
Introduction
Merkel cell carcinoma (MCC) is a relatively rare cancer, with roughly 400 cases per 100,000 persons each year in the United States [1]. Before the age of immunotherapy, there was significant room for improvement in survival rates, with five-year survival of 60% overall and 14–21% among patients with distant disease [2, 3]. Now, immune checkpoint inhibitors (CPIs) in the metastatic setting are associated with often durable response rates approaching 70% and three-year overall survival (OS) of up to 64% [4–6]. However, knowledge of predictors of response is lacking. Clinicians require a greater understanding of predictive markers for therapy selection, while researchers require an improved understanding of underlying mechanisms to inform drug development and trial design. Identification of molecular underpinnings of disease and clinical benefit from CPI is a promising avenue for addressing these unmet needs. Knepper and colleagues recently explored the correlation of clinical and molecular characteristics with benefit from immunotherapy in MCC [7]. In the present study, we build on this prior approach by incorporating multivariable analysis techniques, studying patients over a longer follow-up time period, and by studying a homogeneous MCC population all treated with CPIs. Additionally, we set out to include a greater number of CPI-treated patients in our work and statistically analyze the correlations of specific single nucleotide variants (SNVs) and copy number variations (CNVs) with response to CPIs.
Results
Clinical characteristics
The cohort was comprised of approximately two thirds men and one third women with a median age of 71 (Table 1). Seven (16%) were immunosuppressed, including three patients with hematological malignances (acute myeloid leukemia, smoldering multiple myeloma, and chronic myeloid leukemia), two patients with autoimmune disease (with history of anti-tumor necrosis factor treatment but no active treatment during CPI), one with history of kidney transplant (receiving everolimus and prednisone), and one with recently diagnosed, active tuberculosis receiving rifamycin/isoniazid/pyrazinamide/ethambutol treatment. Primary site of disease was most commonly the skin of the limbs (63%) and head and neck (33%), and less frequently involved the trunk (7%). For eight patients (18%), the primary site of disease was unknown. The majority of the patients had pathologic stage III disease at diagnosis (62%, including 11 IIIA and 17 IIIB) (Supplementary Table 1). The majority of cases were either cN0 (12, 27%) or cN1 (20, 44%) at diagnosis. Pathologic nodal staging was similar: pN0 (11, 24%) and pN1 (20, 44%). Five patients (11%) had distant metastatic disease at diagnosis. Surgery with adjuvant radiation was the most common initial therapy for primary disease (44%), while 36% of patients received a chemotherapy-containing regimen as part of initial therapy.
Table 1: Demographics, clinical, and survival characteristics of patients with Merkel cell carcinoma treated with an immune checkpoint inhibitor (univariate analyses)
| Characteristic | All (%)A, N = 45 | ResponseB (%), N = 17 | No response (%), N = 22 | p-value |
|---|---|---|---|---|
| Median age in years (range) | 71 (39–89) | 71 (50–89) | 71 (50–89) | 0.90 |
| Sex | ||||
| Male | 31 (69) | 12 (71) | 14 (64) | 0.27 |
| Female | 14 (31) | 5 (29) | 8 (36) | |
| Immune suppressionC | ||||
| Yes | 7 (16) | 2 (12) | 4 (18) | 0.29 |
| No | 38 (84) | 15 (88) | 18 (82) | |
| Primary site of disease | ||||
| Limb | 19 (42) | 8 (47) | 10 (45) | - |
| Head and neck | 15 (33) | 5 (29) | 8 (36) | |
| Unknown | 8 (18) | 3 (18) | 4 (18) | |
| Trunk | 3 (7) | 1 (6) | 0 (0) | |
| Initial staging at diagnosisD | ||||
| Stage I | 9 (20) | 5 (29) | 2 (9) | - |
| Stage II | 3 (7) | 0 (0) | 3 (14) | |
| Stage III | 28 (62) | 11 (65) | 14 (64) | |
| Stage IV | 5 (11) | 1 (6) | 3 (14) | |
| Initial treatment regimen | ||||
| Surgery and RT | 20 (44) | 7 (41) | 10 (45) | 0.435D |
| ChemoRT | 10 (22) | 4 (24) | 4 (18) | |
| RT alone | 6 (13) | 2 (12) | 4 (18) | |
| Surgery + ChemoRT | 3 (7) | 1 (6) | 2 (9) | |
| Surgery alone | 2 (4) | 2 (12) | 0 (0) | |
| Chemotherapy alone | 2 (4) | 0 (0) | 1 (5) | |
| Surgery + Chemotherapy | 1 (2) | 1 (6) | 0 (0) | |
| None | 1 (2) | 0 (0) | 1 (5) | |
| Number experiencing recurrence | 36 (80) | 13 (76) | 18 (82) | 0.19 |
| Medianime to recurrence (months) | 4.8 (0.6–21.1) | 3.7 (0.6–12.2) | 6.3 (0.9–21.1) | 0.10 |
| Initial staging at diagnosis of R/M diseaseE | ||||
| Stage III | 13 (29) | 7 (41) | 6 (27) | - |
| Stage IV | 32 (71) | 10 (59) | 16 (73) | |
| CPI line of therapy | ||||
| Adjuvant | 2 (4) | 2 (12) | 0 (0) | - |
| 1 | 25 (56) | 11 (65) | 13 (59) | |
| 2 | 13 (31) | 3 (18) | 7 (32) | |
| 3 | 2 (4) | 0 (0) | 1 (5) | |
| 4 | 2 (4) | 1 (6) | 1 (5) | |
| CPI therapy | ||||
| PD-1 inhibitor monotherapy | 23 (51) | 9 (53) | 10 (45) | 0.22 F |
| PD-L1 inhibitor monotherapy | 21 (47) | 7 (41) | 12 (55) | |
| PD-1/CTLA-4 dual therapy | 1 (2) | 1 (6) | 0 (0) | |
| Median baseline ALC (× 106/L) (range) | 780 (0–16,660) | 680 (390–16,660) | 875 (310–2,030) | 0.30 |
| Immune-related adverse eventsG | ||||
| Grade 2 or lower | 37 (82) | 12 (71) | 20 (91) | 0.09 |
| Grade 3 or above | 8 (18) | 5 (29) | 2 (9) | |
Thirty-six (80%) patients experienced disease recurrence, which occurred at a median of 4.8 months (range: 0.6–21) following the end of definitive therapy. The majority of patients were stage IV at the time of recurrence or metastasis (71%), and the remaining were stage III (29%). Nearly all patients received either anti-programmed cell death-1 (PD-1) monotherapy with pembrolizumab or nivolumab (23, 51%) or anti-programmed death-ligand 1 (PD-L1) monotherapy with avelumab (21, 47%), while one patient received combined anti-PD-1/anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) therapy. Most patients received CPI as first-line therapy in the metastatic setting (60%). Median absolute lymphocyte count (ALC) prior to administration of CPI was 780 × 106 cells/liter (650 for patients who had received chemotherapy as part of initial therapy versus 970 for those who had not, p = 0.33). Eight (18%) patients experienced grade 3 or above immune-related adverse events (irAEs). These were autoimmune disorders of the respiratory system, gastrointestinal system, thyroid gland, joints, meninges, liver, adrenal gland, and heart.
Response and survival outcomes
With a median follow-up time of 25.2 months, duration of therapy ranged from zero (one dose) to 26.3 months. Nine patients were still on therapy at the time of data collection. Among 39 evaluable patients (six patients did not undergo restaging scans following CPI initiation), confirmed objective responses to CPI were achieved in 17/39 (43%) patients (Supplementary Table 2). Of these, thirteen (33%) experienced a complete response (CR) and four (10%) experienced a partial response (PR). The median time to response was 2.1 months (range 0.1–12.0). Median duration of response was 24.2 months (95% confidence interval [CI] 8.8-not reached [NR]), with 86.6% of responders experiencing an ongoing response at six months and 70.0% of responders with an ongoing response at twelve months. Among the non-responders (56%), five (13%) experienced stable disease (SD) and seventeen (44%) progressive disease (PD). Response rate was numerically lower (but not to the degree of statistical significance) among patients who had distant metastatic disease at initial diagnosis compared to those initially diagnosed with local or locoregional disease (25% vs. 45%, p = 0.62). Having received any prior cytotoxic chemotherapy, whether in the curative or palliative setting, did not impact response rate (42% for chemotherapy-naïve vs. 46% chemotherapy-expereinced, p = 1.00). Response rate did not vary by specific CPI agent (47% for PD-1 inhibition, 37% for PD-L1 inhibition, and 0% for the single patient on dual PD-1/CTLA-4 inhibition, p = 0.23 by Fisher’s exact test for PD-1 vs. PD-L1). The clinical benefit rate (CBR), which combines the CRs, PR and SD rates, was 56%. Two patients received treatment through initial progression. Of these, one never experienced a response, while the other went on to experience a sustained CR starting two months after observed PD and lasting for over two years. Of the patients who experienced SD as best response, two were still on therapy at the time of data collection (one for over a year). The duration of treatment for the other three patients who experienced SD as best response were 4.2 months, 15.4 months, and 22.2 months.
Median OS for the entire cohort was 15.5 months (95% CI 9.0–28.7). Six-month OS was 80.0% (95% CI 65.0–89.0), 1-year OS was 59.6% (95% CI 43.0–72.7), and 2-year OS was 36.7% (95% CI 21.1–52.8) (Figure 1). Median OS was not reached among responders, while median OS was 13.0 months among non-responders (HR 0.08; 95% CI 0.03–0.21, p < 0.01). Estimated 6-month OS was 100% among responders and 81.8% among non-responders, and estimated 2-year OS was 85.3 vs. 8.2%, respectively.
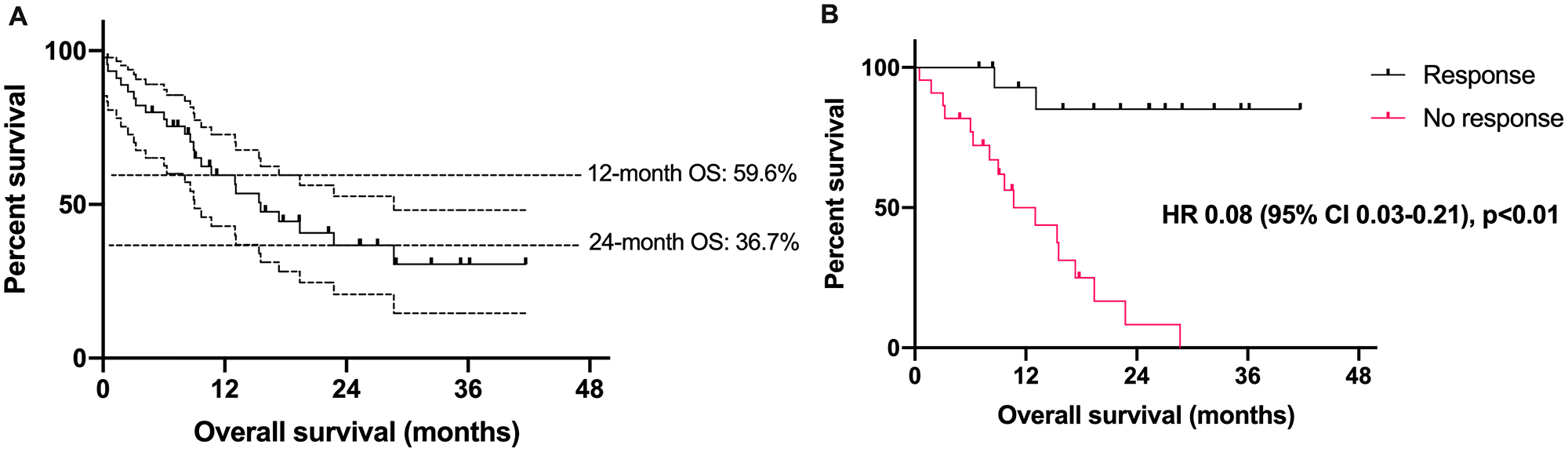
Figure 1: Survival outcomes in patients with Merkel cell carcinoma treated with an immune checkpoint inhibitor. (A) Overall survival (in months) among 45 patients with Merkel cell carcinoma treated with an immune checkpoint inhibitor. Dotted lines represent 95% confidence intervals. (B) Overall survival among patients with Merkel cell carcinoma based on response to immune checkpoint inhibitor. CI = confidence interval, HR = hazard ratio.
Clinical predictors of outcomes
On multivariable analysis, patients with a higher stage at initial diagnosis of primary disease were less likely to respond to CPI therapy (odds ratio [OR] 0.06, p = 0.04, Table 2). Patients with a longer interval between the completion of initial treatment and recurrence (disease-free interval, DFI) were less likely to respond to CPI therapy (OR 0.75, p = 0.05). All other clinical, pathological and genetic factors failed to independently impact CPI response. No clinical, pathological or genetic features independently predicted survival outcomes on CPI therapy (Table 3) at this cohort size and for this duration of follow-up.
Table 2: The effect of clinical, pathologic, and genetic features on CPI response in Merkel cell carcinoma patients (multivariate logistic regression modeling)
| Variable | Total (n = 39) | ||
|---|---|---|---|
| OR | [95% CI] | p-value | |
| Female sex | 3.00 | 0.05–163.94 | 0.59 |
| Older age at diagnosis | 0.91 | 0.78–1.06 | 0.24 |
| Higher stage at diagnosis | 0.06 | < 0.01–0.92 | 0.04 |
| Smoking history (current or former) | 0.71 | 0.03–14.63 | 0.83 |
| Immunosuppressed | 0.05 | < 0.01–2.87 | 0.15 |
| Longer time to recurrence | 0.75 | 0.56–0.99 | 0.05 |
| Greater CPI line of therapy | 1.21 | 0.35–4.24 | 0.76 |
| Higher absolute lymphocyte count | 1.12 | 0.69–1.82 | 0.64 |
| Grade 3 or greater adverse eventsA | 12.39 | 0.67–229.27 | 0.09 |
| Higher total mutational burden | 0.94 | 0.83–1.07 | 0.35 |
| MCCP | 0.28 | 0.01–13.39 | 0.52 |
Table 3: The effect of clinical, pathologic, and genetic features on overall survival in patients with Merkel cell carcinoma treated with immune checkpoint inhibition (multivariate Cox proportional hazard modeling)
| Variable | Total (n = 45) | ||
|---|---|---|---|
| HR | [95% CI] | p-value | |
| Female sex | 2.38 | 0.16–34.61 | 0.53 |
| Older age at diagnosis | 1.04 | 0.97–1.13 | 0.28 |
| Higher stage at diagnosis | 2.88 | 0.69–12.08 | 0.15 |
| Smoking history (current or former) | 3.19 | 0.55–18.57 | 0.20 |
| Immunosuppressed | 7.49 | 0.59–95.51 | 0.12 |
| Longer time to recurrence | 1.12 | 0.97–1.30 | 0.13 |
| Greater CPI line of therapy | 1.09 | 0.45–2.61 | 0.85 |
| Higher absolute lymphocyte count | 0.39 | 0.06–2.53 | 0.33 |
| Grade 3 or greater adverse events A | 0.05 | <0.01–1.08 | 0.05 |
| Higher total mutational burden | 1.02 | 0.93–1.11 | 0.68 |
| MCCP | 1.65 | 0.18–15.28 | 0.66 |
Molecular analysis
Of the 35/37 tumor samples whose Merkel cell polyomavirus (MCPyV) status could be inferred based on the genetic information from our panel, 16 (43%) were MCPyV-positive (MCCP) (Figure 2). Among responders, 5 (33%) were MCCP and 10 (67%) were MCPyV-negative (MCCN). Rates of MCPyV positivity were not significantly different among CPI responders and non-responders (p = 0.10 by univariable analysis) and did not correlate significantly with survival outcomes (Tables 2 and 3). Total mutational burden (TMB) was not significantly different among CPI responders and non-responders (median 19.7 versus 4.8 mutations per megabase, p = 0.11), and did not correlate significantly with survival outcomes. Rates of UV-related mutations did not correlate with outcomes.
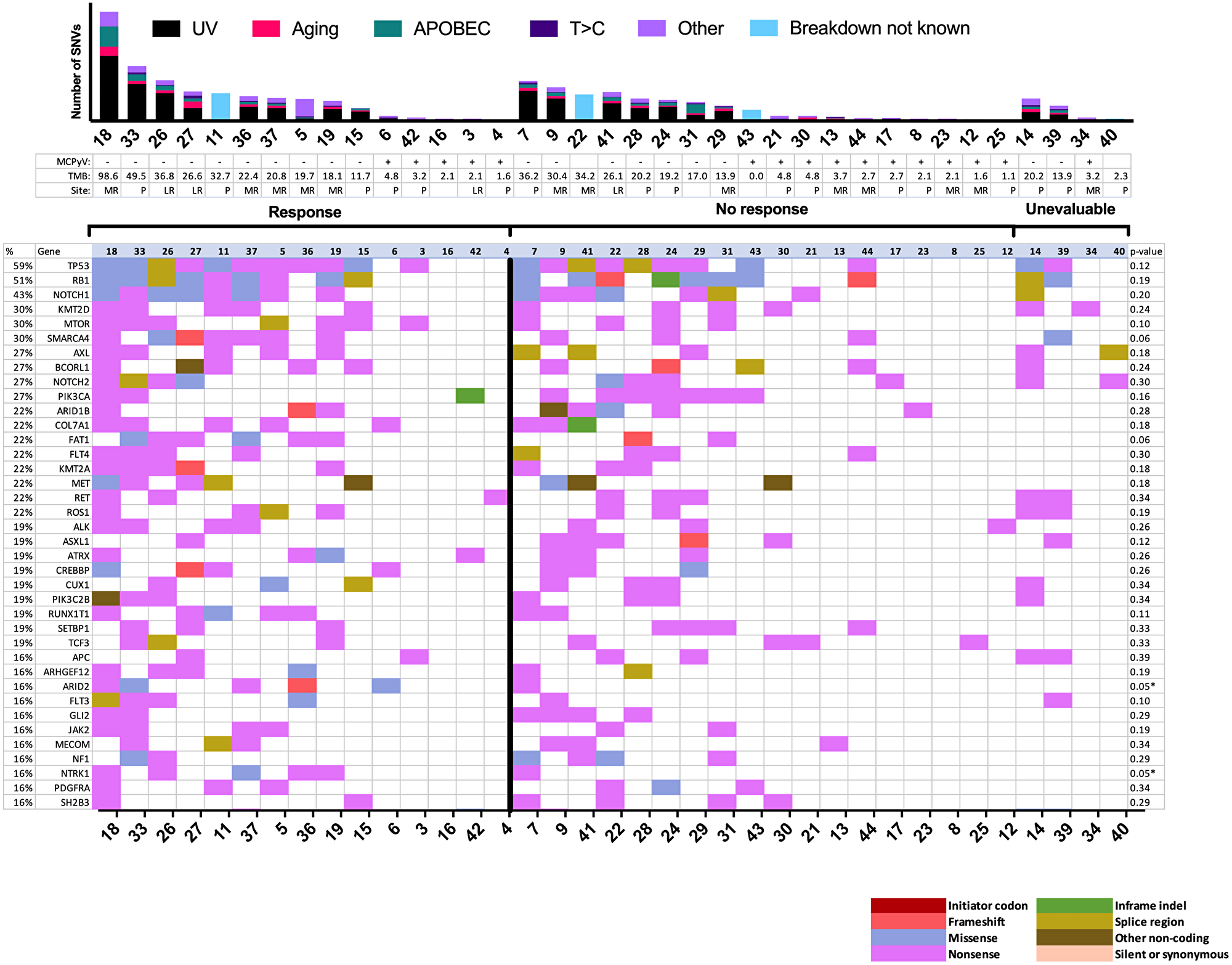
Figure 2: Mutational landscape by response to immune checkpoint inhibitor. Mutational plot showing the most frequently mutated genes (top-to-bottom, ≥15%) ordered by response and by total number of SNVs, with gene frequency listed at left (%), and Fisher exact test p values (response versus no response) at right. Asterisks denote values less than 0.05 (significant before Bonferroni correction, for which cutoff for significance is 0.0001 for our panel of 447 genes). The bar graph at top shows the total number of panel single nucleotide variants detected per sample by mutation signature. Blank MCPyV and TMB denote unknown values. LR = locoregional recurrence; MCPyV = Merkel cell polyomavirus status; MR = metastatic recurrence; P = primary site; SNV = single nucleotide variant; TMB = total mutational burden in mutations per Mb.
The most common SNVs among the entire sequenced cohort were those in TP53 (59%) and RB1 (51%). Among responders, there were significantly more mutations in ARID2 (p = 0.05) and NTRK1 (p = 0.05) by traditional analysis, although these findings were not significant when considering the stricter Bonferroni cutoff for multiple testing (p < 1.1 × 10-4 required). ARID2 and NTRK1 SNVs were not significantly correlated with time to recurrence. Mutations in SMARCA4 (p = 0.06) and FAT1 (p = 0.06) trended towards being more frequent among responders. CNVs were analyzed among 33 patients (Figure 3). CNVs occurred at a median rate of 47 per sample (range: 0–184) and did not associate significantly with outcomes.
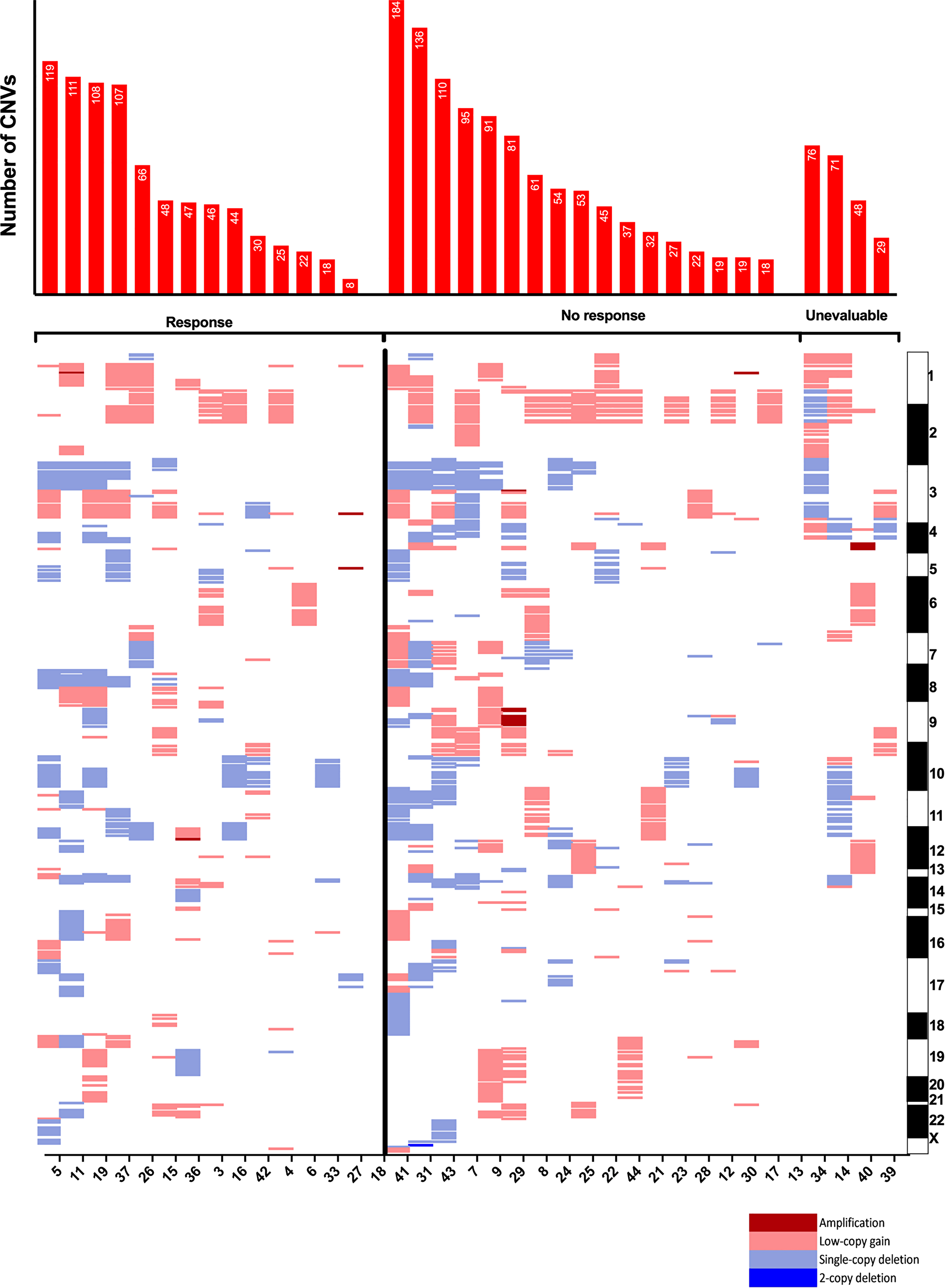
Figure 3: Copy-number variants by response to immune checkpoint inhibitor. Copy-number variants arranged by chromosomal band loci. Each column represents an individual tumor and corresponding gene loci. Chart at top shows total number of genes with copy number variants per sample. Patient samples presented by response and in order of descending total number of CNVs. CNV = copy-number variant.
DISCUSSION
Overall, our cohort of patients with CPI-treated MCC demonstrated the strong efficacy (60% 12-month OS and 37% 24-month OS) that clinicians have come to expect with immunotherapy in this setting. As anticipated, individuals who experienced a response to CPIs also experienced longer OS. The demographics of our cohort appears to largely match those of the U.S. MCC population: median age of 71 years in our cohort versus 75–79 in epidemiologic studies, roughly two-thirds men [1]. Our cohort deviates from expected prevalence of MCCP disease (43% in our cohort, versus roughly 80% expected across all individuals with MCC) [8]. This discrepancy may reflect the advanced, aggressive disease of patients in our cohort, as MCCP tumors tend to have better prognoses [9]. The disparity is also likely due, to some degree, to chance.
We observed a similar response rate to those recorded by Knepper and colleagues in their previous study that explored genomics and clinical benefit from CPIs (43% versus 44%) [7]. To our knowledge, there was no overlap between the patient sample in the Knepper study and our study. As the studies used different platforms for genomic testing, the genomic data between the two studies are distinct. As was the case in the previous study, we did not observe a correlation between either TMB or MCPyV status and clinical benefit from CPIs. Both studies identified TP53 and RB1 as the most commonly altered genes across each full cohort. Unlike the prior study, we included analysis of DFI, time to response, duration of response, survival time, and statistical tests related to the correlation of SNVs and CNVs with response.
After controlling for other clinical parameters, longer time to recurrence was associated with lower odds of response to CPI. We speculate this finding may be related to prior radiotherapy, as patients with node-positive disease are both more likely to recur sooner and are more likely to have received radiotherapy (and may thus be more likely to benefit from CPIs) [10]. Data in non-small cell lung cancer has indicated that patients with prior exposure to radiotherapy tend to have disease that is more likely to respond to immunotherapy [11]. The potential mechanisms through which immunotherapy and radiotherapy may synergize (e.g., radiotherapy-induced activation of immune cells and antigen presentation) have been widely studied and described [12–17]. Since only six patients in our study did not receive radiotherapy as part of initial treatment, we cannot make any definitive statements about radiotherapy’s association with CPI response. On multivariable analysis, patients with less advanced (earlier stage) disease at diagnosis were also more likely to experience a response to CPI. This observation reflects the impressive 71% CR rate to CPIs among radiographically evaluable patients who had originally been diagnosed with stage I disease, received definitive therapy, then experienced disease recurrence. Similarly, our previous work has demonstrated an association between lower total tumor burden and greater likelihood of response to CPIs in squamous cell carcinoma of the head and neck [18]. However, given that only a small number of patients in our cohort had stage I or stage II disease, and given the wide 95% CI for this covariable, we defer any conclusions related to this observation for the current study.
Response rates in our study did not vary significantly by presence of metastatic disease at diagnosis, by any prior chemotherapy exposure, or by line of therapy for advanced disease. These observations are largely in line with previous studies, which have demonstrated trends towards improved response in patients who have received fewer prior systemic agents, but not to a degree of statistical significance [4, 6, 19]. Such trends may reflect the rarity of this disease and no study being sufficiently powered to detect differences of this variety. The present cohort, for example, included only four radiographically-evaluable patients with metastatic disease at initial diagnosis, only one of whom experienced a radiographic response. The 25% response rate among those with metastatic disease at diagnosis indeed could, in larger studies, differ significantly from the 45% response rate among those with local or locoregional disease at diagnosis. Our study is limited by its retrospective nature and, given the rarity of the disease, small sample size.
The genomic landscape of the tumors in our trial were largely concordant with existing literature. Our study shared the top three most commonly mutated genes, TP53, RB1, and NOTCH1, with a recent retrospective study of a similar-sized cohort [7]. In line with this previous study, we observed a predominantly ultraviolet-associated mutational pattern in nearly all samples. Degree of ultraviolet signature predominance in a given sample did not associate with clinical benefit from CPIs. Also consistent with the prior work, TMB did not associate significantly with response, likely due to small sample size. SNVs in the ARID2 and NTRK1 genes correlated with CPI response, although this association was not significant after Bonferroni correction. All but one patient with ARID2-mutated tumors in our cohort experienced response to CPIs. The association between ARID2 and immune function has previously been established in melanoma [20]. The ARID2 protein is involved in the regulation of chromatin remodeling and its loss of function is associated with increased tumor cell sensitivity to interferon-γ and expression of T-cell cytotoxicity genes. Previous work has identified ARID2 as a commonly mutated gene in MCC [7].
NTRK1 codes for tropomyosin receptor kinase A (TrkA), a tyrosine receptor kinase that feeds into the MAPK pathway. TrkA expression may be characteristic of MCC: In one MCC case series, all 36 specimens exhibited cytoplasmic TrkA, although staining was generally weak (only 2 (6%) samples stained strongly for TrkA). [21] Its ligand, nerve growth factor (NGF) is expressed by tumor-associated interdigitating cells in roughly 70% of MCC cases – a sensical observation for a tumor of neuroendocrine origin [22]. A small study (n = 10) revealed that MCCP tumors may tend to display activating alternative splicing of NTRK1 mRNA. [23] Notably, none of the samples with NTRK1 SNVs in our cohort were MCCP. If TrkA activity is a key oncogenic pathway for MCCP tumors, we could speculate that NTRK1 alterations may confer an “MCCP-like” phenotype onto MCCN tumors by activating this pathway.
Specific protein changes in NTRK1 may be particular to MCC, as none of the amino acid changes observed in our cohort (Supplementary Table 3) have been seen in The Cancer Genome Atlas program [24]. As such, it is difficult to determine the significance of the specific amino acid changes in our cohort. One specific amino acid change, D668N, has been studied previously and was shown to activate tyrosine kinase activity of TrkA [25]. Evidence from lung cancer models, in which NTRK1 activity has been shown to inhibit the effect of CPIs via several mechanisms, including the promotion of T-cell exhaustion, offer some rational for some interaction between the two pathways [26]. In particular, repressing NTRK1 signalling could enhance tumoral benefit from CPIs. Our finding may provide support for the study of Trk inhibitors such as larotrectinib in advanced MCC. Furthermore, ARID2 and NTRK1 alterations could serve as predictive markers, in the context of other clinical and pathologic findings, as clinicians make management decisions related to CPIs in patients with MCC. Our study is limited by not including gene expression analysis – future work that includes such methods can help elucidate underlying driving mechanisms.
Our study points to several factors for clinicians to consider in the context of other clinical and pathologic findings when making treatment decisions. In particular, patients with ARID2 mutations, NTRK1 mutations, or shorter time to recurrence may be expected to have a higher likelihood of benefit from CPIs. These findings present potential areas for future basic scientific research related to molecular mechanisms. Future clinical study may explore the potential for Trk inhibition in combination or sequence with immunotherapies.
Materials and Methods
Study cohort
Forty-five patients with biopsy-proven MCC treated with an immune checkpoint inhibitor-containing regimen at the Dana-Farber Cancer Institute from 2013 to 2020 were retrospectively identified following expedited institutional review board approval. We obtained tumor tissue samples from 37 (82%) patients for molecular testing (other samples were either exhausted or not available). Among these, 17 (46%) samples came from a primary tumor site, four (11%) came from an area of locoregional recurrence, 13 (35%) were derived from a distant metastatic focus, and site classification was unknown for three (8%). Demographics, clinical, and survival characteristics, along with clinical and pathological features were collected from patient electronic health medical records. Response data was estimated from clinical radiology reports and physician notes based on Response Evaluation Criteria in Solid Tumors, RECIST v1.1 [27].
Molecular analysis
All patients sequenced in this study were consented individually to the Dana-Farber institutional Cancer Research Study (protocols 11-104 and 17-000) and a clinical laboratory improvement amendments (CLIA)-certified laboratory performed molecular testing. The molecular testing was performed on tissue samples with at least 50% viable malignant cell quality, and with at least 20% tumor on hematoxylin and eosin slide review. As previously described, the most updated iteration of this molecular testing (version 3) analyzes tumor tissue for a panel of known cancer-related mutations within the protein-coding portions of 447 genes [28, 29]. TMB was calculated as the number of non-synonymous somatic mutations per megabase of exonic sequence data across all genes on the panel. Tumors were assessed for likely MCPyV status based on genetic information, including data gathered using ViroPanel, an expansion of our institutional OncoPanel platform that captures oncogenic viral sequences, where available [30, 31]. Mechanistic origin of single nucleotide and dinucleotide variants was determined as described previously [30]. Sequencing data is available from the authors upon request.
Statistical analysis
Student t-test was used for continuous variables and a Fisher exact was used for categorical variables to compare clinical characteristics by response. The Kaplan-Meier method and log-rank testing were used for survival analyses. All tests were two-sided. All tests were univariate except for multivariate analysis of odds ratio of response and hazard ratio for death by response to immunotherapy. We compared the results of statistical tests with and without a Bonferroni correction, when appropriate. Our study had a 6% power to detect a 10% difference in odds ratio based on response. Data were analyzed using Stata/IC (version 16.1).
CONFLICTS OF INTEREST
AJK receives research support from the Pritzker School of Medicine and from the American Society of Hematology. Dr. Silk has received research funding (to the institution) from Biohaven Pharmaceuticals, Merck, Clinagen, and consulting fees from Bristol-Meyers Squibb, EMD Serono, Merck, and Sanofi Genzyme. JAD has received honoraria for advisory board participation with Merck & Co., Inc. and EMD Serono, Inc. JAD has received research funding from Constellation Pharmaceuticals, Inc. GJH has received research funding to institution from BMS, Exicure, GSK, Regeneron, Sanofi Genzyme, Kartos; consulting/honoraria from Regeneron, Sanofi Genzyme, BMS, Maverick, Merck, Kura, Bicara, and Exicure.
FUNDING
MT has received consulting fees from EMD Serono and Merck.
References
1. Paulson KG, Park SY, Vandeven NA, Lachance K, Thomas H, Chapuis AG, Harms KL, Thompson JA, Bhatia S, Stang A, Nghiem P. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J Am Acad Dermatol. 2018; 78:457–63.e2. https://doi.org/10.1016/j.jaad.2017.10.028. [PubMed].
2. Freeman MB, Holman DM, Qin J, Lunsford NB. Merkel cell carcinoma incidence, trends, and survival rates among adults aged >/=50 years from United States Cancer Statistics. J Am Acad Dermatol. 2019; 80:1154–6. https://doi.org/10.1016/j.jaad.2018.10.045. [PubMed].
3. Harms KL, Healy MA, Nghiem P, Sober AJ, Johnson TM, Bichakjian CK, Wong SL. Analysis of Prognostic Factors from 9387 Merkel Cell Carcinoma Cases Forms the Basis for the New 8th Edition AJCC Staging System. Ann Surg Oncol. 2016; 23:3564–71. https://doi.org/10.1245/s10434-016-5266-4. [PubMed].
4. Kaufman HL, Russell JS, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, Shih KC, Lebbe C, Milella M, Brownell I, Lewis KD, Lorch JH, von Heydebreck A, et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after >/=1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J Immunother Cancer. 2018; 6:7. https://doi.org/10.1186/s40425-017-0310-x. [PubMed].
5. Nghiem P, Bhatia S, Lipson EJ, Sharfman WH, Kudchadkar RR, Brohl AS, Friedlander PA, Daud A, Kluger HM, Reddy SA, Boulmay BC, Riker AI, Burgess MA, et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J Clin Oncol. 2019; 37:693–702. https://doi.org/10.1200/JCO.18.01896. [PubMed].
6. Topalian SL, Bhatia S, Hollebecque A, Awada A, Boer JPD, Kudchadkar RR, Goncalves A, Delord JP, Martens UM, Picazo JML, Oaknin A, Spanos WC, Aljumaily R, et al. Abstract CT074: Non-comparative, open-label, multiple cohort, phase 1/2 study to evaluate nivolumab (NIVO) in patients with virus-associated tumors (CheckMate 358): Efficacy and safety in Merkel cell carcinoma (MCC). Cancer Res. 2017; 77. https://doi.org/10.1158/1538-7445.AM2017-CT074.
7. Knepper TC, Montesion M, Russell JS, Sokol ES, Frampton GM, Miller VA, Albacker LA, McLeod HL, Eroglu Z, Khushalani NI, Sondak VK, Messina JL, Schell MJ, et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin Cancer Res. 2019; 25:5961–71. https://doi.org/10.1158/1078-0432.CCR-18-4159. [PubMed].
8. Schadendorf D, Lebbe C, Zur Hausen A, Avril MF, Hariharan S, Bharmal M, Becker JC. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. Eur J Cancer. 2017; 71:53–69. https://doi.org/10.1016/j.ejca.2016.10.022. [PubMed].
9. Sihto H, Kukko H, Koljonen V, Sankila R, Bohling T, Joensuu H. Clinical factors associated with Merkel cell polyomavirus infection in Merkel cell carcinoma. J Natl Cancer Inst. 2009; 101:938–45. https://doi.org/10.1093/jnci/djp139. [PubMed].
10. Frohm ML, Griffith KA, Harms KL, Hayman JA, Fullen DR, Nelson CC, Wong SL, Schwartz JL, Bichakjian CK. Recurrence and Survival in Patients With Merkel Cell Carcinoma Undergoing Surgery Without Adjuvant Radiation Therapy to the Primary Site. JAMA Dermatol. 2016; 152:1001–7. https://doi.org/10.1001/jamadermatol.2016.1428. [PubMed].
11. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017; 18:895–903. https://doi.org/10.1016/s1470-2045(17)30380-7. [PubMed].
12. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015; 16:e498–e509. https://doi.org/10.1016/s1470-2045(15)00007-8. [PubMed].
13. Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, Deweese TL, Drake CG. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer Immunol Res. 2015; 3:345–55. https://doi.org/10.1158/2326-6066.CIR-14-0196. [PubMed].
14. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, Schwendener R, Pruschy M, Knuth A, van den Broek M. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012; 189:558–66. https://doi.org/10.4049/jimmunol.1200563. [PubMed].
15. Liao YP, Wang CC, Butterfield LH, Economou JS, Ribas A, Meng WS, Iwamoto KS, McBride WH. Ionizing radiation affects human MART-1 melanoma antigen processing and presentation by dendritic cells. J Immunol. 2004; 173:2462–9. https://doi.org/10.4049/jimmunol.173.4.2462. [PubMed].
16. Larmonier N, Merino D, Nicolas A, Cathelin D, Besson A, Bateman A, Solary E, Martin F, Katsanis E, Bonnotte B. Apoptotic, necrotic, or fused tumor cells: an equivalent source of antigen for dendritic cell loading. Apoptosis. 2006; 11:1513–24. https://doi.org/10.1007/s10495-006-8765-0. [PubMed].
17. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005; 174:7516–23. https://doi.org/10.4049/jimmunol.174.12.7516. [PubMed].
18. Sridharan V, Rahman RM, Huang RY, Chau NG, Lorch JH, Uppaluri R, Haddad RI, Hanna GJ, Schoenfeld JD. Radiologic predictors of immune checkpoint inhibitor response in advanced head and neck squamous cell carcinoma. Oral Oncol. 2018; 85:29–34. https://doi.org/10.1016/j.oraloncology.2018.08.005. [PubMed].
19. D’Angelo SP, Bhatia S, Brohl AS, Hamid O, Mehnert JM, Terheyden P, Shih KC, Brownell I, Lebbe C, Lewis KD, Linette GP, Milella M, Georges S, et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J Immunother Cancer. 2020; 8:e000674. https://doi.org/10.1136/jitc-2020-000674. [PubMed].
20. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, Long HW, Yuan GC, Doench J, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018; 359:770–5. https://doi.org/10.1126/science.aao1710. [PubMed].
21. Wehkamp U, Stern S, Kruger S, Hauschild A, Rocken C, Egberts F. Tropomyosin Receptor Kinase A Expression on Merkel Cell Carcinoma Cells. JAMA Dermatol. 2017; 153:1166–9. https://doi.org/10.1001/jamadermatol.2017.2495. [PubMed].
22. Wehkamp U, Stern S, Kruger S, Weichenthal M, Hauschild A, Rocken C, Egberts F. Co-expression of NGF and PD-L1 on tumor-associated immune cells in the microenvironment of Merkel cell carcinoma. J Cancer Res Clin Oncol. 2018; 144:1301–8. https://doi.org/10.1007/s00432-018-2657-x. [PubMed].
23. Cappabianca L, Guadagni S, Maccarone R, Sebastiano M, Chiominto A, Farina AR, Mackay AR. A pilot study of alternative TrkAIII splicing in Merkel cell carcinoma: a potential oncogenic mechanism and novel therapeutic target. J Exp Clin Cancer Res. 2019; 38:424. https://doi.org/10.1186/s13046-019-1425-3. [PubMed].
24. TCGA Research Network. https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga.
25. Miranda C, Zanotti G, Pagliardini S, Ponzetto C, Pierotti MA, Greco A. Gain of function mutations of RTK conserved residues display differential effects on NTRK1 kinase activity. Oncogene. 2002; 21:8334–9. https://doi.org/10.1038/sj.onc.1206052. [PubMed].
26. Konen JM, Rodriguez BL, Fradette JJ, Gibson L, Davis D, Minelli R, Peoples MD, Kovacs J, Carugo A, Bristow C, Heffernan T, Gibbons DL. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/p53 Mutant Lung Cancer. Cancers (Basel). 2019; 11:462. https://doi.org/10.3390/cancers11040462. [PubMed].
27. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009; 45:228–47. https://doi.org/10.1016/j.ejca.2008.10.026. [PubMed].
28. Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, Ligon AH, Sholl LM, Kuo FC, MacConaill LE, Lindeman NI, Dong F. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer. Arch Pathol Lab Med. 2017; 141:751–8. https://doi.org/10.5858/arpa.2016-0527-OA. [PubMed].
29. Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, Pugh TJ, van Hummelen P, Thorner AR, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016; 1:e87062. https://doi.org/10.1172/jci.insight.87062. [PubMed].
30. Starrett GJ, Thakuria M, Chen T, Marcelus C, Cheng J, Nomburg J, Thorner AR, Slevin MK, Powers W, Burns RT, Perry C, Piris A, Kuo FC, et al. Clinical and molecular characterization of virus-positive and virus-negative Merkel cell carcinoma. Genome Med. 2020; 12:30. https://doi.org/10.1186/s13073-020-00727-4. [PubMed].
31. Slevin MK, Wollison BM, Powers W, Burns RT, Patel N, Ducar MD, Starrett GJ, Garcia EP, Manning DK, Cheng J, Hanna GJ, Kaye KM, Van Hummelen P, et al. ViroPanel: Hybrid Capture and Massively Parallel Sequencing for Simultaneous Detection and Profiling of Oncogenic Virus Infection and Tumor Genome. J Mol Diagn. 2020; 22:476–87. https://doi.org/10.1016/j.jmoldx.2019.12.010. [PubMed].