
Introduction
Breast cancer (BC) is the most frequently diagnosed cancer among females and accounts for most of the cancer-related incidences and mortalities [1] Globally, statistics have shown massive increase in incidence and mortalities from 1.7 million and 521,900 to 8.6 million and 4.2 million in BC, respectively [1, 2]. Approximately one in eight women will be diagnosed with invasive BC in their lifetime and one in 39 women will die from BC [3]. There are increasing attempts in cancer research to correlate molecular subtypes of BC in order to tailor treatment and develop new therapies [4]. Based on BC molecular subtypes, luminal A tumor appears to present better prognosis, have high survival rate and reduced chances of recurrence [5–7]. Currently used markers in the management and prognostic of BC do not equate accurate prediction of how individuals will respond to prescribed treatment of each subtype and their survival thereof. The poor survival and high mortality rates in BC are in part due to lack of effective BC prognostic biomarkers. It is therefore necessary to identify novel biomarkers that can serve as targets for BC therapeutic intervention.
More so, genomic exploration has been employed to identify novel gene regulators or roles of pre-existing gene members in signaling pathways [8]. In the same fashion, this advancement can be deployed in cancer research to sort out genetic alteration in order to tailor therapeutic responses most especially in BC [9].
The human protein kinases are fascinating targets for the discovery of novel cancer therapy due to their crucial role in cancer development and progression, and other related processes such as metabolic diseases [10, 11]. Aurora A kinase (officially: AURKA; aliases: Aurora-2, RKI, and STK15) is a conserved serine/threonine kinase which belong to the superfamily aurora kinase (AK). The AK primarily functions by regulating key cellular functions including mitosis and signaling pathways. The dysfunction of AK may result in mitotic arrest, aneuploidy, and apoptosis. Increased expression or amplification of AURKA is common in most human cancers including BC [12–18]. AURKA has been established as a legit oncogene and thus, a vital therapeutic target in cancer [19]. Its expression has been shown to be modulated by small molecules including microRNAs in cancers [20–23].
Non-coding RNAs (ncRNAs) have been thought to regulate the expression of target genes at post-translational level. The relevance of these ncRNAs particularly microRNAs and ncRNAs as therapeutic targets and delivery strategies in clinical translation were investigated for their involvement in cancer and related diseases. Diverse approaches have both in the past and recently been employed by researches to comprehend the complexity of BC.
Previous studies indicated that miR-32 may be tumor suppressive in nature [24, 25] and its regulatory effect on AURKA have been shown in various cancer subtypes [19, 26–29]. Due to their regulatory role in the expression of tumor associated genes at post-translational level [30, 31], we proposed that human argonaute protein (hAgo2) may regulate the expression of AURKA via hsa-miR-32-3p. Although the complexity of BC subtypes is correlated with molecular and genetic information from tumor cells, prognosis and therapeutic managements are monitored mainly by tumor stage, grade, hormone receptor status and HER2 status. Therefore, in silico and molecular docking studies were used to investigate the expression of AURKA gene in relation to its clinicopathological characteristics and the mechanism of regulation through hsa-miR-32-3p assisted hAgo2 in BC.
Results
Expression of AURKA in cancers
Using Oncomine database, AURKA expression was identified in various human cancers, in both hematological malignancies and solid tumors (Figure 1A). The mRNA levels of AURKA were significantly upregulated in BC patients in 11 datasets and down-regulated in 1 dataset. Furthermore, the GEPIA dataset revealed that the mRNA expression level of AURKA was significantly higher in BC tissues than in normal breast tissues (Figure 1B). In Curtis Breast Statistics’ dataset, AURKA was overexpressed compared to normal samples in all breast cancer types (Figure 2). In Zhao Breast Statistics’ dataset, AURKA was overexpressed in invasive ductal breast cancer and lobular BC (Figure 2). In TCGA Statistics’ dataset, AURKA was overexpressed in all the samples (Figure 2) while down-regulation was observed in the Finak Breast Statistics’ dataset in invasive BC with fold change of -11.071 compared to the normal samples. As shown in Table 1, the transcription levels of AURKA in different BC types were also higher than those in normal breast tissues at P < 0.05.
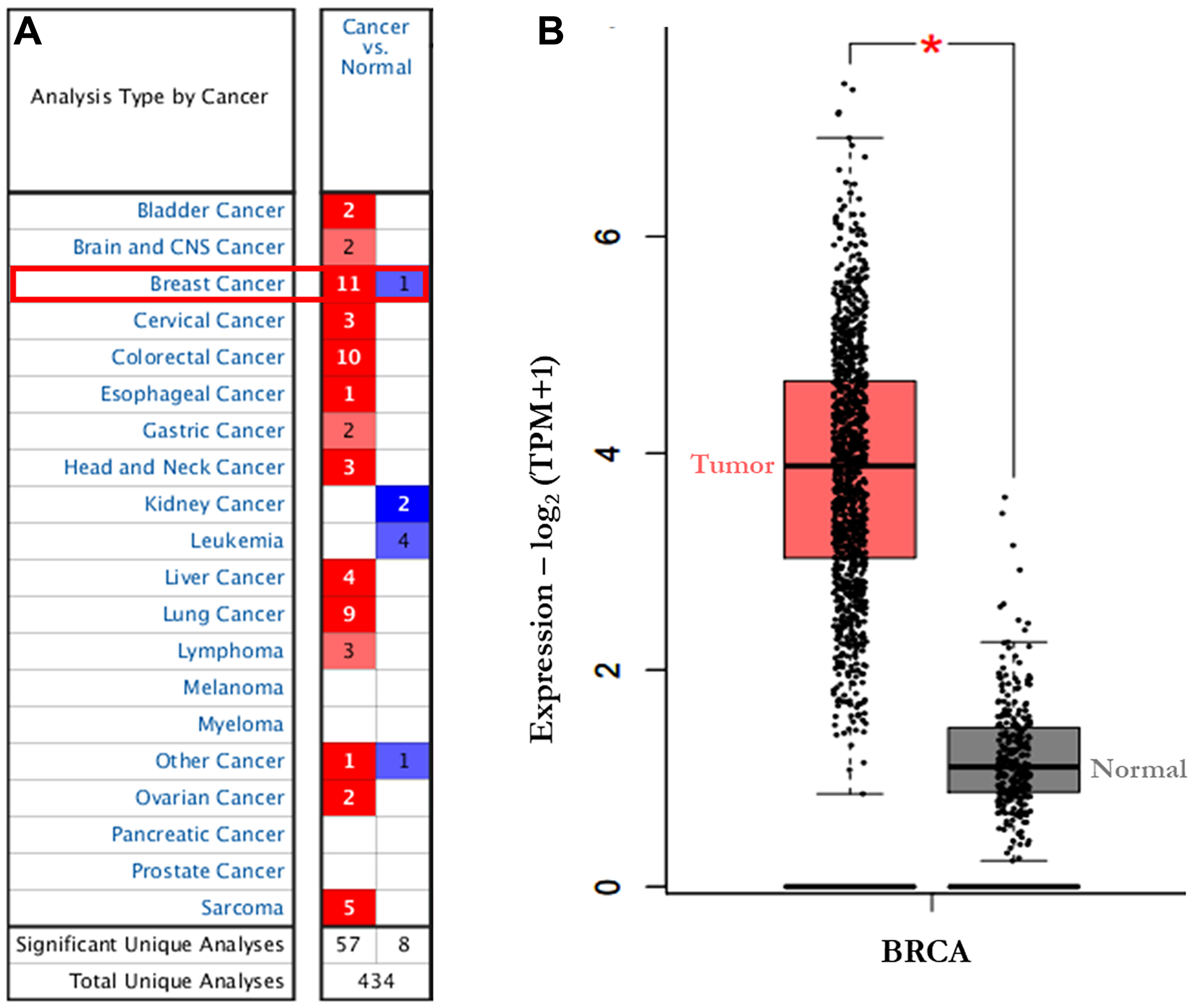
Figure 1: The transcription levels of AURKA in different types of cancers (Oncomine). The graphic demonstrated the numbers of datasets with statistically significant mRNA over-expression (red) or down-expression (blue) of the target gene. (A) the expression of AURKA in 20 datasets (Oncomine). (B) the expression of AURKA in BC (GEPIA). The threshold was designed with following parameters: Threshold (p-value): 1.0 × 10-6; threshold (fold change) × 2; and gene rank of top 5%; TPM: Transcript per million. Abbreviations: GEPIA, Gene Expression Profiling Interactive Analysis.
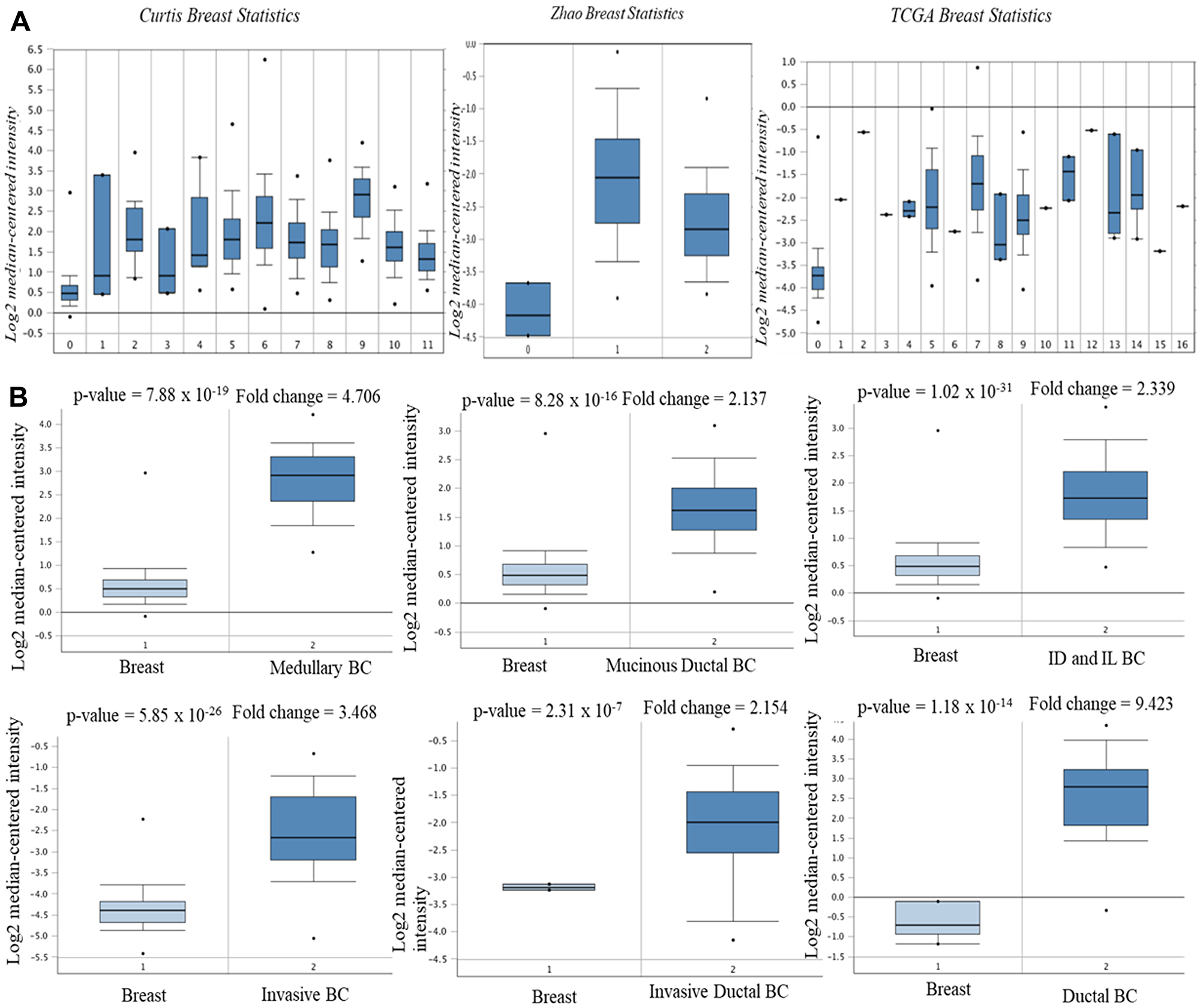
Figure 2: mRNA expression of AURKA in BC subtypes (Oncomine database). The three datasets (A) and the BC subtypes (B). Note: Curtis Breast Statistics: 0. Breast (B); 1. Benign Breast Neoplasm (BBN); 2. Breast Carcinoma (BC); 3. Breast Phyllodes Tumor (BPT); 4. Ductal Breast Carcinoma in situ (DBCi); 5. Invasive Breast Carcinoma (IBC); 6. Invasive Ductal Breast Carcinoma (IDBC); 7. Invasive ductal and invasive lobular breast carcinoma; 8. invasive lobular Breast Carcinoma; 9. Medullary Breast Carcinoma (MBC); 10. Mucinous Breast Carcinoma (MuBC); 11. Tubular Breast Carcinoma (TCB). Zhao Breast Statistics: 0. Breast; 1. Invasive ductal BC; 2. Lobular BC. TCGA Breast Statistics: 0. Breast; 1. Apocrine Breast Carcinoma; 2. Breast Large Cell Neuroendocrine Carcinoma; 3. Ductal Breast Carcinoma; 4. Intraductal Cribriform Breast Carcinoma; 5. Invasive Breast Carcinoma; 6. Invasive Cribriform Breast Carcinoma; 7. Invasive Ductal Breast Carcinoma; 8. Invasive Ductal and Lobular Carcinoma; 9. Invasive Lobular Carcinoma; 10. Invasive Papillary Breast Carcinoma; 11. Male Breast Carcinoma; 12. Metastatic Breast Carcinoma; 13. Mixed Lobular and Ductal Breast Carcinoma; 14. Mucinous Breast Carcinoma; 15. Papilary Breast Carcinoma; 16. Pleomorphic Breast Carcinoma.
Table 1: AURKA mRNA expression level in BC compared to normal tissues (Oncomine) in 12 datasets
| BC Subtype | Fold-change | P-value | Rank (%) | Sample | Source |
|---|---|---|---|---|---|
| Ductal BC | 9.423 | 1.18 × 10-14 | 1 | 47 | (Richardson et al., 2006) |
| Medullary BC | 4.706 | 7.88 × 10-19 | 1 | 2136 | (Curtis et al., 2012) |
| Invasive Ductal BC | 3.168 | 4.88 × 10-120 | 1 | 2136 | (Curtis et al., 2012) |
| Invasive Lobular BC | 2.115 | 5.80 × 10-44 | 2 | 2136 | (Curtis et al., 2012) |
| Invasive Ductal & Invasive Lobular BC | 2.339 | 1.02 × 10-31 | 2 | 2136 | (Curtis et al., 2012) |
| Invasive BC | 2.586 | 9.96 × 10-7 | 3 | 2136 | (Curtis et al., 2012) |
| Mucinous BC | 2.137 | 8.28 × 10-16 | 3 | 593 | TCGA, 2011 |
| Invasive Ductal BC | 4.702 | 5.39 × 10-53 | 1 | 593 | TCGA, 2011 |
| Invasive BC | 3.468 | 5.83 × 10-26 | 1 | 593 | TCGA, 2011 |
| Invasive Lobular BC | 2.351 | 7.20 × 10-14 | 2 | 64 | TCGA, 2011 |
| Invasive Ductal BC | 2.154 | 2.34 × 10-7 | 4 | 64 | (Zhao et al., 2004) |
| Invasive Breast Carcinoma Stroma | –11.071 | 2.11 × 10-30 | 2 | 59 | (Finak et al., 2008) |
Survival analysis of AURKA in BC
The prognostic significance of AURKA was assessed in all BC datasets using KM-plotter (Figure 3). The increased AURKA mRNA level was strongly associated with poor overall survival (OS), post-progression survival (PPS), distant metastasis free survival (DMFS) and recurrence-free survival (RFS). To validate this result, PrognoScan database was employed. The PrognoScan revealed that high expression of AURKA was significantly associated with poor OS, DFS, higher risk of distance metastasis and relapse at p < 0.05 (Figure 4A and 4B).
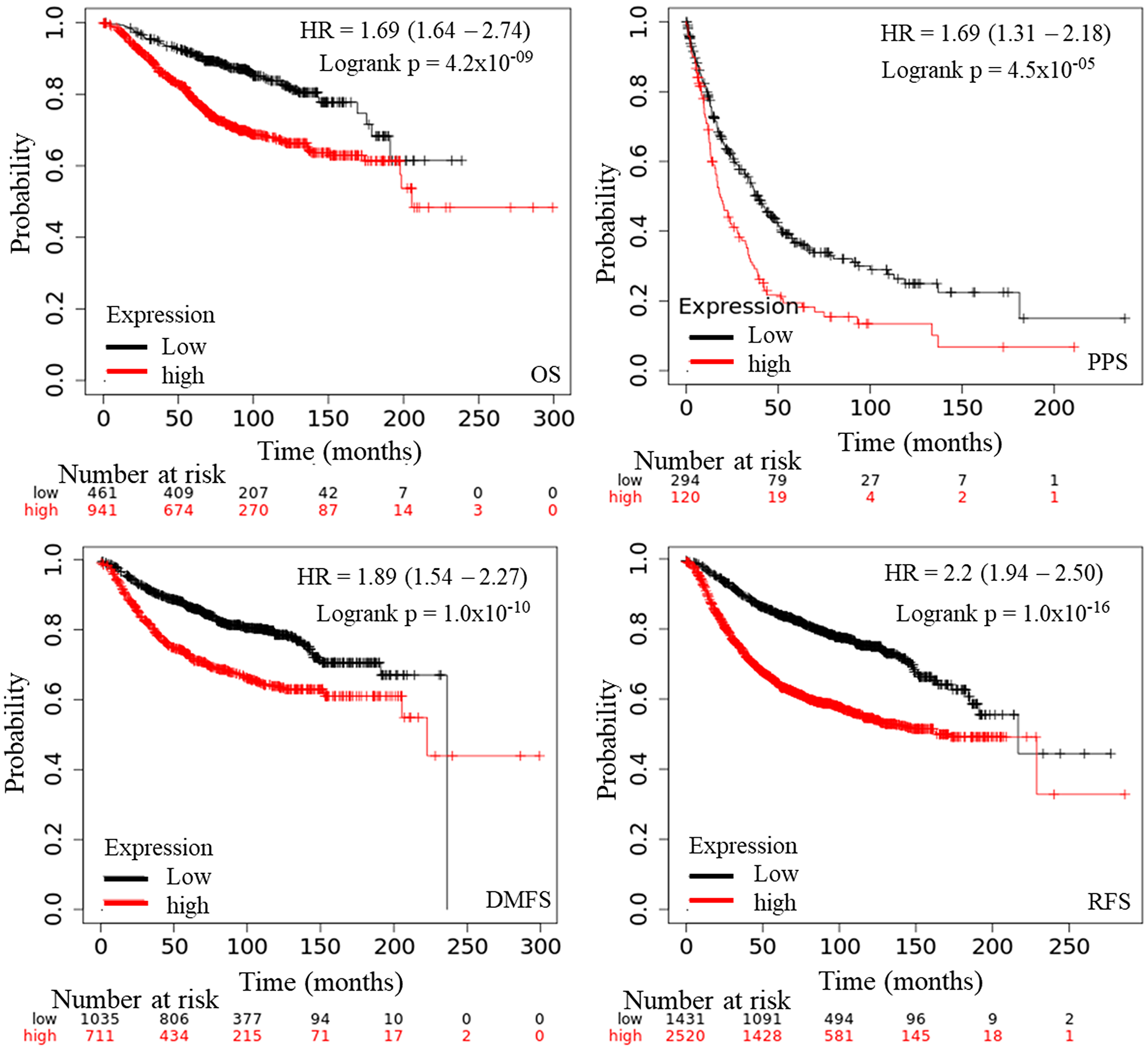
Figure 3: The prognostic value of mRNA level of AURKA in BC patients (Kaplan–Meier plotter). Notes: The OS, PPS, DMFS, and RFS survival curve comparing the patient with high (red) and low (black) AURKA expression in BC were plotted from Kaplan–Meier plotter database as the threshold of P-value < 0.05, respectively. Endpoints Affymetrix IDs: 208079_s_at. Abbreviations: OS: overall survival; PPS: progression free survival; RFS: relapses free survival; DMFS: distance metastasis free survival.
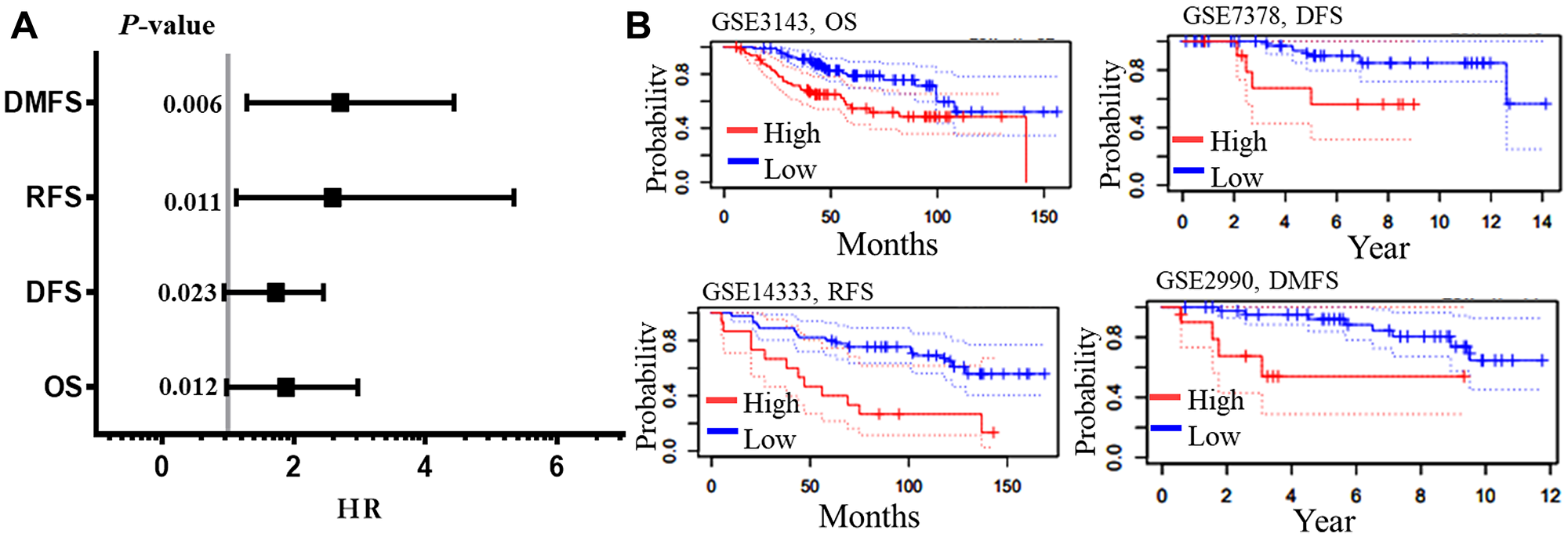
Figure 4: Survival analysis of AURKA in BC. The meta-analysis of the hazard ration with their p-values (A), and the four end points pictorial images from PrognoScan database. (B) The survival curves depict the high (Red) and low (blue) expressions with the datasets and endpoints. Abbreviations: OS: overall survival; DFS: disease free survival; RFS: relapses free survival; DMFS: distance metastasis free survival; HR: hazard ratio.
Prognostic values of AURKA in BC patients with different clinicopathological features
The association of AURKA gene with other clinicopathological features (such as; ER, PR, HERS2, lymph node status, TP53 status, as well as the BC’s pathological and clinical grades), and its correlation with these clinicopathological features was assessed in BC (Table 2). The mRNA expression level of AURKA was correlated with OS and DMFS of all patients with BC. In terms of ER status, increased mRNA expression of AURKA was associated with longer DMFS in ER-negative, but high mRNA expression of AURKA was linked to poor OS and DMFS in ER-positive and OS in ER-negative in BC patients. For ER status, increased expression was associated with poor prognosis both positive-ER in OS and DMFS and in negative-ER in OS. Only the ER-positive was related to better survival in DMFS. Conversely, the mRNA levels of AURKA is significantly higher in human epidermal growth factor receptor 2 (HER2) positive BC patients compared with the HER2-negative ones. Although there is significant correlation between the two subtypes, increased expression was associated with good prognosis in HER2-positive in both OS and DMFS while poor survival was observed in HER2-negative in OS and DMFS. For clinical lymph node status in BC patients, high mRNA expression of AURKA was associated with poor OS and DMFS in the subgroups (positive and negative nodal status). The high expression of AURKA mRNA was associated with improved OS and poor DMFS in pathological grade I BC patients; and poor OS and DMFS in pathological grade II BC patients. However, there is no significant association of AURKA with prognosis in grade III BC patients. Mutation of TP53 in cancers, including BC, is very common [32]. Its wild type codes for p53 which is involved in a number of physiological functions such as cellular senescence, metabolism, DNA repair, cell cycle arrest, programmed cell death, and many other processes following cellular stress [33]. Mutated TP53 may cause BC to metastasize as a result of altered p53 protein, which in turn fails to recognize and trigger apoptosis in cells with mutated genes. TP53 mutations are a negative prognostic factor in BC (Varna et al., 2011). Tumors with TP53 mutations are more likely to be aggressive (triple-negative (basal-like) or HER-2-positive (Luminal-B) BC) [34–36]. High mRNA expression level of AURKA was associated with poor OS and DMFS in wide type TP53 BC. However, high expression levels of this gene were not correlated with mutated-TP53-type BC. The welch’s test (bc-GenExMiner) was further used to analyze the subtypes of each clinicopathological features.
Table 2: The prognostic values of AURKA in BC patients with different clinicopathological features (Kaplan–Meier plotter: dataset 208079_s_at)
| AURKA | Clinicopathological | OS | DMFS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| features | Case | HR | 95% CI | p-value | Case | HR | 95%CI | p-value | |
| Overall | 1402 | 2.12 | 1.64–2.74 | 4.2 × 10–9 | 1746 | 1.87 | 1.54–2.27 | 1.0 × 10-10 | |
| ER stat | ER (+) | 548 | 2.2 | 1.54–3.13 | 7.7 × 10–6 | 664 | 2.74 | 1.96–3.82 | 8.4 × 10-10 |
| ER (–) | 251 | 1.33 | 0.83–2.12 | 0.23 | 218 | 0.54 | 0.34–0.85 | 0.0072 | |
| PR stat | PR (+) | 83 | 4.1 | 0.1–5.99 | 0.02 | 192 | 4.41 | 1.13–14.83 | 0.0088 |
| PR (–) | 89 | 2.7 | 0.96–7.57 | 0.05 | 154 | 0.66 | 0.34–1.27 | 0.21 | |
| HER2 | HER2 (+) | 129 | 0.37 | 0.15–0.9 | 0.023 | 126 | 0.42 | 0.21–0.83 | 0.0096 |
| HER2 (–) | 130 | 8.33 | 1.12–62.17 | 0.013 | 150 | 3.11 | 1.31–7.38 | 0.0068 | |
| LN stat | LN (+) | 313 | 1.59 | 1.05–2.42 | 0.029 | 382 | 1.78 | 1.21–2.63 | 0.0031 |
| LN (–) | 594 | 2.23 | 1.53–3.25 | 1.7 × 10–5 | 988 | 2.3 | 1.74–3.02 | 1.0 × 10-9 | |
| Grade | I | 161 | 4.888 | 1.41–16.9 | 0.0057 | 188 | 4.5 | 1.66–12.26 | 0.0013 |
| II | 387 | 2.02 | 1.31–3.11 | 0.0012 | 546 | 2.2 | 1.56–3.12 | 4.8 × 10-6 | |
| III | 503 | 1.35 | 0.97–1.87 | 0.074 | 458 | 1.22 | 0.86–1.73 | 0.27 | |
| TP53 | Mutated | 111 | 0.42 | 0.12–1.39 | 0.14 | 83 | 0.37 | 0.11–1.25 | 0.095 |
| Wide type | 187 | 3.36 | 1.31–8.63 | 0.0075 | 109 | 2.8 | 1.28–6.12 | 0.0071 | |
Genetic alterations of AURKA and clinicopathological parameters in BC patients
As depicted in Table 3, the transcription levels of AURKA were compared by bc-GenExMiner using the welch’s test among BC patients associated with some clinicopathological parameters [37]. These parameters include the age of the patient, together with statuses on the disease nodal, absence or presence of BC receptors, TP53, and so on. For age criterion, high expression of AURKA was significantly associated with patients above 51 years compared to patients below the age of 51 years. Also, BC patients with positive nodal status, mutated-TP53, basal-like and triple negative BC (TNBC) status had higher AURKA mRNA compared to those with negative-nodal, wild type-TP53, not-basal-like and not-TNBC statuses (Table 3). Receptor statuses (PR and ER) were negatively correlated with the AURKA expression. On the contrary, the AURKA mRNA levels were significantly increased in BC patients with higher expression levels of HER2 compared to the HER2-negative subtype (Table 3). In Sorlie’s intrinsic molecular subtypes, the components were associated with the higher mRNA level of AURKA but the basal-like and luminal B has no difference with respect to elevated level of AURKA (Figure 5).
Table 3: The relationship between mRNA expression of AURKA and clinicopathological features of BC
| Variables | AURKA | ||
|---|---|---|---|
| Case | mRNA | p-value | |
| Age | 0.0001 | ||
| ≥ 51 | 1387 | ↑ | |
| ≤ 51 | 1974 | — | |
| Nodal status | 0.0221 | ||
| + | 1574 | ↑ | |
| – | 2215 | — | |
| ER | 0.0001 | ||
| + | 3255 | ↓ | |
| – | 1177 | — | |
| PR | 0.0001 | ||
| + | 1335 | ↓ | |
| – | 917 | — | |
| HER2 | 0.0001 | ||
| + | 286 | ↑ | |
| – | 1711 | — | |
| TP53 Status | 0.0001 | ||
| Mutation | 247 | ↑ | |
| Wild Type | 523 | — | |
| Basal-Like Status | 0.0001 | ||
| Basal-Like | 962 | ↑ | |
| Not Basal-Like | 3549 | — | |
| Triple-Negative Status | 0.0001 | ||
| TNBC | 416 | ↑ | |
| Not | 3439 | — | |
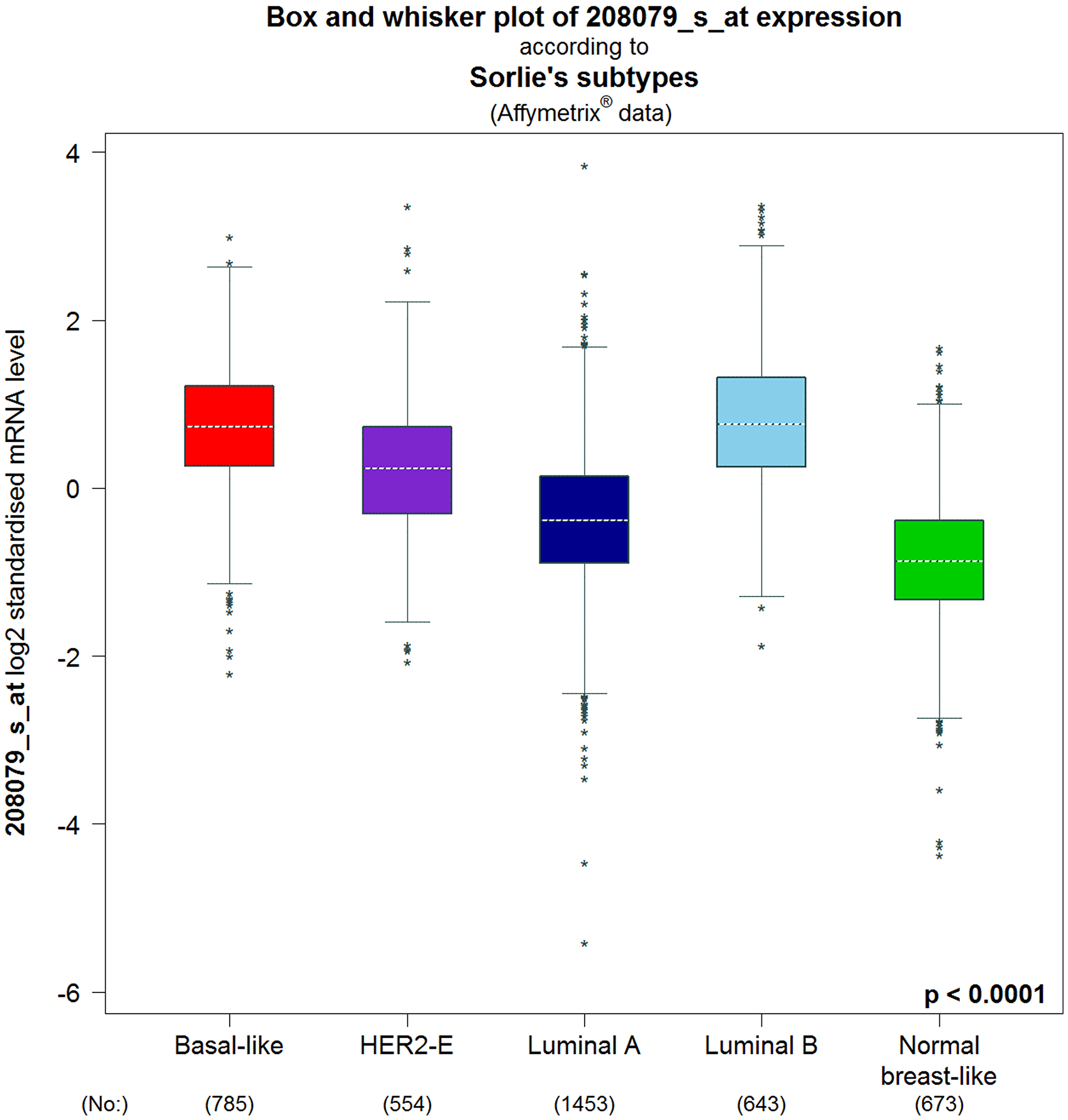
Figure 5: Intrinsic molecular subtype of AURKA in BC patients (bc-GenExMiner v4.4). The box plots are based on Intrinsic molecular subtype of AURKA. Correlation between AURKA expression and genetic information in BC patients. The significant different between groups was assessed by Welch’s test to generate p value, along with Dunnett-Tukey-Kramer’s and the P-value was set at 0.05.
Gene association of AURKA
The importance of PPis have been reported in biological processes and cellular function in both prokaryotes and eukaryotes including humans (normal and disease states) [38]. Identification of protein partners for AURKA can be an important lead to unravel the mechanisms of action for diseases typically cancer and further generate therapeutic drugs and treatment advancement. Numerous experimental approaches have been used for the discovery of interacting genes but the processes are demanding with limited outputs. However, computational approach has been increasingly used to validate and predict hidden protein partners. This study identified 10 proteins associated with AURKA genes through curated database entries, experimental validation, text mining, co-expression and or protein homology since proteins are constantly regulated and rarely function in isolation (Figure 6). These genes are crucial to the roles and specific molecular network of AURKA in disease, most importantly BC. The 10 associated proteins identified for AURKA at p < 0.05 include: Polo like kinase 1 (PLK1), centromere protein A (CENPA), DLG associated protein 5 (DLGAP5), TPX2, microtubule nucleation factor (TPX2), aurora kinase A (AURKA), baculoviral IAP repeat containing 5 (BIRC5), cell division cycle 20 (CDC20), cyclin B2 (CCNB2), cyclin dependent kinase 1 (CDK1), transforming acidic coiled-coil containing protein 3 (TACC3), ubiquitin conjugating enzyme E2 C (UBE2C), have a PPI enrichment p-value of 2.74 × 10-13, average node degree of 10 and average local clustering coefficient of 1.0. Further studies to give insights into the relationship of each of the identified genes with respect to AURKA expression in BC, is highly recommended.
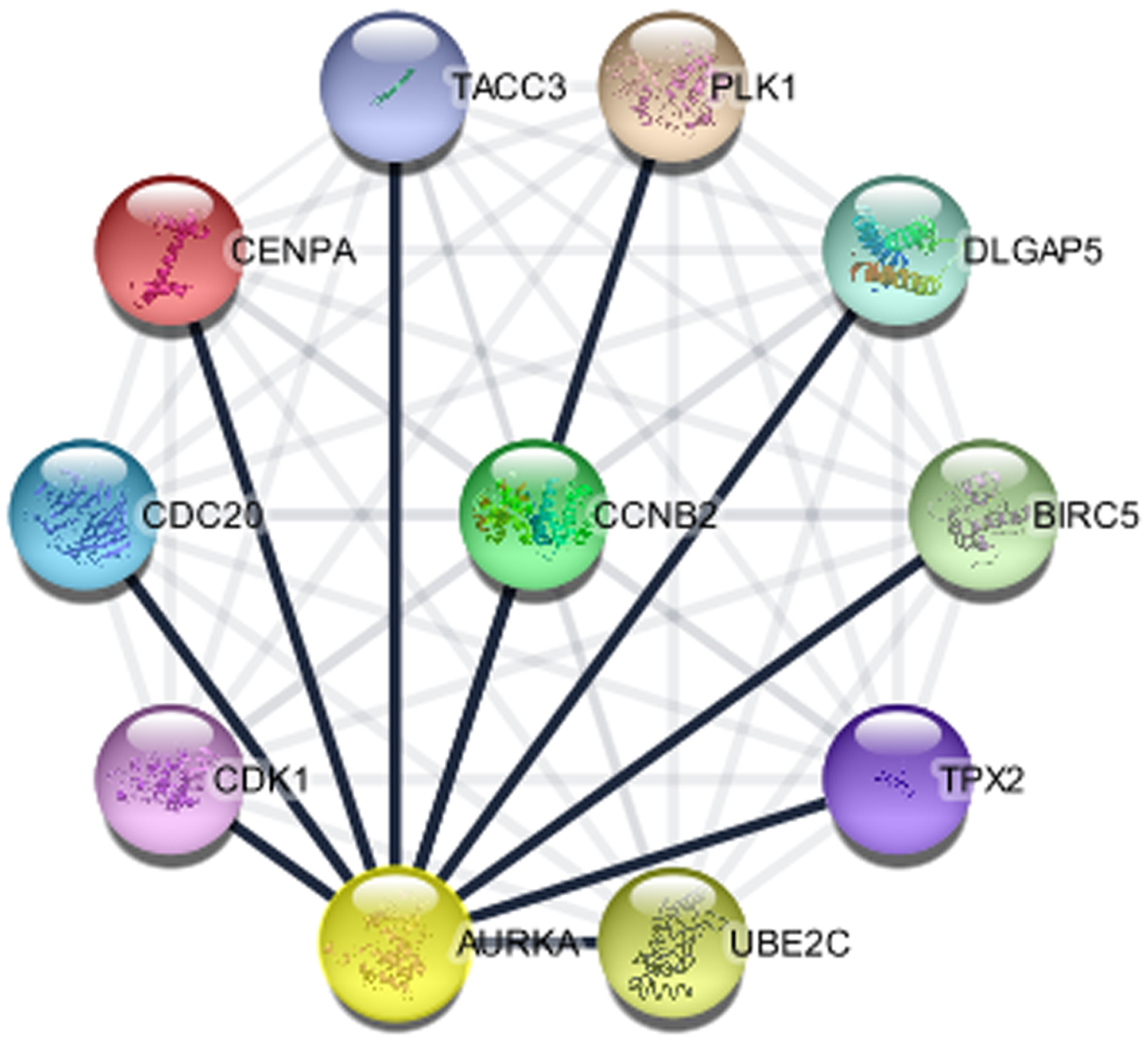
Figure 6: The protein interaction network of genes associated with AURKA (STRING in Cytoscape).
Functional enrichment and pathway analysis
The discovery of pathways and specific processes that are significant with factors regulating activities to genes of interest is important in cancer research. To identify functional categories and characteristic biological attributes of associated genes, gene oncology (GO) enrichment analysis was performed using DAVID database. DAVID gives a high-throughput and attractive data collection condition, and merge the functional genomic annotations with intuitive graphical representations encouraging the transition between genomic information and the biological meaning. The GENETIC_ASSOIATION_DB_DISEASE analysis was employed to evaluate AURKA gene targets associated with BC from the setup installation file of the genes from STRING database. Additionally, GO provides classifications of genes in relation to their molecular and cellular structures and functions [39].
The three categories of GO terms include the biological process (BP), cellular component (CC), and molecular function (MF). The KEGG pathway in DAVID also contains adequate information of known metabolic and regulatory pathways and accelerates the mapping of genes to KEGG pathways for systemic analysis of gene functions [40].
To explore the functions as well as the pathways crucial to the associated genes in relation to BC, GO and KEGG pathway enrichment analyses were carried out for the protein partners as presented in Table 4. The p-value is the probability that the chosen gene for any of the three GO term categories occurred by chance. Therefore, a low p-value corresponds to a greater likelihood of significance set at the false discovery rate (FDR) <0.01. The AURKA associated proteins are mainly involved in all three GO term categories in BP ontology (cell division), MF ontology (protein binding), and CC ontology (spindle). KEGG pathway indicated that the AURKA and associated genes were mainly enriched in pathways such as cell cycle, progesterone-mediated oocyte maturation, oocyte meiosis, and p53 signaling pathway.
Table 4: Functional and pathway enrichment analysis of AURKA and its associated genes (DAVID)
| Category | Term | Count | % | p-value |
|---|---|---|---|---|
| GOTERM_BP_DIRECT | cell division | 8 | 72.7 | 1.80 × 10-10 |
| GOTERM_BP_DIRECT | mitotic nuclear division | 7 | 63.6 | 2.00 × 10-09 |
| GOTERM_BP_DIRECT | G2/M transition of mitotic cell cycle | 6 | 54.5 | 8.20 × 10-09 |
| GOTERM_BP_DIRECT | anaphase-promoting complex-dependent catabolic process | 5 | 45.5 | 9.30 × 10-08 |
| GOTERM_BP_DIRECT | regulation of ubiquitin-protein ligase activity involved in mitotic cell cycle | 4 | 36.4 | 2.70 × 10-07 |
| GOTERM_CC_DIRECT | spindle | 5 | 45.5 | 3.80 × 10-07 |
| GOTERM_CC_DIRECT | microtubule cytoskeleton | 5 | 45.5 | 6.20 × 10-07 |
| GOTERM_CC_DIRECT | spindle microtubule | 4 | 36.4 | 1.60 × 10-06 |
| GOTERM_CC_DIRECT | nucleoplasm | 9 | 81.8 | 9.90 × 10-06 |
| GOTERM_CC_DIRECT | spindle pole | 4 | 36.4 | 2.40 × 10-05 |
| GOTERM_MF_DIRECT | protein binding | 11 | 100 | 1.50 × 10-03 |
| GOTERM_MF_DIRECT | anaphase-promoting complex binding | 2 | 18.2 | 3.50 × 10-03 |
| GOTERM_MF_DIRECT | ATP binding | 5 | 45.5 | 8.30 × 10-03 |
| GOTERM_MF_DIRECT | protein kinase activity | 3 | 27.3 | 1.80 × 10-02 |
| GOTERM_MF_DIRECT | protein serine/threonine kinase activity | 3 | 27.3 | 2.00 × 10-02 |
| KEGG_PATHWAY | Oocyte meiosis | 5 | 45.5 | 2.20 × 10-06 |
| KEGG_PATHWAY | Cell cycle | 4 | 36.4 | 1.90 × 10-04 |
| KEGG_PATHWAY | Progesterone-mediated oocyte maturation | 3 | 27.3 | 3.20 × 10-03 |
| KEGG_PATHWAY | p53 signaling pathway | 2 | 18.2 | 6.60 × 10-02 |
Receptor and ligand structure preparation and docking
The hAgo2, microRNA, and microRNA-AURKA were considered for the molecular docking analysis in order to evaluate the mechanism of regulation of AURKA in BC (Figure 7). The concept of ligand preparation was to produce corresponding low energy 3D structures from the format generated from RNA-composer with the option to expand each input structures by generating variation on the ionization states, tautomers, stereochemistry and ring conformation (Figure 7A and 7B). The ligands (hsa-miR-32-3p and microRNA-AURKA duplex) were prepared using LigPrep, a module in Maestro (Figure 7C). The receptor, hAgo2 was prepared and verified by Schrodinger suite and PDBSum PROCHEK, respectively (Figure 7D and 7E). Briefly, Protein preparation module in Schrodinger (Maestro v12.2) was employed to ascertain the readiness of the protein for docking by the following steps: (a) Pre-processing (basic task): Assignment of bond orders, filling of missing side chains and loops by prime, and deletion of water molecules beyond 5 Å from het groups; (b) Review and modification: inspection and correction of ionization and the tautomeric state of the het groups present in the receptor using Epik at a target pH range of 7.0+/–2.0; (c) Refinement: Optimization of the orientation of hydrogen bond network (PROPKA at pH 7.0) and structural restrained minimization by force field Optimized Potentials for Liquid Simulations (OPLS_2005) at root mean square deviation (RMSD) of 0.30 Å. As shown in Figure 7D, the crystal structure of hAgo-2 which was used in this study was stable and obeyed theoretical predication.
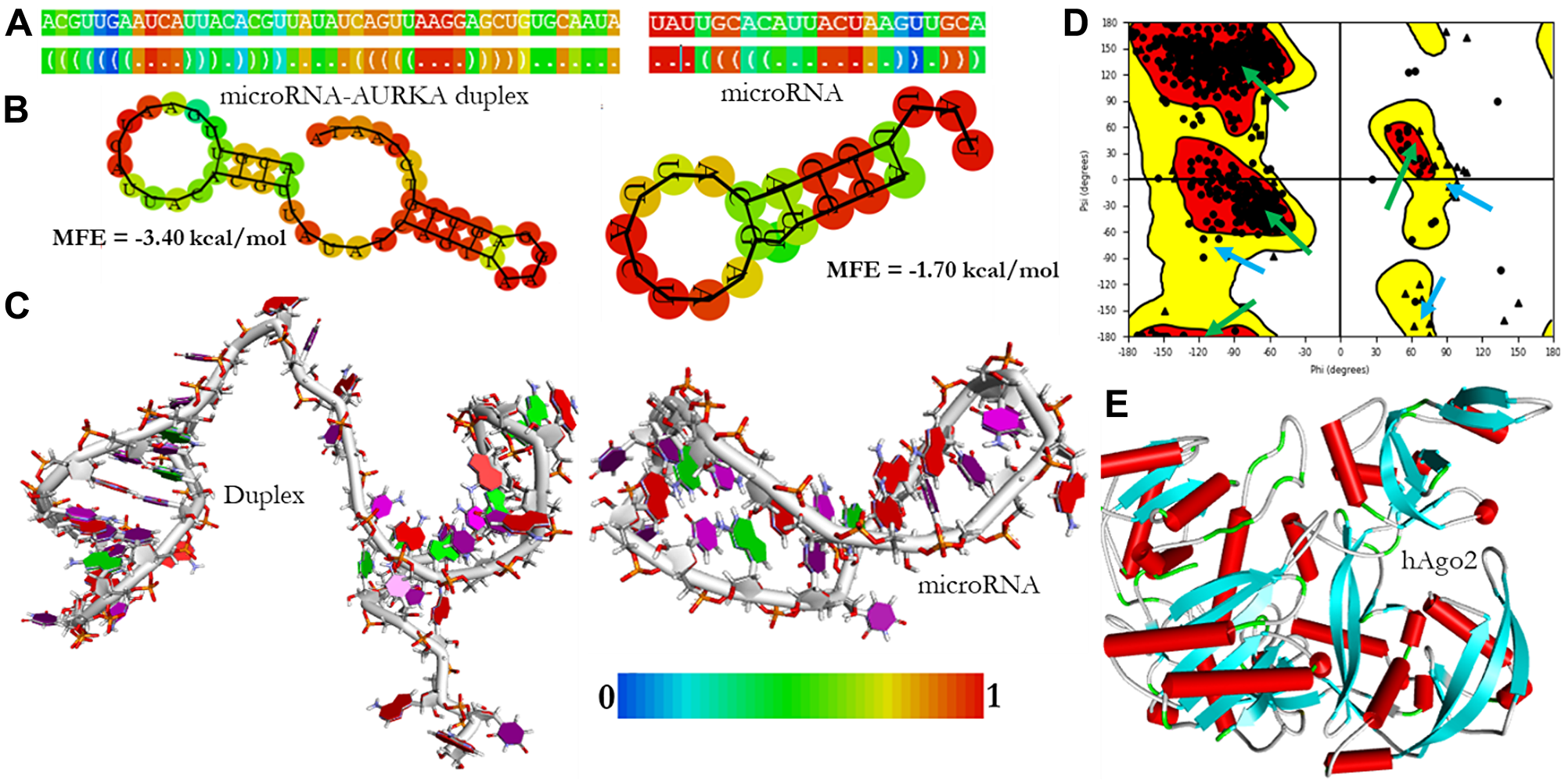
Figure 7: The structural models of microRNA and microRNA-AURKA duplex. The dot-bracket notations are colored by base-pairing probability (A), the secondary structures are colored by positional entropy (B), the 3D structures were modeled and visualized by DSV v19 (C), the Ramachandra plot of the prepared hAgo2 receptor was verified by PDBSum PROCHEK. The area marked with green arrows are residues in the most favored region while the regions marked with teal arrows are residues in additional allowed region (D) and the 3D model of the prepared hAgo2 was done by Maestro v12.2 (E).
Molecular docking analysis
Docking analysis
PatchDock web-based docking algorithm was employed for the docking profile of AURKA and hAgo-2 alongside miR-32-3p. The PDB file of hAgo-2 protein and the ligands were used as inputs with clustering RMSD of 1.5 Å. The generated complexes were based on their geometric shape complementarity score. The results with the highest score were chosen for each of the ligands and hAgo-2 (Table 5). The binding affinity of the complex was assessed through their binding scores, and interacting amino acid residues between the ligands and the receptor protein (Tables 6 and 7 and Figures 8 and 9). The result revealed that hAgo-2 protein formed 10 pairs of hydrogen bonds and 3 pairs of Pi interactions with miR-32-3p-AURKA duplex with 59 amino acids involved in the binding cavity of the hAgo-2 protein compared to the 40 amino acids residues found in the pocked of hAgo-2 protein when interacting with miR-32-3p and six pains of hydrogen bonds which demonstrated the complexes are highly stable.
Table 5: The docking scores between microRNA, microRNA-AURKA duplex and hAGO-2 protein
| Category | Score | Area | ACE | No of AAs |
|---|---|---|---|---|
| miR-32-hAgo2 | 16038 | 3113.20 | –434.34 | 40 |
| Duplex-hAgo2 | 24104 | 4130.40 | –379.12 | 59 |
Table 6: Molecular docking analysis results of the ligands and receptor with participating aa residues (3.5 Å)
| Category | AA involved | Aromatic AA | Hydrophobic AA | AA-H-bonds |
|---|---|---|---|---|
| microRNA-hAgo2 | 40a | PHE156 | LEU265 | SER828 |
| TYR174 | ILE353 | GLU821 | ||
| THR357 | VAL237 | THR830 | ||
| TYR225 | LEU238 | ASP823 | ||
| VAL256 | LYS550 | |||
| ILE353 | ASP499 | |||
| LEU356 | HIS829 | |||
| ALA603 | ||||
| ALA825 | ||||
| Duplex-hAgo2 | 59a | PHE156 | ILE159 | ARG69 |
| THR526 | ILE353 | ARG287 | ||
| THR285 | VAL206 | GLU826 | ||
| TYR338 | VAL284 | ARG286 | ||
| ALA825 | ASP252 | |||
| LYS65 | ||||
| SER828 | ||||
| SER824 | ||||
| GLU821 | ||||
| GLU821 | ||||
| PRO523 |
Table 7: Hydrogen bonds and their respective residues between the ligands and receptor within the distance of 2 Å
| Complex | AA residue | Atoms | Nucleic acid residue | Distance |
|---|---|---|---|---|
| MicroRNA-hAgo2 | SER828 | HG - O2 | C21 | 1.9 |
| GLU821 | O - H3 | U4 | 1.9 | |
| THR830 | H - OP2 | A22 | 1.3 | |
| ASP823 | O - H22 | G5 | 1.9 | |
| LYS550 | HE2 - O5′ | A15 | 1.9 | |
| ASP499 | OD1 - H5′′ | U4 | 2.0 | |
| HIS829 | O - H8 | A22 | 1.6 | |
| Duplex-hAgo2 | ARG69 | H – O4 | U39 | 1.7 |
| ARG287 | HE - O2 | U22 | 2.0 | |
| GLU826 | O - HO2′ | G18 | 1.9 | |
| ARG286 | HD2 - O3′ | U22 | 1.4 | |
| ASP252 | OD2 - H1′ | U19 | 1.7 | |
| LYS65 | O – H5 | U37 | 2.0 | |
| SER828 | HB2 - O2′ | G18 | 1.8 | |
| SER824 | OG - H1′ | U4 | 2.0 | |
| GLU821 | OE1 - H2′ | U4 | 1.9 | |
| GLU821 | OE2 - H5′ | U5 | 1.6 | |
| PRO523 | O - H8 | A7 | 1.9 |
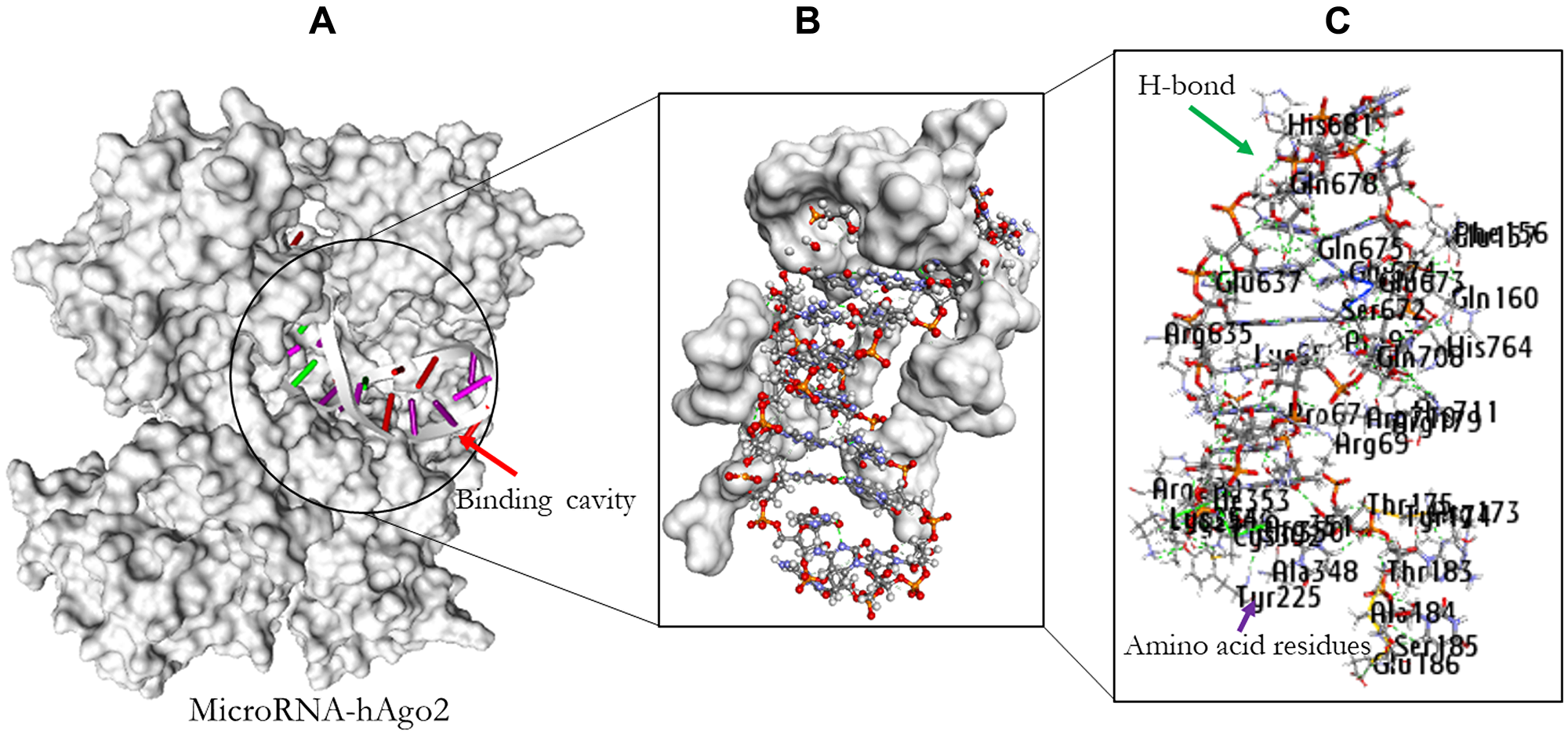
Figure 8: Molecular docking result of miR-32 and hAgo2. The binding position of the miR-32 in the pocket of hAgo2 (A); Residual amino acids participating in their interaction (B); Amino acid residues (C).
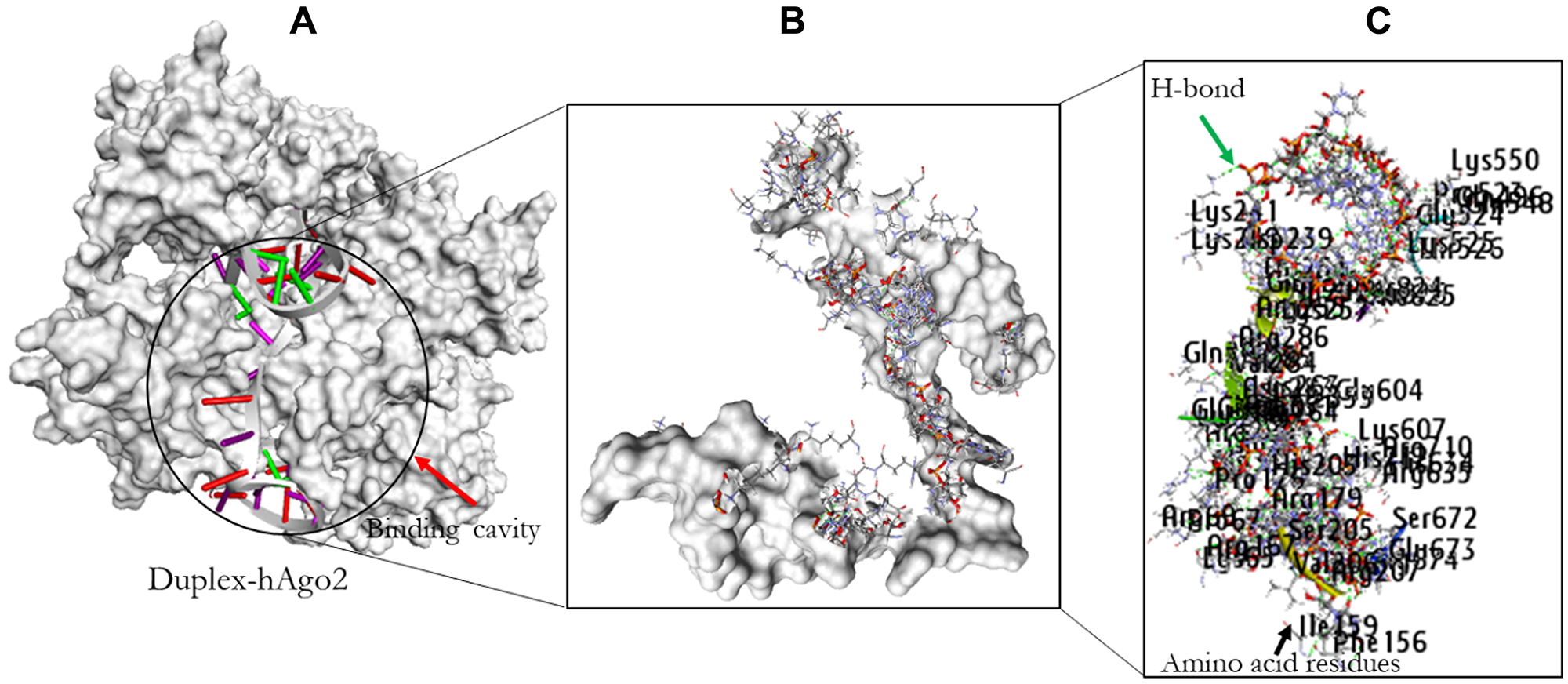
Figure 9: The docking result of miR-32-AURKA and hAgo2. The binding position of the complex in the pocket of hAgo2 (A); Residual amino acids participating in their interaction (B); Amino acid residues (C).
DISCUSSION
The process of carcinogenesis and therapeutic responses present a terrific challenge to favorable therapeutic outcome [41]. However, identifying cancer specific targets can pave the way for effective treatment options against specific cancer types [42]. Computational approaches are extensively used to investigate molecular and genetic mechanism of cancer progression by identifying lead genes and the abnormal regulatory pathways of disease [43]. In cancer research, these methods have revealed molecular targets for therapeutic intervention and cancer mechanisms (development, progression, or metastasis) in a systematical, accurate and effective manner [44]. Additionally, due to cancer complexity, it is crucial to investigate the molecular basis and identify significant therapeutic targets of this disease. Recently, researches aimed at identifying the mode of action of BC have increased. Numerous therapeutic interventions have been developed [45]. Nevertheless, these strategies induce a range of therapeutic responses and resistance can develop in BC patients, therefore novel therapeutic intervention is desirable.
Due to the importance of microRNAs in carcinogenesis, they suggest potential alternative therapeutic targets for cancer [46]. Specifically, Polini et al. [47] revealed the tumor suppressive role of miR-193a and further suggested that their expression could be attributed to novel treatment for cutaneous melanoma patients.
In this study, the role of AURKA gene expression with clinicopathological details was investigated using in silico and molecular docking study and proposed that the expression of AURKA gene may be regulated by miR-32-3p through RNA induced silencing complex in BC. AURKA is a serine/threonine kinase involved in the regulation of mitotic chromosomal segregation and centrosome function. The conservation of AURKA gene has been previously reported in various species including human [48]. Based on its critical role in cancer biology, it may be of great importance to evaluate the status for selecting the most effective therapeutic options most especially in BC.
To evaluate whether a gene have therapeutic target potential, it often needs to be uniquely expressed or highly expressed in tumors than in most normal tissues. Gene expression profiles of 434 total unique analyses in Oncomine were employed to perform bioinformatics analysis in this study. The expression of AURKA was significantly over-expressed in 11 out of 12 BC datasets. The overexpression could be correlated with high occurrence of BC [49, 50]. This result was validated by GEPIA and the mRNA expression level of AURKA was found to be significantly upregulated in BC than normal breast tissues. Studies have shown that AURKA is involved in multiple mechanisms-associated with cancer initiation [51]. AURKA overexpression was also suggested to increase the expression of MMP-2, MMP-7, MMP-9, leading to tumor metastasis by degrading extracellular matrix proteins [52–54]. Zhong et al. [55] identified AURKA as potential therapeutic targets in glioblastoma-bearing rats. Therefore, AURKA is an attractive target for cancer therapy.
The transcription levels of AURKA in different BC types were remarkably higher than those in normal tissues, and associated with poorer OS, PPS, DMFS, RFS, and DFS. As evident, the overall clinicopathological features in this study shows that overexpression of this gene was correlated with poor survival. Interestingly, high levels of AURKA was associated with poor OS and DMFS in ER status, PR status, HER2 status, grade stage I and II, and in wide-TP53-type BC patients.
Protein-protein interaction (PPi) network analysis demonstrated other 10 significant genes correlated with AURKA. To further clarify the mechanism of AURKA, a network was constructed for AURKA and the 10 neighboring genes. GO and KEGG analyses indicated that these genes were mainly enriched in a number of GO terms. The functional and pathway enrichment analyses were highly consistent with the findings that p53 signaling, cell cycle and cell division ontology functions were regulated abnormally in BC [56]. This study contributed to the growing evidence regarding the correlated signaling pathway of AURKA which could offer great opportunity into the development of biomarkers for the diagnosis and prognosis of BC.
Phosphorylation/dephosphorylation have been reported as the two predominant mechanisms regulating AURKA [57]. The gene was phosphorylated at three amino acid residues, Serine-53, Threonine-295, and Serine-349. Mutation in the amino acid residues Threonine-295 and Serine-349 is associated with reduced activity. Phosphorylation of Threonine-295 is required for protein kinase (PKA) activation which then phosphorylate (Threonine-295) and activate the kinase in vitro [58]. Protein phosphatase-1 negatively regulates Aurora A by dephosphorylating T288 [59]. Additionally, studies show that ncRNAs regulate gene expression by guiding Argonaute proteins to complementary sites on target RNA molecules [60–63]. The expression of microRNAs has been reported in cancers [64–69]. Specifically, microRNAs such are miR-346 and miR-361-3p were suggested to modulate the expression of PSA, TMPRSS2 and DRG1 in prostate cancer [70].
The first microRNA to be reported in the dysregulation of BC was in 2005 [71]. The miR-155 expression was upregulated in BC, its expression was associated with clinicopathological markers, tumor subtype, and poor survival rates [72, 73]. MiR-665 expression was also reported to predicts poor survival and promotion of tumor metastasis by targeting NR4A3 in BC [74]. In addition, deletion of CRISPR/Cas9 suggested the oncogenic roles of miR-23b and miR-27b in BC [75]. Mature microRNAs assist AGO by guiding the complex to target sites in mRNAs that are partially complementary to the microRNA sequence (the seed region) [76], and induce repression of gene expression at the level of mRNA stability or translation [77]. Several proteins interfere with mRNA degradation and translational repression, some of them are necessary components of the RNA induced silencing complex (RISC) that transports those microRNAs to complementary sites within mRNA [78]. The human AGO-2 is a member of the AGO subfamily that is commonly expressed and associate with microRNAs. Although different classes of ncRNAs have different biogenesis pathways and exert diverse functions, all of them must associate with any AGO subfamily for activity [77].
Expression of AURKA among other genes was directly inhibited by miR-186 in neuroblastoma cells [79]. The epigenetic regulation of AURKA by miR-4715-3p has also been reported in gastrointestinal cancers using in silico prediction [23]. MicroRNA-490-3p suppresses hepatocellular carcinoma cell proliferation and migration by targeting AURKA [80]. Importantly, the expression of miR-32-3p has been reported to suppress AURKA but the mechanism of action deserves closer attention at both atomic and molecular levels [19]. The possible regulation of AURKA expression was investigated at both atomic and molecular level by evaluating the binding interaction between miR-32-hAgo-2 complex and miR-32-AURKA duplex bound hAgo-2 using molecular docking study. The pool of microRNAs targeting AURKA was assessed by TargetScan. Furthermore, their minimum folding energy as well as their scores were carried out by miRTarBase database. The result showed that miR-32 has the highest score of 164.00 and the lowest minimum energy of -14.20 kcal/mol. This microRNA was further evaluated for its binding activity against AURKA gene using molecular docking analysis. For the secondary folding analysis, determined by the RNAfold, the minimum folding energy (MFE) of the microRNA and microRNA-AURKA duplex were -1.70 kcal/mol and -3.40 kcal/mol, respectively. Three properties (composition, sequence length, and conformation) have been reported to be significant to RNA molecule [81]. Trotta [81] reported that longer sequences are more stable due to their ability to form stacking and hydrogen bond interactions. Sequence arrangement is also another determining factor that affects the folding structure stability. The low MFE of the microRNA was justified by the sequence length of 22 nucleotides and the number of loops formed. The duplex sequence length of 45 nucleotides and extension of loops, are great advantages to confer stability for possible regulation.
In nature, the stability of molecular binding interactions between ligand and its corresponding receptor depends largely on specific amino acids involved (hydrophobic and aromatic amino acid). The interaction between miR-32 and miR-32-AURKA duplex and the human argonaute protein were evaluated through these aforementioned amino acid residues observed in the binding cavity within specific distance. The result revealed the presence of specific interactive residues of hAgo-2 involved in binding of the ligands. Also hydrogen bonds were observed between the interaction atoms of the receptor and the ligands. These interactions taken together, may be important and thus confer regulation of AURKA gene most especially in BC. Furthermore, the number of hydrogen bonds involved in the interacting atoms of hAgo-2 and miR-32-AURKA complex together with the amino acid residues involved in binding, and number of important amino acids such as hydrophobic and aromatic amino in the cavity of hAgo-2 are significantly higher compared to the binding interaction observed in miR-32-hAgo-2 complex. This could therefore, confer stability to the binding of hAgo-2 to AURKA for better regulation.
MATERIALS AND METHODS
Expression analysis of AURKA
Web-based data-mining platforms were used to evaluate the expression (Oncomine) of AURKA and further validated using Gene Expression Profiling Interactive Analysis (GEPIA-2) database. Oncomine is a database that contains cancer microarrays from genome-wide expression analyses [82], available at https://www.oncomine.org/. Expression of AURKA was assessed in twenty cancer types and their normal counterparts, and also compared among BC subtypes relative to normal clinical mRNA datasets. Only expression that showed a 2-fold difference between cancer and normal tissues, a p-value of 1.0 × 10-6, and a priority of 5% were considered.
The mRNA levels of AURKA in BC at a p-value less than 0.05 were validated by GEPIA-2 database at http://gepia2.cancer-pku.cn/#analysis. TCGA and GTEx datasets were used to match the tumor as normal data at the cutoff of |Log2FC|of 1.
Prognosis and expression correlation of AURKA
The statistical exploratory database (Breast cancer Gene expression Miner v4.5) assessed at http://bcgenex.centregauducheau.fr/BC-GEM/GEM-requete.php consists of BC transcriptomic data (10, 0001 DNA microarrays and 4, 712 RNA-seq) [37, 83]. This software offers the possibility to explore expression of genes of interest in BC and the statistical analyses are categorized as correlation, expression and prognosis. This tool was used to study the AURKA expression in BC, the correlation between AURKA mRNA levels and clinicopathological features in BC clinical dataset (Probe set ID: 208079_s_at).
Survival analysis of AURKA in BC
The Kaplan-Meier database (KM-plot) is a manually curated web-based tool handled by a PostgreSQL server assessed at https://kmplot.com/analysis/index.php?p=service&cancer=breast. This plotter is composed of gene expression and clinical data, and is used to evaluate the prognostic value of a biomarker. The expression of AURKA gene in BC patients in relation to survival rate was analyzed by KM-plot as a function of log-rank p-value, and the hazard ratio of 95% confidence intervals. The database can assess 54, 000 cancer-linked genes from 21 cancer types, with breast as the largest dataset [84]. This database is a repository for the meta-analysis based on the discovery and validation of biomarkers from the cancer survivors [85].
Prognostic and meta-analysis of AURKA in BC (PrognoScan)
PrognoScan is a web-based tool for meta-analysis of prognostic value of genes. With its relatively simple user interface, it correlates gene expression with patient prognosis on the available cancer microarray datasets. Advantages of this tool include its large collection of publicly available cancer microarray datasets with clinical annotation and the assessment of the biological relationship between gene expression and prognosis. Furthermore, it utilizes the minimum p-value approach for classifying patients for survival analysis that evaluates the optimal cutpoint in continuous gene expression estimation without prior biological knowledge or assumption and therefore, enables systematic meta-analysis of numerous datasets [86]. This tool assessed at http://dna00.bio.kyutech.ac.jp/PrognoScan/ was used to evaluate AURKA as a potential biomarker and a possible BC therapeutic target.
AURKA interaction analysis (STRING and Cytoscape)
The Search Tool for the Retrieval of Interacting Genes (STRING) database accessed at http://www.string-db.org/ provides extensive view of pre-existing, predicted interactions, and their associated proteins. To explore the co-expressed protein alongside AURKA gene, the proteins that associates with the AURKA gene was predicted and visualized by Cytoscape at http://www.cytoscape.org/. The output of this analysis is an interaction network showing various interacted genes as nodes. The AURKA node was taken as the “hub” genes in the protein network at a confidence level of 0.90. Additionally, the visual display was then adjusted (nodes, edges, and network) using appropriate layout and plugins to achieve a less dense/ clustered image. An additional interaction was further produced to evaluate their specificity to BC.
Functional enrichment and pathway analysis
Standard gene IDs for a list of genes identified by STRING associated with AURKA were used as input in database for annotation, visualization and integrated discovery (DAVID database) in order to examine the gene functions and disease pathways by annotation of the protein partners alongside AURKA gene. This database was accessed at https://david.ncifcrf.gov/summary.jsp. The web-based tool displays the annotation summary results with several options. For the link gene-disease associations, the GAD_DISEASE was prioritized. The observed result ranked the AURKA-associated genes in different cancer types and processes with the special emphasis on the BC for this study. The gene ontology and pathway analysis were further analysed using this database.
Dataset selection and docking study
Datasets
MiRTarBase v8.0 at http://mirtarbase.mbc.nctu.edu.tw/ is a freely available web-based tool for microRNA-gene interactions. These interactions were experimentally validated through molecular assays; MiRTarBase contains the largest microRNA to targets interactions (MTIs) datasets. Additionally, this tool provides the up to date MTIs collection which are cross referenced with other related databases [87]. The target microRNA sequence of AURKA was predicted by miRanda v10. This tool was used to retrieve the microRNA, and microRNA-target interaction sequences for downstream analysis.
The TargetScanHuman v7.2 is a microRNA target prediction tool that predicts the molecular targets for microRNAs that matches the 8mer, 7mer, and 6mer conserved regions in the seed region of each microRNA (http://www.targetscan.org/vert_72/) [88]. TargetScanHuman was used to confirm the target of the microRNA of interest.
RNAfold database at http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi was used to predict the secondary structures, the minimum folding energy and doc-bracket notations of the microRNA and microRNA-AURKA duplex with the command line: RNAfold -p -d2 --noLP < sequence1.fa > sequence1.out. Furthermore, RNA-composer, a fully automated RNA structure modeling server at http://rnacomposer.cs.put.poznan.pl/ was used to generate a 3D PDB file format of the doc-bracket notations [89]. The crystal structure of (hAgo2) (PDB ID: 4F3T; resolution: 2.25 Å) was retrieved through the Protein Databank (PDB) for docking analysis.
Protein preparation, docking, and visualization analyses
The molecular docking analysis of AURKA and microRNA of interest was performed by Schrodinger suite, PATCHDOCK, and Discovery Studio Visualizer (DSV) following the approach described by Fadaka et al. [90].
Statistical analysis
The graph pad prism 6.0 software was used to analyze all the data in this study. Specifically, ANOVA and Student’s t-test were utilized to compute the clinical data between tests and controls. The ratio of the groups was correlated by the chi-square test. The odds ratio and 95% confidence interval were determined to assess the relationship between the gene of interest and BC risk through logistic analysis. All values were considered statistical significant at p < 0.05.
Conclusions
The expression level of AURKA was significantly upregulated in patients with BC. Aberrant AURKA expression was found to be associated with poor prognosis of BC. The interactions observed within the distance of 3.5 Å and the hydrogen bonds within the distance of 2.0 Å were supportive that AURKA regulation through the AGO protein could be driven by miR-32-3p. These observed interactions are crucial to protein folding, stability and in the binding of targets. Based on the past and recent research on the roles of ncRNAs in the field of cancer, it can be inferred that therapeutics associated with microRNAs could provide desired therapeutic outcomes in cancer patients and related diseases. This study therefore, provides insight into the mode of AURKA regulation by miR-32-3p in BC. Formulations with ncRNAs can be more effective as therapeutic approaches and may represent a novel therapeutic intervention in BC and other cancer subtypes.
Abbreviations
BC: Breast cancer; OS: Overall survival; PPS: Post-progression survival; DMSF: Distant metastasis free survival; RFS: Recurrence-free survival; hAGO2: Human Argonaute protein-2; AA: Amino acids; RISC: RNA induced silencing complex; H-bond: Hydrogen bond; BP: Biological process; MF: Molecular function; CC: Cellular component.
ACKNOWLEDGMENTS
The study was supported by the UWC Biolabels Node. Furthermore, authors would like to acknowledge the National Integrated Cyberinfrastructure system, Center for High Performance Computing (CHPC), Department of Science and Technology, Republic of South Africa for the license to the Lengau cluster and other modules under the Schrodinger suite.
Availability of data and material
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492. [PubMed].
2. Azubuike SO, Muirhead C, Hayes L, McNally R. Rising global burden of breast cancer: the case of sub-Saharan Africa (with emphasis on Nigeria) and implications for regional development: a review. World J Surg Oncol. 2018; 16:63. https://doi.org/10.1186/s12957-018-1345-2. [PubMed].
3. von Allmen D. Pediatric surgical oncology: A brief overview of where we have been and the challenges we face. Semin Pediatr Surg. 2019; 28:150864. https://doi.org/10.1016/j.sempedsurg.2019.150864. [PubMed].
4. Ojo OA, Ajiboye B, Fadaka A, Taro P, Shariati MA. Nrf2-Keap1 Activation, A Promising Strategy in the Prevention of Cancer. Free Radicals & Antioxidants. 2017; 7:1–7. https://doi.org/10.5530/fra.2017.1.1. http://www.antiox.org/sites/default/files/10.5530fra.2017.1.1.pdf.
5. Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. Int J Clin Oncol. 2010; 28:1684–91. https://doi.org/10.1200/JCO.2009.24.9284. [PubMed].
6. Foukakis T, Bergh J. Prognostic and predictive factors in early, non-metastatic breast cancer. UpToDate. 2016. https://www.uptodate.com/contents/prognostic-and-predictive-factors-in-early-non-metastatic-breast-cancer.
7. McGuire A, Lowery AJ, Kell MR, Kerin MJ, Sweeney KJ. Locoregional recurrence following breast cancer surgery in the trastuzumab era: A systematic review by subtype. Ann Surg Oncol. 2017; 24:3124–32. https://doi.org/10.1245/s10434-017-6021-1. [PubMed].
8. Golov AK, Kondratyev NV, Kostyuk GP, Golimbet AVE. Novel Approaches for Identifying the Molecular Background of Schizophrenia. Cells. 2020; 9:246. https://doi.org/10.3390/cells9010246. [PubMed].
9. Lee JS. Exploring cancer genomic data from the cancer genome atlas project. BMB Rep. 2016; 49:607–611. https://doi.org/10.5483/bmbrep.2016.49.11.145. [PubMed].
10. Cohen P. Protein kinases - the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002; 1:309–15. https://doi.org/10.1038/nrd773. [PubMed].
11. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001; 411:355–65. https://doi.org/10.1038/35077225. [PubMed].
12. Liang X, Wang D, Wang Y, Zhou Z, Zhang J, Li J. Expression of aurora kinase A and B in chondrosarcoma and its relationship with the prognosis. Diagn Pathol. 2012; 7:84. https://doi.org/10.1186/1746-1596-7-84. [PubMed].
13. Liu ZG, Yi W, Tao YL, Chan HC, Zeng MS, Xia YF. Aurora-A is an efficient marker for predicting poor prognosis in human nasopharyngeal carcinoma with aggressive local invasion: 208 cases with a 10-year follow-up from a single institution. Oncol Lett. 2012; 3:1237–44. https://doi.org/10.3892/ol.2012.660. [PubMed].
14. Ali H, Dawson S, Blows F, Provenzano E, Pharoah P, Caldas C. Aurora kinase A outperforms Ki67 as a prognostic marker in ER-positive breast cancer. Br J Cancer. 2012; 106:1798–806. https://doi.org/10.1038/bjc.2012.167. [PubMed].
15. Lehman NL, O'Donnell JP, Whiteley LJ, Stapp RT, Lehman TD, Roszka KM, Schultz LR, Williams CJ, Mikkelsen T, Brown SL. Aurora A is differentially expressed in gliomas, is associated with patient survival in glioblastoma and is a potential chemotherapeutic target in gliomas. Cell Cycle. 2012; 11:489–502. https://doi.org/10.4161/cc.11.3.18996. [PubMed].
16. Dotan E, Meropol N, Zhu F, Zambito F, Bove B, Cai K, Godwin A, Golemis E, Astsaturov I, Cohen S. Relationship of increased aurora kinase A gene copy number, prognosis and response to chemotherapy in patients with metastatic colorectal cancer. Br J Cancer. 2012; 106:748–55. https://doi.org/10.1038/bjc.2011.587. [PubMed].
17. Wang J, Yang S, Zhang H, Song Y, Zhang X, Qian H, Han X, Shi Y. Aurora-A as an independent molecular prognostic marker in gastric cancer. Oncol Rep. 2011; 26:23–32. https://doi.org/10.3892/or.2011.1250. [PubMed].
18. Yang F, Guo X, Yang G, Rosen DG, Liu J. AURKA and BRCA2 expression highly correlate with prognosis of endometrioid ovarian carcinoma. Mod Pathol. 2011; 24:836–45. https://doi.org/10.1038/modpathol.2011.44. [PubMed].
19. Ma ZL, Zhang BJ, Wang DT, Li X, Wei JL, Zhao BT, Jin Y, Li YL, Jin YX. Tanshinones suppress AURKA through up-regulation of miR-32 expression in non-small cell lung cancer. Oncotarget. 2015; 6:20111–20. https://doi.org/10.18632/oncotarget.3933. [PubMed].
20. Levinson NM. The multifaceted allosteric regulation of Aurora kinase A. Biochem J. 2018; 475:2025–42. https://doi.org/10.1042/BCJ20170771. [PubMed].
21. Yuan Q, Sun T, Ye F, Kong W, Jin H. MicroRNA-124-3p affects proliferation, migration and apoptosis of bladder cancer cells through targeting AURKA. Cancer Biomark. 2017; 19:93–101. https://doi.org/10.3233/CBM-160427. [PubMed].
22. Zhang H, Bao J, Zhao S, Huo Z, Li B. MicroRNA-490-3p suppresses hepatocellular carcinoma cell proliferation and migration by targeting the aurora kinase A gene (AURKA). Arch Med Sci. 2019; 16:395–406. https://doi.org/10.5114/aoms.2019.91351. [PubMed].
23. Gomaa A, Peng D, Chen Z, Soutto M, Abouelezz K, Corvalan A, El-Rifai W. Epigenetic regulation of AURKA by miR-4715-3p in upper gastrointestinal cancers. Sci Rep. 2019; 9:16970. https://doi.org/10.1038/s41598-019-53174-6. [PubMed].
24. Zhang J, Kuai X, Song M, Chen X, Yu Z, Zhang H, Mao Z. microRNA-32 inhibits the proliferation and invasion of the SGC-7901 gastric cancer cell line in vitro. Oncol Lett. 2014; 7:270–4. https://doi.org/10.3892/ol.2013.1667. [PubMed].
25. Dacic S, Kelly L, Shuai Y, Nikiforova MN. miRNA expression profiling of lung adenocarcinomas: correlation with mutational status. Mod Pathol. 2010; 23:1577–82. https://doi.org/10.1038/modpathol.2010.152. [PubMed].
26. Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J. 2012; 31:2207–21. https://doi.org/10.1038/emboj.2012.63. [PubMed].
27. Eterno V, Zambelli A, Villani L, Tuscano A, Manera S, Spitaleri A, Pavesi L, Amato A. AurkA controls self-renewal of breast cancer-initiating cells promoting wnt3a stabilization through suppression of miR-128. Sci Rep. 2016; 6:1–13. https://doi.org/10.1038/srep28436. [PubMed].
28. Wu W, Yang J, Feng X, Wang H, Ye S, Yang P, Tan W, Wei G, Zhou Y. MicroRNA-32 (miR-32) regulates phosphatase and tensin homologue (PTEN) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol Cancer. 2013; 12:30. https://doi.org/10.1186/1476-4598-12-30. [PubMed].
29. Xia W, Zhou J, Luo H, Liu Y, Peng C, Zheng W, Ma W. MicroRNA-32 promotes cell proliferation, migration and suppresses apoptosis in breast cancer cells by targeting FBXW7. Cancer Cell Int. 2017; 17:14. https://doi.org/10.1186/s12935-017-0383-0. [PubMed].
30. Liu J, Zheng M, Tang YL, Liang XH, Yang Q. MicroRNAs, an active and versatile group in cancers. Int J Oral Sci. 2011; 3:165–75. https://doi.org/10.4248/IJOS11063. [PubMed].
31. Bartels CL, Tsongalis GJ. [MicroRNAs: novel biomarkers for human cancer]. Ann Biol Clin (Paris). 2010; 68:263–72. https://doi.org/10.1684/abc.2010.0429. [PubMed].
32. Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. https://doi.org/10.1038/nature11412. [PubMed].
33. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017; 170:1062–78. https://doi.org/10.1016/j.cell.2017.08.028. [PubMed].
34. Varna M, Bousquet G, Plassa LF, Bertheau P, Janin A. TP53 status and response to treatment in breast cancers. J Biomed Biotechnol. 2011; 2011:284584. https://doi.org/10.1155/2011/284584. [PubMed].
35. Wang Y, Helland Å, Holm R, Skomedal H, Abeler V, Danielsen H, Tropé CG, Børresen-Dale A, Kristensen G. TP53 mutations in early-stage ovarian carcinoma, relation to long-term survival. Br J Cancer. 2004; 90:678–85. https://doi.org/10.1038/sj.bjc.6601537. [PubMed].
36. Langerød A, Zhao H, Borgan Ø, Nesland JM, Bukholm IR, Ikdahl T, Kåresen R, Børresen-Dale AL, Jeffrey SS. TP53mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007; 9:R30. https://doi.org/10.1186/bcr1675. [PubMed].
37. Jézéquel P, Frénel JS, Campion L, Guérin-Charbonnel C, Gouraud W, Ricolleau G, Campone M. bc-GenExMiner 3.0: new mining module computes breast cancer gene expression correlation analyses. Database (Oxford). 2013; 2013:bas060. https://doi.org/10.1093/database/bas060. [PubMed].
38. Shatnawi M. Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology Emerging Trends in Computer Science and Applied Computing 2015, pp 99–121. Chapter 6 - Review of Recent Protein-Protein Interaction Techniques. https://doi.org/10.1016/B978-0-12-802508-6.00006-5. https://www.researchgate.net/profile/Maad_Shatnawi/publication/283822046_Review_of_Recent_Protein-Protein_Interaction_Techniques/links/5c0645d0299bf169ae307ba2/Review-of-Recent-Protein-Protein-Interaction-Techniques.pdf.
39. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. Gene ontology: tool for the unification of biology. Nat Genet. 2000; 25:25–9. https://doi.org/10.1038/75556. [PubMed].
40. Fadaka AO, Klein A, Pretorius A. In silico identification of microRNAs as candidate colorectal cancer biomarkers. Tumour Biol. 2019; 41:1010428319883721. https://doi.org/10.1177/1010428319883721. [PubMed].
41. Fadaka A, Ajiboye B, Ojo O, Adewale O, Olayide I, Emuowhochere R. Biology of glucose metabolization in cancer cells. J Oncol Sci. 2017; 3:45–51. https://doi.org/10.1016/j.jons.2017.06.002.
42. Fadaka AO, Bakare OO, Pretorius A, Klein A. Genomic profiling of microRNA target genes in colorectal cancer. Tumour Biol. 2020; 42:1010428320933512. https://doi.org/10.1177/1010428320933512.
43. Dimitrakopoulos CM, Beerenwinkel N. Computational approaches for the identification of cancer genes and pathways. Wiley Interdiscip Rev Syst Biol Med. 2017; 9:e1364. https://doi.org/10.1002/wsbm.1364. [PubMed].
44. Otoukesh B, Boddouhi B, Moghtadaei M, Kaghazian P, Kaghazian M. Novel molecular insights and new therapeutic strategies in osteosarcoma. Cancer Cell Int. 2018; 18:158. https://doi.org/10.1186/s12935-018-0654-4. [PubMed].
45. Gurses HE, Hatipoglu O, Gunduz M, Gunduz E. A concise review of molecular pathology of breast cancer. Chapter 4, MicroRNAs as therapeutic targets in human breast cancer. 2015. https://doi.org/10.5772/59428.
46. Fadaka AO, Pretorius A, Klein A. Biomarkers for Stratification in Colorectal Cancer: MicroRNAs. Cancer Control. 2019; 26:1073274819862784. https://doi.org/10.1177/1073274819862784. [PubMed].
47. Polini B, Carpi S, Doccini S, Citi V, Martelli A, Feola S, Santorelli FM, Cerullo V, Romanini A, Nieri P. Tumor Suppressor Role of hsa-miR-193a-3p and-5p in Cutaneous Melanoma. Int J Mol Sci. 2020; 21:6183. https://doi.org/10.3390/ijms21176183. [PubMed].
48. Guo XG, Zheng L, Feng WB, Xia Y. The AURKA gene rs2273535 polymorphism contributes to breast carcinoma risk-meta-analysis of eleven studies. Asian Pac J Cancer Prev. 2014; 15:6709–14. https://doi.org/10.7314/apjcp.2014.15.16.6709. [PubMed].
49. Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X, Howe C, O'Brien S, Maglaty M, Ecsedy JA. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014; 33:539–49. https://doi.org/10.1038/onc.2012.632. [PubMed].
50. Yang N, Wang C, Wang Z, Zona S, Lin S, Wang X, Yan M, Zheng F, Li S, Xu B. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene. 2017; 36:3428–40. https://doi.org/10.1038/onc.2016.490. [PubMed].
51. Rong R, Jiang L, Sheikh M, Huang Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene. 2007; 26:7700–8. https://doi.org/10.1038/sj.onc.1210575. [PubMed].
52. Wang R, Song Y, Liu X, Wang Q, Wang Y, Li L, Kang C, Zhang Q. UBE2C induces EMT through Wnt/β-catenin and PI3K/Akt signaling pathways by regulating phosphorylation levels of Aurora-A. Int J Oncol. 2017; 50:1116–26. https://doi.org/10.3892/ijo.2017.3880. [PubMed].
53. Liu X, Li Z, Song Y, Wang R, Han L, Wang Q, Jiang K, Kang C, Zhang Q. AURKA induces EMT by regulating histone modification through Wnt/β-catenin and PI3K/Akt signaling pathway in gastric cancer. Oncotarget. 2016; 7:33152–64. https://doi.org/10.18632/oncotarget.8888. [PubMed].
54. Dere R, Perkins AL, Bawa-Khalfe T, Jonasch D, Walker CL. β-catenin links von Hippel-Lindau to aurora kinase A and loss of primary cilia in renal cell carcinoma. J Am Soc Nephrol. 2015; 26:553–64. https://doi.org/10.1681/ASN.2013090984. [PubMed].
55. Zhong S, Bai Y, Wu B, Ge J, Jiang S, Li W, Wang X, Ren J, Xu H, Chen Y, Zhao G. Selected by gene co-expression network and molecular docking analyses, ENMD-2076 is highly effective in glioblastoma-bearing rats. Aging. 2019; 11:9738–66. https://doi.org/10.18632/aging.102422. [PubMed].
56. Thu KL, Soria-Bretones I, Mak TW, Cescon DW. Targeting the cell cycle in breast cancer: towards the next phase. Cell Cycle. 2018; 17:1871–85. https://doi.org/10.1080/15384101.2018.1502567. [PubMed].
57. Kollareddy M, Dzubak P, Zheleva D, Hajduch M. Aurora kinases: structure, functions and their association with cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008; 152:27–33. https://doi.org/10.5507/bp.2008.004. [PubMed].
58. Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV. Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci U S A. 2002; 99:15440–5. https://doi.org/10.1073/pnas.202606599. [PubMed].
59. Walter AO, Seghezzi W, Korver W, Sheung J, Lees E. The mitotic serine/threonine kinase Aurora2/AIK is regulated by phosphorylation and degradation. Oncogene. 2000; 19:4906–16. https://doi.org/10.1038/sj.onc.1203847. [PubMed].
60. Wei K, Wu L, Chen Y, Lin Y, Wang Y, Liu X, Xie D. Argonaute protein as a linker to command center of physiological processes. Chin J Cancer Res. 2013; 25:430–41. https://doi.org/10.3978/j.issn.1000-9604.2013.08.13. [PubMed].
61. Moser JJ, Fritzler MJ. The microRNA and messengerRNA profile of the RNA-induced silencing complex in human primary astrocyte and astrocytoma cells. PLoS One. 2010; 5:e13445–e. https://doi.org/10.1371/journal.pone.0013445. [PubMed].
62. Pratt AJ, MacRae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009; 284:17897–901. https://doi.org/10.1074/jbc.R900012200. [PubMed].
63. Zhang R, Jing Y, Zhang H, Niu Y, Liu C, Wang J, Zen K, Zhang CY, Li D. Comprehensive Evolutionary Analysis of the Major RNA-Induced Silencing Complex Members. Sci Rep. 2018; 8:14189. https://doi.org/10.1038/s41598-018-32635-4. [PubMed].
64. Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016; 1:1–9. https://doi.org/10.1038/sigtrans.2015.4. [PubMed].
65. Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol. 2014; 24:R762–R76. https://doi.org/10.1016/j.cub.2014.06.043. [PubMed].
66. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol. 2009; 4:199–227. https://doi.org/10.1146/annurev.pathol.4.110807.092222. [PubMed].
67. Jansson MD, Lund AH. MicroRNA and cancer. Mol Oncol. 2012; 6:590–610. https://doi.org/10.1016/j.molonc.2012.09.006.
68. Tavakolian S, Goudarzi H, Torfi F, Faghihloo E. Evaluation of microRNA-9 and-192 expression levels as biomarkers in patients suffering from breast cancer. Biomed Rep. 2020; 12:30–4. https://doi.org/10.3892/br.2019.1257. [PubMed].
69. Fadaka AO, Ojo BA, Adewale OB, Esho T, Pretorius A. Effect of dietary components on miRNA and colorectal carcinogenesis. Cancer Cell Int. 2018; 18:130. https://doi.org/10.1186/s12935-018-0631-y. [PubMed].
70. Fletcher CE, Sulpice E, Combe S, Shibakawa A, Leach DA, Hamilton MP, Chrysostomou SL, Sharp A, Welti J, Yuan W, Dart DA, Knight E, Ning J, et al. Androgen receptor-modulatory microRNAs provide insight into therapy resistance and therapeutic targets in advanced prostate cancer. Oncogene. 2019; 38:5700–24. https://doi.org/10.1038/s41388-019-0823-5. [PubMed].
71. Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007; 67:8980–4. https://doi.org/10.1158/0008-5472.CAN-07-0895. [PubMed].
72. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005; 65:7065–70. https://doi.org/10.1158/0008-5472.CAN-05-1783. [PubMed].
73. Mattiske S, Suetani RJ, Neilsen PM, Callen DF. The oncogenic role of miR-155 in breast cancer. Cancer Epidemiol Biomarkers Prev. 2012; 21:1236–43. https://doi.org/10.1158/1055-9965.EPI-12-0173. [PubMed].
74. Zhao XG, Hu JY, Tang J, Yi W, Zhang MY, Deng R, Mai SJ, Weng NQ, Wang RQ, Liu J, Zhang HZ, He JH, Wang HY. miR-665 expression predicts poor survival and promotes tumor metastasis by targeting NR4A3 in breast cancer. Cell Death Dis. 2019; 10:479. https://doi.org/10.1038/s41419-019-1705-z. [PubMed].
75. Hannafon BN, Cai A, Calloway CL, Xu YF, Zhang R, Fung KM, Ding WQ. miR-23b and miR-27b are oncogenic microRNAs in breast cancer: evidence from a CRISPR/Cas9 deletion study. BMC Cancer. 2019; 19:642. https://doi.org/10.1186/s12885-019-5839-2. [PubMed].
76. Fadaka AO, Bakare OO, Sibuyi NRS, Klein A. Gene Expression Alterations and Molecular Analysis of CHEK1 in Solid Tumors. Cancers. 2020; 12:662. https://doi.org/10.3390/cancers12030662. [PubMed].
77. Ender C, Meister G. Argonaute proteins at a glance. J Cell Sci. 2010; 123:1819–23. https://doi.org/10.1242/jcs.055210. [PubMed].
78. Höck J, Weinmann L, Ender C, Rüdel S, Kremmer E, Raabe M, Urlaub H, Meister G. Proteomic and functional analysis of Argonaute-containing mRNA–protein complexes in human cells. EMBO Rep. 2007; 8:1052–60. https://doi.org/10.1038/sj.embor.7401088. [PubMed].
79. Neviani P, Wise PM, Murtadha M, Liu CW, Wu CH, Jong AY, Seeger RC, Fabbri M. Natural killer–derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019; 79:1151–64. https://doi.org/10.1158/0008-5472.CAN-18-0779. [PubMed].
80. Zhang H, Bao J, Zhao S, Huo Z, Li B. MicroRNA-490-3p suppresses hepatocellular carcinoma cell proliferation and migration by targeting the aurora kinase A gene (AURKA). Arch Med Sci. 2019; 16:395–406. https://doi.org/10.5114/aoms.2019.91351. [PubMed].
81. Trotta E. On the normalization of the minimum free energy of RNAs by sequence length. PLoS One. 2014; 9:e113380. [PubMed].
82. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004; 6:1–6. https://doi.org/10.1016/s1476-5586(04)80047-2. [PubMed].
83. Jézéquel P, Campone M, Gouraud W, Guérin-Charbonnel C, Leux C, Ricolleau G, Campion L. bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat. 2012; 131:765–75. https://doi.org/10.1007/s10549-011-1457-7. [PubMed].
84. Li D, Wei P, Peng Z, Huang C, Tang H, Jia Z, Cui J, Le X, Huang S, Xie K. The critical role of dysregulated FOXM1–PLAUR signaling in human colon cancer progression and metastasis. Clin Cancer Res. 2013; 19:62–72. https://doi.org/10.1158/1078-0432.CCR-12-1588. [PubMed].
85. Mihály Z, Kormos M, Lánczky A, Dank M, Budczies J, Szász MA, Győrffy B. A meta-analysis of gene expression-based biomarkers predicting outcome after tamoxifen treatment in breast cancer. Breast Cancer Res Treat. 2013; 140:219–32. https://doi.org/10.1007/s10549-013-2622-y. [PubMed].
86. Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics. 2009; 2:18. https://doi.org/10.1186/1755-8794-2-18. [PubMed].
87. Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018; 46:D296–D302. https://doi.org/10.1093/nar/gkx1067. [PubMed].
88. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4:e05005. https://doi.org/10.7554/eLife.05005. [PubMed].
89. Antczak M, Popenda M, Zok T, Sarzynska J, Ratajczak T, Tomczyk K, Adamiak RW, Szachniuk M. New functionality of RNAComposer: application to shape the axis of miR160 precursor structure. Acta Biochim Pol. 2016; 63:737–44. https://doi.org/10.18388/abp.2016_1329. [PubMed].
90. Fadaka AO, Pretorius A, Klein A. MicroRNA Assisted Gene Regulation in Colorectal Cancer. Int J Mol Sci. 2019; 20:4899. https://doi.org/10.3390/ijms20194899. [PubMed].