients’ clinicopathological characteristics
The 3- and 5-years overall survival rates of 58 patients with HCC were 82.6% and 76.8%, respectively. Cutoff value of TBX15 mRNA for patients’ death was decided by receiver operating characteristics (ROC) curve (AUC 0.727), and Youden’s index was calculated as highest value of 0.4610 at TBX15 mRNA expression with 0.45457 fold. Therefore, we next allocated the patients according to the levels of TBX15 mRNA detected in tumor tissues (cutoff value = 0.5-fold) as follows: TBX15 high-expression group (n = 41) and TBX15 low-expression group (n = 17). Table 2 showed the comparison of clinicopathological characteristics according to TBX15 mRNA expression in tumor tissue. Low TBX15 expression significantly correlated with higher serum DCP levels, and there were no significant differences between any of the other variables. Notably, there was no significant association regarding the presence or absence of hepatitis virus infection (p = 0.8797). The TBX15 low-expression group experienced significantly worse prognosis compared with that of the TBX15 high-expression group (p = 0.0010), and the 5-year overall survival rates were 43.3% and 86.2%, respectively (Figure 3A). Moreover, the disease-free survival rate of the TBX15 low-expression group was significantly worse, and the 5-year disease-free survival rates were 23.5% and 58.0%, respectively (Figure 3B).
Table 2: Clinicopathological variables and TBX15 mRNA expression in HCC tissues
| Factors | TBX15 | p-value | ||
|---|---|---|---|---|
| Low | High | |||
| (n = 17) | (n = 41) | |||
| Age (years) | Median ± SD | 66 ± 8.07 | 68 ± 8.4 | 0.3836 |
| Gender | Male/Female | 16/1 | 29/12 | 0.0825 |
| Diabetes mellitus | Absence/Presence | 11/6 | 30/11 | 0.5403 |
| Hepatic viral infection | Negative/HBV/HCV | 5/ 6/6 | 12/17/12 | 0.8798 |
| Background liver | NL/CH/LC | 3/10/4 | 7/26/8 | 0.9339 |
| ICG R15 (%) | ≦10 / > 10 | 12/5 | 29/12 | > 0.9999 |
| Stage | I, II/III, IV | 7/10 | 26/15 | 0.1511 |
| Maximum tumor size | < 5 cm / ≥ 5 cm | 8/9 | 29/11* | 0.0780 |
| Number | Single/Multiple | 10/7 | 31/10 | 0.2211 |
| Growth type | Expandable/Invasive | 14/3 | 35/6 | > 0.9999 |
| Differentiation | Well/Others | 2/14* | 6/34* | > 0.9999 |
| Portal invasion | Negative/Positive | 12/5 | 32/9 | 0.7396 |
| Venous invasion | Negative/Positive | 14/3 | 38/3 | 0.3450 |
| AFP (ng/ml) | ≦100 / >100 | 13/4 | 26/15 | 0.3775 |
| DCP (IU/L) | ≦300 / > 300 | 5/12 | 20/21 | 0.041 |
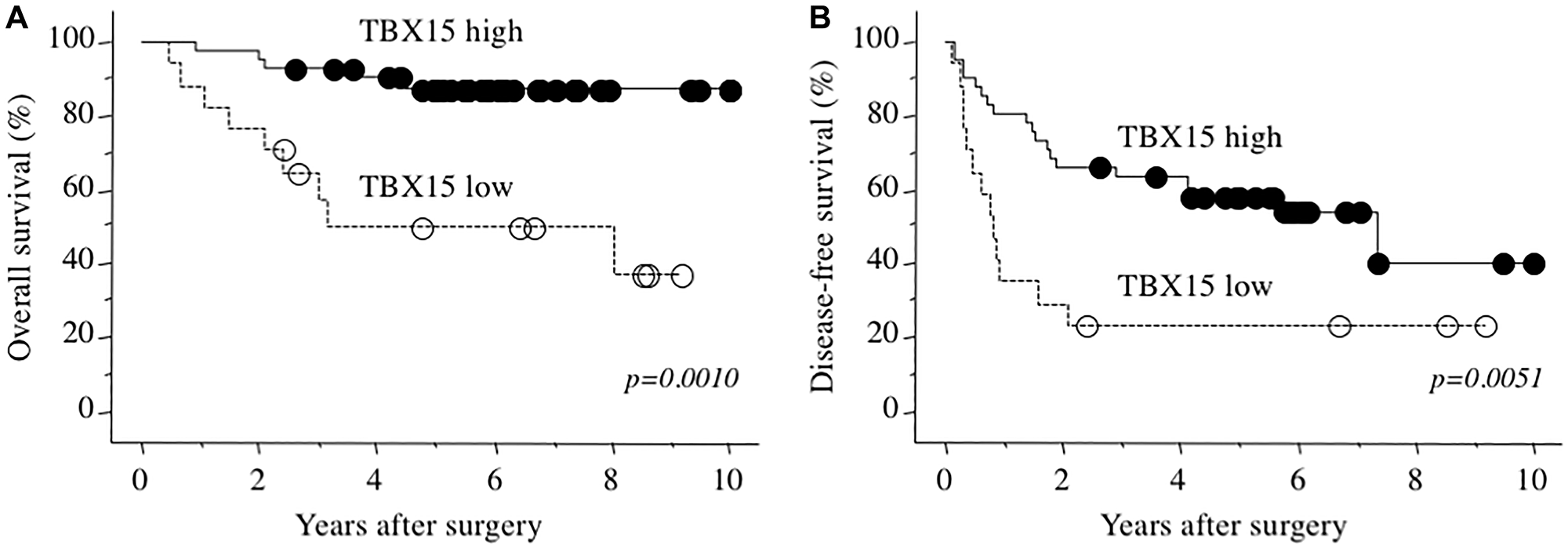
Figure 3: (A) Overall survival as a function of TBX15 mRNA expression. (B) Disease-free survival as a function of TBX15 mRNA expression.
Prognostic factors identified using univariate and multivariate analyses
Univariate analysis identified the number of tumors (multiple) (p = 0.0461), growth type (invasive) (p = 0.0057), venous invasion (positive) (p < 0.0001), serum DCP levels (> 300 IU/L) (p < 0.0001), and TBX15 mRNA levels (Low) (p = 0.0010) as significant poor prognostic factors for overall survival (Table 3). When TBX15 mRNA expression was entered into the proportional hazards model along with number of tumors (multiple), growth type (invasive), venous invasion (positive), and serum DCP levels (> 300 IU/L), low TBX15 mRNA expression in tumor tissue was identified as one of the independent predictors of poor prognosis (Table 3). Also, in disease-free survival, growth type (invasive) (p = 0.0012), venous invasion (positive) (p < 0.0001), and low TBX15 mRNA expression (p = 0.0064) were identified as significant independent predictors of poor prognosis (Table 4).
Table 3: Prognostic factors of overall survival after curative resection
| Factors | 5-years survival (%) | p-value | Hazard ratio | 95% CI | p-value | |
|---|---|---|---|---|---|---|
| Age (years) | ≦70 / >70 | 81.5/80.1 | 0.4698 | |||
| Gender | Male / Female | 74.9/83.1 | 0.4241 | |||
| Diabetes mellitus | Absence/Presence | 80.0/69.1 | 0.5892 | |||
| Hepatic viral infection | Negative/HBV/HCV | 70.9/76.5/81.9 | 0.8087 | |||
| Stage | I, II/III, IV | 87.8/62.6 | 0.0607 | |||
| ICG R15 (%) | ≦10 / >10 | 74.8/81.9 | 0.9584 | |||
| Tumor size (cm) | ≦5 / >5 | 83.4/63.5 | 0.1775 | |||
| Number | Single / Multiple | 85.2/57.4 | 0.0461 | 1.730 | 0.452–4.651 | 0.4657 |
| Growth type | Expandable/Invasive | 83.0/41.7 | 0.0057 | 11.765 | 1.984–90.909 | 0.0078 |
| Differentiation | Well/Others | 87.5/78.3 | 0.9703 | |||
| Portal invasion | Negative/Positive | 84.0/55.0 | 0.0507 | |||
| Venous invasion | Negative/Positive | 84.3/16.7 | < 0.0001 | 1.634 | 0.309–6.667 | 0.6451 |
| AFP (ng/ml) | ≦100 / > 100 | 78.7/72.9 | 0.6964 | |||
| DCP (IU/L) | ≦300 / > 300 | 100/59.7 | < 0.0001 | 40.681 | 2.783–594.731 | 0.0068 |
| TBX15 mRNA | High/Low | 87.2/49.9 | 0.0006 | 8.475 | 2.398–38.462 | 0.0014 |
Table 4: Prognostic factors of disease-free survival after curative resection
| Factors | 5-years survival (%) | p-value | Hazard ratio | 95% CI | p-value | |
|---|---|---|---|---|---|---|
| Age (years) | ≦70 / > 70 | 48.1/47.7 | 0.5105 | |||
| Gender | Male/Female | 46.1/53.8 | 0.6279 | |||
| Diabetes mellitus | Absence/Presence | 46.0/52.9 | 0.7841 | |||
| Hepatic viral infection | Negative/HBV/HCV | 55.6/47.1/43.5 | 0.8369 | |||
| Stage | I, II/III, IV | 57.1/35.2 | 0.1491 | |||
| ICG R15 (%) | ≦10 / > 10 | 48.2/47.1 | 0.4302 | |||
| Tumor size (cm) | ≦5 / > 5 | 50.8/39.4 | 0.5735 | |||
| Number | Single/Multiple | 55.9/27.5 | 0.0868 | |||
| Growth type | Expandable/Invasive | 52.4/22.2 | 0.0012 | 3.106 | 1.350–6.849 | 0.0073 |
| Differentiation | Well/Others | 50.0/49.3 | 0.8424 | |||
| Portal invasion | Negative/Positive | 49.5/42.9 | 0.6086 | |||
| Venous invasion | Negative/Positive | 53.3/0 | < 0.0001 | 4.608 | 1.745–11.364 | 0.0018 |
| AFP (ng/ml) | ≦100 / >100 | 43.1/57.4 | 0.2920 | |||
| DCP (IU/L) | ≦300 / >300 | 55.5/42.0 | 0.3295 | |||
| TBX15 mRNA | High/Low | 58.0/23.5 | 0.0064 | 2.079 | 1.079–4.505 | 0.0301 |
DISCUSSION
The present study presents an array analysis of DNA methylation that identified TBX15 as the most hypermethylated gene tumor tissue of patients with HCC. These findings indicate that TBX15 is a candidate regulatory gene that contributes to tumor progression. TCGA data also revealed that TBX15 hypermethylation was associated with poor prognosis in HCC patients. In our validation set comprising 58 patients with HCC, we found that TBX15 mRNA expression was significantly reduced in tumor tissues compared with that of nontumor tissues, likely because of TBX15 hypermethylation. Further, TBX15 expression did not significantly correlate with tumor status, except for serum DCP levels. In contrast, a significant finding was that relatively low expression of TBX15 in tumor tissue was an independent prognostic factor for overall and disease-free survival.
The T-box gene family that encodes transcription factors plays diverse, critical roles during embryogenesis [17]. Further, TBX family members contribute to the regulation of the proliferation and differentiation of tissue-specific stem and progenitor cells during organogenesis [17, 18]. Moreover, evidence indicates the association of diverse T-box genes in oncogenesis, tumor invasion, and metastasis of certain cancers [19, 20]. Brachyury, a member of the T-box gene family, induces stemness markers such as NANOG, CD133, CD166, and CD44 in a colorectal cancer cell line by regulating the β-catenin oncogene and contributing to the epithelial-to-mesenchymal transition (EMT) that mediates invasion by tumor cells and metastasis [21]. Regarding the relationships of each subfamily of T-box gene and cancer, TBX2 and TBX3 over-expressions were observed in several human cancers, including ovarian, cervical, mammary, liver and pancreatic carcinomas, and current findings revealed its association with malignant behavior [20]. And TBX2 and TBX3 over-expressions has been elucidated to induce the tumor growth and metastasis, functioning as downstream target gene of Wnt/beta-catenin and pRb pathway through the suppression of p14ARF, p21CIP1, NDRG1 and E-cadherin [22, 23]. In contrast, TBX5 induces apoptosis and inhibits the growth of human osteosarcomas and lung carcinomas [24]. Further, TBX5 may act as a novel tumor suppressor gene, which is inactivated by promoter methylation in colon cancer cells [25]. Detection of TBX5 methylation may therefore serve as a potential biomarker of prognosis [25].
TBX15 was originally shown to be involved in mesodermal differentiation of the limb skeleton and face as well as in the impairment of adipocyte differentiation [17, 18]. Mutations of TBX15 are associated with the congenital morphological abnormalities of patients with Cousin syndrome [26, 27]. Recently, the association of TBX15 and cancer was demonstrated by the detection of breast cancer tissue-specific down-regulation of TBX15, suggesting the use of TBX15 expression as a biomarker to facilitate accurate diagnosis and prognosis or as a predictive marker for treatment efficiency [28]. Further, the TBX15 promoter is hypermethylated in histological subtypes of ovarian cancer, and the methylation levels and expression of TBX15 inversely correlate [29]. Thus, hypermethylated TBX15 may serve as a potential biomarker for early detection of the induction and progression of ovarian cancer [29].
In respect to HCC, a study using optimized liquid hybridization capture-based bisulfite sequencing detected tumor-specific hypermethylation of the TBX15 promoter, suggesting its contribution to hepatocarcinogenesis [30]. Similarly, genome-wide DNA methylation analysis revealed that the promoter region of TBX15 is hypermethylated and may therefore serve as marker to classify tumor and nontumor tissues [31]. In TCGA data, TBX15 mRNA expression was significantly lower in tumor tissue of HCC compared to nontumor tissue in HCC patients, as well. However, these reports did not investigate the malignant potential of TBX15.
The activity of TBX15 that may be associated with tumor progression and oncogenesis is unknown, and a few studies have addressed this question. For example, TBX15 is a methylation marker of prostate cancer associated with advanced stage and ETS-related gene (ERG) expression, which regulates cell proliferation, angiogenesis, and apoptosis [32]. Further, a combination of TBX15 and other hypermethylated genes is a useful biomarker for ERG expression and recurrence of prostate cancer [30]. TBX15 acts as an anti-apoptotic factor in thyroid cancer by inhibiting the expression of proapoptotic BAX and increasing the expression of antiapoptotic BCL2 and BCL-XL regulators [33]. TBX15 is up-regulated by TNF-α activation of the NF-κB pathway [34], and the EMT-related gene BMI1 directly represses the expression of TBX15 in myeloid progenitor cells [35]. Further, TBX15 may interact with corepressors of the Groucho family during mesodermal differentiation [36], which regulate several signaling mechanisms such as the Wingless/Wnt, transforming growth factor-β and NOTCH signaling pathways [37]. These findings suggest an association of TBX15 with oncogenesis in which TBX15 functions as a tumor suppressor related to tumor progression and resistance to treatment through the EMT, which was regulated by signaling pathways such as Wingless/Wnt [38]. However, TCGA pan cancer analysis of mRNA expression revealed that TBX15 expressions indicated a various trend according to the types of cancer. Actually, TBX15 of breast cancer and cholangiocarcinoma were down-regulated compared to normal tissue as well as HCC, meanwhile this gene expressions of other cancer were up-regulated. Hence, this gene might have the pivotal role for tumor malignancy according to the types of cancer, which was possibly related to embryological property.
To further investigate the possible regulatory mechanism of TBX15 in tumor malignancy of HCC, we performed GSEA according to TBX15 expression. Notably, the TBX15 expression-associated signatures “REACTIVE OXYGEN SPECIES (ROS) PATHWAY” was negatively correlated to TBX15 expression with significant displayed considerable normalized enrichment scores (Supplementary Figure 4). To the best of our knowledge, there was no reports in respect with the relationship between TBX15 and ROS in tumor malignancy. Many reports have demonstrated the pivotal role of ROS in biological processes [39]. Particular in tumor microenviroment, moderate ROS concentration induces cancer cell survival, angiogenesis and metastasis via several cell survival signaling cascade such as MMPs and VEGF, meanwhile high ROS concentration leads to cancer cell apoptosis [39]. In HCC with low TBX15 expression, ROS production could be moderate and contribute to the worse tumor malignancy. Therefore, ROS targeted therapy might be considered in clinical settings of HCC, because anti-TBX15 therapy was not established in both basic and clinical researches. Further study is still needed to discover the true mechanism for tumor malignancy of TBX15 focused on ROS concentration and its signaling pathway in each cancer. Also, those new findings might be one of clues for the discrepancy which several cancers indicated various TBX15 expressions.
Together, the present and previous findings support the importance of conducting further investigations to determine the role of TBX15 in cancer, particularly in tumor progression. However, several limitations must be considered in our study. First, the patient population was very small. Second, selection bias was possible, because of the retrospective one. Third, we did not determine the actual mechanism of TBX15 for tumor malignancy in basic experiment. Those issues should be solved in several cancers, to clarify the true function of TBX15 for tumor malignancy in each cancer. In conclusion, reduced expression of TBX15 may serve as a potential biomarker for predicting tumor progression and poor survival as well as a target for antitumor therapy in HCC.
Materials and Methods
Patients and microarray analysis of DNA methylation
We enrolled 15 patients with NBNC-HCC who underwent hepatectomy at Tokushima University Hospital. The Institutional Review Board of the University of Tokushima Graduate School authorized this study in advance (approved no.: 1800-3), and all patients provided written informed consent.
The DNeasy Blood & Tissue Kit (QIAGEN, USA) was used to extract DNA extraction. Genome-wide DNA methylation status was assessed using an Infinium HumanMethylation450 BeadChip Kit (Illumina, USA) containing 485,764 CpG sites. The DNA methylation profiles of autosomal CpGs in tumor tissue were compared with those of nontumor liver tissues. All analyses were performed according to the procedure provided by the manufacturer. The methylation score assigned to each CpG site is represented by a Beta-value calculated according to the normalized probe fluorescence intensity ratios of methylated to unmethylated CpG sites. Beta-values vary between 0 (fully unmethylated) and 1 (fully methylated). The mean methylation level of each gene (stratified according to upstream region/gene body and CpG island/shore/shelf/nonisland) was calculated as described previously [14].
Patient selection for gene-expression analysis
For the establishment of our study design, we firstly investigated and compared TBX15 mRNA expression among NBNC, HBV and HCV patients with HCC who underwent radical resection. Consequently, there were no significant difference TBX15 mRNA in both tumor and non-tumor tissue among those patients (Supplementary Figure 5). Therefore, in next validation study, we enrolled other 58 patients including NBNC, HBC and HCV patients with HCC without detectable distant organ metastases who underwent radical resection at Tokushima University Hospital, Japan, between April 2004 and March 2011. The 58 patients underwent liver resection as their initial treatment, and curative resection was confirmed according to the findings of pathological examinations. We excluded patients with intrahepatic cholangiocarcinoma such as mixed-type liver cancer. Tumor status, stage and curability were defined according to the Classification of Primary Liver Cancer by the Liver Cancer Study Group of Japan [40]. The 35 men and 13 women ranged in age from 40–81 years (mean, 66.8 years). The age, sex, liver function, hepatitis virus infection, and tumor pathological status were determined.
The patients were followed in the outpatient clinic according to a standard protocol. Briefly, patients were examined every 3 months during the first postoperative year and at least every 4 months thereafter. The levels of α-fetoprotein (AFP) and des-γ-carboxy prothrombin (DCP) were determined during each visit. Dynamic computed tomography of the abdomen was performed every 3 months during the first postoperative year. Recurrence was diagnosed according to computed tomography, magnetic resonance imaging, or both. The mean follow-up of patients was 85.3 months (range, 29.2–140.7 months).
Immunohistochemistry
Immunohistochemical staining was performed in surgical sections. Tissue specimens were fixed in 10% formaldehyde, embedded in paraffin, and cut into 4-μm-thick sections. We used a rabbit polyclonal anti-TBX15 antibody (1:100, ab115576; Abcam, Cambridge, UK) as a primary antibody. Tissue section was overlaid with 2nd antibody (Dako Real EnVision Kit/HRP, DAKO, Glostrup, Denmark). Peroxidase labeling was developed by incubating in 3,3´ deaminobenzidine tetrahydrochloride. Finally, nuclear counterstaining was completed using Meyer’s hematoxylin solution. Then, sections were dehydrated in alcohol and xylene and enclosed in synthetic resin.
Quantitative real-time reverse transcription-PCR (qRT-PCR)
The mRNA levels of TBX15 in tumor and nontumor liver tissues from 58 patients were evaluated using qRT-PCR. Total RNA was extracted using an RNeasy Mini Kit (74106, QIAGEN, Hilden, Germany). Reverse transcription-PCR was performed using a GeneAmp PCR System 9700 and a High Capacity cDNA Reverse Transcription Kit (4368813, Applied Biosystems, Carlsbad, CA, USA). Quantitative RT-PCR was performed using the StepOne Plus Real-Time PCR System and TaqMan Gene Expression Assay (Applied Biosystems). The assay identification number was TBX15 (Hs00537087_ml, Applied Biosystems), and TaqMan human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA served as the control (4326317E, Applied Biosystems). The mRNA expression level was calculated as the ratio of a test sample to that of GAPDH.
TCGA methylation analysis of HCC
We obtained the methylation array datasets of 371 hepatocellular patients from the Broad Institute’s Firehose. The array contained 59 probe sites in TBX15 genomic region. The probe sites were clustered into 3 groups by the methylation beta values, and the methylation score of TBX15 was calculated as the mean of the beta values in each cluster.
TCGA pan cancer analysis of mRNA expression
We obtained mRNA expression data of 371 hepatocellular patients from the Broad Institute’s Firehose (http://gdac.broadinstitute.org/). We also obtained mRNA expression data of 520 head and neck cancer patients (Head and neck ca.), 501 thyroid carcinoma patients (Thyroid ca.), 1093 breast cancer patients (Breast ca), 176 lung adenocarcinoma patients (Lung adenoca.), 184 esophageal cancer patients (Esophageal ca.), 415 gastric cancer patients (Gastric ca.), 285 colorectal cancer patients (Colorectal ca.), 36 cholangiocarcinoma patients (Cholangio ca.), 178 pancreatic cancer patients (Pancreas ca.), 533 kidney clear cell carcinoma patients (Clear cell renal ca.), 497 Prostate cancer patients (Prostate ca.), 304 cervical cancer patients (cervical), 176 Endometrial carcinoma (Endometrial ca.), 303 ovarian cancer patients (Ovarian ca.) from the Broad Institute’s Firehose. The mRNA expression data was normalized with quantile normalization [41].
Gene set enrichment analysis (GSEA)
The associations between TBX15 expression and previously defined gene sets were analyzed by gene set enrichment analysis (GSEA) using HCC expression profiles from TCGA dataset. The biologically defined gene sets were obtained from the Molecular Signatures Database v5.2 (http://software.broadinstitute.org/gsea/msigdb/index.jsp).
Statistical analysis
Statistical analyses were performed using JMP 11.2.0. software (SAS, Campus Drive, Cary, NC, USA). Data are presented as the median ± standard deviation (SD). Statistical significance was defined as p < 0.05. Cutoff value of TBX15 mRNA expression was assessed using Youden’s J statistic with ROC curve. The significance of the relationships between TBX15 expression and clinicopathological variables were analyzed using the Mann–Whitney and Fisher’s exact tests. Comparisons between more than 3 groups were calculated using one-way ANOVA with the Turkey-Kramer’s test. Survival curves were calculated using the Kaplan–Meier method, and the differences were compared using the log-rank test. To identify independent factors that influenced overall and disease-free survival, variables identified as significant by univariate analysis were included in the multivariate analysis employing the Cox proportional hazards model.
Abbreviations
HCC: hepatocellular carcinoma; miRNA: microRNA; LINE-1: long interspersed nuclear element-1; NBNC-HCC: HCC patients without HCV and HBV; TBX15: T-Box 15; NL normal liver; CH: chronic hepatitis; LC: liver cirrhosis; AFP: α-fetoprotein; DCP: des-γ-carboxy prothrombin; ROC: receiver operating characteristics; MMP: matrix metalloproteinases; VEGF: vascular endothelial growth factor.
ACKNOWLEDGMENTS
This study was a cooperative research on Japanese Society of Gastrointestinal Carcinogenesis.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This research was partly supported by Japan Agency for Medical Research and Development (AMED), Project code: 15fk0210018h0003, JP18fk0210001, JP20fk0210048, during the conduct of the study.
References
1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012; 379:1245–55. https://doi.org/10.1016/S0140-6736(11)61347-0. [PubMed].
2. Venook AP, Papandreou C, Furuse J, de Guevara LL. The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist. 2010; 15:5–13. https://doi.org/10.1634/theoncologist.2010-S4-05. [PubMed].
3. Wubetu GY, Utsunomiya T, Ishikawa D, Yamada S, Ikemoto T, Morine Y, Iwahashi S, Saito Y, Arakawa Y, Imura S, Kanamoto M, Zhu C, Bando Y, et al. High STAT4 expression is a better prognostic indicator in patients with hepatocellular carcinoma after hepatectomy. Ann Surg Oncol. 2014; 21:S721–728. https://doi.org/10.1245/s10434-014-3861-9. [PubMed].
4. Imura S, Tovuu LO, Utsunomiya T, Morine Y, Ikemoto T, Arakawa Y, Kanamoto M, Iwahashi S, Saito Y, Takasu C, Yamada S, Ishikawa D, Bando Y, et al. Role of Fbxw7 expression in hepatocellular carcinoma and adjacent non-tumor liver tissue. J Gastroenterol Hepatol. 2014; 29:1822–1829. https://doi.org/10.1111/jgh.12623. [PubMed].
5. Davaadorj M, Imura S, Saito YU, Morine Y, Ikemoto T, Yamada S, Takasu C, Hiroki T, Yoshikawa M, Shimada M. Loss of SFRP1 Expression Is Associated with Poor Prognosis in Hepatocellular Carcinoma. Anticancer Res. 2016; 36:659–664. [PubMed].
6. Iwahashi S, Shimada M, Utsunomiya T, Imura S, Morine Y, Ikemoto T, Takasu C, Saito Y, Yamada S. Epithelial-mesenchymal transition-related genes are linked to aggressive local recurrence of hepatocellular carcinoma after radiofrequency ablation. Cancer Lett. 2016; 375:47–50. https://doi.org/10.1016/j.canlet.2016.02.041. [PubMed].
7. Iizuka N, Oka M, Yamada-Okabe H, Nishida M, Maeda Y, Mori N, Takao T, Tamesa T, Tangoku A, Tabuchi H, Hamada K, Nakayama H, Ishitsuka H, et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. Lancet. 2003; 361:923–929. https://doi.org/10.1016/S0140-6736(03)12775-4. [PubMed].
8. Yoshioka S, Takemasa I, Nagano H, Kittaka N, Noda T, Wada H, Kobayashi S, Marubashi S, Takeda Y, Umeshita K, Dono K, Matsubara K, Monden M. Molecular prediction of early recurrence after resection of hepatocellular carcinoma. Eur J Cancer. 2009; 45:881–889. https://doi.org/10.1016/j.ejca.2008.12.019. [PubMed].
9. Kurokawa Y, Matoba R, Takemasa I, Nagano H, Dono K, Nakamori S, Umeshita K, Sakon M, Ueno N, Oba S, Ishii S, Kato K, Monden M. Molecular-based prediction of early recurrence in hepatocellular carcinoma. J Hepatol. 2004; 41:284–291. https://doi.org/10.1016/j.jhep.2004.04.031. [PubMed].
10. Utsunomiya T, Ishikawa D, Asanoma M, Yamada S, Iwahashi S, Kanamoto M, Arakawa Y, Ikemoto T, Morine Y, Imura S, Ishibashi H, Takasu C, Shimada M. Specific miRNA expression profiles of non-tumor liver tissue predict a risk for recurrence of hepatocellular carcinoma. Hepatol Res. 2014; 44:631–638. https://doi.org/10.1111/hepr.12164. [PubMed].
11. Kim JW, Ye Q, Forgues M, Chen Y, Budhu A, Sime J, Hofseth LJ, Kaul R, Wang XW. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology. 2004; 39:518–527. https://doi.org/10.1002/hep.20053. [PubMed].
12. Weisenberger DJ. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J Clin Invest. 2014; 124:17–23. https://doi.org/10.1172/JCI69740. [PubMed].
13. Zhu C, Utsunomiya T, Ikemoto T, Yamada S, Morine Y, Imura S, Arakawa Y, Takasu C, Ishikawa D, Imoto I, Shimada M. Hypomethylation of long interspersed nuclear element-1 (LINE-1) is associated with poor prognosis via activation of c-MET in hepatocellular carcinoma. Ann Surg Oncol. 2014; 21:S729–735. https://doi.org/10.1245/s10434-014-3874-4. [PubMed].
14. Utsunomiya T, Shimada M, Morine Y, Tajima A, Imoto I. Specific molecular signatures of non-tumor liver tissue may predict a risk of hepatocarcinogenesis. Cancer Sci. 2014; 105:749–754. https://doi.org/10.1111/cas.12431. [PubMed].
15. Ammerpohl O, Pratschke J, Schafmayer C, Haake A, Faber W, von Kampen O, Brosch M, Sipos B, von Schönfels W, Balschun K, Röcken C, Arlt A, Schniewind B, et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma. Int J Cancer. 2012; 130:1319–1328. https://doi.org/10.1002/ijc.26136. [PubMed].
16. Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, Santella RM. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics. 2013; 8:34–43. https://doi.org/10.4161/epi.23062. [PubMed].
17. Showell C, Binder O, Conlon FL. T-box genes in early embryogenesis. Dev Dyn. 2004; 229:201–218. https://doi.org/10.1002/dvdy.10480. [PubMed].
18. Takashima Y, Suzuki A. Regulation of organogenesis and stem cell properties by T-box transcription factors. Cell Mol Life Sci. 2013; 70:3929–3245. https://doi.org/10.1007/s00018-013-1305-5. [PubMed].
19. Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest. 2010; 120:533–544. https://doi.org/10.1172/JCI38379. [PubMed].
20. Papaioannou VE. The T-box gene family: emerging roles in development, stem cells and cancer. Development. 2014; 141:3819–3833. https://doi.org/10.1242/dev.104471. [PubMed].
21. Sarkar D, Shields B, Davies ML, Müller J, Wakeman JA. BRACHYURY confers cancer stem cell characteristics on colorectal cancer cells. Int J Cancer. 2012; 130:328–337. https://doi.org/10.1002/ijc.26029. [PubMed].
22. Lu J, Li XP, Dong Q, Kung HF, He ML. TBX2 and TBX3: the special value for anticancer drug targets. Biochim Biophys Acta. 2010; 1806:268–274. https://doi.org/10.1016/j.bbcan.2010.07.001. [PubMed].
23. Renard CA, Labalette C, Armengol C, Cougot D, Wei Y, Cairo S, Pineau P, Neuveut C, de Reyniès A, Dejean A, Perret C, Buendia MA. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 2007; 67:901–910. https://doi.org/10.1158/0008-5472.CAN-06-2344. [PubMed].
24. He ML, Chen Y, Peng Y, Jin D, Du D, Wu J, Lu P, Lin MC, Kung HF. Induction of apoptosis and inhibition of cell growth by developmental regulator hTBX5. Biochem Biophys Res Commun. 2002; 297:185–192. https://doi.org/10.1016/s0006-291x(02)02142-3. [PubMed].
25. Yu J, Ma X, Cheung KF, Li X, Tian L, Wang S, Wu CW, Wu WK, He M, Wang M, Ng SS, Sung JJ. Epigenetic inactivation of T-box transcription factor 5, a novel tumor suppressor gene, is associated with colon cancer. Oncogene. 2010; 29:6464–6474. https://doi.org/10.1038/onc.2010.370. [PubMed].
26. Lausch E, Hermanns P, Farin HF, Alanay Y, Unger S, Nikkel S, Steinwender C, Scherer G, Spranger J, Zabel B, Kispert A, Superti-Furga A. TBX15 mutations cause craniofacial dysmorphism, hypoplasia of scapula and pelvis, and short stature in Cousin syndrome. Am J Hum Genet. 2008; 83:649–655. https://doi.org/10.1016/j.ajhg.2008.10.011. [PubMed].
27. Dikoglu E, Simsek-Kiper PO, Utine GE, Campos-Xavier B, Boduroglu K, Bonafé L, Superti-Furga A, Unger S. Homozygosity for a novel truncating mutation confirms TBX15 deficiency as the cause of Cousin syndrome. Am J Med Genet A. 2013; 161A:3161–3165. https://doi.org/10.1002/ajmg.a.36173. [PubMed].
28. Li WX, He K, Tang L, Dai SX, Li GH, Lv WW, Guo YC, An SQ, Wu GY, Liu D, Huang JF. Comprehensive tissue-specific gene set enrichment analysis and transcription factor analysis of breast cancer by integrating 14 gene expression datasets. Oncotarget. 2017; 8:6775–6786. https://doi.org/10.18632/oncotarget.14286. [PubMed].
29. Gozzi G, Chelbi ST, Manni P, Alberti L, Fonda S, Saponaro S, Fabbiani L, Rivasi F, Benhattar J, Losi L. Promoter methylation and downregulated expression of the TBX15 gene in ovarian carcinoma. Oncol Lett. 2016; 12:2811–2819. https://doi.org/10.3892/ol.2016.5019. [PubMed].
30. Gao F, Liang H, Lu H, Wang J, Xia M, Yuan Z, Yao Y, Wang T, Tan X, Laurence A, Xu H, Yu J, Xiao W, et al. Global analysis of DNA methylation in hepatocellular carcinoma by a liquid hybridization capture-based bisulfite sequencing approach. Clin Epigenetics. 2015; 7:86. https://doi.org/10.1186/s13148-015-0121-1. [PubMed].
31. Zheng Y, Huang Q, Ding Z, Liu T, Xue C, Sang X, Gu J. Genome-wide DNA methylation analysis identifies candidate epigenetic markers and drivers of hepatocellular carcinoma. Brief Bioinform. 2018; 19:101–108. https://doi.org/10.1093/bib/bbw094. [PubMed].
32. Kron K, Liu L, Trudel D, Pethe V, Trachtenberg J, Fleshner N, Bapat B, van der Kwast T. Correlation of ERG expression and DNA methylation biomarkers with adverse clinicopathologic features of prostate cancer. Clin Cancer Res. 2012; 18:2896–2904. https://doi.org/10.1158/1078-0432.CCR-11-2901. [PubMed].
33. Arribas J, Giménez E, Marcos R, Velázquez A. Novel antiapoptotic effect of TBX15: overexpression of TBX15 reduces apoptosis in cancer cells. Apoptosis. 2015; 20:1338–1346. https://doi.org/10.1007/s10495-015-1155-8. [PubMed].
34. Arribas J, Cajuso T, Rodio A, Marcos R, Leonardi A, Velázquez A. NF-κB Mediates the Expression of TBX15 in Cancer Cells. PLoS One. 2016; 11:e0157761. https://doi.org/10.1371/journal.pone.0157761. [PubMed].
35. Yuan J, Takeuchi M, Negishi M, Oguro H, Ichikawa H, Iwama A. Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia. 2011; 25:1335–1343. https://doi.org/10.1038/leu.2011.85. [PubMed].
36. Farin HF, Bussen M, Schmidt MK, Singh MK, Schuster-Gossler K, Kispert A. Transcriptional repression by the T-box proteins Tbx18 and Tbx15 depends on Groucho corepressors. J Biol Chem. 2007; 282:25748–2559. https://doi.org/10.1074/jbc.M703724200. [PubMed].
37. Buscarlet M, Stifani S. The ‘Marx’ of Groucho on development and disease. Trends Cell Biol. 2007; 17:353–361. https://doi.org/10.1016/j.tcb.2007.07.002. [PubMed].
38. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017; 36:1461–1473. https://doi.org/10.1038/onc.2016.304. [PubMed].
39. Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Khan MA, Sethi G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules. 2019; 9:735. https://doi.org/10.3390/biom9110735. [PubMed].
40. The Liver Cancer Study Group of Japan: The General Rules for the Clinical and Pathological Study of Primary Liver Cancer, ed 5, Rivised Version. Tokyo, Kanehara. 2009.
41. Bolstad BM, Irizarry RA, Astrand M, Speed T. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics. 2003; 19:185–193. https://doi.org/10.1093/bioinformatics/19.2.185. [PubMed].