
Introduction
Sleep disorders in general, and more particularly obstructive sleep apnea (OSA), have been associated with accelerated cancer progression, aggressiveness, and mortality [1, 2]. OSA is a highly prevalent disorder that occurs in all age groups and both sexes with an estimated prevalence of 4 % to 10 % in adults [3]. This disorder is characterized by repetitive obstructions of the upper airway during sleep that result in intermittent hypoxia (IH), increased inspiratory efforts, repetitive arousals from sleep to reestablish respiration leading to sleep fragmentation (SF) and episodic elevations in the concentrations of blood CO2 [4, 5]. In the last 2 decades, it has become apparent that numerous and serious end-organ morbidities are associated with OSA and virtually affect all organ systems, including cancer [1-10]. The mechanisms potentially leading to the cardiovascular [11-14], cognitive [15-17], and metabolic [18-20] morbidities of OSA have been extensively studied in human and animal models, with IH being proposed as a major determinant of the processes involving tumor invasion and metastasis [21, 22]. Indeed, we have recently shown that tumor growth, cell proliferation, migration and invasiveness are all increased in murine models of OSA, being selectively exposed to either IH [23] or SF [24].
Fragmented DNA is released into the bloodstream during the growth and expansion of tumors [25, 26]. Increased plasma cirDNA concentrations have been observed in several types of cancer, leading to the assumption that the concentrations of plasma cirDNA may serve as biomarker for early cancer detection and diagnosis, or for prognosis by monitoring or potentially predicting the response to therapies [27-31]. Increased plasma cirDNA concentration have also been reported in many other non-oncogenic pathologies as well (e.g., trauma, sepsis, myocardial infarction, etc.; for review see [32]). One major challenge in the assessment of cirDNA is the complex origin of the nucleic acids that are present in circulation. Nucleic acids are released from tumors as well as from normal cells through several cellular mechanisms such as apoptosis, necrosis, exosome-mediated release, and shedding from macrophages after the absorption of necrotic cells [33-36]. The use of animal models injected with tumor cells (“xenografts”) enables the concomitant study of cirDNA in bodily fluids and tissue samples, and has been suggested as a useful model system to examine the origin and variations of cirDNA upon experimental interventions [37-40]. Notably, increasing concentrations of cirDNA in OSA patients have been identified and were positively correlated with disease severity, suggesting that cirDNA may reflect pathogenic changes that may be relevant to disease severity or to its associated morbidities such as cancer [41].
As tumors develop, cells undergo major epigenetic changes [42]. Epigenetic aberrations can be also used to detect and characterize malignant growth [43], particularly by characterizing the scope and magnitude of epigenetic DNA alterations in plasma, serum and other bodily fluids [44]. In this study, we describe the first comprehensive cirDNA analysis in cancer associated to intermittent hypoxia in an animal model, as a hallmark of OSA in humans. Using xenografted mice exposed to IH and control conditions, we here show a significant increase on plasma cirDNA amounts, which is associated with the severity of the tumor. Moreover, we provide a large-scale epigenetic profiling of plasma cirDNA in xenografted animals exposed to intermittent hypoxia. We used system biology approaches to genomic variations associated with IH exposure. Lastly, we applied single-locus qPCR strategies to study candidate loci in plasma cirDNA and genomic DNA from tissue and blood samples.
Results
Tumor growth and invasion in xenografted mice exposed to intermittent hypoxia
Tumor size and weight in the XenoIH group were higher compared to the XenoRA group (Figures 1A and 1B, respectively). We found significant differences for tumor size (median sizes were 745.78 mm3 and 2211.43 mm3 for XenoRA and XenoIH, respectively; t = -2.66, df = 8.61, p-value = 0.026; Welch Two Sample t-test), and tumor weight (median weights were 0.72 mg and 1.45 mg for XenoRA and XenoIH, respectively; t = -2.27, df = 13.16, p-value = 0.040). Invasion towards the skeletal muscle was observed in all tumors in the XenoIH group (n=8), but only in 3 out of 8 tumors in the XenoRA group (p=0.025, Fisher’s Exact test) (Figure 1C).
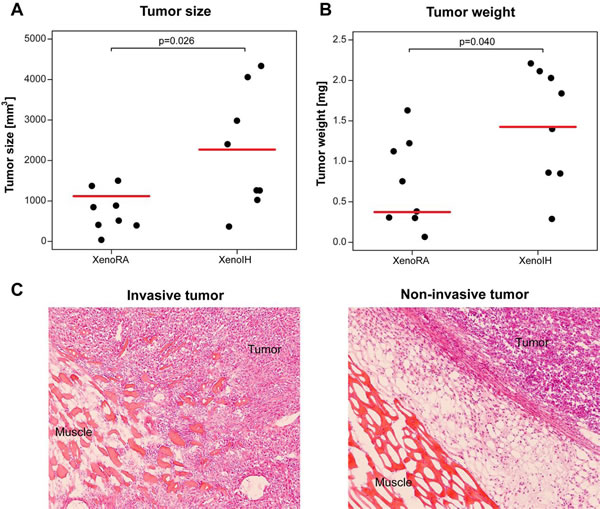
Figure 1: Effects of IH exposure on xenografted tumors. Tumors in animals exposed to IH grew significantly larger than those in control mices. Y-axis depicts the volume (panel A) and weight (panel B) of the tumors assessed at the time of sacrifice (4 weeks after injection). Horizontal red lines correspond to the median size for each group. Panel C illustrates tumor invasiveness as observed upon IH exposures (left panel) and non- invasive tumor, as observed mainly in RA conditions (right panel).
Quantification of circulating DNA (cirDNA) in plasma
Mean cirDNA amounts were highest in IH-exposed mice, particularly in XenoIH mice, with a significant group effect (F (3, 32) = 6.89, p=0.001; one-way ANOVA) (Figure 2A). Tukey post-hoc comparisons indicated significant differences between the XenoRA and XenoIH groups (M=-510.62, 95% CI (-940.83, -80.42), p=0.015). Pairwise comparison showed that mice bearing the tumors (XenoRA and XenoIH groups together, mean cirDNA concentration = 591.28 ng/mL plasma) had significantly higher plasma cirDNA concentration than those not carrying the tumors (CtrlRA and CtrlIH together, mean cirDNA concentration = 271.44 ng/mL plasma) (t = -2.47, df = 21.05, p-value = 0.022, Welch Two sample t-test). Exposure to IH resulted in increased plasma cirDNA concentrations in xenografted mice (mean cirDNA concentrations: XenoIH=846.59 ng/mL plasma, XenoRA=335.96 ng/mL plasma; t = -2.53, df = 7.30, p-value = 0.038), and in mice not carrying the tumors, although the latter differences were not statistically significant (mean cirDNA concentrations: CtrlIH=352.76 ng/mL plasma, CtrlRA=190.12 ng/mL plasma; t = -1.59, df = 12.14, p-value = 0.138).
Significant correlations emerged between the concentration of plasma cirDNA and tumor weight (R2=0.580, p=0.029; Pearson’s product-moment correlation test) (Figure 2B) and tumor size (R2=0.765, p=0.001) (Figure 2C), but not with the weight of the mice (R2=-0.134, p=0.437) (Figure S1). Furthermore, we found that mice bearing invasive tumors (mean concentration 718.65 ng/mL plasma) had significantly higher plasma cirDNA concentrations than those bearing non-invasive tumors (mean concentration 311.06 ng/mL plasma) (t = 2.53, df = 10.79, p-value = 0.028; Welch Two Sample t-test) (Figure 2D).
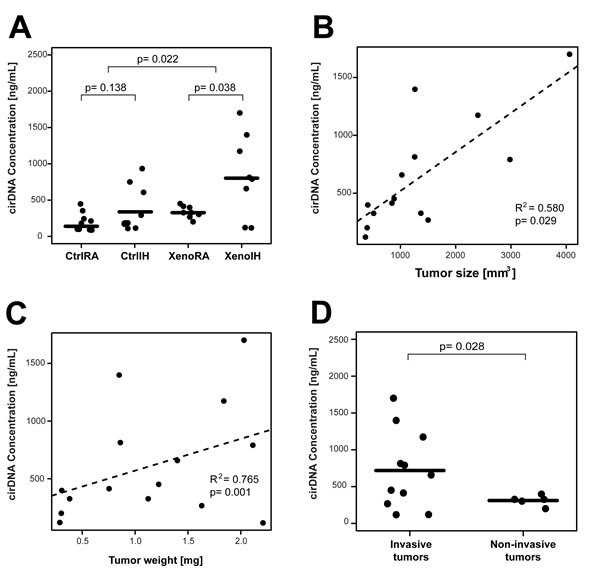
Figure 2: Plasma cirDNA concentration in xenografted and control mice under IH and RA conditions. A) Mouse bearing the tumors showed significantly increased plasma cirDNA amounts. IH exposures are associated with elevated plasma cirDNA concentrations in xenografted (XenoIH and XenoRA groups) and control (CtrlIH and CtrlRA groups). Horizontal lines correspond to the mean size for each group. B) and C) Plasma cirDNA showed a significant positive correlation with tumor size (Panel B) and weight (Panel C). Dashed lines depict the trend line for each correlation. D) Plasma cirDNA is significantly elevated in invasive tumors compared to non-invasive tumors. Plasma cirDNA concentration were assessed by qPCR. p-values < 0.05 are considered significant (t-test).
Epigenomic profiling of plasma cirDNA
We selected 3 samples that best represented each group according to the biological variables (Table S1) and interrogated these samples using microarrays. Data quality control showed that all microarray results were robust, with equivalent densities of signal intensity. All microarrays were included in posterior analyses (n=3 per group) (Figure S2).
Of the 4,106,240 mouse-genome features on the microarray, 46,589 features showed statistically significant differences (p-value<0.05; One-way ANOVA) and fold changes higher than 2 (Figure 3A). Analysis of chromosomal distribution showed that features showing differential microarray signal were overrepresented on chromosomes 7 (p=0.009, OR=0.96, 95% CI = 0.92-0.98, Fisher’s exact test), 13 (p=0.005, OR=1.07, 95% CI = 1.02-1.12), 14 (p=0.04, OR=1.05, 95% CI = 1.00-1.10) and X (p=0.002, OR=0.93, 95% CI = 0.88-0.97) (Figure 2B, Table S2). Figure 2C shows the distribution of the candidate regions across those chromosomes. We identified two chromosomal bands containing significantly overrepresented clusters of features with decreased microarray signal in the Xeno IH group compared to the XenoRA group: 7qB3 (p=0.002, OR=0.00, 95% CI = 0.00-0.50) and XqF5 (p=0.039, OR=0.00, 95% CI=0.00-1.13)(Table S2).
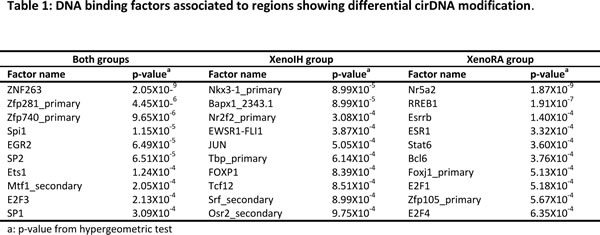
To detect genomic regions showing differential cirDNA modification, adjacent probes showing equivalent differential cirDNA modification were grouped. We identified 2,094 differentially modified regions, with 1,053 and 1,041 regions showing higher cDNA modification in XenoIH and XenoRA groups, respectively (Table S3). We did not detect significant differences in the length (996.25 ± 320.84 and 1002.27 ± 578.88 nucleotides, respectively) or number of features (26.51 ± 8.82 and 26.61 ± 12.66 features, respectively) between regions displaying higher cirDNA modifications in the XenoIH or XenoRA groups (p=0.557; Wilcoxon rank sum test) (Figure 3A and 3B). Among the differentially modified regions, 1,568 were associated to annotated RNA transcripts, with 1,406 associated to mRNA transcripts and 107 to non-coding RNAs (ncRNA). Regions were mapped associated to the Transcription Start Site (TSS) (n=231; association was defined as TSS ± 2 kb) or to the gene coding portion (n=1,337) of the gene (Table S3). We found significant differences in the distribution of the distance to the TSS between regions with higher cirDNA modifications in the XenoIH (n=723; mean distance to TSS=48.6 ± 84.4 kbp) and the XenoRA groups (n=845; mean distance to TSS=68.8 ± 115.0 kbp) (p=6.41 x 10-6; Wilcoxon rank sum test) (Figure 4C). The number of TSS-associated regions was significantly overrepresented among the regions with higher cirDNA modification in the XenoIH group (n=131/723) compared to those in the XenoRA group (n=100/845) (p=5.8 x 10-4 OR=1.65, 95% CI = 1.23-2.21, Fisher’s exact test). To identify the putative regulatory elements located within those regions, we selected the top 10 motifs in each group and compared them against databases of DNA binding factors. We identified 53 DNA binding factors, of which 13 were unique for the regions with high cirDNA modification in the XenoIH group, 28 for those in the XenoRA group and 15 were shared between the candidate lists. Table 1 shows the top 10 transcription binding site regions found in each group.
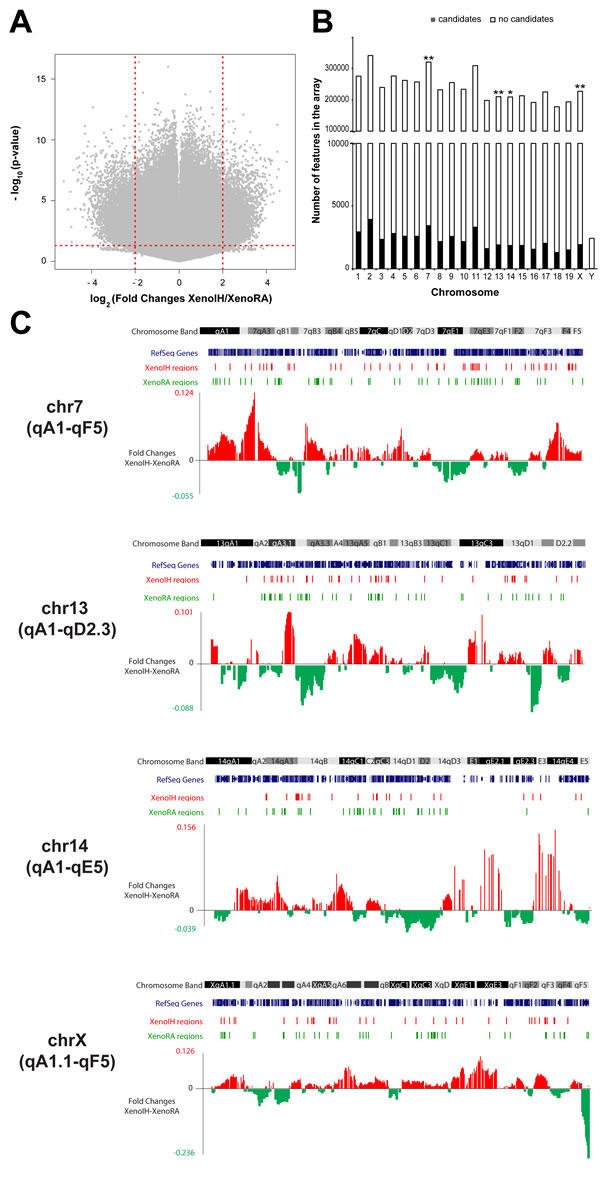
Figure 3: Large-scale cirDNA modification profiling using promoter microarrays. A) Volcano plot of microarray data. The x-axis represents fold changes differences between the groups, with coefficients expressed in the log2 scale. Samples with increased microarray signals in XenoIH and XenoRA groups had positive and negative coefficients, respectively. The y-axis represents the -log10-transformed p-values. The horizontal dashed red line depicts the cutoff value for the p-value (-log10(p< 0.05)=1.3). The vertical dashed red lines depict the cutoff vallues for the fold changes (log2(4)=2). B) Chromosomal distribution of microarray features showing differential cirDNA modification between the groups. Stacked bars show the number of features that showed (black) or not showed (white) differential cirDNA modification per chromosome. Significance level have been determined by Fisher test (**: p<0.01; *: p<0.05) C) Genome browser images showing the distribution of differential cirDNA modification in chromosomes 7, 13, 14 and X. Chromosome bands are shown in black, grey and white scale. RefSeq genes are shown in blue. Regions of differential cirDNA modification in XenoIH and XenoRA groups are shown as red and green bars, respectively. Red and green peaks correspond to fold changes higher in the XenoIH and XenoRA groups, respectively.
We further assessed the Gene Ontology (GO) groups overrepresented in regions displaying differential cirDNA modifications (Table S4). Among the regions showing higher cirDNA modification in the XenoIH group, we observed enrichment in categories associated to glutamate metabolism (i.e. GO:0014065~phosphoinositide 3-kinase cascade; 45.9 FC) and transport mechanisms (i.e. GO:0051938~L-glutamate import; 23.0 FC; GO:0032983~kainate selective glutamate receptor complex; 23.2 FC). Conversely, in regions with cirDNA modification that were lower in XenoIH, we observed high enrichment in categories associated to establishment of location (i.e. GO:0010159~specification of organ position; 24.8 FC), cyclic nucleotide metabolism (i.e. GO:0004115~3’,5’-cyclic-AMP phosphodiesterase activity; 18.7 FC) and cellular organization (i.e. GO:0016010~dystrophin-associated glycoprotein complex; 9.5 FC; GO:0005605~basal lamina with 8.8 FC).
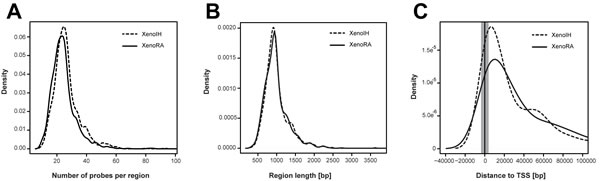
Figure 4: Characterization of regions showing differential cirDNA modifications. Adjacent microarray features showing equivalent differences between the XenoIH and XenoRA groups were combined, according to the MAT algorithm. A) Density plot of the number of features per region in each group. B) Density plot of the length of the candidate region in each group. C) Distribution of the distance to TSS in each group. The distance from the begining of each region to the closest TSS are shown in the X-axis. Gray-shaded area depicts the segment designed as “TSS-associated”(± 2 kbp from TSS). Dashed and solid lines represent the XenoIH and XenoRA groups, respectively.
Single locus DNA modification analysis of candidate regions
We selected 6 loci (Table 2, Figure 5A) displaying significant differential cirDNA modification in the microarray analysis (Rab3a, Atp6v0c, B4galnt1, Slc1a1, Ttl and Krt15) and whose corresponding genes have been reported as associated to lung cancer phenotypes (Table 2). The qMSRE-PCR assays contained, at least, one site for the restriction enzyme used in the microarray analysis. We observed high intragroup variation in the cirDNA modification enrichment across all studied loci. Noteworthy, while the MATscores show the cumulative DNA modification effects at the restriction sites of the three enzymes across extended DNA fragments captured by adjacent probes in the tiling microarray, qMSRE-PCR assays cover much shorter DNA fragments (around 100 bp) enabling the assessment of DNA modification only at one restriction site. Therefore, the precise CG positions driving the cirDNA modifications observed by microarray might have not been targeted in the verification effort. Despite the biological and methodological caveats, we detected one locus (Rab3a) showing significant cirDNA modification differences between the XenoRA and XenoIH groups (mean enrichment: XenoRA=0.7 ± 0.3 FC, XenoIH=9.83 ± 5.2 FC; p=0.008, F-test) (Table 2; Figure 5A).
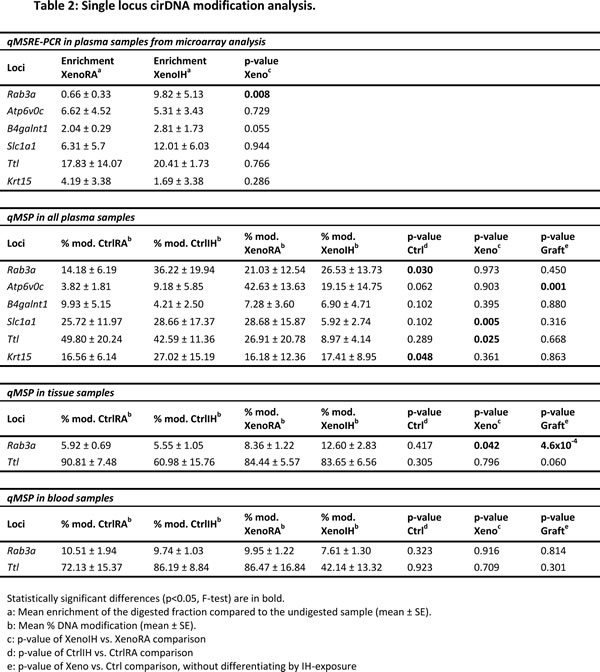
Next, we extended the analysis to all mice included in the study. We quantified the cirDNA modification in the 6 loci in plasma cirDNA (Table 2 and Figure 5B) as well as genomic DNA samples from tumor tissues and peripheral blood cells (PBC) (Table 2 and Figures 5C and D). Quantitative methylation specific PCR (qMSP) assays contained at least one restriction site for the enzymes used in the microarray and qMSRE-PCR assays. Similarly to the observations by qMSRE-PCR, intragroup variation in plasma cirDNA samples was high. We detected two loci (Slc1a1 and Ttl) showing significant cirDNA modification differences between the groups (Slc1a1 locus: mean cirDNA modification: XenoRA= 28.7 ± 15.9 %, XenoIH= 5.9 ± 2.8 %; p=0.005; Ttl locus: mean cirDNA modification: XenoRA= 26.9 ± 20.8 %, XenoIH= 9.0 ± 4.1 %; p=0.025) (Figure 5B). We quantified the DNA modification values in two loci (Rab3a and Ttl; Table 2 and Figure 5C and D, respectively) in genomic DNA from tissue and PBC samples. The observed intragroup variation was lower than in plasma cirDNA. For the Rab3a locus, we detected significant DNA modification differences in tissue genomic DNA concordant with those observed in plasma cirDNA (mean cirDNA modification: XenoRA= 8.4 ± 1.2 %, XenoIH= 12.6 ± 2.8 %; p=0.042), but no differences were detected in PBC genomic DNA (mean cirDNA modification: XenoRA= 9.9 ± 1.2 %, XenoIH= 7.6 ± 1.3 %; p=0.916) (Figure 5C). Conversely, DNA modification percentages in the Ttl locus were equivalent for the XenoRA and XenoIH groups in tissue genomic DNA (mean cirDNA modification: XenoRA= 84.4 ± 5.6 %, XenoIH= 83.6 ± 6.5 %; p=0.796), but DNA modification in PBC genomic DNA was higher in XenoRA than in XenoIH (mean cirDNA modifications: XenoRA= 86.5 ± 16.8 %, XenoIH= 42.1 ± 13.3 %; p=0.709) in concordance with plasma cirDNA results, though the evident differences did not reach statistical significance (Figure 5D).
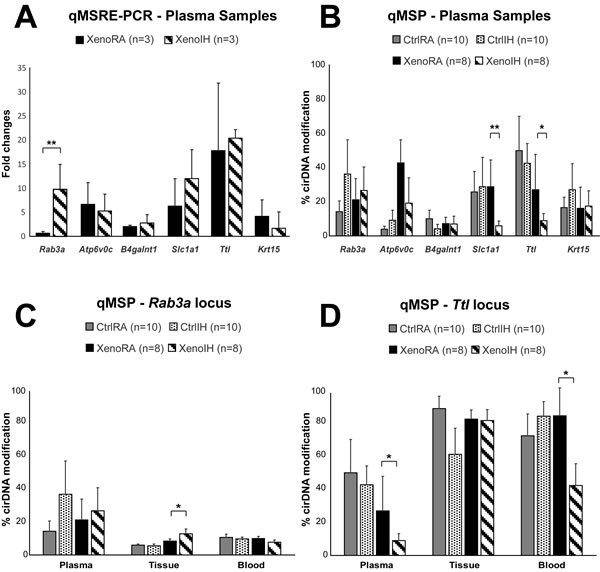
Figure 5: Single locus cirDNA modification analysis. A) Plasma cirDNA modifications were assessed in six loci by qMSRE-PCR in the same samples used in the microarray. Y-axis shows the XenoIH/XenoRA fold changes. B) cirDNA modification by qMSP results in all plasma samples. Y-axis represents the % of cirDNA modification. C) and D) qMSP results in plasma, tissue and blood samples for the Rab3a and Ttl loci, respectively. Solid black, dashed bars, solid gray and dotted bars represent the XenoRA, XenoIH, CtrlRA and CtrlIH groups, respectively. The height of the bars corresponds to the mean values. Error bars are SE. Significance level was determined by F-test (**: p<0.01; *: p<0.05).
Discussion
In this study, we combined the benefits of a murine xenograft model with sensitive detection using real-time PCR methods and epigenetic profiling using high-density microarrays to study cirDNA in tumors exposed to IH, a hallmark of OSA.
Although elevated amounts of plasma cirDNA have been widely reported in the majority of cancer types, their application as biomarkers has been questioned, primarily because of the high inter-patient variation within cases and controls [45, 46]. We found that the amount of cirDNA in plasma was significantly increased in xenografted mice when compared to those not bearing the tumors (Figure 2A). We observed some intra-group variation, even when our experimental setup enabled the control of phenotypic variables that can covariate with shedding of DNA to circulation (i.e. age, sex, genetic background, etc.) or technical variables for the cirDNA handling (i.e. time to cirDNA isolation and cirDNA isolation batches), which could not be readily controlled in many studies using clinical samples. When analyzing possible covariates, we only found significant correlation of plasma cirDNA concentration with tumor size, weight and invasiveness, but not with the weight of the animal bearing the tumor or technical parameters. Our findings suggest that inter-individual variation in cirDNA shedding might be rather related to biological features of the tumor upon IH exposures. In particular, we found that exposure to IH during sleep was associated to increased plasma cirDNA in both xenografted and control mice (Figure 2A). These findings concur with reports elevated plasma cirDNA amount in OSA patients [41]. Furthermore, we have recently reported that two of the major components of OSA – sleep fragmentation and IH – promote more aggressive tumor biological characteristics [23, 24]. While the results of the present study consolidate these previous findings, further studies with clinical samples are warranted to investigate a putative biomarker application for cirDNA quantitation among cancer patients with and without concurrent sleep disorders, particularly considering the strong emerging epidemiological evidence linking adverse cancer outcomes in the presence of OSA [1, 2, 47].
Epigenetic DNA modifications (mainly, cytosine methylation and hydroxylmethylation), histone modifications and non-coding RNAs have been demonstrated as fundamental molecular mechanisms for the establishment of oncogenic phenotypes and tumor progression [42]. Furthermore, sensitive detection of epigenetic marks in cirDNA have been shown as potential biomarkers [44] and some of them are already being applied in clinical diagnostic assays (e.g., SEPT9 DNA methylation for early detection and screening of colorectal cancer [48, 49]). Large-scale cirDNA modification analysis using high density microarrays or deep-sequencing enables the evaluation of thousands of loci in parallel to generate molecular signatures for diagnostics [50], but also enables the evaluation of variation at the epigenomic level. Here, we identified more than 2,000 regions showing differential cirDNA modifications between xenografted tumors exposed to IH or RA conditions (Figure 3A and Table S3). These regions were associated with more than 1,400 annotated mRNA transcripts and over 100 ncRNAs, suggesting a major role of epigenetic processes in the modulation of the tumor phenotype by IH-exposure.
We consequently applied system biology approaches to identify possible associations of these regions to major molecular mechanisms of genome regulation. Regions with higher cirDNA modification upon IH exposure were preferentially associated with regulatory elements proximal to TSS, while regions with decreased cirDNA modification were preferentially associated to distal regulatory elements (Figure 4C and Table 1). Several molecular models have been proposed in which the interplay between DNA modification and the binding of transcription factors (TFs) at promoter and enhancer regions regulate the expression of the cognate genes (reviewed in [51]). Our findings support further investigation on the precise epigenetic regulation of TSS-associated and distal regulatory elements in sleep disorders. Gene ontology analysis revealed an enrichment of genes involved in glutamate metabolism and transport among genes associated to regions gaining cirDNA modification upon IH-exposure during sleep. Concordant with our findings, it has been shown that cells grown under hypoxia depend on the reductive carboxylation of glutamine-derived α-ketoglutarate for de novo lipogenesis and tumor growth [52]. Moreover, it has been demonstrated that the glutamate-transporter SLC1A5 is essential for cell growth in lung cancer [53] and that the intake of L-glutamine regulates mTOR signalling [54].
We detected clusters of variation of microarray signals in the XenoIH group in chromosomes 7, 13, 14 and X (Figures 2B and 2C). In the microarray-based method, decrease of signals in large regions may be due to a loss of cirDNA modification, but also to genome loss by deletions of chromosome re-arrangements [50]. Partial losses in chromosomes 7 and 14 have been reported in mouse lung adenocarcinoma cell lines [55]. By analyzing signal clustering in chromosomal bands, we identified regions of significant loss of signal at 7qB3 and XqF5 loci. Although there is no information available on chromosomal rearrangements in murine lung tumor cell lines, synteny analysis revealed orthologous regions in human (15q15.3 and Xp22.2 for 7qB3 and XqF5, respectively), which are associated to malignant phenotypes in the lung. For example, loss of heterozygosity (LOH) at 15q15 has been reported in lung carcinomas [56]. In turn, the Xq22 region harbors several putative tumors suppressor genes which are inactivated by mutations in lung cancer cell lines and primary tumors (i.e. GRPR [57], VEGFD [58],and MID1 [59]).
Analysis at single loci using quantitative approaches revealed high heterogeneity in cirDNA modification levels among plasma samples belonging to the same group (Figure 5). This is a common trend in epigenetic studies using cirDNA, which may hinder the transfer of basic research findings to clinical applications [60-62]. There are two main factors that may explain the observed heterogeneity. First, plasma cirDNA has a complex origin [35, 63, 64], since not only tumor cells, but also epithelial cells and circulating cells in blood can shed their DNA into circulation. Second, different cellular types and cells at different degree of differentiation have different epigenetic profiles, even when they belong to the same tissular structure (i.e. organ or tumor) [65]. The use of the xenografted model enabled the verification in tissue as well as blood samples from the same animal to further investigate the origin of the variation. Two loci showing significant differences in plasma cirDNA modifications illustrate the complexity of the plasma cirDNA profiles. Differences in the Rarb3a locus were also detected in tissue samples, but the difference was not present in PBC samples (Figure 5C), suggesting that the cirDNA profile of this locus represents that found in the tumor and DNA modification profiles in blood cells will not mask the results. On the contrary, differences in the Ttl locus did not represent those in the tissue, but rather reflected those found in the blood samples (Figure 5D).
In summary, we have shown that exposures to IH during sleep mimicking those routinely encountered in moderately severe OSA patients are accompanied by increases in the shedding of cirDNA into circulation, in both tumor and non-tumor-injected mice, with the presence of tumor further increasing cirDNA. Futhermore, our large-scale profiling approach revealed that cirDNA carries epigenetic modifications that may characterize specific cell populations. Furthermore, the intrinsic high variability of cirDNA methylation within the tumor suggests that some tumor cell populations may preferentially release their DNA upon IH exposures.
Methods
Animals, hypoxic exposures, and epithelial lung tumor model
C57BL/6J male mice (7-week old) were acquired from Jackson Laboratories (Bar Harbor, ME). IH exposures and tumor characterization were performed as previously described [23]. In brief, mice were subjected to IH with alternating cycles of 90 seconds (6% FIO2 followed by 21% FIO2, 20 cycles/hour) for 12 hours/day during daylight followed by 21% FIO2 for the remaining 12 hours. The control group was exposed to continuous circulating 21% FIO2 (RA). Mice were pre-exposed during 2 weeks to either RA or IH, and half of the mice were then randomly selected and injected with 1 x 105 TC1 murine lung tumor cells in the left flank. Every 3 days, tumor volume was estimated. After 4 weeks from tumor injection, mice were sacrificed and tumors excised and weighed. All experimental procedures were approved by The Institutional Animal Care and Use Committee of the University of Chicago.
Plasma cirDNA and genomic DNA isolation
Blood and tissue samples were collected after sacrifice and immediately processed. The plasma fraction was separated by centrifugation and cirDNA was isolated using the QIAmp Nucleic Acid isolation kit (Qiagen, Valencia, CA). Blood and tissue samples were lysed using Proteinase K digestion and genomic DNA isolated using standard phenol/chloroform extraction.
cirDNA quantification
cirDNA amount in each plasma sample was quantified using SYBR-green based quantitative PCR (qPCR) [38]. A 150 bp assay was designed located in the intron 2 of the Kras gene. The qPCR reaction consisted of 10 uL plasma cirDNA, 1× ABI master mix containing Taq polymerase, dNTPs, SYBR green dye and ROX as passive dye (Life Technologies,Carlsbad, CA, USA) and 200 nM of specific primers (Table S5). Amplification and analyses were performed using the 7500 System (Applied Biosystems, Foster City, CA). cirDNA quantity was calculated by CT extrapolation to a calibration curve built with murine DNA.
cirDNA modification profiling
Several biochemical modifications have been reported at cytosine residues, namely methylation (5-mC), hydroxymethylation (5-hmC), formylation (5-fmC) and carboxylation (5-cmC) [66], with 5-mC being the most widely studied, and commonly referred as “DNA methylation”. Although multiple methods of epigenetic analysis have been developed in the last decades (reviewed in [67]), not all approaches are suitable for cirDNA epigenetic analysis due to the small amount of starting material. Whereas antibody-based detection methods (i.e., MeDIP and hMeDIP) are specific for 5-mC or 5-hmC, they require relatively large starting DNA amounts, and are therefore unfeasible for plasma cirDNA analysis. Therefore, we used a bisulfite-based and methylation-sensitive enzymatic restriction approach that recognizes modified Cs, but does not differentiate between 5-mC and 5-hmC [68]. To maintain a precise terminology, we will therefore apply the term ‘DNA modification’ in the description of the experiments.
Large-scale cirDNA epigenetic modification profiles were assessed in plasma cirDNA samples from xenografted mice exposed to IH (XenoIH group, n=3) or to RA (XenoRA, n=3) conditions, according to previously described methods [50]. Briefly, universal DNA adaptors were ligated to the ends of cirDNA fragments, followed by digestion with DNA modification-sensitive enzymes (HpaII, HinP1 and HpyCH4IV). cirDNA fragments that survived enzymatic hydrolysis were amplified by adaptor-mediated PCR. The enriched differentially cirDNA modified fraction was fragmented, biotin-labeled, and hybridized on Affymetrix GeneChip Mouse Promoter Array 1.0R (Affymetrix, Santa Clara, CA) and scanned, according to manufacturer’s protocol. The array consisted of over 4.6 million probes tiled to interrogate over 28,000 mouse promoter regions.
Microarray data analysis
GCOS 1.3 software (Affymetrix) was used to produce .cel files. Microarray raw data were deposited in NCBI’s Gene Expression Omnibus (GEO) database (accession number: GSE61070). Data quality control was performed using the STARR package [69] in the R statistical environment (version 3.0.2) [70]. Data were analyzed using the Partek Genomic Suite Software (PGS) (St. Louis, MO). Signals were adjusted according to probe sequence and background corrected using the Robust Microarray method (RMA) [71]. One-way ANOVA was used to detect probes showing differential cirDNA modification between the groups. The significance level was set on p<0.05 and fold changes higher than 2 (Figure 3). Model-based analysis of tiling-arrays (MAT) [72] was used to identify regions of differential cirDNA modification by combining adjacent probes showing significant differences between the groups. A sliding window of 500 bp was set, according to the average size of the fragments produced in the amplicon preparation step [50]. Data were visualized by importing it to the UCSC Genome Browser (http://genome.ucsc.edu, NCBI36/mm8 assembly, which corresponded to the building of the microarray). Transcript association and chromosomal distribution were determined using the LiftOver tool to the latest available mouse genome annotation (GRCm38/mm10 assembly; last accessed August 2014). Genomic motifs were identified using the Multiple Em for Motif Elicitation tool (MEME) [73] and compared them against databases of DNA binding factors using the TOMTOM motif comparison tool [74]. Gene ontology analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) version 6.7 [75].
Single locus cirDNA epigenetic analysis by DNA modification-sensitive restriction quantitative PCR (qMSRE-PCR)
Fifteen nanograms of purified amplicon were subjected to real-time PCR, using specific primers (Table S5) and the same SYBR-green conditions used for cirDNA quantification. Adaptor-mediated PCR products from the undigested samples were used as reference to calculate the enrichment (ΔCttarget). A fragment within the Gapdh locus, which did not contain any CG position, was used as calibrator (ΔCtcalibrator). Fold change (FC) enrichment was calculated using the equation: FC=(2^(ΔCttarget -ΔCtcalibrator)).
Single locus cirDNA epigenetic analysis by quantitative Methylation Specific PCR analysis (qMSP)
cirDNA and genomic DNA samples were bisulfite-treated using the Epitect kit (Qiagen) and amplified using the whole bisulfitome amplification kit (Qiagen). Bisulfite treated and amplified cirDNA (10 ng) was subjected to locus-specific amplification using qMSP primers [76] (Table S5) and the same SYBR-green conditions used for cirDNA quantification. Fragment surrounding each qMSP assay in which primers did not contain any CG site were used as reference (ΔCtsample). Sssi methylated DNA was used as a calibrator (ΔCtmethylated). Percentage of methylation was calculated using the equation: %Me=(2^(ΔCtsample-ΔCtmethylated))*100).
Acknowledgements
This work was supported in part by the Herbert T. Abelson Chair in Pediatrics.
Funding
This work was supported in part by the Herbert T. Abelson Chair in Pediatrics.
Conflict of interest
The authors declare no conflict of interest.
References
1. Martinez-Garcia MA, Martorell-Calatayud A, Nagore E, Valero I, Selma MJ, Chiner E, Landete P, Montserrat JM, Carrera C, Perez-Gil A, Campos-Rodriguez F, and Farre R. Association between sleep disordered breathing and aggressiveness markers of malignant cutaneous melanoma. Eur Respir J. 2014; 43:1661-1668.
2. Nieto FJ, Peppard PE, Young T, Finn L, Hla KM, and Farre R. Sleep-disordered breathing and cancer mortality: results from the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med. 2012; 186:190-194.
3. Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008; 5:136-143.
4. Arnardottir ES, Mackiewicz M, Gislason T, Teff KL, and Pack AI. Molecular signatures of obstructive sleep apnea in adults: a review and perspective. Sleep. 2009; 32:447-470.
5. Dempsey JA, Veasey SC, Morgan BJ, and O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010; 90:47-112.
6. Marcus CL, Brooks LJ, Draper KA, Gozal D, Halbower AC, Jones J, Schechter MS, Ward SD, Sheldon SH, Shiffman RN, Lehmann C, and Spruyt K. Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics. 2012; 130:e714-755.
7. Raynes-Greenow CH, Hadfield RM, Cistulli PA, Bowen J, Allen H, and Roberts CL. Sleep apnea in early childhood associated with preterm birth but not small for gestational age: a population-based record linkage study. Sleep. 2012; 35:1475-1480.
8. Piteo AM, Lushington K, Roberts RM, van den Heuvel CJ, Nettelbeck T, Kohler MJ, Martin AJ, and Kennedy JD. Prevalence of snoring and associated factors in infancy. Sleep Med. 2012; 12:787-792.
9. Kaushal N, Ramesh V, and Gozal D. Human apolipoprotein E4 targeted replacement in mice reveals increased susceptibility to sleep disruption and intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2012; 303:R19-29.
10. Vijayan VK. Morbidities associated with obstructive sleep apnea. Expert Rev Respir Med. 2012; 6:557-566.
11. Gozal D, Kheirandish-Gozal L, Bhattacharjee R, Molero-Ramirez H, Tan HL, and Bandla HP. Circulating adropin concentrations in pediatric obstructive sleep apnea: potential relevance to endothelial function. J Pediatr. 2013; 163:1122-1126.
12. Kim J, Gozal D, Bhattacharjee R, and Kheirandish-Gozal L. TREM-1 and pentraxin-3 plasma levels and their association with obstructive sleep apnea, obesity, and endothelial function in children. Sleep. 2013; 36:923-931.
13. Kheirandish-Gozal L, Khalyfa A, Gozal D, Bhattacharjee R, and Wang Y. Endothelial dysfunction in children with obstructive sleep apnea is associated with epigenetic changes in the eNOS gene. Chest. 2013; 143:971-977.
14. Vrints H, Shivalkar B, Hilde H, Vanderveken OM, Hamans E, Van de Heyning P, De Backer W, and Verbraecken J. Cardiovascular mechanisms and consequences of obstructive sleep apnoea. Acta Clin Belg. 2013; 68:169-178.
15. Kheirandish-Gozal L, Yoder K, Kulkarni R, Gozal D, and Decety J. Preliminary functional MRI neural correlates of executive functioning and empathy in children with obstructive sleep apnea. Sleep. 2014; 37:587-592.
16. Nair D, Ramesh V, Li RC, Schally AV, and Gozal D. Growth hormone releasing hormone (GHRH) signaling modulates intermittent hypoxia-induced oxidative stress and cognitive deficits in mouse. J Neurochem. 2013; 127:531-540.
17. Gagnon K, Baril AA, Gagnon JF, Fortin M, Decary A, Lafond C, Desautels A, Montplaisir J, and Gosselin N. Cognitive impairment in obstructive sleep apnea. Pathol Biol (Paris). 2014.
18. Kheirandish-Gozal L, Peris E, Wang Y, Tamae Kakazu M, Khalyfa A, Carreras A, and Gozal D. Lipopolysaccharide-binding protein plasma levels in children: effects of obstructive sleep apnea and obesity. J Clin Endocrinol Metab. 2014; 99:656-663.
19. Gozal D, Capdevila OS, and Kheirandish-Gozal L. Metabolic alterations and systemic inflammation in obstructive sleep apnea among nonobese and obese prepubertal children. Am J Respir Crit Care Med. 2008; 177:1142-1149.
20. Bonsignore MR, Borel AL, Machan E, and Grunstein R. Sleep apnoea and metabolic dysfunction. Eur Respir Rev. 2013; 22:353-364.
21. Lee SL, Rouhi P, Dahl Jensen L, Zhang D, Ji H, Hauptmann G, Ingham P, and Cao Y. Hypoxia-induced pathological angiogenesis mediates tumor cell dissemination, invasion, and metastasis in a zebrafish tumor model. Proc Natl Acad Sci U S A. 2009; 106:19485-19490.
22. Rofstad EK, Gaustad JV, Egeland TA, Mathiesen B, and Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer. 2010; 127:1535-1546.
23. Almendros I, Wang Y, Becker L, Lennon FE, Zheng J, Coats BR, Schoenfelt KS, Carreras A, Hakim F, Zhang SX, Farre R, and Gozal D. Intermittent hypoxia-induced changes in tumor-associated macrophages and tumor malignancy in a mouse model of sleep apnea. Am J Respir Crit Care Med. 2014; 189:593-601.
24. Hakim F, Wang Y, Zhang SX, Zheng J, Yolcu ES, Carreras A, Khalyfa A, Shirwan H, Almendros I, and Gozal D. Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling. Cancer Res. 2014; 74:1329-1337.
25. Mandel P and Metais P. Les acides nucliques du plasma sanguin chez l’homme. C R Seances Soc Biol Fil. 1948; 142:241-243.
26. Chan KC and Lo YM. Circulating nucleic acids as a tumor marker. Histol Histopathol. 2002; 17:937-943.
27. Szpechcinski A, Chorostowska-Wynimko J, Kupis W, Maszkowska-Kopij K, Dancewicz M, Kowalewski J, and Orlowski T. Quantitative analysis of free-circulating DNA in plasma of patients with resectable NSCLC. Expert Opin Biol Ther. 2012; 12 Suppl 1:S3-9.
28. Yoon KA, Park S, Lee SH, Kim JH, and Lee JS. Comparison of circulating plasma DNA levels between lung cancer patients and healthy controls. J Mol Diagn. 2009; 11:182-185.
29. Cherepanova AV, Tamkovich SN, Bryzgunova OE, Vlassov VV, and Laktionov PP. Deoxyribonuclease activity and circulating DNA concentration in blood plasma of patients with prostate tumors. Ann N Y Acad Sci. 2008; 1137:218-221.
30. Sai S, Ichikawa D, Tomita H, Ikoma D, Tani N, Ikoma H, Kikuchi S, Fujiwara H, Ueda Y, and Otsuji E. Quantification of plasma cell-free DNA in patients with gastric cancer. Anticancer Res. 2007; 27:2747-2751.
31. Catarino R, Ferreira MM, Rodrigues H, Coelho A, Nogal A, Sousa A, and Medeiros R. Quantification of free circulating tumor DNA as a diagnostic marker for breast cancer. DNA Cell Biol. 2008; 27:415-421.
32. Butt AN and Swaminathan R. Overview of circulating nucleic acids in plasma/serum. Ann N Y Acad Sci. 2008; 1137:236-242.
33. Stroun M, Anker P, Lyautey J, Lederrey C, and Maurice PA. Isolation and characterization of DNA from the plasma of cancer patients. Eur J Cancer Clin Oncol. 1987; 23:707-712.
34. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Jr., Goodman SN, David KA, Juhl H, Kinzler KW, and Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102:16368-16373.
35. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, and Diaz LA, Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985-990.
36. Schorey JS and Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008; 9:871-881.
37. Gorges TM, Schiller J, Schmitz A, Schuetzmann D, Schatz C, Zollner TM, Krahn T, and von Ahsen O. Cancer therapy monitoring in xenografts by quantitative analysis of circulating tumor DNA. Biomarkers. 2012; 17:498-506.
38. Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C, and Thierry AR. High fragmentation characterizes tumour-derived circulating DNA. PLoS One. 2011; 6:e23418.
39. Rago C, Huso DL, Diehl F, Karim B, Liu G, Papadopoulos N, Samuels Y, Velculescu VE, Vogelstein B, Kinzler KW, and Diaz LA, Jr. Serial assessment of human tumor burdens in mice by the analysis of circulating DNA. Cancer Res. 2007; 67:9364-9370.
40. Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, and Molina F. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Res. 2010; 38:6159-6175.
41. Ye L, Ma GH, Chen L, Li M, Liu JL, Yang K, Li QY, Li N, and Wan HY. Quantification of circulating cell-free DNA in the serum of patients with obstructive sleep apnea-hypopnea syndrome. Lung. 2010; 188:469-474.
42. Baylin SB and Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11:726-734.
43. Sandoval J and Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev. 2012; 22:50-55.
44. Marzese DM, Hirose H, and Hoon DS. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Expert Rev Mol Diagn. 2013; 13:827-844.
45. El Messaoudi S, Rolet F, Mouliere F, and Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta. 2013; 424:222-230.
46. Zeerleder S. The struggle to detect circulating DNA. Crit Care. 2006; 10:142.
47. Gharib SA, Seiger AN, Hayes AL, Mehra R, and Patel SR. Treatment of obstructive sleep apnea alters cancer-associated transcriptional signatures in circulating leukocytes. Sleep. 2014; 37:709-714, 714A-714T.
48. Potter NT, Hurban P, White MN, Whitlock KD, Lofton-Day CE, Tetzner R, Koenig T, Quigley NB, and Weiss G. Validation of a Real-Time PCR-Based Qualitative Assay for the Detection of Methylated SEPT9 DNA in Human Plasma. Clin Chem. 2014; 60:1183-1191.
49. Church TR, Wandell M, Lofton-Day C, Mongin SJ, Burger M, Payne SR, Castanos-Velez E, Blumenstein BA, Rosch T, Osborn N, Snover D, Day RW, and Ransohoff DF. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut. 2014; 63:317-325.
50. Cortese R, Kwan A, Lalonde E, Bryzgunova O, Bondar A, Wu Y, Gordevicius J, Park M, Oh G, Kaminsky Z, Tverkuviene J, Laurinavicius A, Jankevicius F, et al. Epigenetic markers of prostate cancer in plasma circulating DNA. Hum Mol Genet. 2012; 21:3619-3631.
51. Blattler A and Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013; 288:34287-34294.
52. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011; 481:380-384.
53. Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, and Massion PP. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013; 19:560-570.
54. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009; 136:521-534.
55. Sargent LM, Senft JR, Lowry DT, Jefferson AM, Tyson FL, Malkinson AM, Coleman AE, and Reynolds SH. Specific chromosomal aberrations in mouse lung adenocarcinoma cell lines detected by spectral karyotyping: a comparison with human lung adenocarcinoma. Cancer Res. 2002; 62:1152-1157.
56. Girard L, Zochbauer-Muller S, Virmani AK, Gazdar AF, and Minna JD. Genome-wide allelotyping of lung cancer identifies new regions of allelic loss, differences between small cell lung cancer and non-small cell lung cancer, and loci clustering. Cancer Res. 2000; 60:4894-4906.
57. Xiao D, Wang J, Hampton LL, and Weber HC. The human gastrin-releasing peptide receptor gene structure, its tissue expression and promoter. Gene. 2001; 264:95-103.
58. Katoh Y and Katoh M. Comparative integromics on VEGF family members. Int J Oncol. 2006; 28:1585-1589.
59. Arigoni M, Barutello G, Riccardo F, Ercole E, Cantarella D, Orso F, Conti L, Lanzardo S, Taverna D, Merighi I, Calogero RA, Cavallo F, and Quaglino E. miR-135b coordinates progression of ErbB2-driven mammary carcinomas through suppression of MID1 and MTCH2. Am J Pathol. 2013; 182:2058-2070.
60. Mabert K, Cojoc M, Peitzsch C, Kurth I, Souchelnytskyi S, and Dubrovska A. Cancer biomarker discovery: Current status and future perspectives. Int J Radiat Biol. 2014; 90:659-677.
61. Gonzalez-Masia JA, Garcia-Olmo D, and Garcia-Olmo DC. Circulating nucleic acids in plasma and serum (CNAPS): applications in oncology. Onco Targets Ther. 2014; 6:819-832.
62. Crowley E, Di Nicolantonio F, Loupakis F, and Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013; 10:472-484.
63. Fleischhacker M and Schmidt B. Circulating nucleic acids (CNAs) and cancer--a survey. Biochim Biophys Acta. 2007; 1775:181-232.
64. Delgado PO, Alves BC, Gehrke Fde S, Kuniyoshi RK, Wroclavski ML, Del Giglio A, and Fonseca FL. Characterization of cell-free circulating DNA in plasma in patients with prostate cancer. Tumour Biol. 2013; 34:983-986.
65. Esteller M. Epigenetics in cancer. N Engl J Med. 2008; 358:1148-1159.
66. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, and Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011; 333:1300-1303.
67. Rivera CM and Ren B. Mapping human epigenomes. Cell. 2013; 155:39-55.
68. Nestor C, Ruzov A, Meehan R, and Dunican D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques. 2010; 48:317-319.
69. Zacher B, Torkler P, and Tresch A. Analysis of Affymetrix ChIP-chip data using starr and R/Bioconductor. Cold Spring Harb Protoc. 2011; 2011:pdb top110.
70. Team RC, R: A language and environment for statistical computing. 2014, R Foundation for Statistical Computing: Vienna, Austria.
71. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249-264.
72. Johnson WE, Li W, Meyer CA, Gottardo R, Carroll JS, Brown M, and Liu XS. Model-based analysis of tiling-arrays for ChIP-chip. Proc Natl Acad Sci U S A. 2006; 103:12457-12462.
73. Bailey TL, Williams N, Misleh C, and Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006; 34:W369-373.
74. Gupta S, Stamatoyannopoulos JA, Bailey TL, and Noble WS. Quantifying similarity between motifs. Genome Biol. 2007; 8:R24.
75. Huang da W, Sherman BT, and Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44-57.
76. Lo PK, Watanabe H, Cheng PC, Teo WW, Liang X, Argani P, Lee JS, and Sukumar S. MethySYBR, a novel quantitative PCR assay for the dual analysis of DNA methylation and CpG methylation density. J Mol Diagn. 2009; 11:400-414.