
Introduction
Cancer immunotherapy relies on the activation of certain leukocytes (most typically, T cells and/or natural killer cells), with the aim to induce a selective biocidal activity against tumor cells. Immunotherapy approaches, which may include the use of recombinant cytokines [1–4], immune checkpoint inhibitors [5–7] and immune cells [8, 9], are increasingly being used in the clinical practice. In 1992, interleukin (IL)-2 was approved as the first cancer immunotherapy, providing a long-term survival benefit to a small proportion of patients with metastatic melanoma or renal cell carcinoma [10–12]. However, high-dose IL2 causes substantial toxicity, thus limiting this treatment modality to younger and physically fit patients. The modest accumulation of IL2 at the site of disease [13] and its short half-life [14, 15] represent additional limitations for a broader applicability of this immunostimulatory agent in cancer treatment.
Recently, there has been a growing interest in the engineering of novel cytokine products with improved anticancer properties and better tolerability. Molecular strategies have included the generation of antibody fusion proteins, the introduction of mutations in the cytokine moiety and/or the conjugation to polymers [3, 16–27]. IL2 muteins may alter the interaction of the cytokine with one or more of the IL2 receptor subunits [20], thus altering the selectivity towards the intermediate affinity receptor [consisting of CD122 (IL2Rβ) and CD132 (γC)] or towards the high affinity receptor [consisting of CD25 (IL2Rα), CD122, and CD132] [28]. Since regulatory T cells (Tregs) predominantly express the high affinity receptor [28], the selective activation of the intermediate affinity receptor (expressed by resting and memory lymphocytes including CD8+ T cells) [28] has attracted considerable research efforts [29–35]. In one approach, the strength of the interaction of IL2 with the cognate intermediate affinity receptor was increased through mutations, which also stabilized folding [34]. Treating tumor-bearing mice with such a mutein expanded both Treg and CD8+ T cells to a lesser extent than memory CD8+ T, compared to the administration of wild-type IL2. The increased CD8+: Treg ratio correlated with an improved therapeutic effect [34]. In a second approach, an increased selectivity towards the intermediate affinity IL2 receptor was achieved by blocking the interaction with CD25 either through IL2 mutations or by antibody blockade of the IL2 epitope involved in CD25 binding [31–33, 35]. Interestingly, a decreased IL2 toxicity was observed in CD25 knock-out mice [36].
In 2019, Silva et al. described a series of engineered cytokines, which displayed biological features intermediate between those of IL2 and of IL15. IL15 interacts with CD122 (IL2Rβ) and CD132 (γC) like IL2, but recognizes a different alpha subunit (CD215, rather than CD25). One of the new computationally designed cytokine variants, termed by the authors Neoleukin™-2/15, exhibited an increased affinity towards the receptor components shared by IL2 and IL15, with a complete absence of CD25 binding. Moreover, the new engineered cytokine was much more stable compared to wild-type IL2 [37]. Treatment of mice with recombinant preparations of the cytokine mimetic led to an increased CD8+: Treg ratio and improved therapeutic effects compared to murine IL2 in mouse cancer models, suggesting that the new cytokine may be more suitable for pharmaceutical applications. However, the protein has a small size (approximately 12 kDa), clears rapidly from circulation and lacks a tumor targeting moiety.
While the half-life extension of IL2 has been addressed by PEGylation [21] and albumin-fusion [38], selective tumor accumulation has generally been achieved by fusion to antibodies forming “immunocytokines” [3, 16]. The choice of a suitable antibody format may heavily influence biodistribution properties and performance [16, 39, 40]. Antibody fragments are cleared more rapidly than full-length IgG’s and may lead to superior tumor: blood ratios in vivo [16]. Fusion proteins based on the L19 antibody (specific to the alternatively-spliced extra-domain B (EDB) of fibronectin, a marker of angiogenesis) and on IL2 or TNF are being investigated in Phase III clinical trials for the treatment of patients with melanoma (https://clinicaltrials.gov/ NCT02938299, EudraCT 2015–002549-72) or with soft-tissue sarcoma (https://clinicaltrials.gov/ NCT03420014, EudraCT 2016-003239-38) [41]. The EDB of fibronectin represents an attractive tumor-associated antigen, which is expressed in most malignancies but is undetectable in most normal tissues except for some structures in female reproductive organs [42].
In this work, we have fused the L19 antibody in diabody format to Neoleukin™-2/15 (termed “L19-Neo™” and “Neo™-L19”). The new fusion proteins were able to selectively localize at the tumor site (as evidenced by quantitative biodistribution experiments with radiolabeled protein preparations). Neo™-L19 displayed a potent anticancer activity in two immunocompetent mouse models of cancer.
Results
Cloning, expression and in vitro characterization of fusion proteins
The engineered cytokine Neoleukin™-2/15 moiety was genetically fused at the N- or C-terminus of the L19 antibody in diabody format (Figure 1A and Supplementary Sequence Information). The linker connecting the two moieties comprised 15 amino acids (Figure 1A, 1F), while a short linker (5 amino acids) between VH and VL ensured a stable diabody formation [43]. The fusion proteins (termed L19-Neo™ and Neo™-L19) were expressed in CHO cells and purified by protein A affinity chromatography, utilizing the engineered protein A binding site on L19 [44]. The molecular weight of the fusion proteins was confirmed by LC/MS. SDS-PAGE and size exclusion chromatography analyses indicated a high purity and homogeneity (Figure 1). Avid binding of the fusion proteins to EDB was confirmed by surface plasmon resonance (Figure 1E, 1J).
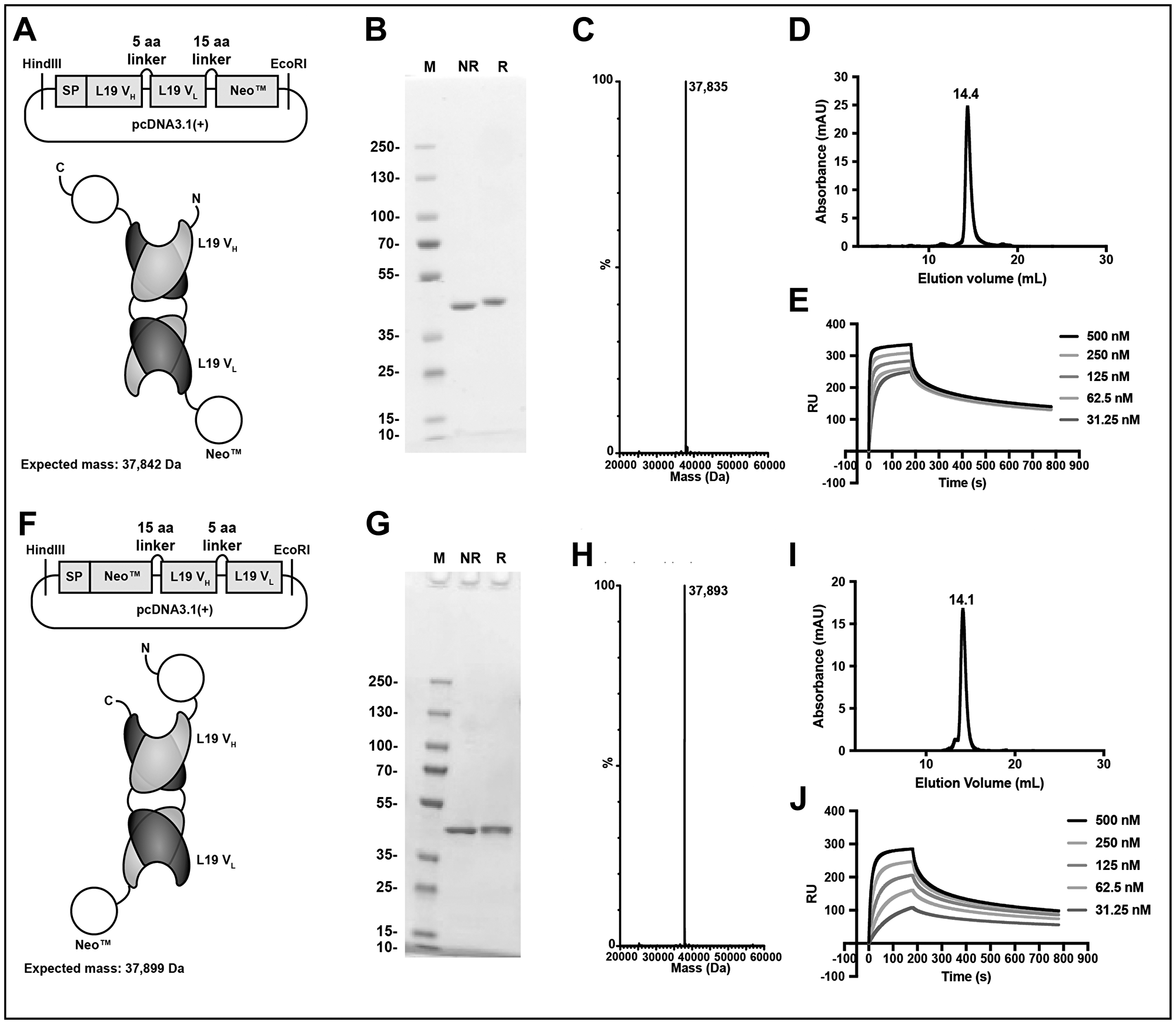
Figure 1: Design and in vitro characterization of the immunocytokine fusion proteins. Top, L19-Neo™. Bottom, Neo™-L19. (A, F) Cloning scheme, expected protein structure and expected mass. (B, G) SDS-PAGE analysis stained for protein by Coomassie Blue. M: PageRuler™ Plus Prestained Protein Ladder, NR: non-reducing, R: reducing. (C, H) MS profile. (D, I) Size exclusion chromatography analysis. (E, J) Surface plasmon resonance experiment using a serial dilution of the fusion protein and surface immobilized EDB of fibronectin.
We tested cytokine activity using a recently reported NF-κB reporter assay in transduced murine CTLL-2 cells [45] (Figure 2A) and a proliferation assay with murine CTLL-2 cells (Supplementary Figure 1). Both assays indicated that the fusion proteins had similar cytokine activities, which was comparable to the one of the recombinant cytokine. The binding of the cytokine moiety to its cognate receptor was investigated by flow cytometry on murine CTLL-2 cells using fluorescein-modified immunocytokines. Surprisingly, L19-Neo™ and Neo™-L19 did not show binding to the very same cells that were successfully used for the proliferation assay, while the L19-IL2 fusion protein [13], used as positive control, showed a concentration-dependent increase in average fluorescence signal, as expected (Figure 2B). The discrepancies might be a result of differences in binding kinetics, since the experiment requires a long-lasting interaction for binding to be observed. We confirmed that L19-IL2 was able to bind to hCD25 by surface plasmon resonance, while L19-Neo™ and Neo™-L19 did not interact with the receptor subunit (Figure 2C and Supplementary Figure 2).
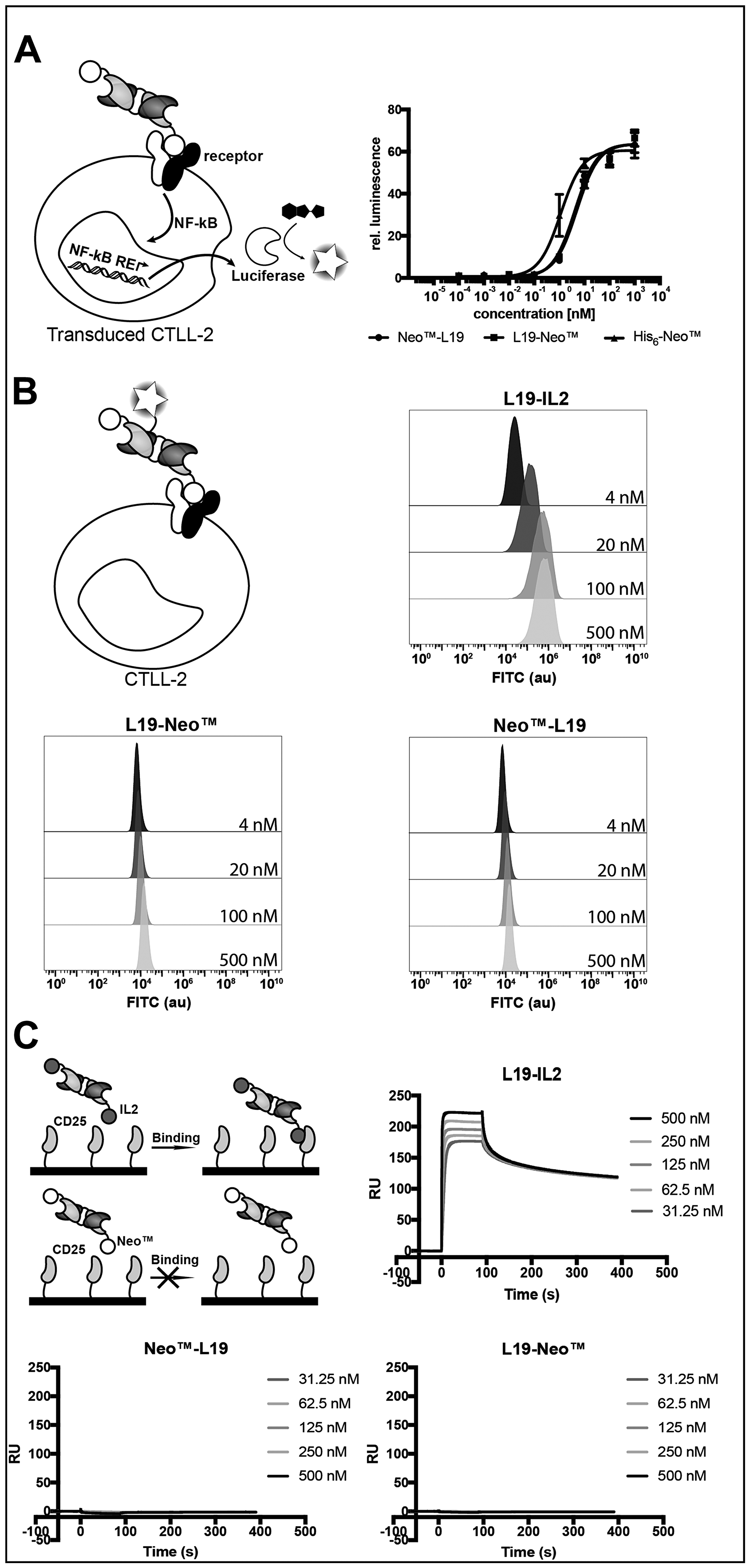
Figure 2: Characterization of the cytokine moiety in the fusion proteins. (A) NF-κB coupled luciferase reporter assay comparing the fusion proteins with His6-Neo™. EC50 (mean (n = 3) ± SD): L19-Neo™ 5.01 ± 0.57 nM, Neo™-L19 4.14 ± 0.55 nM, His6-Neo™ 1.14 ± 0.21 nM. (B) Binding investigating of FITC-modified fusion proteins on murine CTLL-2 cells by flow cytometry. (C) SPR experiment using a serial dilution of fusion protein in the solution phase and surface immobilized hCD25. Zoomed images of the sensograms for the fusion proteins are shown in Supplementary Figure 2.
In vivo biodistribution and therapy studies
The tumor homing potential of the L19-Neo™ and Neo™-L19 immunocytokines was investigated by a quantitative biodistribution experiment using radiolabeled protein preparations. The products were injected intravenously into immunocompetent mice bearing F9 teratocarcinoma tumors, which were sacrificed 24 hours later. A selective tumor accumulation was observed for both fusion proteins, but Neo™-L19 showed a slightly improved percent of injected dose per gram of tumor (%ID/g; 8.3 ± 2.2% versus 3.9 ± 2.1%, respectively) and, for this reason, was chosen for subsequent therapy experiments. Both products have low levels in blood (~1%ID/g) and in other tissues (Figure 3). The biodistribution profiles were better than the ones observed using the KSF antibody moiety, specific to hen egg lysozyme and serving as negative control of irrelevant specificity in the mouse (Supplementary Figures 3 and 4).
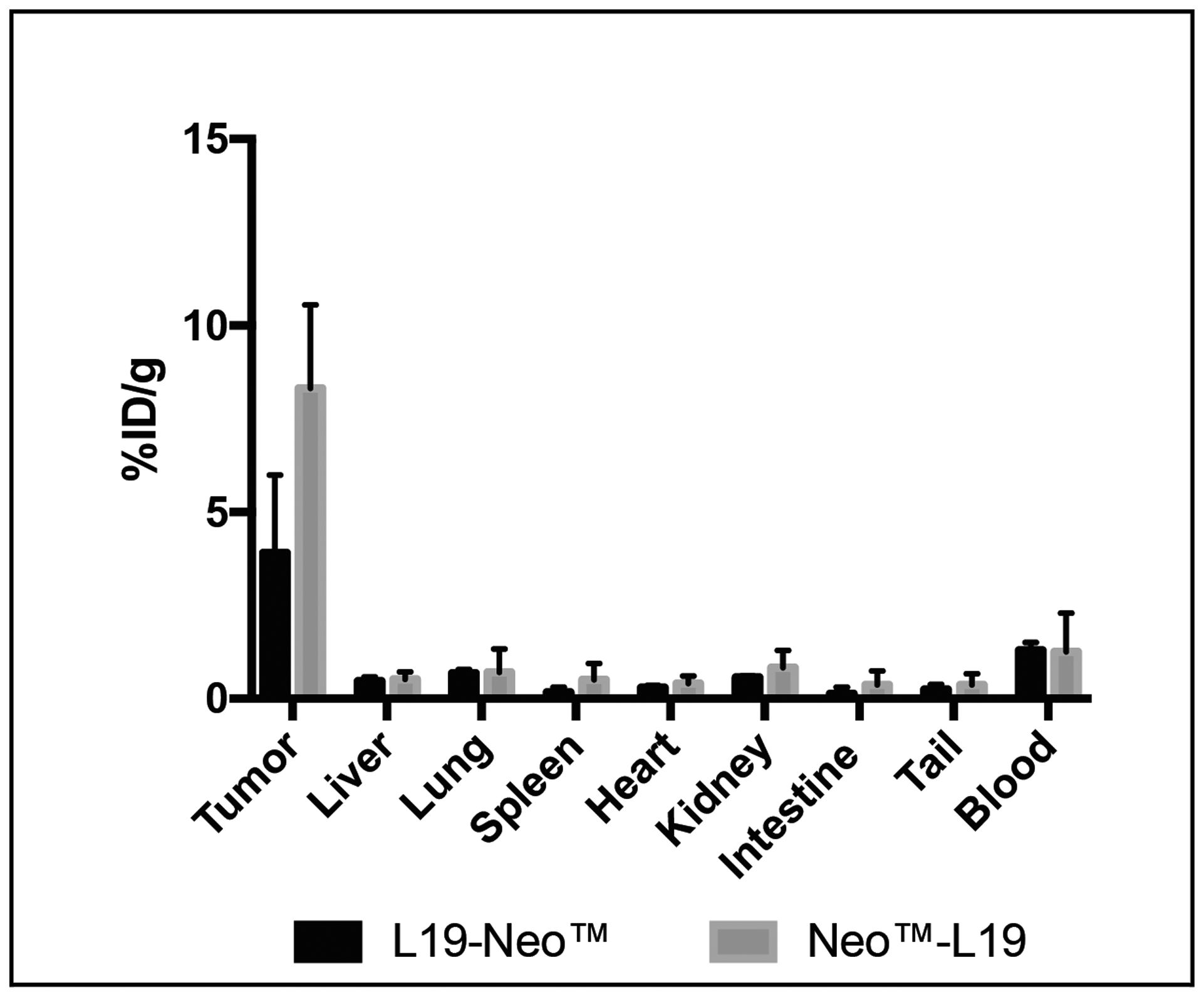
Figure 3: Investigation of the tumor homing potential of the fusion proteins in vivo. A quantitative biodistribution experiment using intravenously injected radioiodinated fusion proteins. The analysis was performed after 24 h and is expressed in percentage of the injected dose per gram of tissue (%ID/g ± SD, n = 3).
Therapy experiments were performed in immunocompetent mice bearing subcutaneous F9 teratocarcinomas. In an initial therapy experiment, Neo™-L19 and Neo™-KSF were administered intravenously (60 μg per injection) on days 7, 9, and 11. Both products inhibited tumor growth compared to saline treatment, but Neo™-L19 was more potent and gave a 50% cure rate (Supplementary Figure 5). In a second therapy experiment with mice bearing F9 teratocarcinomas, Neo™-L19 was injected at doses of 10 and 100 μg per injection on days 9, 11, and 13. The treatment resulted in a potent tumor-growth inhibiting activity compared to saline (Figure 4 and Supplementary Figure 6), with 2/5 durable complete responses in the Neo™-L19 group, while the nontargeted Neo™-KSF fusion did not cure any mouse (0/5). Mice with complete responses were observed for 60 days after last tumor indication, and in all cases the tumor eradication persisted. The treatments were in general well tolerated indicated by the small changes in body weights (Figure 4).
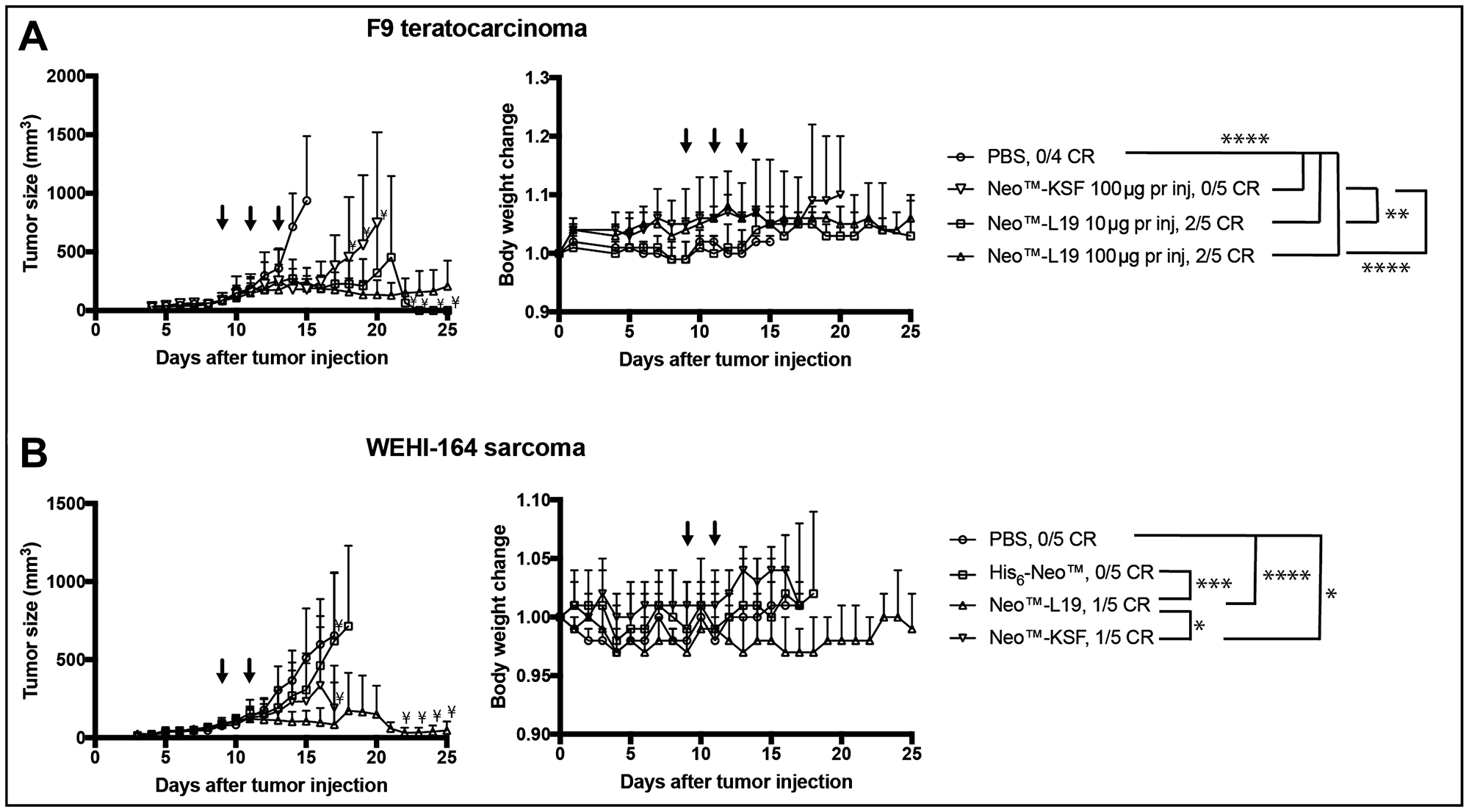
Figure 4: Activity of Neo™-L19 in F9 teratocarcinoma or WEHI-164 fibrosarcoma tumor bearing mice. (A) F9 teratocarcinoma. Left: Tumor bearing mice received 3 injections (↓) of Neo™-L19, Neo™-KSF or saline (PBS) when the tumors reached an average size of 90 mm3. The data is represented as the mean ± SD. Statistical analysis was performed by two-way ANOVA with post Bonferroni test (Not significant P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). All the immunocytokine treatments were statistically different from the saline treatment on day 14 (****P < 0.0001). On day 20, Neo™-L19 10 μg per inj (**P < 0.01) and Neo™-L19 100 μg per inj (****P < 0.0001) were different from the Neo™-KSF (100 μg per inj) group. Right: Body weight changes during the treatment represented as the mean ± SD. (B) WEHI-164 fibrosarcoma. Left: Tumor bearing mice received 2 equimolar injections (↓) of Neo™-L19, Neo™-KSF, His6-Neo™ or saline (PBS) when the tumors reached an average size of 90 mm3. The data is represented as the mean ± SD. On day 16, the Neo™-L19 (****P < 0.0001) and Neo™-KSF (*P < 0.05) groups showed a significant difference to the saline group. On day 16, Neo™-L19 was different from the His6-Neo™ (***P < 0.001) and Neo™-KSF (*P < 0.05) groups. On day 17, Neo™-L19 and Neo™-KSF was not significantly different (P > 0.05). Right: Body weight changes during the treatment represented as the mean ± SD. Only significant differences between treatment groups have been indicated. ¥only two mice remaining in the group. CR: complete response.
We also performed therapy experiments in immunocompetent BALB/c mice bearing subcutaneous WEHI-164 fibrosarcomas. A preliminary parallel investigation indicated that a third injection of 100 μg immunocytokine was not well tolerated, since it resulted in body weight loss (Supplementary Figure 7). Neo™-L19, Neo™-KSF and His6-Neo™ were administered in equimolar doses on days 9 and 11. The Neo™-L19 (****P < 0.0001) and Neo™-KSF (*P < 0.05) fusion proteins showed a potent tumor-growth inhibition (Figure 4 and Supplementary Figure 8), while the His-tagged recombinant cytokine, in our hands, had a modest tumor growth inhibitory effect. Collectively, the therapy experiments indicate that the tumor-homing immunocytokine Neo™-L19 potently inhibited tumor growth in two independent immunocompetent mouse models of cancer.
DISCUSSION
High-dose IL2 is considered the first effective cancer immunotherapy, since the treatment can lead to complete responses for melanoma or renal cell carcinoma patients with only a few patients experiencing recurrence [4]. However, administered systemically at high doses, IL2 can cause severe toxicity including life-threatening side effects reducing the number of suitable patients and narrowing the therapeutic window. Different IL2 muteins [20] and fusion proteins [16] have addressed the limitations of high dose IL2 treatment in attempts to improve the therapeutic index. The in silico designed IL2 mimic, Neoleukin™-2/15, was developed by Baker and coworkers and is being explored by Neoleukin™ Therapeutics Inc. for its antitumor potential [37]. However, the therapeutic potential of the cytokine mimic might be limited by rapid renal clearance due to its small size and its lack of tumor-homing potential.
In this study, we have investigated if the therapeutic potential of the engineered cytokine can be improved by facilitating selective tumor accumulation. We have described the production of novel fusion proteins (L19-Neo™ and Neo™-L19) that bind EDB of fibronectin present on the tumor extracellular matrix. The activities of the diabody and cytokine moieties were characterized in vitro, and selective tumor accumulation was demonstrated in F9 tumor bearing mice. Since Neo™-L19 had superior tumor accumulation, it was investigated in two syngeneic mouse cancer models (F9 and WEHI-164) showing potent antitumor activity as monotherapy.
The targeting of cytokines to the tumor microenvironment by the means of antibody fusion can lead to selective tumor accumulation as observed for Neo™-L19 [13, 16, 46–48]. As expected, a fusion protein of irrelevant specificity in the mouse (Neo™-KSF) showed a reduced tumor uptake, confirming the role played by the L19 antibody in the active delivery of the payload (Supplementary Figure 4).
Bispecific antibodies carrying high-affinity binding moieties towards a tumor-associated antigen and the CD3 antigen on T cells exhibit an in vivo localization both to tumors and to secondary lymphoid organs [49]. It has previously been claimed that interactions between the cytokine moieties and their cognate receptors on circulating lymphocytes or in secondary lymphoid organs might hamper the tumor targeting properties of fusion proteins [40]. This feature has not been seen for many immunocytokines based on the L19 antibody [13, 50–52] and was also not observed for Neo™-L19. Indeed, the fusion protein exhibited a preferential uptake in the tumor (8.3 ± 2.2% ID/g) and a tumor: spleen ratio of approximately 16 twenty-four hours after intravenous administration. The absence of immunocytokine trapping on lymphocytes may be due to the low number of cognate receptors on T cells and NK cells, or to an insufficient kinetic stability of the cytokine: receptor complex. The diabody format, chosen in our work, may be preferable to the more conventional IgG format, since the reduced molecular weight and the absence of FcRn binding may lead to better penetration into the neoplastic mass and faster clearance from circulation [16]. Even though EDB (+)-fibronectin expression is typically associated with the tumor neo-vasculature, the antigen is predominantly found on the abluminal aspect of tumor blood vessels [53].
Mechanistic investigations into tumor targeted IL2 based treatments have indicated that immunological responses are correlated with an increased intratumoral presence of NK and CD8+ T cells [17, 54]. While importance of NK and CD8+ T cells have also been indicated for IL15 based treatments [55–57], these treatments have received considerable interest for their increased Effector: Treg ratio [58, 59], which is related to improved cancer therapies. Neoleukin™-2/15 cancer treatments have also been shown to increase the CD8+: Treg suggesting a mechanism similar to IL2 and IL15 [37]. We therefore believe that the mechanism for the antitumor activity of the immunocytokines is similar to these other cytokines, but must be further investigated in future studies.
Different immunocytokines containing IL2 or IL2 muteins are currently being investigated in the clinic to treat varies malignancies [41, 60–63]. For certain cytokines (such as IL2, IL12 and TNF) the fusion with a tumor-homing antibody allows the creation of novel biopharmaceuticals, which display a preferential uptake in tumor lesions and which perform substantially better than the corresponding nontargeted cytokine in immunocompetent mouse models [50, 51, 64, 65]. The binding of L19-Neo™ and Neo™-L19 to surface immobilized EDB indicated differences in the binding kinetics under circumstances that allowed for avid binding (Figure 1E, 1J). While the differences might impact the biodistribution of the immunocytokines, other factors will also have a major impact on the localization in vivo. We and others have reported that not all cytokine payloads can be efficiently delivered to the tumor by fusion with antibodies. Cytokine trapping at low dose has been reported for fusion proteins based on interferon-gamma [66, 67], but also for IL15 fusions [55, 68]. In other cases, aberrant glycosylation may lead to suboptimal biodistribution performance [2, 69, 70]. Neo™-L19 is not glycosylated and we were pleased to see how the L19-based targeted delivery of the engineered IL2 and IL15 mimic led to an improved therapeutic effect for the F9 teratocarcinoma model. The contribution of L19-based targeting to an increased therapeutic index was less striking in the WEHI-164 model. Collectively, the results presented indicate that a new fusion protein could be created, which displayed an excellent performance in biodistribution studies and which mediated a potent anticancer activity in two immunocompetent mouse models of cancer when used as single agent. The results suggest that this product, or similar fusion proteins featuring an N-terminal fusion in the diabody format, may deserve to be investigated in clinical trials.
Materials and Methods
Design and cloning of the immunocytokines
The sequences for the constructs can be found in the Supplementary Information. The diabodies consist of a VH and a VL domain that are connected by a 5 amino acid (15 bp) linker. The diabody and the novel cytokine were connected by a 15 amino acid (45 bp) linker. In the 5′ of the protein gene, the sequence was linked to a mammalian signal peptide that allows for transient gene expression in CHO-S cells. The gene for L19-Neo™ was purchased from GenScript in a pcDNA3.1(+) vector, while the Neo™-L19 sequence cloned from the previous sequence and double digested with HindIII and EcoRI, and subsequently ligated into digested pcDNA3.1(+). The KSF construct was cloned by Gibson Isothermal assembly (NewEnglandBiolabs #E2621S) replacing the L19 moiety.
Protein expression by transient gene expression
Transient gene expression was used to expressed all of the proteins. For L19-Neo™, Neo™-L19, His6-Neo™ and Neo™-KSF, CHO-S cells were diluted to a concentration of 4 × 106 cells/mL in Pro-CHO medium (Lonza) containing 8 mM Ultraglutamine (Gibco), 4 mM HT supplement (Gibco), and 1% antibiotic-antimycotic (Gibco). The plasmid (0.8 μg/106 cells) and PEI (2.5 μg/106 cells) were added to the cells, and the mixture was incubated at 31 °C for 6 days for protein production. For hCD25, the plasmid DNA (1.25 μg/106 cells) was diluted in sterile NaCl solution (150 mM, 25 μL/106 cells). This solution was mixed with PEI (5 μg/106 cells) in sterile NaCl solution (150 mM, 25 μL/106 cells) and incubated at room temperature for 10 min. The PEI-DNA solution was mixed with CHO-S cells (2 × 106 cells/mL) in Pro-CHO medium (Lonza) and incubated at 37°C for 4 hours. The culture was diluted 1:1 with PowerCHO medium (Lonza) and incubated at 31°C for 6 days.
The cells were removed by centrifugation, and the supernatant was filtered. The proteins were captured by protein A affinity (L19-Neo™, Neo™-L19, and Neo™-KSF) or Ni-NTA (His6-Neo™ and hCD25) chromatography. Proteins from protein A columns were eluted with TEA (1.4% V/V%) into tubes containing a neutralizing solution (Tris 1 M, pH 7.4), while the proteins from His-tag purification were eluted with imidazole (500 mM, PBS, pH 7.4). The products were dialyzed into PBS (pH 7.4) at 4°C. Biochemical analysis was performed by non-reducing and reducing SDS-PAGE with Coomassie Blue staining, size exclusion chromatography using a Superdex 200 Increased 10/300GL column (Flow rate: 0.75 mL/min, GE Healthcare) on an Äkta Pure FPLC system, and LC-MS (Column: 2.1 × 50 mm Acquity BEH300 C4 1.7 μm (Waters), Instrument: Waters Acquity UPLC H-Class 147 coupled with a Waters Xevo G2-X2 Qtof).
Cell cultures
CHO-S cells were cultured in Power-CHO medium (Lonza), containing 8 mM Ultraglutamine (Lonza), 4 mM HT-supplement and 1× antibiotic-antimycotic (Gibco) at 37°C. The cells were maintained at densities between 0.5 and 8.0 × 106 cells/mL in a shaking incubator (150 rpm).
In general, the remaining cancer cells were cultured under a 5% CO2 atmosphere at 37°C. CTLL-2 and transduced reporter CTLL-2 cells [45] were cultured in RPMI (Gibco) containing 10% FBS (Gibco), 1× antibiotic-antimycotic (Gibco), 2 mM Ultraglutamine (Lonza), 25 mM HEPES (Gibco), 50 μM β-mercaptoethanol (Sigma) and 60 U/mL IL-2 (Proleukin, Roche Diagnostics). F9 teratocarcinoma cells were cultured in flasks coated with 0.1% gelatin (Type B from Bovine Skin, Sigma), and they were cultured in DMEM (Gibco) containing 10% FBS (Gibco) and 1× antibiotic-antimycotic (Gibco). WEHI-164 fibrosarcoma cells were cultured in RPMI (Gibco) containing 10% FBS (Gibco) and 1× antibiotic-antimycotic (Gibco).
Surface plasmon resonance
All surface plasmon resonance experiments were performed on a Biacore S200 instrument at 25°C. The binding events were investigated in PBS buffer (Gibco) containing 0.005% Tween-20. The proteins were immobilized on CM-5 S-series (GE Healthcare) sensor chips by activation of the surface with EDC and NHS. The remaining activated esters were capped with ethanolamine.
For EDB binding, EDB was captured on the NHS activated surface to a level of approximately 620 RU. The reference flow was capped with ethanolamine. A 2-fold dilution series of the immunocytokines (500–31.25 nM) were injected for 180 sec (association) and followed by 600 sec of dissociation at a flow rate of 20 μL/min. The surface was regenerated with five 15 sec injections of 10 mM HCl.
For the CD25 binding, CD25 was captured on the NHS activated surface to a level of approximately 550 RU. The immunocytokines were injected in a 2-fold dilution series (500 nM–31.25 nM) with 90 sec association and 300 sec dissociation at a flow rate of 30 μL/min. The surface was regenerated with a 15 sec injection of 1 M MgCl2 in 10 mM sodium acetate pH 5.5.
Binding analysis by flow cytometry
CTLL-2 cells were washed twice with HBSS (Gibco) and incubated in culturing medium without IL2 for 24 h. The starved CTLL-2 cells were centrifugated and resuspended in FACS buffer (0.5% BSA, 2 mM EDTA in PBS) to a density of 3 × 106 cells/mL. The cells were seeded in a 96 well plate containing 300,000 cells/well.
For staining procedures involving FITC-modified immunocytokines, proteins were incubated with cells on ice for 1 h. The cells were then washed twice with FACS buffer, resuspended in FACS buffer, and analyzed by flow cytometry (CytoFLEX, Beckman Coulter). The data was analyzed by the software FlowJo (v3, Tree Star).
Proliferation assay
CTLL-2 cells were washed twice with HBSS (Gibco) and incubated in culturing medium without IL2 for 24 h. The IL2-starved cells were seeded in a 96-well plate with a density of 25,000 cells/well. The cells were incubated in medium, containing the immunocytokines at varying concentration, at 37°C under 5% CO2 for 48 h. 20 μL CellTiter 96 Aqueous One Solution (Promega) was added to each well and the plate incubated for 1–2 hours at 37°C. The absorbance was measured at 490 nm and 620 nm. The relative proliferation was calculated as: relative proliferation = (OD490-620treated-OD490-620medium)/(OD490-620untreated-OD490-620medium). The data was fitted in GraphPad Prism 7 using a [Agonist] vs response (three parameters) model.
NF-κB activity assay
The NF-κB reporter cell line was used according to the previous report [45]. Briefly, the reporter cells (transduced reporter CTLL-2) were starved for IL2 6-9 hours prior to use. The cells were seeded in 96-well plates to a density of 50,000 cells/well. The cells were incubated in growth medium (200 μL), containing the immunocytokines in varying concentrations, at 37°C under a 5% CO2 atmosphere for several hours. The luciferase production was assessed by transferring 20 μL of the solution to an opaque 96-well plate (Optiplate-96, white, Perkin Elmer). To each well was added 80 μL 1 μg/mL Coelenterazine (Carl Roth AG) in PBS, and the luminescence (595 nm) was measured immediately after the addition of the reagent. The relative luminescence was calculated by division with values obtained for cells cultured without inducer added to the medium. The data was fitted in GraphPad Prism 7 using a [Agonist] vs response (three parameters) model.
FITC labelling of fusion proteins
The proteins were buffer exchanged into a carbonate buffer (100 mM, pH 9.0) by washing in Amicon Ultra ultracentrifugation filters (MWCO 10k, 11000 g, 10 min - Sigma) three times. To the solution of protein was added 50 molar equivalents of FITC (Sigma), and the reaction mixture was incubated at 4°C overnight. The conjugate was purified using a PD SpinTrap™ G-25 (Sigma) according to Manufacturer’s instructions.
Animal experiments
7–8 weeks old immunocompetent female BALB/c mice and 7–8 weeks old immunocompetent female 129/Sv were purchased from Janvier (France). On the day of tumor injections, the exponentially growing tumor cells (F9 teratocarcinoma or WEHI-164 fibrosarcoma) were harvested, washed and resuspended in Hank’s Balanced Salt Solution, HBSS, (Gibco). The tumor cells (F9 cells; 10 × 106 cells per mouse for 129/Sv mice. WEHI-164 cells; 2.5 × 106 cells per mouse for BALB/c mice) were injected subcutaneously in the right flank. The animals were monitored daily and the tumor sizes were measured with a caliper. Tumor volume was calculated as: Tumor volume [mm3] = length [mm] × width [mm] × width [mm] × 0.5. The animals were euthanized when the longest diameter of the tumor exceeded 15 mm, weight loss was greater than 15% body weight, tumors ulcerated, or 24 h after the last injection in biodistribution experiments. For the biodistribution experiments, tumor, lung, liver, spleen, heart, kidney, intestine, tail, and blood were collected.
All animal experiments were performed under the project license ZH04/2018 granted by the Veterinäramt des Kanton Zürich, Switzerland, in agreement with Swiss regulations. The animals were housed (groups of 2–5 mice per cage with food and water ad libitum) in an open hygiene barrier (reduced pathogen exposure) facility at the Swiss Federal Institute of Technology (ETH Zurich).
Quantitative biodistribution studies
Chloramine T hydrate (5 μL, 5 mg/mL in MQ water) was added to a solution of immunocytokines (100 μg) and iodine-125 (200 μCi, Hartmann Analytic). After 1 min and 45 sec, the radiolabeled proteins were purified by size exclusion chromatography using PD-10 columns (GE Healthcare) blocked with BSA (1 mL of 1 mg/mL in sterile filtered PBS) and equilibrated with sterile filtered PBS (25 mL). Immunocompetent 129/Sv mice (n = 3) were injected subcutaneously in the right flank with 10 × 106 F9 teratocarcinoma cells. The mice were monitored daily, and the tumor sizes were measured with a caliper. Tumor volume was determined as previously described. When the tumors had a volume of approximately 200–300 mm3, 10 μg of radioiodinated immunocytokine was injected intravenously into the lateral tail vein. The mice were sacrificed after 24 h, and the tumor, lung, liver, spleen, heart, kidney, intestine, tail, and blood were collected. The radioactivity was quantified by a Packard Cobra γ-counter. The radioactivity was expressed as a percentage of the injected dose per gram of tissue (%ID/g) ± SD.
Therapy experiments
On the day of tumor injections, the exponentially growing tumor cells (F9 teratocarcinoma or WEHI-164 fibrosarcoma) were harvested by trypsin digestion, washed and resuspended in HBSS (Gibco). The tumor cells (F9 cells; 10 × 106 cells per mouse for 129/Sv mice. WEHI-164 cells; 2.5 × 106 cells per mouse for BALB/c) were injected subcutaneously in the right flank. The animals were monitored daily and the tumor sizes were measured with a caliper. Tumor volumes were calculated as previously described. When the tumors were clearly detectable with an average size of about 90 mm3, the mice were randomly divided into the different treatment groups (n = 4–5). The treatments were initiated at an average tumor size of approximately 90 mm3 and were administered intravenously into the lateral tail vein. For the F9 teratocarcinoma therapies, the mice received three injections of 10, 60, or 100 μg of Neo™-L19, 60 or 100 μg Neo™-KSF or phosphate buffered saline at intervals of 48 h. For the WEHI-164 fibrosarcoma, the mice received two injects of 100 μg Neo™-L19, 100 μg Neo™-KSF or 37 μg (equimolar with the immunocytokines) His6-Neo™ or phosphate buffered saline with a 48 h interval. Animals were euthanized according to the conditions previously described. Animals with complete responses were monitored for an additional 60 days after last tumor measurement to investigate tumor regrowth.
ACKNOWLEDGMENTS
The authors would like to thank Sheila Dakhel and Samuele Cazzamali for their inputs to the study.
CONFLICTS OF INTEREST
D.N. is a cofounder, shareholder and board member at Philogen. The other authors declare no competing interests.
FUNDING
We gratefully acknowledge the financial support by the Carlsberg Foundation through Internationalisation Fellowships for M.R.M. (Grants: CF18-0132 and CF19-0060), ETH Zurich, the European Research Council (ERC) under the program European Union’s Horizon 2020 research and innovation program (Grant: 670603), the Swiss National Science Foundation (Grant: 310030_182003/1), and the Federal Commission for Technology and Innovation (KTI, Grant: 12803.1 VOUCH-LS).
References
1. Punnonen J, Rosen D, Zuniga L, Sprogøe K, Tabrizi M. Cytokine Therapeutics in Cancer Immunotherapy: Design and Development. Curr Pharmacol Rep. 2019; 5:377–390. https://doi.org/10.1007/s40495-019-00193-6.
2. Neri D. Antibody–Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol Res. 2019; 7:348–354. https://doi.org/10.1158/2326-6066.CIR-18-0622. [PubMed].
3. Murer P, Neri D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N Biotechnol. 2019; 52:42–53. https://doi.org/10.1016/j.nbt.2019.04.002. [PubMed].
4. Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol. 2014; 192:5451–5458. https://doi.org/10.4049/jimmunol.1490019. [PubMed].
5. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018; 50:1–11. https://doi.org/10.1038/s12276-018-0191-1. [PubMed].
6. Callahan MK, Kluger H, Postow MA, Segal NH, Lesokhin A, Atkins MB, Kirkwood JM, Krishnan S, Bhore R, Horak C, Wolchok JD, Sznol M. Nivolumab Plus Ipilimumab in Patients With Advanced Melanoma: Updated Survival, Response, and Safety Data in a Phase I Dose-Escalation Study. J Clin Oncol. 2018; 36:391–398. https://doi.org/10.1200/JCO.2017.72.2850. [PubMed].
7. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. 2010; 363:711–723. https://doi.org/10.1056/NEJMoa1003466. [PubMed].
8. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008; 8:299–308. https://doi.org/10.1038/nrc2355. [PubMed].
9. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019; 10:2250. https://doi.org/10.3389/fimmu.2019.02250. [PubMed].
10. Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-Dose Recombinant Interleukin 2 Therapy for Patients With Metastatic Melanoma: Analysis of 270 Patients Treated Between 1985 and 1993. J Clin Oncol. 1999; 17:2105–16. https://doi.org/10.1200/JCO.1999.17.7.2105. [PubMed].
11. Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995; 13:688–696. https://doi.org/10.1200/JCO.1995.13.3.688. [PubMed].
12. Klapper JA, Downey SG, Smith FO, Yang JC, Hughes MS, Kammula US, Sherry RM, Royal RE, Steinberg SM, Rosenberg S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma. Cancer. 2008; 113:293–301. https://doi.org/10.1002/cncr.23552. [PubMed].
13. Carnemolla B, Borsi L, Balza E, Castellani P, Meazza R, Berndt A, Ferrini S, Kosmehl H, Neri D, Zardi L. Enhancement of the antitumor properties of interleukin-2 by its targeted delivery to the tumor blood vessel extracellular matrix. Blood. 2002; 99:1659–1665. https://doi.org/10.1182/blood.V99.5.1659. [PubMed].
14. Konrad MW, Hemstreet G, Hersh EM, Mansell PWA, Mertelsmann R, Kolitz JE, Bradley EC. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res. 1990; 50:2009–2017. [PubMed].
15. Bindon C, Czerniecki M, Ruell P, Edwards A, McCarthy WH, Harris R, Hersey P. Clearance rates and systemic effects of intravenously administered interleukin 2 (IL-2) containing preparations in human subjects. Br J Cancer. 1983; 47:123–133. https://doi.org/10.1038/bjc.1983.15. [PubMed].
16. Hutmacher C, Neri D. Antibody-cytokine fusion proteins: Biopharmaceuticals with immunomodulatory properties for cancer therapy. Adv Drug Deliv Rev. 2019; 141:67–91. https://doi.org/10.1016/j.addr.2018.09.002. [PubMed].
17. Gillies SD, Reilly EB, Lo KM, Reisfeld RA. Antibody-targeted interleukin 2 stimulates T-cell killing of autologous tumor cells. Proc Natl Acad Sci U S A. 1992; 89:1428–1432. https://doi.org/10.1073/pnas.89.4.1428. [PubMed].
18. Gillies SD. A new platform for constructing antibody-cytokine fusion proteins (immunocytokines) with improved biological properties and adaptable cytokine activity. Protein Eng Des Sel. 2013; 26:561–569. https://doi.org/10.1093/protein/gzt045. [PubMed].
19. Niethammer AG, Xiang R, Ruehlmann JM, Lode HN, Dolman CS, Gillies SD, Reisfeld RA. Targeted interleukin 2 therapy enhances protective immunity induced by an autologous oral DNA vaccine against murine melanoma. Cancer Res. 2001; 61:6178–6184. https://cancerres.aacrjournals.org/content/61/16/6178. [PubMed].
20. Tang A, Harding F. The challenges and molecular approaches surrounding interleukin-2-based therapeutics in cancer. Cytokine X. 2019; 1:100001. https://doi.org/10.1016/j.cytox.2018.100001.
21. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, Ali CF, Chang TK, Konakova M, et al. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clin Cancer Res. 2016; 22:680–690. https://doi.org/10.1158/1078-0432.CCR-15-1631. [PubMed].
22. Hu P, Hornick JL, Glasky MS, Yun A, Milkie MN, Khawli LA, Anderson PM, Epstein AL. A chimeric Lym-1/interleukin 2 fusion protein for increasing tumor vascular permeability and enhancing antibody uptake. Cancer Res. 1996; 56:4998–5004. [PubMed].
23. Dela Cruz JS, Trinh KR, Chen HW, Ribas A, Morrison SL, Penichet ML. Anti-HER2/neu IgG3–(IL-2) and anti-HER2/neu IgG3–(GM-CSF) promote HER2/neu processing and presentation by dendritic cells: Implications in immunotherapy and vaccination strategies. Mol Immunol. 2006; 43:667–676. https://doi.org/10.1016/j.molimm.2005.04.007. [PubMed].
24. Dela Cruz JS, Huang TH, Penichet ML, Morrison SL. Antibody-cytokine fusion proteins: innovative weapons in the war against cancer. Clin Exp Med. 2004; 4:57–64. https://doi.org/10.1007/s10238-004-0039-y. [PubMed].
25. Yoo EM, Trinh KR, Tran D, Vasuthasawat A, Zhang J, Hoang B, Lichtenstein A, Morrison SL. Anti-CD138-Targeted Interferon Is a Potent Therapeutic Against Multiple Myeloma. J Interferon Cytokine Res. 2015; 35:281–291. https://doi.org/10.1089/jir.2014.0125. [PubMed].
26. Gillies S, Lan Y, Brunkhorst B, Wong WK, Li Y, Lo KM. Bi-functional cytokine fusion proteins for gene therapy and antibody-targeted treatment of cancer. Cancer Immunol Immunother. 2002; 51:449–460. https://doi.org/10.1007/s00262-002-0302-6. [PubMed].
27. Gillies SD, Young D, Lo KM, Roberts S. Biological activity and in vivo clearance of anti-tumor antibody/cytokine fusion proteins. Bioconjug Chem. 1993; 4:230–235. https://doi.org/10.1021/bc00021a008. [PubMed].
28. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012; 12:180–190. https://doi.org/10.1038/nri3156. [PubMed].
29. Heaton KM, Ju G, Grimm EA. Human interleukin 2 analogues that preferentially bind the intermediate-affinity interleukin 2 receptor lead to reduced secondary cytokine secretion: implications for the use of these interleukin 2 analogues in cancer immunotherapy. Cancer Res. 1993; 53:2597–2602. [PubMed].
30. Vazquez-Lombardi R, Loetsch C, Zinkl D, Jackson J, Schofield P, Deenick EK, King C, Phan TG, Webster KE, Sprent J, Christ D. Potent antitumour activity of interleukin-2-Fc fusion proteins requires Fc-mediated depletion of regulatory T-cells. Nat Commun. 2017; 8:15373. https://doi.org/10.1038/ncomms15373. [PubMed].
31. Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia-Martínez K, Fuente D, León K. Human IL-2 Mutein with Higher Antitumor Efficacy Than Wild Type IL-2. J Immunol. 2013; 190:6230–6238. https://doi.org/10.4049/jimmunol.1201895. [PubMed].
32. Arenas-Ramirez N, Zou C, Popp S, Zingg D, Brannetti B, Wirth E, Calzascia T, Kovarik J, Sommer L, Zenke G, Woytschak J, Regnier CH, Katopodis A, et al. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci Transl Med. 2016; 8:367ra166. https://doi.org/10.1126/scitranslmed.aag3187. [PubMed].
33. Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, Xu L, Liang Y, Guo J, Bian Y, Xu H, Shi J, Wang F, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nat Commun. 2019; 10:3874. https://doi.org/10.1038/s41467-019-11782-w. [PubMed].
34. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012; 484:529–533. https://doi.org/10.1038/nature10975. [PubMed].
35. Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, Alvarez JC, Losey HC. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J Immunother Cancer. 2020; 8:e000673. https://doi.org/10.1136/jitc-2020-000673. [PubMed].
36. Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010; 107:11906–11911. https://doi.org/10.1073/pnas.1002569107. [PubMed].
37. Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labão-Almeida C, Ali LR, Quijano-Rubio A, Ruterbusch M, Leung I, Biary T, Crowley SJ, Marcos E, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019; 565:186–191. https://doi.org/10.1038/s41586-018-0830-7. [PubMed].
38. Yao Z, Dai W, Perry J, Brechbiel MW, Sung C. Effect of albumin fusion on the biodistribution of interleukin-2. Cancer Immunol Immunother. 2004; 53:404–410. https://doi.org/10.1007/s00262-003-0454-z. [PubMed].
39. Borsi L, Balza E, Bestagno M, Castellani P, Carnemolla B, Biro A, Leprini A, Sepulveda J, Burrone O, Neri D, Zardi L. Selective targeting of tumoral vasculature: Comparison of different formats of an antibody (L19) to the ED-B domain of fibronectin. Int J Cancer. 2002; 102:75–85. https://doi.org/10.1002/ijc.10662. [PubMed].
40. Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A. 2015; 112:3320–3325. https://doi.org/10.1073/pnas.1416159112. [PubMed].
41. Weide B, Neri D, Elia G. Intralesional treatment of metastatic melanoma: a review of therapeutic options. Cancer Immunol Immunother. 2017; 66:647–656. https://doi.org/10.1007/s00262-016-1952-0. [PubMed].
42. Carnemolla B, Balza E, Siri A, Zardi L, Nicotra MR, Bigotti A, Natali PG. A tumor-associated fibronectin isoform generated by alternative splicing of messenger RNA precursors. J Cell Biol. 1989; 108:1139–1148. https://doi.org/10.1083/jcb.108.3.1139. [PubMed].
43. Holliger P, Prospero T, Winter G. “Diabodies”: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci U S A. 1993; 90:6444–6448. https://doi.org/10.1073/pnas.90.14.6444. [PubMed].
44. Pini A, Viti F, Santucci A, Carnemolla B, Zardi L, Neri P, Neri D. Design and Use of a Phage Display Library. J Biol Chem. 1998; 273:21769–21776. https://doi.org/10.1074/jbc.273.34.21769. [PubMed].
45. Mock J, Pellegrino C, Neri D. A universal reporter cell line for bioactivity evaluation of engineered cytokine products. Sci Rep. 2020; 10:3234. https://doi.org/10.1038/s41598-020-60182-4. [PubMed].
46. De Luca R, Soltermann A, Pretto F, Pemberton-Ross C, Pellegrini G, Wulhfard S, Neri D. Potency-matched Dual Cytokine–Antibody Fusion Proteins for Cancer Therapy. Mol Cancer Ther. 2017; 16:2442–2451. https://doi.org/10.1158/1535-7163.MCT-17-0211. [PubMed].
47. De Luca R, Gouyou B, Ongaro T, Villa A, Ziffels B, Sannino A, Buttinoni G, Galeazzi S, Mazzacuva M, Neri D. A Novel Fully-Human Potency-Matched Dual Cytokine-Antibody Fusion Protein Targets Carbonic Anhydrase IX in Renal Cell Carcinomas. Front Oncol. 2019; 9:1228. https://doi.org/10.3389/fonc.2019.01228. [PubMed].
48. Ongaro T, Matasci M, Cazzamalli S, Gouyou B, De Luca R, Neri D, Villa A. A novel anti-cancer L19-interleukin-12 fusion protein with an optimized peptide linker efficiently localizes in vivo at the site of tumors. J Biotechnol. 2019; 291:17–25. https://doi.org/10.1016/j.jbiotec.2018.12.004. [PubMed].
49. Crawford A, Haber L, Kelly MP, Vazzana K, Canova L, Ram P, Pawashe A, Finney J, Jalal S, Chiu D, Colleton CA, Garnova E, Makonnen S, et al. A Mucin 16 bispecific T cell–engaging antibody for the treatment of ovarian cancer. Sci Transl Med. 2019; 11:eaau7534. https://doi.org/10.1126/scitranslmed.aau7534. [PubMed].
50. Puca E, Probst P, Stringhini M, Murer P, Pellegrini G, Cazzamalli S, Hutmacher C, Gouyou B, Wulhfard S, Matasci M, Villa A, Neri D. The antibody-based delivery of interleukin-12 to solid tumors boosts NK and CD8 + T cell activity and synergizes with immune checkpoint inhibitors. Int J Cancer. 2020; 146:2518–2530. https://doi.org/10.1002/ijc.32603. [PubMed].
51. Borsi L, Balza E, Carnemolla B, Sassi F, Castellani P, Berndt A, Kosmehl H, Birò A, Siri A, Orecchia P, Grassi J, Neri D, Zardi L. Selective targeted delivery of TNFα to tumor blood vessels. Blood. 2003; 102:4384–4392. https://doi.org/10.1182/blood-2003-04-1039. [PubMed].
52. Trachsel E, Bootz F, Silacci M, Kaspar M, Kosmehl H, Neri D. Antibody-mediated delivery of IL-10 inhibits the progression of established collagen-induced arthritis. Arthritis Res Ther. 2007; 9:R9. https://doi.org/10.1186/ar2115. [PubMed].
53. Niesner U, Halin C, Lozzi L, Günthert M, Neri P, Wunderli-Allenspach H, Zardi L, Neri D. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjug Chem. 2002; 13:729–736. https://doi.org/10.1021/bc025517+. [PubMed].
54. Hutmacher C, Gonzalo Núñez N, Liuzzi AR, Becher B, Neri D. Targeted Delivery of IL2 to the Tumor Stroma Potentiates the Action of Immune Checkpoint Inhibitors by Preferential Activation of NK and CD8 + T Cells. Cancer Immunol Res. 2019; 7:572–583. https://doi.org/10.1158/2326-6066.CIR-18-0566. [PubMed].
55. Kaspar M, Trachsel E, Neri D. The Antibody-Mediated Targeted Delivery of Interleukin-15 and GM-CSF to the Tumor Neovasculature Inhibits Tumor Growth and Metastasis. Cancer Res. 2007; 67:4940–4948. https://doi.org/10.1158/0008-5472.CAN-07-0283. [PubMed].
56. Kermer V, Baum V, Hornig N, Kontermann RE, Müller D. An Antibody Fusion Protein for Cancer Immunotherapy Mimicking IL-15 trans -Presentation at the Tumor Site. Mol Cancer Ther. 2012; 11:1279–1288. https://doi.org/10.1158/1535-7163.MCT-12-0019. [PubMed].
57. Kim PS, Kwilas AR, Xu W, Alter S, Jeng EK, Wong HC, Schlom J, Hodge JW. IL-15 superagonist/IL-15RαSushi-Fc fusion complex (IL-15SA/IL-15RαSu-Fc; ALT-803) markedly enhances specific subpopulations of NK and memory CD8+ T cells, and mediates potent anti-tumor activity against murine breast and colon carcinomas. Oncotarget. 2016; 7:16130–16145. https://doi.org/10.18632/oncotarget.7470. [PubMed].
58. Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, Riddell SR. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009; 114:2417–2426. https://doi.org/10.1182/blood-2008-12-189266. [PubMed].
59. Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci. 2012; 33:35–41. https://doi.org/10.1016/j.tips.2011.09.004. [PubMed].
60. Lansigan F, Nakamura R, Quick DP, Vlock D, Raubitschek A, Gillies SD, Bachanova V. DI-Leu16-IL2, an Anti-CD20-Interleukin-2 Immunocytokine, Is Safe and Active in Patients with Relapsed and Refractory B-Cell Lymphoma: A Report of Maximum Tolerated Dose, Optimal Biologic Dose, and Recommended Phase 2 Dose. Blood. 2016; 128:620. https://doi.org/10.1182/blood.V128.22.620.620.
61. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, Dunn C, Bolijn M, Benz J, Stihle M, Lang S, Roemmele M, Hofer T, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017; 6:e1277306. https://doi.org/10.1080/2162402X.2016.1277306. [PubMed].
62. Albertini MR, Yang RK, Ranheim EA, Hank JA, Zuleger CL, Weber S, Neuman H, Hartig G, Weigel T, Mahvi D, Henry MB, Quale R, McFarland T, et al. Pilot trial of the hu14.18-IL2 immunocytokine in patients with completely resectable recurrent stage III or stage IV melanoma. Cancer Immunol Immunother. 2018; 67:1647–1658. https://doi.org/10.1007/s00262-018-2223-z. [PubMed].
63. Catania C, Maur M, Berardi R, Rocca A, Di Giacomo AM, Spitaleri G, Masini C, Pierantoni C, González-Iglesias R, Zigon G, Tasciotti A, Giovannoni L, Lovato V, et al. The tumor-targeting immunocytokine F16-IL2 in combination with doxorubicin: dose escalation in patients with advanced solid tumors and expansion into patients with metastatic breast cancer. Cell Adh Migr. 2015; 9:14–21. https://doi.org/10.4161/19336918.2014.983785. [PubMed].
64. Gutbrodt KL, Schliemann C, Giovannoni L, Frey K, Pabst T, Klapper W, Berdel WE, Neri D. Antibody-Based Delivery of Interleukin-2 to Neovasculature Has Potent Activity Against Acute Myeloid Leukemia. Sci Transl Med. 2013; 5:201ra118. https://doi.org/10.1126/scitranslmed.3006221. [PubMed].
65. Pasche N, Wulhfard S, Pretto F, Carugati E, Neri D. The Antibody-Based Delivery of Interleukin-12 to the Tumor Neovasculature Eradicates Murine Models of Cancer in Combination with Paclitaxel. Clin Cancer Res. 2012; 18:4092–4103. https://doi.org/10.1158/1078-0432.CCR-12-0282. [PubMed].
66. Ebbinghaus C, Ronca R, Kaspar M, Grabulovski D, Berndt A, Kosmehl H, Zardi L, Neri D. Engineered vascular-targeting antibody-interferon-γ fusion protein for cancer therapy. Int J Cancer. 2005; 116:304–313. https://doi.org/10.1002/ijc.20952. [PubMed].
67. Hemmerle T, Neri D. The Dose-Dependent Tumor Targeting of Antibody-IFN Fusion Proteins Reveals an Unexpected Receptor-Trapping Mechanism In Vivo. Cancer Immunol Res. 2014; 2:559–567. https://doi.org/10.1158/2326-6066.CIR-13-0182. [PubMed].
68. Liu B, Jones M, Kong L, Noel T, Jeng EK, Shi S, England CG, Alter S, Miller JS, Cai W, Rhode PR, Wong HC. Evaluation of the biological activities of the IL-15 superagonist complex, ALT-803, following intravenous versus subcutaneous administration in murine models. Cytokine. 2018; 107:105–112. https://doi.org/10.1016/j.cyto.2017.12.003. [PubMed].
69. Venetz D, Hess C, Lin CW, Aebi M, Neri D. Glycosylation profiles determine extravasation and disease-targeting properties of armed antibodies. Proc Natl Acad Sci U S A. 2015; 112:2000–5. https://doi.org/10.1073/pnas.1416694112. [PubMed].
70. Bootz F, Venetz D, Ziffels B, Neri D. Different tissue distribution properties for glycosylation variants of fusion proteins containing the p40 subunit of murine interleukin-12. Protein Eng Des Sel. 2016; 29:445–455. https://doi.org/10.1093/protein/gzw038. [PubMed].