
Introduction
Mutations in the serine/threonine kinase BRAF are found in 40% of melanomas [1]. The most prevalent mutation in melanoma is BRAF V600E, which constitutively activates downstream mitogen-activated protein kinase (MAPK) signalling. Introduction of single agent BRAF mutant inhibitors (BRAFi), such as vemurafenib and dabrafenib, revolutionised the treatment of metastatic melanomas [2, 3]. Furthermore, the combination of BRAF and MEK inhibitors (such as dabrafenib and trametinib, or vemurafenib and cobimetinib) improved tumour response rate and progression-free survival, while attenuating some of the serious adverse events observed with monotherapy [4, 5]. However, even with combination therapies, resistance to MAPK inhibition remains a major challenge in clinical care, with the majority of patients progressing within 10 months [6]. Identification of the mechanism of resistance to targeted therapies in individual patients may offer new insights into strategies for overcoming resistance.
Resistance to BRAF inhibitors usually involves MAPK reactivation, via mutations in NRAS or MAP2K1/2, upregulation of receptor tyrosine kinases, mutant BRAF gene amplification or alternative BRAF splicing [7–10]. In around 30% of resistant tumours, resistance to BRAF inhibition is conferred by alternative splicing via generation of BRAF isoforms lacking the RAS binding domain (RBD) encoded by exons 3–5 [7, 11]. In the absence of the RBD, these BRAF isoforms dimerise even in the presence of low levels of RAS, conferring and conferring resistance to BRAF inhibition [12]. Four BRAF splicing variants, referred to as p61, p55, p48 and p41 were named based on their predicted molecular weight (Figure 1A).
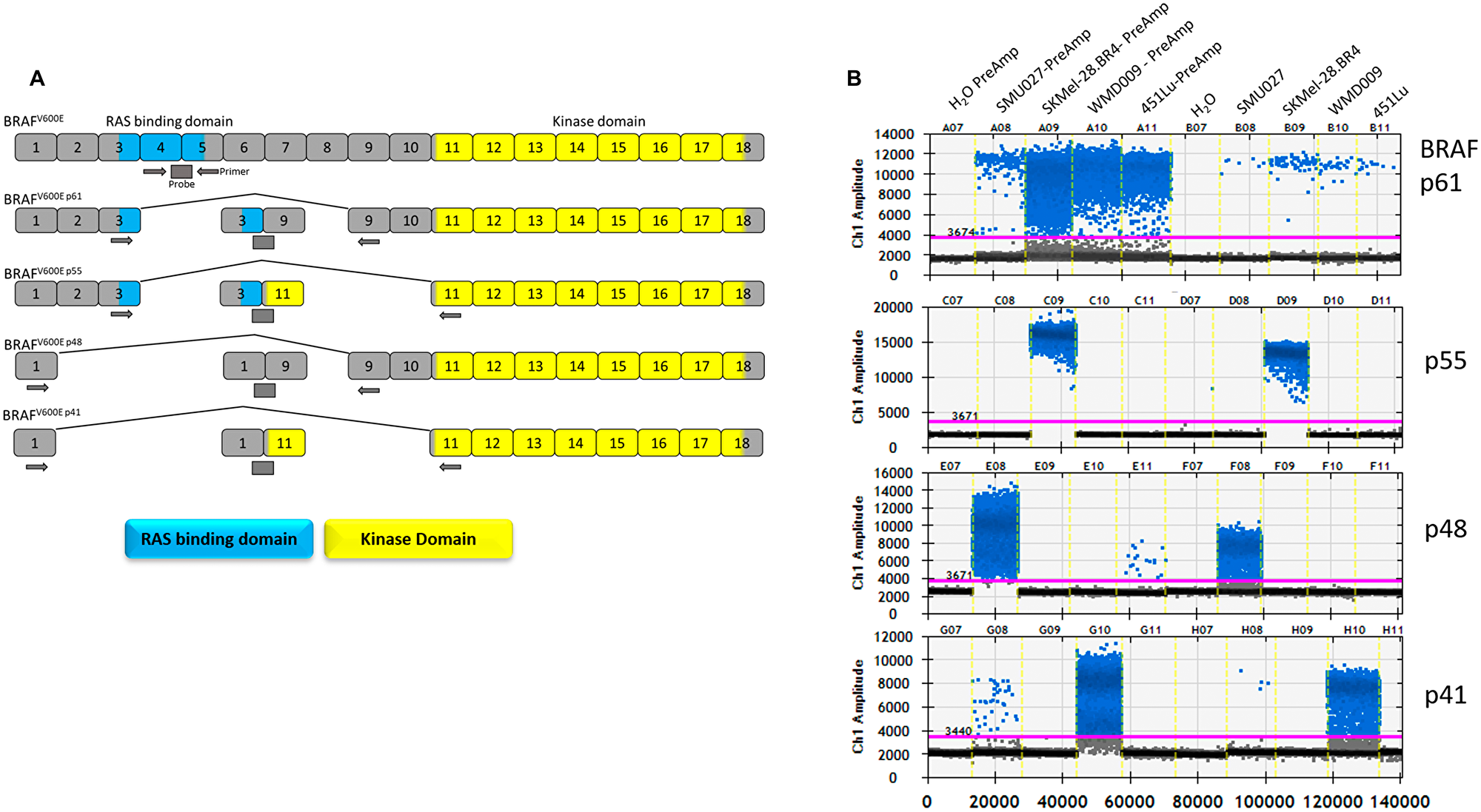
Figure 1: BRAF splice variants detection by droplet digital PCR. (A) BRAF exon organisation of the splicing variants and location of primers and probes used for detection of splicing variants. (B) Droplet digital PCR Ch1,1D plots of each BRAF splicing detection assay of melanoma cell lines carrying BRAF splicing variants, with and without the pre-amplification, (PreAmp) step.
The analysis of tumour-derived biomarkers in blood of cancer patients is transforming clinical cancer pathology. Circulating tumour DNA (ctDNA) has emerged as a non-invasive biomarker to track tumour burden and allow monitoring of the cancer genome in blood across several malignancies including melanoma [13, 14]. We, and others, have previously demonstrated that increased melanoma ctDNA levels and the appearance of circulating mutations associated with of acquired resistance, provide highly specific, early information of relapse during BRAF targeted therapies [15, 16]. The latter supports a clinical model that incorporates the analysis of ctDNA in patients undergoing BRAF-inhibitor therapies as a routine test to detect early relapse and resistance [13, 14]. However, resistance involving aberrant splicing variants, cannot be detected through ctDNA analysis, and can only be identified using RNA.
Circulating tumour-derived cell free RNA or circulating tumour RNA (ctRNA) can be found in plasma from patients with cancer [17–19]. Various studies have also demonstrated the ability to amplify tumour-related mRNAs from sera of patients with melanoma, breast cancer, and other malignancies [20–24]. However, the possibility that extracellular RNA could survive in the blood has not been widely accepted, due to plasma containing potent ribonucleases capable of degrading cell-free RNA [25]. Regardless of this, studies have documented the presence of ctRNA in serum/plasma of cancer patients [26, 27].
Extracellular vesicles (EVs) are known to contain various RNA types, including microRNA, mRNA and circular RNA [28]. EV RNA has been described as having a substantial role in the transfer and movement of epigenetic information and is stable enough for whole transcriptome sequencing [29–31]. It is plausible that plasma cell-free RNA (cfRNA) is a mixture of RNA protected by RNA binding proteins and RNA contained within EVs. Here we explore whether alternative BRAF splicing variants can be detected in melanoma patients who have developed resistance to BRAF inhibitor therapy. We report a new methodological approach based on digital droplet polymerase chain reaction (ddPCR) to assess the presence of BRAF splicing variants in plasma cfRNA and EV-derived RNA. We interrogated plasma from 38 melanoma patients identified as having clinically diagnosed treatment resistance after initial response to BRAF inhibitor monotherapy or BRAF/MEK inhibitor combination. In addition, cfRNA was interrogated from an additional five patients who were confirmed to have tumour specific BRAF splicing variants in their progressing tumours. Finally, we evaluated the presence of BRAF splicing variants in EV RNA isolated from plasma of two melanoma patients failing BRAF inhibition.
Results
Development of a sensitive assay for detection of BRAF splicing variants in plasma
We developed ddPCR assays for the detection of alternative BRAF splicing variants: p61, p55, p48 and p41, which are associated with resistance to BRAF inhibition, as well as the canonical full length BRAF (pFL) (Figure 1A). The assays were validated using three melanoma cell lines with resistance to BRAF inhibition and known to express alternative BRAF splicing variants (Figure 1B). As previously reported, SK-Mel-28. BR4 expresses BRAF p55 (exon 4-10Δ), SMU027 expresses BRAF p48 (exon 2-8Δ) and WMD009 expresses BRAF p41 (exon 2-10Δ) [9, 32]. Surprisingly without amplification all cell lines carry the BRAF p61, albeit at lower concentrations in comparison to the dominant splice variant expressed. Similarly, SMU027 expressed low levels of BRAF p41 compared to WMD009.
To enhance sensitivity of detection of BRAF splicing variants, a pre-amplification step was introduced. This resulted in a significant enhancement of the signal without loss of specificity (Figure 1B). Importantly, the introduced pre-amplification step maintained a linear relation between copies of input cDNA and output BRAF splicing variant copies (R2 = 0.9986, p < 0.0001), with a 6,396 ± 569 fold increase in BRAF splicing variant copies (Supplementary Figure 2).
We tested plasma from nine healthy volunteers, to confirm that the BRAF splicing variants are not commonly found in normal plasma and that the assay does not introduce artefacts or induce false detection of BRAF alternative splicing variants (Supplementary Figure 3).
Analysis of BRAF splicing variants in plasma of melanoma patients
We prospectively collected longitudinal plasma samples from 38 patients: 7 vemurafenib and 31 dabrafenib/trametinib treated cases (Supplementary Figure 4). Of the 7 vemurafenib cases, 2 had baseline plasma for ctDNA analysis, and of these 1 experienced a decrease in mutant BRAF during treatment (MM143), while the other had an increase in mutant BRAF copies during treatment (MM141). Of the 31 dabrafenib/trametinib cases, 25 had assessable baseline plasma, and of these 24 cases had detectable ctDNA (as mutant BRAF copies) at baseline. In 19 of the 24 cases, ctDNA decreased during treatment in comparison to the baseline sample. Analysis of blood samples collected at the time of progressive disease (red points – Supplementary Figure 4), identified 29 patients (5 on vemurafenib and 24 on combination dabrafenib/trametinib therapy) with detectable ctDNA at progression. Of those, seven had additional NRAS Q61 mutations and one had a BRAF gene amplification detectable in plasma (Figure 2 and Supplementary Table 1).
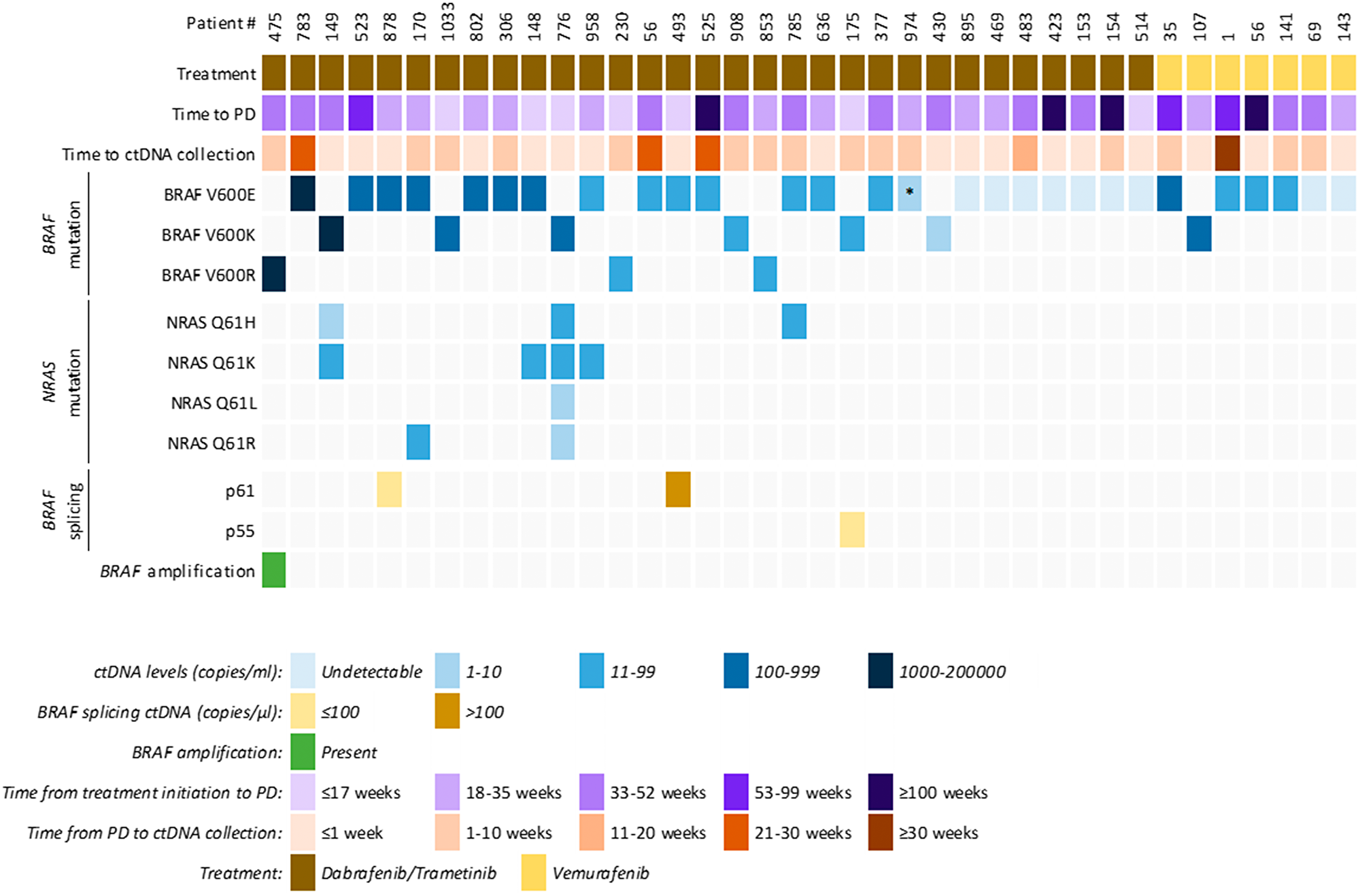
Figure 2: Clinical and ctDNA profile of patients that showed clinical progressive disease to targeted therapy. Overview of ctDNA results (copies/mL plasma) from BRAF, NRAS, BRAF splicing and BRAF amplification in 38 melanoma patients. Each column is an individual patient, showing their clinical characteristics, quantitative ctDNA results (copies/ml or μl of plasma) and presence of BRAF amplification. *BRAF V600E c.1799_1800GT>AA, p. (Val600Glu), also known as BRAF V600E2 mutation.
Three patients (MM175, MM493 and MM878) had detectable BRAF splicing variants in plasma using our assay, indicating the presence of ctRNA. BRAF splicing variants were not detected in the pre-treatment plasma (MM493, MM878) despite clear detectable ctDNA (Figure 3). Unfortunately, no plasma collected at baseline was available from MM175 for BRAF splicing variant assessment.
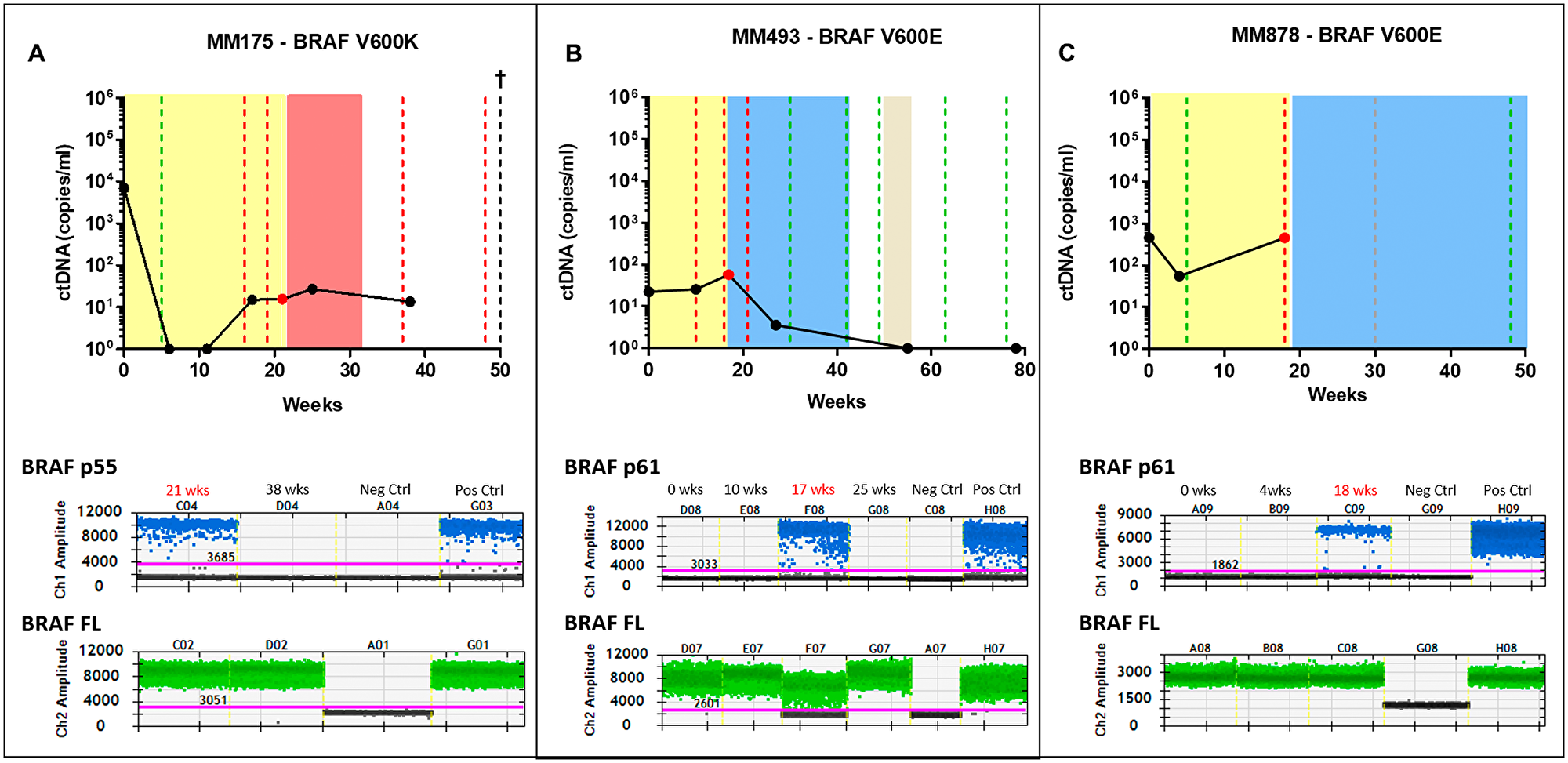
Figure 3: Detection of splice variants in the combination therapy cohort. Three patients undergoing combination BRAFi therapy were positive for a splicing variant. Patients MM175 (A) and patients MM493 (B), were both positive for the p61 variant at the point of progressive disease shown in the droplet digital plots. Patient MM878 (C), was positive for the p55 variant also shown in the droplet digital plot. The longitudinal graphs represent the ctDNA tracking of the activating mutation for the patients. Red, green and grey dotted lines represent clinical response, progressive disease and stable disease respectively. The singular red dot represents the time-initial point tested positive for the splicing variants and neighbouring time-points were tested to confirm the presence or absence of the splicing variants. The coloured area indicates the period during which systemic therapy was administered; colours representing dabrafenib/trametinib (yellow), ipilimumab (red) and pembrolizumab (blue). Droplet digital plot represents the positivity for the indicated BRAF splicing variant (blue dots). BRAF full length (BRAF FL, green dots) was use as positive control for RNA extraction and amplification. No template control and positive control (SK-Mel-28. BR4) were included in each test.
MM175 presented with multiple spine, rib and liver metastases. The patient received dabrafenib/trametinib combination treatment and showed a partial response at 5 weeks, with undetectable ctDNA (Figure 3A). Subsequent scans confirmed progression with the patient being moved onto ipilimumab immunotherapy. Analysis of plasma ctRNA at the final point of dabrafenib/trametinib progression (21 weeks) and after change of therapy to ipilimumab (38 weeks), revealed that the BRAF splicing variant p55 was present only at the time of progression but undetectable while on ipilimumab treatment, despite having sustained BRAF V600K ctDNA levels.
MM493 presented with multiple brain, lung and abdomen metastases. One brain metastasis was resected while and the others were treated with whole brain radiotherapy. This patient received dabrafenib/trametinib combination treatment with partial response at 8 weeks and limited decrease in ctDNA (Figure 3B). BRAF p61, but no other BRAF splicing variants, was detected at the time of progression (17 weeks, Figure 3B). Plasma collected at baseline, during response (10 weeks) or after starting pembrolizumab (25 weeks) did not have detectable BRAF splicing variants. BRAF V600E ctDNA was detectable in this patient at all four-time points analysed but became undetectable by week 54 in response to treatment with pembrolizumab.
The third patient (MM878) presented with widespread melanoma in lung, lymph node, adrenal, bone and gallbladder. The patient was placed on dabrafenib/trametinib combination treatment and a partial metabolic response was observed 5 weeks later which was concordant with a decrease from 458 copies/ml to 56 copies/ml decrease in BRAF V600E ctDNA (Figure 3C). At the time of progression additional skeletal metastases were observed by PET scan 18 weeks into therapy, with ctDNA levels significantly increased to 458 copies/ml, with the PET scan confirming progression of several skeletal metastases. The BRAF p61 variant was detectable in plasma at the point of progression, but was undetectable at baseline or the first follow up sample collected 4 weeks into therapy (Figure 3C).
To validate our findings, we next tested the plasma derived from five melanoma patients that were treated with vemurafenib monotherapy who developed resistance. Tumour samples obtained at the time of progression had confirmed BRAF splicing variants by transcriptome analysis [8] (Figure 4). We analysed plasma samples from these patients collected near the time of resecting the progressed tumour. Only one of these five cases (MIA_10) had a detectable splicing variant (BRAF p61) in plasma which was concordant with the results of the matched tumour sample. BRAF splicing variants were not detectable in the baseline plasmas from theses patient.
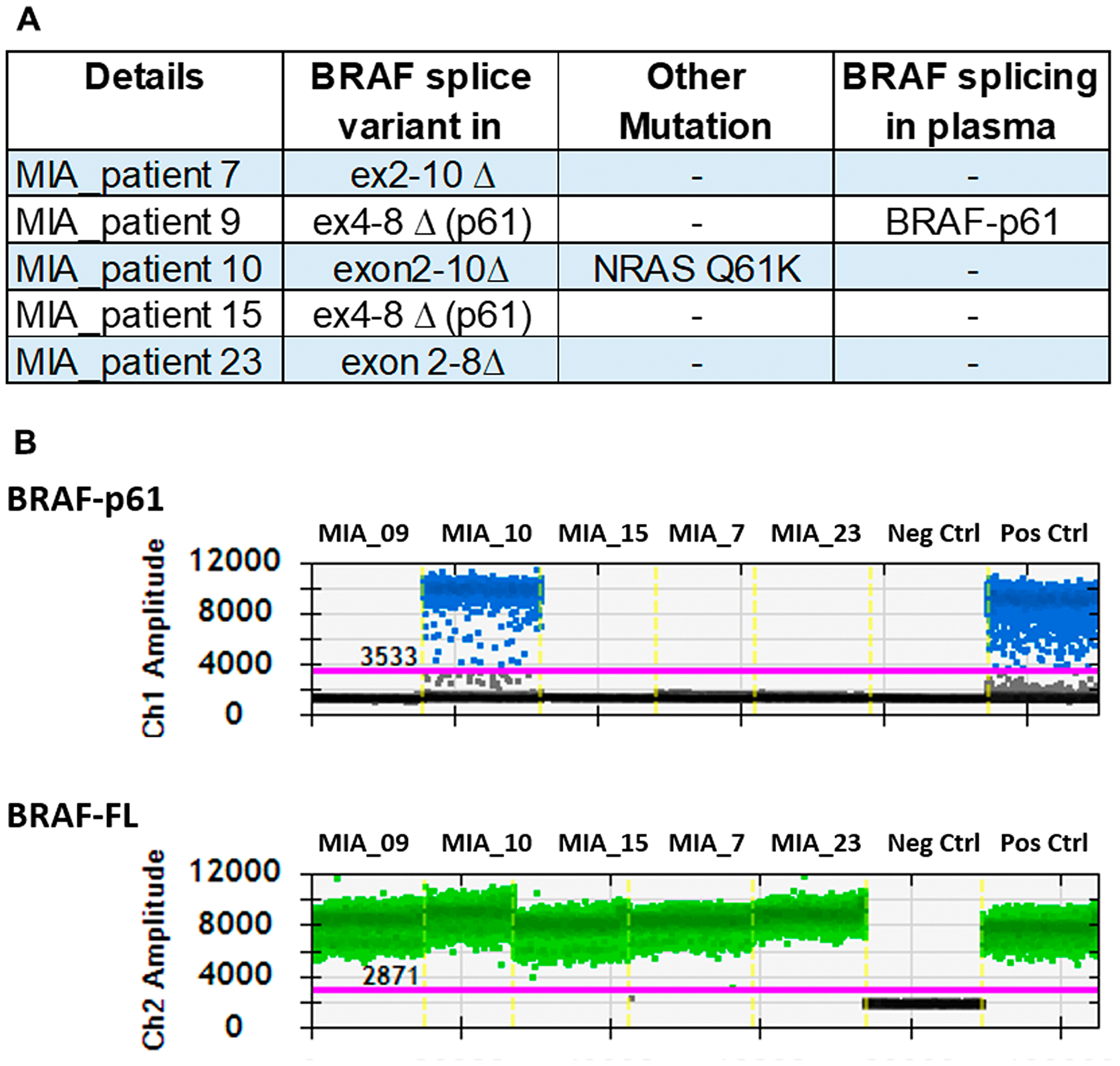
Figure 4: Detection of BRAF splice variants in patients known to have the splice variant. (A) Patients who had confirmation of splicing variants in the tumour were investigated to confirm the presence of the variant in the plasma. (B) The droplet digital plot represents the positivity for p61 in one of the samples.
BRAF splicing variants are associated with extracellular vesicles
Plasma extracellular RNAs are thought to be carried by extracellular vesicles [33]. To evaluate whether BRAF splicing variants are found in EVs, we analysed extracellular vesicles extracted from the plasma of patients MM493 and MM878. EVs isolated from the supernatant of SK-Mel-28. BR4 cell line and from the plasma of a healthy control were used as positive and negative controls, respectively.
The effective isolation of EVs was confirmed by western blot analysis, with positive signal for the tetraspanins CD9, CD63, CD81 and Tumour Susceptibility Gene 101 (TSG101), while negative for the cellular marker calnexin (Figure 5A and Supplementary Figure 5). Transmission electron microscopy (TEM) was used to visualise the extracted EVs, demonstrating the presence of exosomes with a size of 100 nm (Figure 5B). Analysis of EV RNA from SK-Mel-28. BR4 cells confirmed the presence of BRAF p61 and BRAF FL, while EV RNA from healthy volunteer only carried BRAF FL variants. EV RNA from both MM493 and MM878 contained BRAF p61 transcripts (Figure 5C).
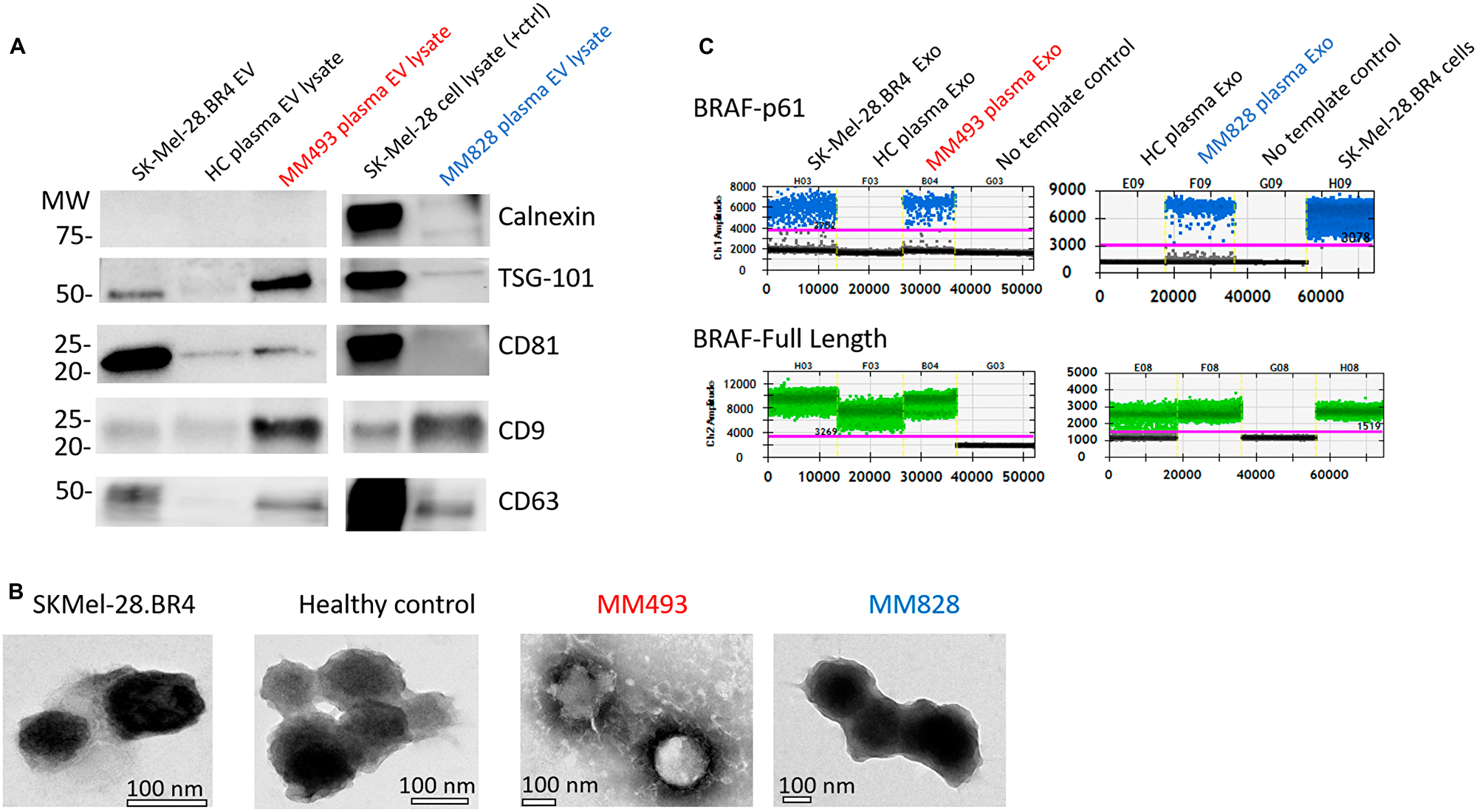
Figure 5: Validation of EV isolation from exo-easy and confirmation of splicing variant presence. (A) EVs isolated using the ExoEasy Kit were tested for the associated markers TSG-101, CD81, CD9 and CD63 and the negative marker Calnexin. Loading was standardised at 4 μg across all EVs preparations. (B) Electron micrographs were used to determine the presence of EV/exosomes in the appropriate size range. (C) Droplet digital testing for BRAF p61 (blue dots) and FL-BRAF (green dots) in EV-derived RNA from the patients, healthy controls and cell line SK-Mel-28. BR4.
DISCUSSION
The emergence of BRAF splicing variants is a common mechanism of escape to BRAF inhibition in melanoma [10, 12]. Elucidation of the molecular escape mechanisms usually requires the sourcing of tumour tissue at the time of progressive disease. Here we show that tumour specific splicing variants, known to mediate treatment resistance, can be detected in plasma.
Our study demonstrated the detection of resistance mediating BRAF splicing variants in ctRNA of melanoma patients failing BRAF inhibiting therapies. Moreover, we showed that these BRAF splicing variants are co-purified or associated with EVs.
The frequency at which we detect splicing variants in this cohort was low. Tumour analyses within cohorts treated with BRAF inhibiting monotherapies suggest a prevalence of approximately 30% for splicing variants [8, 10]. Long et al. suggested that BRAF splicing were unlikely to occur in combination therapy as MAPK signalling driven by such variants will require MEK activity and therefore will remain sensitive to MEK inhibitors [9, 34]. Nevertheless, we did not detect BRAF splicing variants in any of the 7 patients treated with vemurafenib monotherapy. In contrast, we observed BRAF splicing variants in 3 of 31 patients treated with combination BRAF/MEK inhibitors. A study by Wagle et al., reported the detection of BRAF p41 in the post-treatment tumour of 1 out of 5 patients that developed resistance to dabrafenib/trametinib [35].
In this study, we failed to detect BRAF splicing variants in 4 of 5 patients for which the tumours were confirmed to carry BRAF splicing variants by tissue analysis. The isolation and presence of circulating RNA was confirmed in all samples through the presence of BRAF full length. Moreover, we showed that our method had high analytical sensitivity and specificity. However, several technical and biological reasons may explain the low clinical sensitivity of the test. Similar to ctDNA, ctRNA detection is likely to be affected by tumour burden, metabolic activity and location of the tumour [36, 37]. In addition, pre-analytical conditions have not been optimised for the preservation and extraction of ctRNA or EV-derived mRNA in the same way that it has been done for ctDNA [38].
The presence of ctRNA has been previously reported in plasma and other body fluids of cancer patients [17, 39–41]. Early reports published by Lo et al. [42] indicated the presence of tumour derived ctRNA in the plasma of nasopharyngeal carcinoma patients. Beyond this, ctRNA analysis has been expanded primarily into cancer detection cancer or monitoring response [43]. For example, plasma ctRNA expression of the carcinoembryonic antigen has been used for diagnosis of early stage prostate cancer [43] and plasma TERT mRNA levels have demonstrated to be predictive of response to chemoradiotherapy in rectal cancers [44]. Thus, ctRNA shows promise as an additional monitoring tool that can be used in combination with ctDNA and conventional methods. The advantage of ctRNA is implied through the assessment of transcriptional activity of genes.
The effective isolation of EVs was validated using western blot and transmission electron microscopy. Purity was confirmed through negativity of calnexin, along with the positivity of CD9, CD63, CD81 or TSG101 which are EV associated proteins [45]. Patient MM828-EV sample was CD81 negative but positive for the other markers, demonstrating that reliance on one or two markers poses a risk of false negativity. The heterogeneity of EV-associated proteins has been reported previously [46–48], suggesting that a panel of markers is required to validate the presence of EVs.
EVs have been established as carriers of various types of RNA, including mRNA, miRNA and long non-coding RNAs [49–51]. The EV RNA from the patients MM493 and MM878 were found positive for BRAF-p61, matching the results obtained via their plasma ctRNA. This suggests that ctRNA may be carried totally or partially by EVs, and that these vesicles may be protecting RNA from degradation [49, 52, 53].
Alterations on splicing variants have been shown to be associated with cancer progression and treatment resistance [54]. Splicing variants of the androgen receptor, in particular AR-V7, have been found to be significantly higher in hormone refractory prostate cancer and is associated with poor clinical outcomes [55]. Similar to BRAF-splicing variants, AR-V7 can be detected in plasma EV RNA [53, 56]. Altogether, this supports an alternative liquid biopsy modality based on EV RNA or ctRNA.
Liquid biopsies have emerged as a non-invasive approach to assess disease burden and the genetic evolution of tumours in response to therapy [15, 57–59]. Our study provides a foundation for the detection of splicing variants in both ctRNA and EV RNA in melanoma patients failing MAPK inhibition, although its clinical validity requires further evaluation in independent cohorts. This work constitutes an important proof of concept that in addition to plasma ctDNA, ctRNA can provide important tumour specific information related to the development of resistance. However pre-analytical conditions for ctRNA analysis still require optimisation to enable its clinical application.
Materials and Methods
Patient selection
A total of 38 participants with BRAF mutant metastatic melanoma were treated with vemurafenib or with dabrafenib and trametinib combination as per approved label were enrolled (Supplementary Table 1). Patients were required to have a recorded objective response to therapy, either partial or complete confirmed by CT or PET scans. All patients gave their signed informed consent before blood collection and data analysis. The study was performed in accordance with the Declaration of Helsinki and study protocols were approved by the Human Ethics Committees at Edith Cowan University (No.11543) and Sir Charles Gardner Hospital (No.2013-246).
Plasma from another five patients treated recruited at the Melanoma Institute Australia and affiliated hospitals were included in this study (Melanoma Institute Australia Biospecimen Bank for Melanoma Research ×11-0289 & HREC/11/RPAH/444 approved through Sydney Local Health District). These patients were selected as they were previously identified as to have BRAF splicing variants in their progressing tumour after failing treatment with dabrafenib monotherapy.
Cell culture
Melanoma cell line 451Lu was obtained from the Wistar Institute. SK-Mel-28. BR4, SMU027 and WMD009 were previously reported to carry BRAF splicing variants [9, 32]. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific, Carlsbad, CA, USA) supplemented with 10% Foetal Bovine Serum (Thermo Fisher Scientific, Carlsbad, CA, USA) at 37°C and 5% CO2.
RNA extraction from cell lines
RNA was extracted using the All Prep DNA/RNA Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA was eluted in RNAse free water and stored at –80°C until analysis.
Circulating nucleic acid extraction
Blood samples were collected into EDTA vacutainer tubes and plasma was separated within 24 hours by centrifugation at 300 g for 20 minutes, followed by a second centrifugation at 4700 g for 10 minutes and then stored at –80°C until extraction. Plasma cell-free nucleic acids (cfNA) were isolated from healthy donors and AJCC stage IV metastatic melanoma patients using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. cfNA was eluted in 40 μl AVE buffer (Qiagen, Hilden, Germany) and stored at –80°C.
Reverse transcription and preamplification
Extracted RNA (5 μl) from all samples were used for cDNA preparation using the SuperScript® VILO™ cDNA Synthesis Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) followed by a preamplification using the TaqMan® PreAmp Master Mix Kit (Thermo Fisher Scientific, Carlsbad, CA, USA) and all the primers for the BRAF splicing variants at 180 nM, following manufacturer cycling conditions (95°C for 10 minutes, followed by 14 cycles of 95°C for 15 seconds and 60°C for 4 minutes, with a final inactivation step at 99°C for 10 minutes). Linearity of the pre-amplification step was assessed by linear regression of a serial dilution curve (Supplementary Figure 2).
Droplet digital PCR
Primer pairs were designed to amplify over the unique exon-exon junction of each splicing variant, with probes overlapping the junction (Figure 1A and Supplementary Table 2). Probes were custom synthesised by Integrated DNA Technologies (IDT, San Jose, CA, USA). For ctDNA analysis, samples were analysed for the presence of BRAF or NRAS mutant DNA as described previously [15, 60].
Amplifications were performed in a 20 μL reaction containing 1× ddPCR Supermix for Probes (No dUTP, Bio-Rad), 1× Q solution (Qiagen, Hilden, Germany), 250 nM of each probe and 900 nM of each primer plus template. Droplets were generated using the automatic droplet generator QX200 AutoDG (Bio-Rad, Hercules, CA, USA). Amplifications were performed using the following cycling conditions: 1 cycle of 95°C (2.5C/s ramp) for 10 minutes, 40 cycles of 94°C (2.5C/s ramp) for 30 seconds and 55°C for 1 minute, followed by 1 cycle of 98°C (2.5C/s ramp) for 10 minutes. Droplets were analysed through a QX200 droplet reader (Bio-Rad, Hercules, CA, USA). QuantaSoft analysis software (Bio-Rad, Hercules, CA, USA) was used to acquire and analyse data.
BRAF amplification analysis
We optimised a ddPCR assay to assess the presence of BRAF amplifications using plasma-derived cfDNA. BRAF copy number was assessed relative to a reference gene situated in the centromere of chromosome 7 (VOPP1 gene, 7p11.2). For the assessment of BRAF: VOPP1 ratio, a ddPCR assay was performed with the Bio-Rad QX200 system using custom primers/probe sets against BRAF [60] and VOPP1 (Bio-Rad, Hercules, CA, USA). The BRAF: VOPP1 concentration ratio was determined by dividing the BRAF concentration by the VOPP1 concentration offered by the QuantaSoft software (Bio-Rad, Hercules, CA, USA).
We first determined the optimal threshold to define an elevated plasma DNA ddPCR BRAF: VOPP1 ratio using a standard curve of genomic DNA extracted from healthy volunteer WBC combined with different percentages of genomic DNA extracted from a melanoma cell line with known BRAF amplification (Supplementary Figure 1). Plasma-derived cfDNA from six healthy volunteers were used to evaluate specificity. Patient samples with mutant BRAF frequency abundance of 3% or higher were tested for BRAF amplification.
Extracellular vesicle isolation
For EVs isolation, cell lines at 50% confluence were washed three times with PBS and cultured in DMEM supplemented with exosome-depleted FBS (Thermo Fisher Scientific, Carlsbad, CA, USA) for 18 hours at 37°C and 5% CO2. Following this, cell culture media was harvested and centrifuged at 300 g for 5 minutes at 4°C. The supernatant was then transferred to a clean tube and re-centrifuged at 2000 g for 20 minutes. Cleared supernatant was used for EV isolation using the ExoEasy kit (Qiagen, Hilden, Germany) following manufacturer’s instructions. EVs were also isolated from 4 ml of plasma from melanoma patients and heathy volunteers using the ExoEasy kit (Qiagen) RNA was isolated from the extracted EVs using the RNeasy Micro Kit (Qiagen, Hilden, Germany) following manufacturer’s instructions. RNA was eluted in 14 μL.
Western blots
SK-Mel-28 cell lysate was prepared to be used as positive control. Cells were harvested using trypsin EDTA (Thermo Fisher Scientific, Carlsbad, CA, USA). Cell viability was then determined using trypan blue. Cells were then pelleted at 300 g for five minutes, washed twice in PBS and lysed with radioimmunoprecipitation (RIPA) assay buffer (Sigma-Aldrich, St. Louis, Missouri) containing a complete protease inhibitor tablet (Roche, Basel, Switzerland). Cell lysates were incubated for 30 minutes on ice prior to being cleared with a 13,000 g spin for five minutes, with the supernatant being transferred into a clean tube and stored at –20°C until use.
EV pellets were resuspended in PBS prior to mixing 1:1 with RIPA (Sigma-Aldrich, St. Louis, Missouri) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland). EV lysates were incubated for 30 minutes on ice and then stored at –20°C until use. Extracted EV proteins were diluted to 1:4 in Lamelli Buffer (Bio-Rad, Australia), incubated at 95°C for 5 minutes and resolved on a mini TGX 8–16% stain free gel (Bio-Rad). Proteins were transferred onto a nitrocellulose blotting membrane using the Trans-Blot® Turbo™ Transfer System at a constant voltage of 25V for seven minutes (Bio-Rad, Australia). The membranes were blocked in 5% milk tris buffered saline (TBS) for one hour at room temperature before being probed with primary antibody TSG101 (1:1000, clone EPR7130B, Abcam), CD9 (1:500, clone MM2/57, Life-Technologies), CD81 (1:500, clone 1.3.3.22, Life-Technologies). CD63 (1:1000, clone H5C6, BD-Biosciences, calnexin (1:500, clone AF18, Life-Technologies) diluted in 0.5% milk TBS 0.05% Tween 20. The membrane was washed three times for ten minutes with TBS 0.05% tween and subsequently probed with secondary antibodies (sheep anti-mouse IgG-HRP conjugate, polyclonal, 1:2000, GE Healthcare, donkey anti-rabbit IgG-HRP conjugate, polyclonal, 1:2000, GE Healthcare) diluted in 0.5% milk TBS .05% Tween 20. Signals were detected with the GE healthcare Amersham™ ECL™ reagent and were subsequently imaged using the ChemiDoc™ Touch Imaging System (Bio-Rad, Hercules, CA, USA). Images were processed using Image Lab™ software v6.0 (Bio-Rad, Hercules, CA, USA).
Transmission electron microscopy
EVs were resuspended in PBS and fixed in 2% paraformaldehyde prior to transfer onto 200 mesh Formvar-carbon coated copper grids (ProSciTech, Kirwan, QLD). EVs were adsorbed for 15 minutes at room temperature prior to being washed four times in filtered H2O. Samples were then fixed in 1% glutaraldehyde and contrasted with 1% uranyl acetate for 2 minutes before being left for 20 minutes to dry at room temperature. EVs were visualised using JEOL JEM-2100 electron microscope (JEOL, Tokyo, Japan) at an operating voltage of 80 kV. Images were captured using an 11M pixel Gatan Orius digital camera (Gatan, Pleasanton, CA, USA).
ACKNOWLEDGMENTS
The authors thank all the study participants (patients and healthy volunteers) for their assistance with the study.
CONFLICTS OF INTEREST
The following authors have received travel support from: MAK [Merck Sharp and Dohme (MSD), Bristol-Myers Squibb (BMS) and Merck Serono], TMM [BMS, Novartis, AstraZeneca (AZ)] and ESG [MSD]. The following authors sit on advisory boards for: TMM [BMS, MSD, Novartis, AZ]; MSC [Sanofi, Nektar, Merck Serono BMS, MSD, Novartis, Amgen, Pierre Fabre, Ideaya]; AMM [BMS, MSD, Novartis, Roche, Pierre-Fabre], GVL [Aduro, Amgen, Array, BMS, MSD, Novartis, Pierre-Fabre, Oncosec, Roche] and MM [BMS, AZ, Roche, MSD].
FUNDING
MC has received financial support through an “Australian Government Research Training Program” scholarship. EG is supported by fellowships from the Cancer Research Trust and Cancer Council Western Australia (CCWA). This study was funded by a CCWA Collaborative Grant Scheme to EG and KM. Samples collection was supported by a National Health and Medical Research (NHMRC) grant (1119791) to MZ. HR, RAS, and GVL are supported by NHMRC Fellowships. GVL is also supported by the University of Sydney Medical Foundation. AMM is supported by a Cancer Institute NSW Fellowship. ACM is supported by a Western Australian Health Translation Network Early Career Research Fellowship.
References
1. Lyle M, Haydu LE, Menzies AM, Thompson JF, Saw RPM, Spillane AJ, Kefford RF, Mann GJ, Cooper WA, Yu B, Scolyer RA, O’Toole SA, Long GV. The molecular profile of metastatic melanoma in Australia. Pathology. 2016; 48:188–193. https://doi.org/10.1016/j.pathol.2015.12.008. [PubMed].
2. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364:2507–2516. https://doi.org/10.1056/NEJMoa1103782. [PubMed].
3. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380:358–365. https://doi.org/10.1016/S0140-6736(12)60868-X. [PubMed].
4. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, Chiarion Sileni V, Schachter J, Garbe C, Bondarenko I, Gogas H, Mandalá M, Haanen JB, et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N Engl J Med. 2019; 381:626–636. https://doi.org/10.1056/NEJMoa1904059. [PubMed].
5. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined Vemurafenib and Cobimetinib in BRAF-Mutated Melanoma. N Engl J Med. 2014; 371:1867–1876. https://doi.org/10.1056/NEJMoa1408868. [PubMed].
6. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694–1703. https://doi.org/10.1056/NEJMoa1210093. [PubMed].
7. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014; 4:80–93. https://doi.org/10.1158/2159-8290.CD-13-0642. [PubMed].
8. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, Howle J, Saw R, Thompson JF, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014; 20:1965–1977. https://doi.org/10.1158/1078-0432.CCR-13-3122. [PubMed].
9. Long GV, Fung C, Menzies AM, Pupo GM, Carlino MS, Hyman J, Shahheydari H, Tembe V, Thompson JF, Saw RP, Howle J, Hayward NK, Johansson P, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014; 5:5694. https://doi.org/10.1038/ncomms6694. [PubMed].
10. Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, Gogas H, Kefford RF, Thompson JF, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015; 51:2792–2799. https://doi.org/10.1016/j.ejca.2015.08.022. [PubMed].
11. Freeman AK, Ritt DA, Morrison DK. The importance of Raf dimerization in cell signaling. Small GTPases. 2013; 4:180–185. https://doi.org/10.4161/sgtp.26117. [PubMed].
12. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011; 480:387–390. https://doi.org/10.1038/nature10662. [PubMed].
13. Diefenbach RJ, Lee JH, Rizos H. Monitoring Melanoma Using Circulating Free DNA. Am J Clin Dermatol. 2019; 20:1–12. https://doi.org/10.1007/s40257-018-0398-x. [PubMed].
14. Calapre L, Warburton L, Millward M, Ziman M, Gray ES. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017; 404:62–69. https://doi.org/10.1016/j.canlet.2017.06.030. [PubMed].
15. Gray ES, Rizos H, Reid AL, Boyd SC, Pereira MR, Lo J, Tembe V, Freeman J, Lee JH, Scolyer RA, Siew K, Lomma C, Cooper A, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget. 2015; 6:42008–42018. https://doi.org/10.18632/oncotarget.5788. [PubMed].
16. Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, Mandal AK, Lim KH, Saturno G, Furney SJ, Baenke F, Pedersen M, Rogan J, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016; 6:286–299. https://doi.org/10.1158/2159-8290.CD-15-1336. [PubMed].
17. El-Hefnawy T, Raja S, Kelly L, Bigbee WL, Kirkwood JM, Luketich JD, Godfrey TE. Characterization of amplifiable, circulating RNA in plasma and its potential as a tool for cancer diagnostics. Clin Chem. 2004; 50:564–573. https://doi.org/10.1373/clinchem.2003.028506. [PubMed].
18. Beck TN, Boumber YA, Aggarwal C, Pei J, Thrash-Bingham C, Fittipaldi P, Vlasenkova R, Rao C, Borghaei H, Cristofanilli M, Mehra R, Serebriiskii I, Alpaugh RK. Circulating tumor cell and cell-free RNA capture and expression analysis identify platelet-associated genes in metastatic lung cancer. BMC Cancer. 2019; 19:603. https://doi.org/10.1186/s12885-019-5795-x. [PubMed].
19. Yu XM, Wu YC, Liu X, Huang XC, Hou XX, Wang JL, Cheng XL, Mao WM, Ling ZQ. Cell-Free RNA Content in Peripheral Blood as Potential Biomarkers for Detecting Circulating Tumor Cells in Non-Small Cell Lung Carcinoma. Int J Mol Sci. 2016; 17:1845. https://doi.org/10.3390/ijms17111845. [PubMed].
20. Chen XQ, Bonnefoi H, Pelte MF, Lyautey J, Lederrey C, Movarekhi S, Schaeffer P, Mulcahy HE, Meyer P, Stroun M, Anker P. Telomerase RNA as a detection marker in the serum of breast cancer patients. Clin Cancer Res. 2000; 6:3823–3826. https://clincancerres.aacrjournals.org/content/6/10/3823. [PubMed].
21. Kopreski MS, Benko FA, Gocke CD. Circulating RNA as a tumor marker: detection of 5T4 mRNA in breast and lung cancer patient serum. Ann N Y Acad Sci. 2001; 945:172–178. https://doi.org/10.1111/j.1749-6632.2001.tb03882.x. [PubMed].
22. Kopreski MS, Benko FA, Kwak LW, Gocke CD. Detection of tumor messenger RNA in the serum of patients with malignant melanoma. Clin Cancer Res. 1999; 5:1961–1965. https://clincancerres.aacrjournals.org/content/5/8/1961. [PubMed].
23. Silva JM, Rodriguez R, Garcia JM, Munoz C, Silva J, Dominguez G, Provencio M, Espana P, Bonilla F. Detection of epithelial tumour RNA in the plasma of colon cancer patients is associated with advanced stages and circulating tumour cells. Gut. 2002; 50:530–534. https://doi.org/10.1136/gut.50.4.530. [PubMed].
24. Zhang X, Yang X, Zhang Y, Liu X, Zheng G, Yang Y, Wang L, Du L, Wang C. Direct Serum Assay for Cell-Free Bmi-1 mRNA and Its Potential Diagnostic and Prognostic Value for Colorectal Cancer. Clin Cancer Res. 2015; 21:1225–1233. https://doi.org/10.1158/1078-0432.CCR-14-1761. [PubMed].
25. Sorrentino S. The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010; 584:2194–2200. https://doi.org/10.1016/j.febslet.2010.04.018. [PubMed].
26. Yuan T, Huang X, Woodcock M, Du M, Dittmar R, Wang Y, Tsai S, Kohli M, Boardman L, Patel T, Wang L. Plasma extracellular RNA profiles in healthy and cancer patients. Sci Rep. 2016; 6:19413. https://doi.org/10.1038/srep19413. [PubMed].
27. Takahashi K, Ota Y, Kogure T, Suzuki Y, Iwamoto H, Yamakita K, Kitano Y, Fujii S, Haneda M, Patel T, Ota T. Circulating extracellular vesicle-encapsulated HULC is a potential biomarker for human pancreatic cancer. Cancer Sci. 2020; 111:98–111. https://doi.org/10.1111/cas.14232. [PubMed].
28. Colombo M, Raposo G, Théry C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu Rev Cell Dev Biol. 2014; 30:255–289. https://doi.org/10.1146/annurev-cellbio-101512-122326. [PubMed].
29. Turchinovich A, Drapkina O, Tonevitsky A. Transcriptome of Extracellular Vesicles: State-of-the-Art. Front Immunol. 2019; 10:202–202. https://doi.org/10.3389/fimmu.2019.00202. [PubMed].
30. Kim KM, Abdelmohsen K, Mustapic M, Kapogiannis D, Gorospe M. RNA in extracellular vesicles. Wiley Interdiscip Rev RNA. 2017; 8:e1413. https://doi.org/10.1002/wrna.1413. [PubMed].
31. Di Liegro CM, Schiera G, Di Liegro I. Extracellular Vesicle-Associated RNA as a Carrier of Epigenetic Information. Genes (Basel). 2017; 8:240. https://doi.org/10.3390/genes8100240. [PubMed].
32. Gowrishankar K, Snoyman S, Pupo GM, Becker TM, Kefford RF, Rizos H. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J Invest Dermatol. 2012; 132:1850–1859. https://doi.org/10.1038/jid.2012.63. [PubMed].
33. Patton JG, Franklin JL, Weaver AM, Vickers K, Zhang B, Coffey RJ, Ansel KM, Blelloch R, Goga A, Huang B, L’Etoille N, Raffai RL, Lai CP, et al. Biogenesis, delivery, and function of extracellular RNA. J Extracell Vesicles. 2015; 4:27494. https://doi.org/10.3402/jev.v4.27494. [PubMed].
34. Vido MJ, Le K, Hartsough EJ, Aplin AE. BRAF Splice Variant Resistance to RAF Inhibitor Requires Enhanced MEK Association. Cell Reports. 2018; 25:1501–1510.e3. https://doi.org/10.1016/j.celrep.2018.10.049. [PubMed].
35. Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014; 4:61–68. https://doi.org/10.1158/2159-8290.CD-13-0631. [PubMed].
36. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, Le Quesne J, Moore DA, Veeriah S, Rosenthal R, Marafioti T, Kirkizlar E, Watkins TBK, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017; 545:446–451. https://doi.org/10.1038/nature22364. [PubMed].
37. McEvoy AC, Warburton L, Al-Ogaili Z, Celliers L, Calapre L, Pereira MR, Khattak MA, Meniawy TM, Millward M, Ziman M, Gray ES. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer. 2018; 18:726–726. https://doi.org/10.1186/s12885-018-4637-6. [PubMed].
38. Lampignano R, Neumann MHD, Weber S, Kloten V, Herdean A, Voss T, Groelz D, Babayan A, Tibbesma M, Schlumpberger M, Chemi F, Rothwell DG, Wikman H, et al. Multicenter Evaluation of Circulating Cell-Free DNA Extraction and Downstream Analyses for the Development of Standardized (Pre)analytical Work Flows. Clin Chem. 2020; 66:149–160. https://doi.org/10.1373/clinchem.2019.306837. [PubMed].
39. Souza MF, Kuasne H, Barros-Filho MC, Cilião HL, Marchi FA, Fuganti PE, Paschoal AR, Rogatto SR, Cólus IM. Circulating mRNAs and miRNAs as candidate markers for the diagnosis and prognosis of prostate cancer. PLoS One. 2017; 12:e0184094. https://doi.org/10.1371/journal.pone.0184094. [PubMed].
40. Pucciarelli S, Rampazzo E, Briarava M, Maretto I, Agostini M, Digito M, Keppel S, Friso ML, Lonardi S, De Paoli A, Mescoli C, Nitti D, De Rossi A. Telomere-Specific Reverse Transcriptase (hTERT) and Cell-free RNA in Plasma as Predictors of Pathologic Tumor Response in Rectal Cancer Patients Receiving Neoadjuvant Chemoradiotherapy. Ann Surg Oncol. 2012; 19:3089–3096. https://doi.org/10.1245/s10434-012-2272-z. [PubMed].
41. García–Olmo DC, Contreras JD, Picazo MG, López–Torres J, García–Olmo D. Potential clinical significance of perioperative levels of mRNA in plasma from patients with cancer of the larynx or hypopharynx. Head Neck. 2017; 39:647–655. https://doi.org/10.1002/hed.24638. [PubMed].
42. Lo KW, Lo YMD, Leung SF, Tsang YS, Chan LYS, Johnson PJ, Hjelm NM, Lee JCK, Huang DP. Analysis of Cell-free Epstein-Barr Virus-associated RNA in the Plasma of Patients with Nasopharyngeal Carcinoma. Clin Chem. 1999; 45:1292–1294. https://doi.org/10.1093/clinchem/45.8.1292. [PubMed].
43. Papadopoulou E, Davilas E, Sotiriou V, Georgakopoulos E, Georgakopoulou S, Koliopanos A, Aggelakis F, Dardoufas K, Agnantis N, Karydas I, Nasioulas G. Cell-free DNA and RNA in Plasma as a New Molecular Marker for Prostate and Breast Cancer. Ann N Y Acad Sci. 2006; 1075:235–243. https://doi.org/10.1196/annals.1368.032. [PubMed].
44. Rampazzo E, Del Bianco P, Bertorelle R, Boso C, Perin A, Spiro G, Bergamo F, Belluco C, Buonadonna A, Palazzari E, Lonardi S, De Paoli A, Pucciarelli S, et al. The predictive and prognostic potential of plasma telomerase reverse transcriptase (TERT) RNA in rectal cancer patients. Br J Cancer. 2018; 118:878–886. https://doi.org/10.1038/bjc.2017.492. [PubMed].
45. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, Sahoo S, Tahara H, Wauben MH, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014; 3:26913. https://doi.org/10.3402/jev.v3.26913. [PubMed].
46. Willms E, Cabañas C, Mäger I, Wood MJA, Vader P. Extracellular Vesicle Heterogeneity: Subpopulations, Isolation Techniques, and Diverse Functions in Cancer Progression. Front Immunol. 2018; 9:738–738. https://doi.org/10.3389/fimmu.2018.00738. [PubMed].
47. Gyuris A, Navarrete-Perea J, Jo A, Cristea S, Zhou S, Fraser K, Wei Z, Krichevsky AM, Weissleder R, Lee H, Gygi SP, Charest A. Physical and Molecular Landscapes of Mouse Glioma Extracellular Vesicles Define Heterogeneity. Cell Rep. 2019; 27:3972–3987.e6. https://doi.org/10.1016/j.celrep.2019.05.089. [PubMed].
48. Armitage JD, Tan DBA, Cha L, Clark M, Gray ES, Fuller KA, Moodley YP. A standardised protocol for the evaluation of small extracellular vesicles in plasma by imaging flow cytometry. J Immunol Methods. 2019; 468:61–66. https://doi.org/10.1016/j.jim.2019.03.006. [PubMed].
49. Zhou R, Chen KK, Zhang J, Xiao B, Huang Z, Ju C, Sun J, Zhang F, Lv XB, Huang G. The decade of exosomal long RNA species: an emerging cancer antagonist. Mol Cancer. 2018; 17:75. https://doi.org/10.1186/s12943-018-0823-z. [PubMed].
50. Kitagawa T, Taniuchi K, Tsuboi M, Sakaguchi M, Kohsaki T, Okabayashi T, Saibara T. Circulating pancreatic cancer exosomal RNAs for detection of pancreatic cancer. Mol Oncol. 2019; 13:212–227. https://doi.org/10.1002/1878-0261.12398. [PubMed].
51. Yoshii S, Hayashi Y, Iijima H, Inoue T, Kimura K, Sakatani A, Nagai K, Fujinaga T, Hiyama S, Kodama T, Shinzaki S, Tsujii Y, Watabe K, et al. Exosomal microRNAs derived from colon cancer cells promote tumor progression by suppressing fibroblast TP53 expression. Cancer Sci. 2019; 110:2396–2407. https://doi.org/10.1111/cas.14084. [PubMed].
52. Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014; 3:23743. https://doi.org/10.3402/jev.v3.23743. [PubMed].
53. Del Re M, Biasco E, Crucitta S, Derosa L, Rofi E, Orlandini C, Miccoli M, Galli L, Falcone A, Jenster GW, van Schaik RH, Danesi R. The Detection of Androgen Receptor Splice Variant 7 in Plasma-derived Exosomal RNA Strongly Predicts Resistance to Hormonal Therapy in Metastatic Prostate Cancer Patients. Eur Urol. 2017; 71:680–687. https://doi.org/10.1016/j.eururo.2016.08.012. [PubMed].
54. Escobar-Hoyos L, Knorr K, Abdel-Wahab O. Aberrant RNA Splicing in Cancer. Annu Rev Cancer Biol. 2019; 3:167–185. https://doi.org/10.1146/annurev-cancerbio-030617-050407. [PubMed].
55. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009; 69:16–22. https://doi.org/10.1158/0008-5472.CAN-08-2764. [PubMed].
56. Nimir M, Ma Y, Jeffreys SA, Opperman T, Young F, Khan T, Ding P, Chua W, Balakrishnar B, Cooper A, De Souza P, Becker TM. Detection of AR-V7 in Liquid Biopsies of Castrate Resistant Prostate Cancer Patients: A Comparison of AR-V7 Analysis in Circulating Tumor Cells, Circulating Tumor RNA and Exosomes. Cells. 2019; 8:688. https://doi.org/10.3390/cells8070688. [PubMed].
57. Wong SQ, Raleigh JM, Callahan J, Vergara IA, Ftouni S, Hatzimihalis A, Colebatch AJ, Li J, Semple T, Doig K, Mintoff C, Sinha D, Yeh P, et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis Oncol. 2017; 1:1–14. https://doi.org/10.1200/PO.16.00009.
58. Wang R, Li X, Zhang H, Wang K, He J. Cell-free circulating tumor DNA analysis for breast cancer and its clinical utilization as a biomarker. Oncotarget. 2017; 8:75742–75755. https://doi.org/10.18632/oncotarget.20608. [PubMed].
59. Kruger S, Heinemann V, Ross C, Diehl F, Nagel D, Ormanns S, Liebmann S, Prinz-Bravin I, Westphalen CB, Haas M, Jung A, Kirchner T, von Bergwelt-Baildon M, et al. Repeated mutKRAS ctDNA measurements represent a novel and promising tool for early response prediction and therapy monitoring in advanced pancreatic cancer. Ann Oncol. 2018; 29:2348–2355. https://doi.org/10.1093/annonc/mdy417. [PubMed].
60. Reid AL, Freeman JB, Millward M, Ziman M, Gray ES. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin Biochem. 2015; 48:999–1002. https://doi.org/10.1016/j.clinbiochem.2014.12.007. [PubMed].