
Introduction
The World Health Organization (WHO) classification of CNS tumors has started to integrate molecular testing as part of its diagnostic criteria [1]. This led the WHO to classify a new neoplastic entity defined as midline gliomas (e.g., brainstem, thalamic, spinal cord) harboring H3K27M mutations and exhibiting diffuse growth, as H3K27M-mutant diffuse midline gliomas [1–4]. Amongst midline high grade gliomas, Diffuse Intrinsic Pontine Glioma (DIPG) is a highly aggressive and malignant pediatric brain tumor that develops in the brainstem. These tumors mainly arise in children, with peak incidence rates occurring between ages 6 and 9 years [5–7]. DIPGs account for half of childhood high grade gliomas (HGGs) and 85% of gliomas arising in the brainstem [5, 8]. Due to their location, surgical resection is not possible. Transcriptomic analyzes showed that DIPGs have a distinct molecular profile from other high-grade pediatric gliomas. The pathological assessment of DIPG identifies a diffuse pons tumor, sometimes infiltrating the medulla and midbrain [9]. To date, no definitive chemotherapeutic treatment appears to be successful in DIPG. Fractionated focal radiotherapy remains the only treatment capable of reducing tumor progression; however, this treatment is mostly palliative [8, 10, 11]. Additionally, radiation therapy elicits severe cognitive and functional impairments in children, due to damage to the developing brain. Consequently, DIPGs are the leading cause of pediatric brain tumor death [10]. The median survival is around 9 to 12 months, with 2-year survival rates around 10% and 5-year survival rates decreasing to less than 2% [5, 7, 8, 11, 12].
PATHOPHYSIOLOGY AND GENETIC LESIONS
Intense research efforts over the past decade, have led to the discovery of unique mutations driving DIPG. The most frequent mutations affect the N-terminal tail of histone H3.3 and histone H3.1 and result in the change of a lysine to methionine at residue 27 [2–4]. It has been reported that the K27M mutation inhibits Enhancer of Zeste 2 (EZH2) histone methyltransferase activity causing a global hypomethylation at H3K27 displaying low levels of H3K27me3 [13]. DIPGs are also characterized by mutations in ACVR1G328V (mutated in 24% of DIPG cases) [14–17]. ACVR1 encodes a type 1 BMP receptor and the six mutations reported result in constitutive BMP pathway activation [14–17]. Other mutations in DIPG include mutations in targeting tumor protein p53 (TP53), PIK3CA/PIK3R1, and PDGFRα amplifications [17, 18]. These unique set of mutations confer specific biological properties to the cancer cells themselves and they also impact the tumor microenvironment. This leads to the possibility of developing patient specific therapeutic opportunities and also challenges.
PRECLINICAL ANIMAL MODELS OF DIPG
In order to develop and test novel therapeutic modalities for DIPG, it is imperative to have access to accurate preclinical mouse models, in which to test both efficacy and safety. Also, mouse models reflecting the genetic lesions and biology of DIPG are crucial for the development of targeted therapies to improve the prognosis of this devastating brainstem cancer. The first models of DIPG were xenograft models derived from tissue acquired at time of autopsy or biopsy material of DIPG patients [19]. Xenograft models are valuable because they have standardized growth rates, consistent times of death, and desired tumor localization, however, studies must be performed in immunocompromised mice which limits the ability to study the efficacy of immunotherapies. To overcome this limitation, a number of genetically engineered mouse models (GEMMs) of DIPG have been developed. Several GEMMs use a replication-competent avian sarcoma-leukosis virus long terminal repeat splice acceptor (RCAS) in order to achieve specific expression of oncogenes in cells expressing the avian cell surface receptor [19]. This model has been used to model the H3.3K27M mutation by overexpressing PDGF-β, H3.3K27M, and loss of p53 in nestin progenitors in the brainstem [20]. In addition, models of mutated ACVR1 and H3.1K27M and loss of p53 in nestin progentiors in the brainstem have also been generated using the RCAS/tva system [21]. Models using in utero electroporation to deliver PiggyBac DNA transposon plasmids expressing H3.3K27M in neural progenitor cells have also been developed [22, 23]. Marigil et al. recently developed a guide-screw system based DIPG xenograft model, which allows the generation of tumors in a fast and reproducible fashion and allows to deliver the therapeutics via the same screw fixed system route [6, 24, 25]. Smith et al. recently performed a comprehensive histopathological and molecular analysis of 37 novel patient-derived orthotopic xenograft (PDOX) models developed from pediatric brain tumor patients [26], associated datasets can be accessed at (http://pbtp.stjude.cloud). We developed a DIPG model bearing mutated ACVR1G328V or H3.1K27M, using the Sleeping Beauty transposon system to target neural stem cells in either the lateral ventricle or the fourth ventricle zone [27, 28]. Targeting the stem cells that line the fourth ventricle ensures the glioma will develop in the brain stem [27, 28]. From the genetically engineered model we generated neurospheres that enabled the development of an intracranial model of brainstem glioma through implantation of tumor neurospheres into the pons of adult C57BL/6 mice. This implantation model exhibits an intact immune system, it is highly invasive and displays 100% penetrance and short latency enabling it to be used for pre-clinical testing of targeted therapies and immune-mediated strategies for DIPG [27].
IMMUNE-STIMULATORY GENE THERAPY
Due to their location, DIPGs are not resectable and are highly invasive, taking into consideration this we exploited the potential of immunostimulatory gene therapy strategy to implement an immune-mediated therapeutic approach for DIPG, which has the potential to be translated into the clinical arena. This strategy involves the use of adenovirus (Ad) mediated delivery of herpes simplex virus type 1-thymidine kinase (TK) and Fms-like tyrosine kinase 3 ligand (Flt3L), which are injected into the tumor mass. Upon administration of the prodrug ganciclovir (GCV), proliferating tumor cells expressing TK will convert GCV to its phosphorylated active metabolite, which is then further phosphorylated by intracellular kinases, inducing termination of DNA replication leading to immunogenic cell death. Dying tumor cells release damage associated molecular patterns (DAMPs) molecules such as high-mobility group B1 protein (HMGB1), calreticulin, and ATP [29–31]. Flt3L mediates recruitment of dendritic cells (DC) followed by DC activation via Toll-like receptor 2 (TLR-2)-mediated signaling which is stimulated by HMGB1 released by dying tumor cells into the tumor microenvironment (TME). Activated DCs uptake tumor antigens, and transport them to the draining lymph nodes, where they activate T cells, priming a robust anti-tumor cytotoxic and memory T cell response. This in turn leads to tumor regression and long-term immune-mediated survival of brain tumor bearing animals [29, 30]. Figure 1 demonstrates the schematics of TK/Flt3L based immunostimulatory gene therapy and the underlying anti-DIPG immune mechanism. The study by Mendez et al. demonstrated that the combined gene therapy strategy targeting the host tumor immune response inhibits tumor progression and improves median survival of mACVR1 DIPG bearing mice [27]. This combined conditionally cytotoxic immunostimulatory gene therapy approach for newly diagnosed GBM patients, has recently completed its Phase I clinical trial accrual at University of Michigan Medical School (NCT01811992) [29, 30, 32, 33], no severe adverse events were observed in adult patients. Thus, we anticipate that the encouraging survival data and safety profile will enable the translation of this therapeutic approach to treat DIPG patients in the near future.
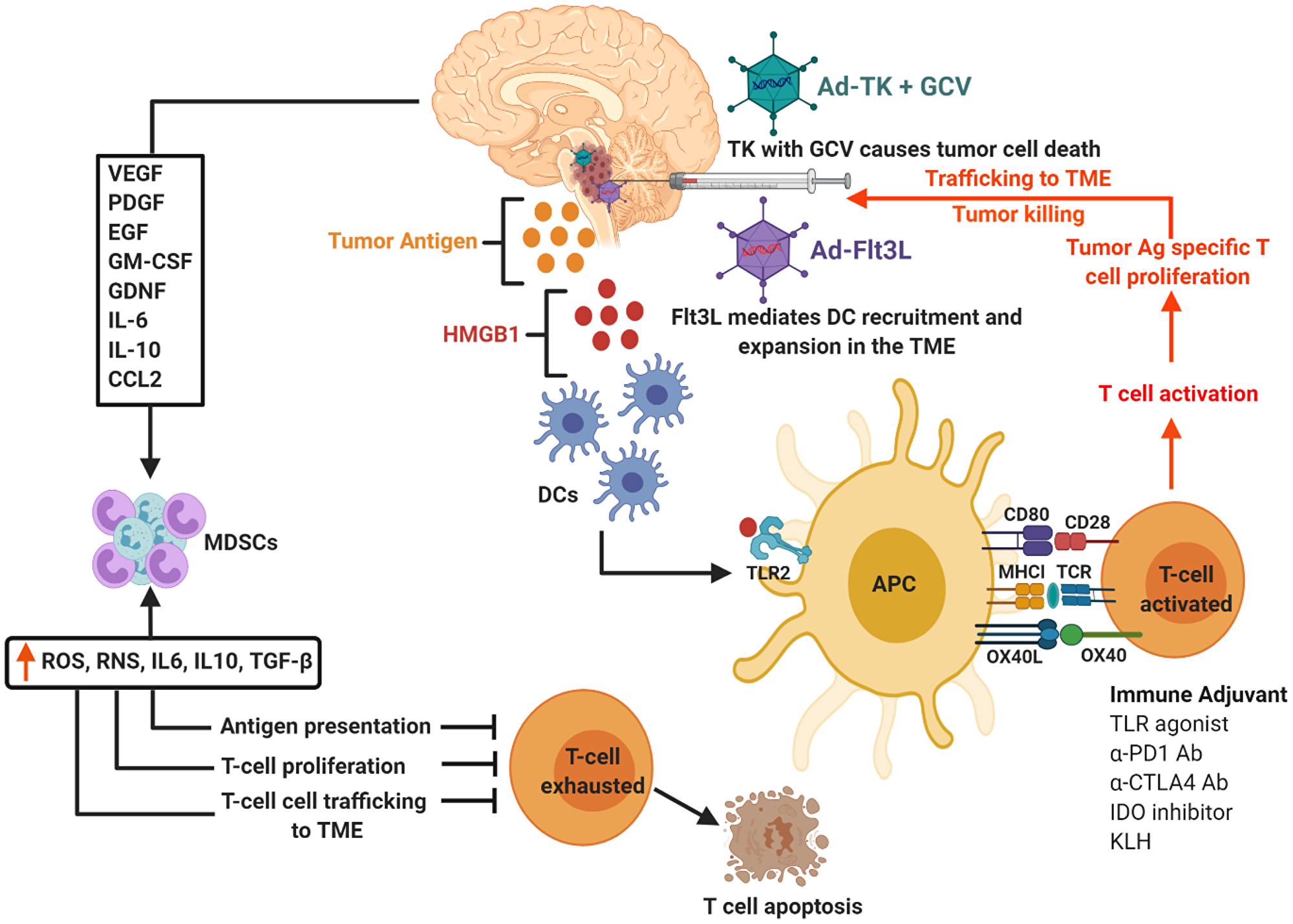
Figure 1: Schematics of TK/Flt3L based immunostimulatory gene therapy and underlying anti-DIPG immune mechanism. Tumor cells transfected with Ad-Flt3L express Flt3L protein enter systemic circulation. In the bone marrow (BM), Flt3L induces the expansion of dendritic cells (DCs), followed by their recruitment and accumulation in the tumor microenvironment (TME). Ganciclovir, which is a prodrug (GCV) is administered systemically. Tumor cells transfected with Ad-TK express TK protein capable of converting GCV to GCV-monophosphate (GCVp), which is further phosphorylated to GCV-diphosphate (GCVpp) by cellular kinase guanylate kinase and to the active antimetabolite GCV-triphosphate (GCVppp) by cellular nucleoside diphosphokinase. GCVppp, is incorporated into the replicating DNA of tumor cells, resulting in DNA replication termination and cell death. This also leads to the concomitant release of damage associated molecular patterns (DAMPs), i.e., HMBG1, Calreticulin, and ATP from dying tumor cells. Recruited DCs uptake the DIPG tumor Ag released from the dying cells. HMGB1 binds to TLR2/4, which facilitates the production of cytokines and tumor antigen cross-presentation. The DCs loaded with tumor antigens migrate to the cervical draining lymph node (dLN) where they present tumor antigens (Ag) to naive T cells, priming tumor specific anti-glioma effector T cells. Primed effector T cells enter the bloodstream from dLN and migrate towards the TME and kill residual tumor cells. Cytokines (VEGF, PDGF, LIF, GDNF, IL-6, IL-10, CCL2) released by glioma cells supporting differentiation and expansion of immune suppressive immature myeloid cells (MDSCs). To block effective anti-tumor immune responses, MDSCs are recruited to the tumor microenvironment and circulate back to lymphoid organs. The differentiation, maturation, activation, and proliferation of T cells are disrupted by these MDSCs, ultimately leading to T cell exhaustion and death.
ONGOING CLINICAL TRIALS
The ineffectiveness of the current standard of care for DIPGs has led to the development of many novel experimental therapies. Currently, immunotherapies stand out as potential treatments due to their minimal invasiveness and comprehensive tumor-eradicating capability [6, 7, 25, 27, 34]. The DIPG microenvironment contains low levels of antigen presenting cells (APCs) and adaptive immune cells, contributing to the tumor’s ability to grow undetected by the immune system [27, 34, 35]. However, unlike other HGGs, DIPGs do not appear to have a highly immunosuppressive or inflammatory microenvironment [35]. These factors make immunotherapies strong candidates for DIPG treatment. Oncolytic adenoviruses have emerged as promising immunotherapies capable of effectively treating DIPGs [6, 7, 25, 34]. One virus in particular, DNX-2401, is currently in Phase I trials (NCT03178032). Replication of this virus is dependent on a defective Rb pathway, a pathway commonly dysregulated in DIPG [6, 7]. This allows the virus to selectively target tumor cells. The high degree of specificity is essential given the sensitive anatomical location of these neoplasms. After infection, the virus replicates and eventually kills the infected cells [6, 7].
As mentioned, the only known effective treatment for DIPG is focal radiation. Although radiation therapy can improve survival and quality of life, ultimately it is not curative and it can have severe adverse effects. It has been shown that DNX-2401 inhibits DNA repair machinery of infected cells [25]. Therefore, DNX-2401 can potentially enhance therapeutic efficacy when combined with radiation therapy. The increased antitumoral effect of combination therapy has been supported using both in vitro and in vivo models [25]. Also, the radiation does not appear to affect replication of DNX-2401, so both therapies can be used concomitantly [25]. This makes the above therapy clinically relevant because has the potential to enhance the efficacy of the current standard of care.
The DIPG TME is relatively immunodeficient, lacking many components necessary to initiate a strong adaptive anti-DIPG immune response [36]. The reduced amount of immune cells and inflammatory signaling in the TME could be partially responsible for the low efficacy of radiation therapy. The combination of both DNX-2401 and radiation has been shown to also increase infiltration of lymphocytes and expression of proinflammatory cytokines in the TME [25]. This immunological shift could contribute to the increased survival observed in vivo when using the combination of DNX-2401 and radiation compared to each individually.
Conclusions and future prospects
The preclinical results by Mendez et al. are promising and indicate that it would be feasible to successfully test our TK/Flt3L-mediated gene therapy in a Phase I clinical trial for DIPG patients. The fact that the Phase I clinical trial for adult glioblastoma (GBM), recently completed at our Institution, using TK/Flt3L immune-stimulatory gene therapy, demonstrated it is a safe therapeutic approach (NCT01811992), provides a strong rationale for working towards its implementation in DIPG patients. The promising results of the immune-mediated gene therapy might be further enhanced by combining it with immune checkpoint inhibitors, opening new avenues worthy of investigation. We hypothesize that a combinatorial approach aiming to activate anti-DIPG immune response used together with immune checkpoint blockade would provide an effective therapeutic strategy against this devastating brainstem high grade glioma. It would therefore be important to test the impact on therapeutic efficacy and long-term survival of combining programmed death ligand 1 (PDL1) or CTLA-4 blockade with TK/Flt3L gene therapy in our preclinical model as a prelude to combinatorial phase I clinical trials. As the phase I clinical trials of TK/Flt3L gene therapy have already been completed and clinical-grade reagents for checkpoint blockade inhibitors are readily available, translating this combinatorial approach from bench to bedside should be feasible in the near future. In conclusion, the highly encouraging results are reported by Mendez et al. have opened an exciting opportunity for DIPG patients that will be tested in clinical trials in the very near future.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
Supported by National Institutes of Health/National Institute of Neurological Disorders & Stroke (NIH/NINDS) Grants R37-NS094804, R01-NS105556, R21-NS107894 to M.G.C.; NIH/NINDS Grants R01-NS076991, R01-NS082311, and R01-NS096756 to P.R.L.; NIH/NIBIB R01-EB022563 and NCI/UO1-CA-224160 to M.G.C. and P.R.L.; the Department of Neurosurgery; Leah’s Happy Hearts Foundation, ChadThough Foundation, Pediatric Brain Tumor Foundation, and Smiles for Sophie Forever Foundation to M.G.C. and P.R.L. RNA Biomedicine Grant F046166 to M.G.C. MGC is supported by a Rogel Cancer Center Scholarship and the Forbes Family Foundation. F.M.M was supported by NIH/NINDS-F31NS103500 and Rackham Pre-Doctoral Fellowship.
References
1. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131:803–20. https://doi.org/10.1007/s00401-016-1545-1. [PubMed].
2. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226–31. https://doi.org/10.1038/nature10833. [PubMed].
3. Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012; 44:251–3. https://doi.org/10.1038/ng.1102. [PubMed].
4. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012; 124:439–47. https://doi.org/10.1007/s00401-012-0998-0. [PubMed].
5. Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, Shaw TI, Zhu X, Larson JD, Easton J, Shao Y, Yergeau DA, Rosencrance C, Boggs K, et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019; 137:637–55. https://doi.org/10.1007/s00401-019-01975-4. [PubMed].
6. Martinez-Velez N, Garcia-Moure M, Marigil M, Gonzalez-Huarriz M, Puigdelloses M, Gallego Perez-Larraya J, Zalacain M, Marrodan L, Varela-Guruceaga M, Laspidea V, Aristu JJ, Ramos LI, Tejada-Solis S, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun. 2019; 10:2235. https://doi.org/10.1038/s41467-019-10043-0. [PubMed].
7. Tejada S, Alonso M, Patino A, Fueyo J, Gomez-Manzano C, Diez-Valle R. Phase I Trial of DNX-2401 for Diffuse Intrinsic Pontine Glioma Newly Diagnosed in Pediatric Patients. Neurosurgery. 2018; 83:1050–6. https://doi.org/10.1093/neuros/nyx507. [PubMed].
8. Misuraca KL, Hu G, Barton KL, Chung A, Becher OJ. A Novel Mouse Model of Diffuse Intrinsic Pontine Glioma Initiated in Pax3-Expressing Cells. Neoplasia. 2016; 18:60–70. https://doi.org/10.1016/j.neo.2015.12.002. [PubMed].
9. Buczkowicz P, Bartels U, Bouffet E, Becher O, Hawkins C. Histopathological spectrum of paediatric diffuse intrinsic pontine glioma: diagnostic and therapeutic implications. Acta Neuropathol. 2014; 128:573–81. https://doi.org/10.1007/s00401-014-1319-6. [PubMed].
10. Siddaway R, Hawkins C. Modeling DIPG in the mouse brainstem. Neuro Oncol. 2020; 22:307–8. https://doi.org/10.1093/neuonc/noaa003. [PubMed].
11. Katagi H, Louis N, Unruh D, Sasaki T, He X, Zhang A, Ma Q, Piunti A, Shimazu Y, Lamano JB, Carcaboso AM, Tian X, Seluanov A, et al. Radiosensitization by Histone H3 Demethylase Inhibition in Diffuse Intrinsic Pontine Glioma. Clin Cancer Res. 2019; 25:5572–83. https://doi.org/10.1158/1078-0432.CCR-18-3890. [PubMed].
12. Carvalho D, Taylor KR, Olaciregui NG, Molinari V, Clarke M, Mackay A, Ruddle R, Henley A, Valenti M, Hayes A, Brandon AH, Eccles SA, Raynaud F, et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun Biol. 2019; 2:156. https://doi.org/10.1038/s42003-019-0420-8. [PubMed].
13. Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013; 340:857–61. https://doi.org/10.1126/science.1232245. [PubMed].
14. Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, Morrison A, Lewis P, Bouffet E, Bartels U, Zuccaro J, Agnihotri S, Ryall S, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014; 46:451–6. https://doi.org/10.1038/ng.2936. [PubMed].
15. Taylor KR, Vinci M, Bullock AN, Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res. 2014; 74:4565–70. https://doi.org/10.1158/0008-5472.CAN-14-1298. [PubMed].
16. Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, Bechet D, Faury D, De Jay N, Ramkissoon LA, Corcoran A, Jones DT, Sturm D, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014; 46:462–6. https://doi.org/10.1038/ng.2950. [PubMed].
17. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014; 46:444–50. https://doi.org/10.1038/ng.2938. [PubMed].
18. Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014; 46:457–61. https://doi.org/10.1038/ng.2925. [PubMed].
19. Misuraca KL, Cordero FJ, Becher OJ. Pre-Clinical Models of Diffuse Intrinsic Pontine Glioma. Front Oncol. 2015; 5:172. https://doi.org/10.3389/fonc.2015.00172. [PubMed].
20. Cordero FJ, Huang Z, Grenier C, He X, Hu G, McLendon RE, Murphy SK, Hashizume R, Becher OJ. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol Cancer Res. 2017; 15:1243–54. https://doi.org/10.1158/1541-7786.MCR-16-0389. [PubMed].
21. Hoeman CM, Cordero FJ, Hu G, Misuraca K, Romero MM, Cardona HJ, Nazarian J, Hashizume R, McLendon R, Yu P, Procissi D, Gadd S, Becher OJ. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun. 2019; 10:1023. https://doi.org/10.1038/s41467-019-08823-9. [PubMed].
22. Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, Henderson S, Mikael LG, Richard-Londt A, Zhang Y, Costa JR, Hebert S, Khazaei S, et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell. 2017; 32:684–700.e9. https://doi.org/10.1016/j.ccell.2017.09.014. [PubMed].
23. Patel SK, Hartley RM, Wei X, Furnish R, Escobar-Riquelme F, Bear H, Choi K, Fuller C, Phoenix TN. Generation of diffuse intrinsic pontine glioma mouse models by brainstem-targeted in utero electroporation. Neuro Oncol. 2020; 22:381–92. https://doi.org/10.1093/neuonc/noz197. [PubMed].
24. Marigil M, Martinez-Velez N, Dominguez PD, Idoate MA, Xipell E, Patino-Garcia A, Gonzalez-Huarriz M, Garcia-Moure M, Junier MP, Chneiweiss H, El-Habr E, Diez-Valle R, Tejada-Solis S, et al. Development of a DIPG Orthotopic Model in Mice Using an Implantable Guide-Screw System. PLoS One. 2017; 12:e0170501. https://doi.org/10.1371/journal.pone.0170501. [PubMed].
25. Martinez-Velez N, Marigil M, Garcia-Moure M, Gonzalez-Huarriz M, Aristu JJ, Ramos-Garcia LI, Tejada S, Diez-Valle R, Patino-Garcia A, Becher OJ, Gomez-Manzano C, Fueyo J, Alonso MM. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol Commun. 2019; 7:64. https://doi.org/10.1186/s40478-019-0714-6. [PubMed].
26. Smith KS, Xu K, Mercer KS, Boop F, Klimo P, DeCupyere M, Grenet J, Robinson S, Dunphy P, Baker SJ, Ellison DW, Merchant TE, Upadayaya SA, et al. Patient-derived orthotopic xenografts of pediatric brain tumors: a St. Jude resource. Acta Neuropathol. 2020; 140:209–25. https://doi.org/10.1007/s00401-020-02171-5. [PubMed].
27. Mendez F, Kadiyala P, Nunez FJ, Carney S, Nunez FM, Gauss JC, Ravindran R, Pawar S, Edwards M, Garcia-Fabiani MB, Haase S, Lowenstein PR, Castro MG. Therapeutic Efficacy of Immune Stimulatory Thymidine Kinase and fms-like Tyrosine Kinase 3 Ligand (TK/Flt3L) Gene Therapy in a Mouse Model of High-Grade Brainstem Glioma. Clinical Cancer Research. 2020; 26:4080. https://doi.org/10.1158/1078-0432.CCR-19-3714. [PubMed].
28. Nunez FM, Gauss JC, Mendez FM, Haase S, Lowenstein PR, Castro MG. Genetically Engineered Mouse Model of Brainstem High-Grade Glioma. Star Protocol. 2020 Nov 20. [Epub ahead of print].
29. Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, Puntel M, Cheng Q, Prieto J, Ribas A, Kupiec-Weglinski J, van Rooijen N, Lassmann H, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005; 65:7194–204. https://doi.org/10.1158/0008-5472.CAN-04-3434. [PubMed].
30. Candolfi M, Yagiz K, Foulad D, Alzadeh GE, Tesarfreund M, Muhammad AK, Puntel M, Kroeger KM, Liu C, Lee S, Curtin JF, King GD, Lerner J, et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin Cancer Res. 2009; 15:4401–14. https://doi.org/10.1158/1078-0432.CCR-09-0155. [PubMed].
31. Candolfi M, Yagiz K, Wibowo M, Ahlzadeh GE, Puntel M, Ghiasi H, Kamran N, Paran C, Lowenstein PR, Castro MG. Temozolomide does not impair gene therapy-mediated antitumor immunity in syngeneic brain tumor models. Clin Cancer Res. 2014; 20:1555–65. https://doi.org/10.1158/1078-0432.CCR-13-2140. [PubMed].
32. Ghulam Muhammad AK, Candolfi M, King GD, Yagiz K, Foulad D, Mineharu Y, Kroeger KM, Treuer KA, Nichols WS, Sanderson NS, Yang J, Khayznikov M, Van Rooijen N, et al. Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin Cancer Res. 2009; 15:6113–27. https://doi.org/10.1158/1078-0432.CCR-09-1087. [PubMed].
33. Mineharu Y, Muhammad AK, Yagiz K, Candolfi M, Kroeger KM, Xiong W, Puntel M, Liu C, Levy E, Lugo C, Kocharian A, Allison JP, Curran MA, et al. Gene therapy-mediated reprogramming tumor infiltrating T cells using IL-2 and inhibiting NF-κB signaling improves the efficacy of immunotherapy in a brain cancer model. Neurotherapeutics. 2012; 9:827–43. https://doi.org/10.1007/s13311-012-0144-7. [PubMed].
34. Schuelke MR, Wongthida P, Thompson J, Kottke T, Driscoll CB, Huff AL, Shim KG, Coffey M, Pulido J, Evgin L, Vile RG. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J Immunother Cancer. 2019; 7:188. https://doi.org/10.1186/s40425-019-0673-2. [PubMed].
35. Lieberman NAP, DeGolier K, Kovar HM, Davis A, Hoglund V, Stevens J, Winter C, Deutsch G, Furlan SN, Vitanza NA, Leary SES, Crane CA. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro Oncol. 2019; 21:83–94. https://doi.org/10.1093/neuonc/noy145. [PubMed].
36. Lin GL, Nagaraja S, Filbin MG, Suva ML, Vogel H, Monje M. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018; 6:51. https://doi.org/10.1186/s40478-018-0553-x. [PubMed].