
Introduction
Bladder cancer is the most frequent cancer of the urinary system in the United States with nearly 82,000 new cases each year and 18,000 deaths, affecting men more often, in a 3:1 ratio [1]. Bladder cancer can be divided broadly into non-muscle invasive bladder cancer (NMIBC) and muscle invasive bladder cancer (MIBC). MIBC can be further sub-divided at the molecular level by the expression of RNA biomarkers between classes, that define basal and luminal characteristics [2–8]. The standard of care for intermediate- to high-risk NMIBC has been Bacille Calmette-Guerin (BCG) since its introduction in 1976, with cystectomy as the recommended standard of care in refractory, high risk disease [9, 10]. While NMIBC makes up 70–80% of total cases, tumor recurrence is frequent and ~30% of cases progress to MIBC [11]. Neoadjuvant chemotherapy prior to radical cystectomy (RC) for MIBC is the standard of care, though the absolute survival benefit is small, and some patients progress during chemotherapy [12]. While progress has been made in the prediction of sensitivity to platinum-based chemotherapies [13, 14], identifying targeted therapies specific to each patient remains a critical need for those patients who progress during chemotherapy and/or after cystectomy.
There have been several large-scale screening efforts in bladder cancer cell lines using 2D cultures. The Broad Institute Cancer Cell Line Encyclopedia (CCLE) has characterized 56 urinary tract carcinomas and screened many of these cell lines against 24 drugs [15, 16]. The Genomics of Drug Sensitivity in Cancer (GDSC) represents one of the largest efforts in total drugs, screening 19 bladder cancer cell lines against 518 drugs [16, 17]. Additional efforts to identify therapeutic targets in bladder cancer include CRISPR screening and epigenetic approaches [18–20]. However, we now know that screening in 3D culture is superior to 2D culture, with improved in vivo relevance [21–26]. Indeed, screening in 3D using ultra-low attachment plates is ideal for bladder cancer cell culture [27], and this method has been utilized in seminal studies for screening patient-derived organoids (PDOs) to predict patient response to drug treatments [28, 29]. While direct screening of patient material is cutting edge and most representative of drug response for that particular patient, such material is typically very limited, which restricts the size of a potential drug screening library. Additionally, bladder cancer cell lines have undergone comprehensive molecular profiling allowing rapid correlational pairing of molecular profile with 3D phenotype [7]. Therefore, there is utility in screening bladder cancer cell lines in large drug screens in 3D cultures to identify novel therapeutic options for future testing in PDOs and, ultimately, patients.
In this work, we treated 17 established bladder cancer cell lines with 652 investigational small-molecules and 3 clinically relevant combinations in 3D cell culture. From this screening, we identified compounds and classes of drugs with promising efficacy in bladder cancer. Then, utilizing established genomic and transcriptomic data for these bladder cancer cell lines, including prioritized mutations, copy number variants, and RNA-based molecular subtyping [7, 15], we correlated these molecular parameters with drug response to identify potentially novel groups of tumors that are vulnerable to specific drugs or classes of drugs. Importantly, we showed that MEK inhibitors are a promising targeted therapy in basal subtype bladder cancer cell lines, and our data indicate that drug screening of 3D cultures provides an important resource for future hypothesis generation.
Results
3D drug screen in bladder cancer cell lines
To examine bladder cell line drug sensitivity, we screened 17 cell lines against 652 investigational small-molecules and 3 clinically relevant combinations in 3D cell culture. From this drug sensitivity data, we calculated a drug sensitivity score 3 (DSS3) for each compound, an advanced drug sensitivity metric that uses the IC50, maximum inhibition, and drug concentration range to score drug sensitivity from 0 (no effect) to 100 (complete effect), Supplementary [30]. We plotted the average and standard deviation for each drug across the 17 cell lines to visualize the DSS3 spread in data, Figure 1. Scores of > 59 are considered “very active”, 30–59 “active”, 21–29 “semi-active”, 9–20 “low active”, and < 9 “inactive” [30]. From our drug screening, we identify 3 drugs (0.5%) as very active, 30 drugs (4.6%) as active, 20 drugs (3.0%) as semi active, 56 drugs (8.5%) as low active, and the remaining 547 (83.4%) as inactive (Supplementary).
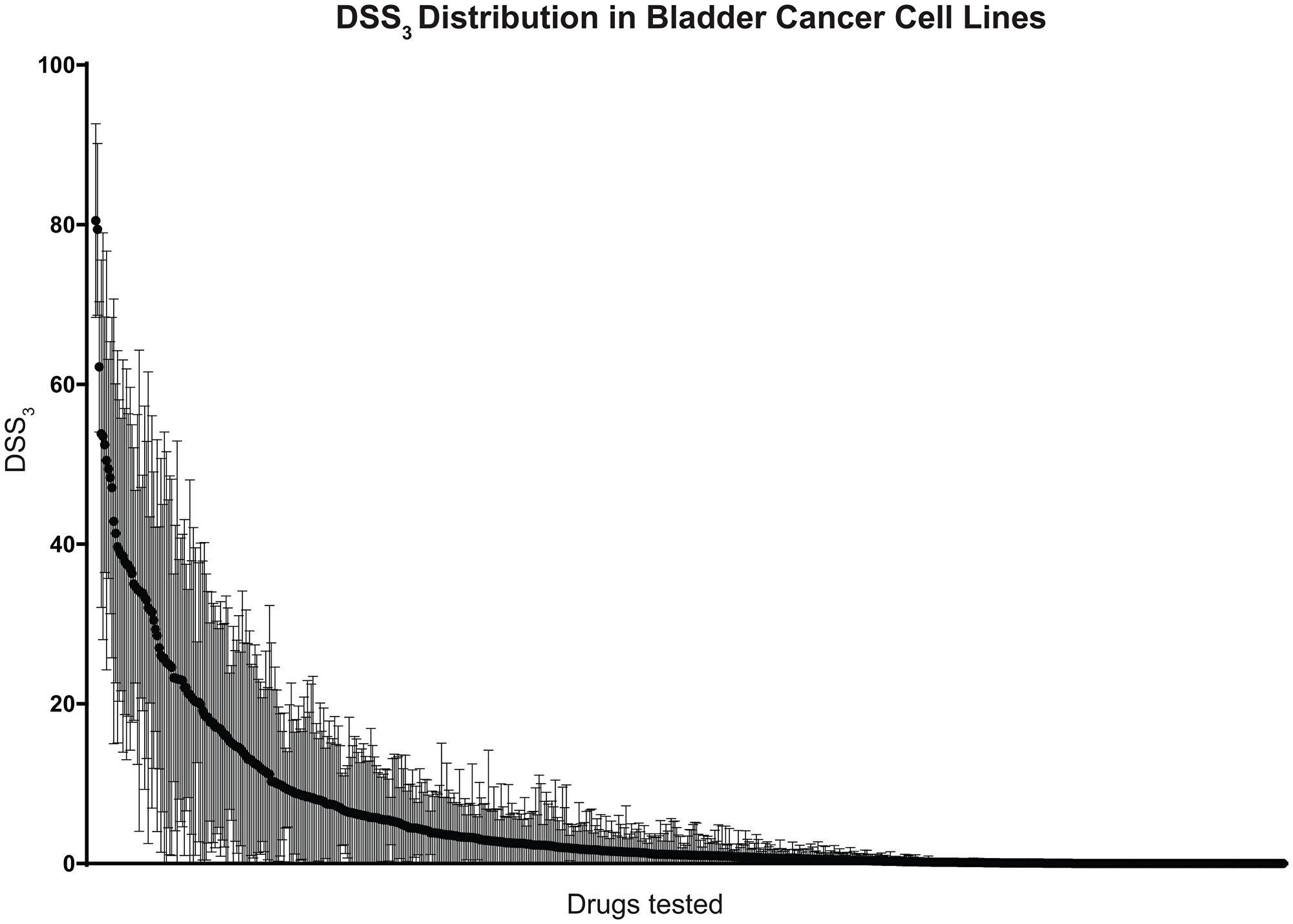
Figure 1: Distribution of drug sensitivities across bladder cancer cell lines. 652 investigational drugs and 3 clinically relevant combinations were tested against 17 bladder cancer cell lines in 3D cell culture. Drugs are ordered along x-axis by average drug sensitivity (DSS3), starting with the most sensitive drug. Black circles indicate average DSS3 and brackets indicate standard deviation across the 17 cell lines.
We identify romidepsin, bortezomib, and triptolide as “very active” compounds across the 17 bladder cancer cell lines, on the basis of their DSS3. Romidepsin is a histone deacetylase (HDAC) inhibitor with an average DSS3 of 80.5 and a standard deviation of 12.1. HDAC inhibitors have been reported previously as potential therapeutic in bladder cancer and our results identify romidepsin and panobinostat (an “active: compound) as active pan-HDAC inhibitors. Bortezomib is a proteasome inhibitor with an average DSS3 of 79.4 and a standard deviation of 10.8. Proteasome inhibitors have been reported as potential therapeutics based on promising pre-clinical data and we identify bortezomib and delanzomib (an “active” compound) as potent proteasome inhibitors. Triptolide is an inhibitor of RNA polymerase I and II–dependent transcription with an average DSS3 of 62.2 and a standard deviation of 8.2. The 30 “active” compounds include both chemotherapeutics and targeted agents, many of which are currently utilized in the treatment of bladder cancer, such as gemcitabine, paclitaxel, vinblastine, and doxorubicin.
We screened three therapeutically relevant combinations with the top dose as the Cmax of each compound, serially diluted 1:5 to generate a dose response curve. For the combination of methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC), a standard of care therapy in first-line therapy in MIBC, we observe a large spread in response, with a standard deviation in the DSS3 of 20.2. Four cell lines have a DSS3 as “active” or “very active” with most responses however, in the range of low active (8/17 cell lines). The overall average DSS3 for MVAC is 19.9. Because the top dose of the combinations is the Cmax, a better measure of therapeutic response for these combinations is the maximum response, which ranges from 52% to 100% in these cell lines (Supplementary), with an average of 81.1. The cisplatin and gemcitabine, alternative first line or second line combination, has a similar spread in data (DSS3 standard deviation = 15.2), but lower DSS3 values (average DSS3 = 8.2), and a wider spread in maximum response (0% to 100%, average = 65.0). Carboplatin and paclitaxel, another common first-line therapy, had the largest average DSS3 value and spread in data (average = 36.9, standard deviation = 22.7), but the lowest average maximum response of 53.9.
Genomic correlates of drug sensitivity in 3D bladder cancer cell line cultures
We utilized DNA and RNA sequencing data previously published by our group, and the CCLE to examine the impact of molecular characteristics on drug sensitivity in these cell lines (see Materials and Methods for details), Figure 2 [7, 15]. As expected, the most prevalent somatic alteration in these bladder cancer cell lines was inactivating TP53 mutations, which were present in 14/17 cell lines, typically accompanied by loss of heterozygosity (LOH). We found that TP53 homozygous-mutant cell lines were more sensitive to onalespib (an Hsp90 inhibitor) and clofarabine (a purine nucleoside anti-metabolite) than TP53 wild-type (WT) cells, Supplementary Figure 1. The next most frequent mutations were oncogenic activating PIK3CA and FGFR3 mutations, which were present in 5/17 and 3/17 cell lines, respectively. We sought to identify drugs where there was a significant difference in DSS3 between groups (mutant and WT) and at least one group had a DSS3 > 10, identifying only AZD-8186 as more effective in PIK3CA WT cell lines, Supplementary Figure 2. If we include PTEN deletion with the PI3K group, because PTEN negatively regulates PI3K, we lose the correlation with AZD-8186 and instead observe a single robust correlate for MLN2238 (a proteasome inhibitor), Supplementary Figure 3. FGFR inhibitor response was not correlated with FGFR3 mutation status for any of the FGFR inhibitors in the panel (erdafitinib, AZD4547, BGJ398, and CH5183284). There were an additional 13 mutations present in 1-2 cell lines, including inactivating CDKN2A, RB1, and PTEN mutations with LOH and activating oncogenic ERBB2, AKT1, HRAS, and KRAS mutations. We did not observe increased ERBB2 inhibitor (lapatinib, canertinib, sapatinib, mubritinib, GW2580, dacomitinib, and WZ4002) sensitivity in ERBB2 activating mutant cell lines, in contrast to similar studies using 2D cell culture [31]. The AKT1 mutated PDX cell line (BC8149) is sensitive to many PI3K/AKT/mTOR inhibitors, while HRAS and KRAS mutated lines (T-24 and UM-UC-3) are largely insensitive to most PI3K/AKT/mTOR inhibitors.

Figure 2: Genomic landscape of bladder cell lines. Bladder cancer subtype is indicated across the top row with each column representing an individual cell line. B-L Score was calculated from normalized RNA sequencing and is shown as a gradient from luminal (yellow) to basal (blue). Average drug sensitivity shows the average DSS3 across all drugs for each cell line. Mutation and genotype are indicated for all prioritized mutations present in 1+ cell line. CNA are indicated for high level amplifications (≥ 2 copy gain) and deep deletions (≥ 2 copy loss) in 1+ cell line.
We integrated the somatic mutation and CNA status to generate mutant groups for comparison. Combining CDKN2A loss-of-function mutants with CDKN2A CNA deep-deletion, we observe significant correlations with CDKN2A loss and poor response to cladribine or clofarabine, both purine analogs, as well as panobinostat and mocetinostat, both HDAC inhibitors, Supplementary Figure 4. If we combine RB1 loss-of-function mutants with RB1 CNA deep-deletion, we observe a significant correlation with RB1 loss and average drug sensitivity, where cells with RB1 loss respond more favorably to targeted agents and chemotherapeutics, Supplementary Figure 5.
MEK inhibition correlates with basal subtype in bladder cancer cell lines
We next wanted to determine if bladder cancer subtype correlated with drug response. Cell lines were classified as luminal, basal, null, or mixed based on RNA sequencing and B-L scoring [7, 32] (see Materials and Methods for details). We observed a strong correlation with MEK response in basal bladder cancer, Figure 3. Across 8 MEK inhibitors, we observed this same trend, with 2 very active inhibitors, 2 semi-active inhibitors, 3 low active inhibitors, and 1 inactive, but with a measurable response. The very active MEK inhibitors, Trametinib and TAK-773, both had a significantly higher MEK DSS3 in basal cell lines vs. other subtype, Figure 4A and 4B. When we normalized the response for all 8 MEK inhibitors, we saw both a significant difference in MEK response in basal vs. other (all remaining subtypes clustered, as well as basal vs. each other subtype, Figure 4C. Average drug response does not correlate with bladder subtype, Figure 4D. In agreement with this finding, we tested another basal bladder cancer cell line, UM-UC-13, and observed excellent anti-proliferative activity with MEK inhibitors (Supplementary Table 1). The only other drug response that we found to correlate with subtype was atuveciclib, a PTEFb/CDK9 inhibitor, with the basal subtype, Supplementary Figure 6.
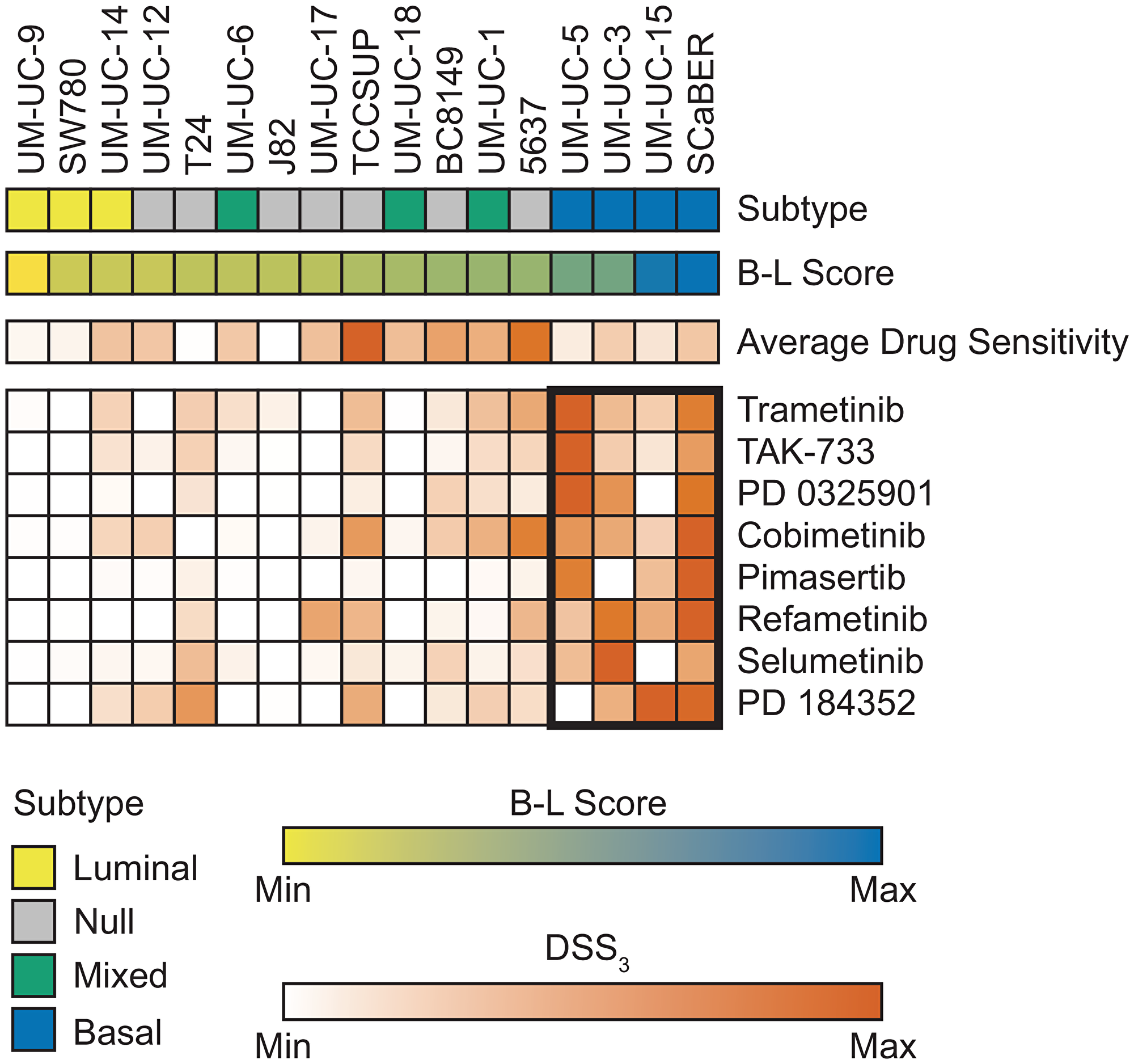
Figure 3: MEK inhibitors show strongest response in basal bladder cell lines. Bladder cancer subtype is indicated across the top row with each column representing an individual cell line. B-L Score was calculated from normalized RNA sequencing and is shown as a gradient from luminal (yellow) to basal (blue). Average drug sensitivity shows the average DSS3 for each cell line. Average DSS3 of MEK inhibitors for each cell line shown with average DSS3 > 0. MEK inhibitors ordered by average basal response with the best response at the top. Basal cell lines marked with bolded rectangle.
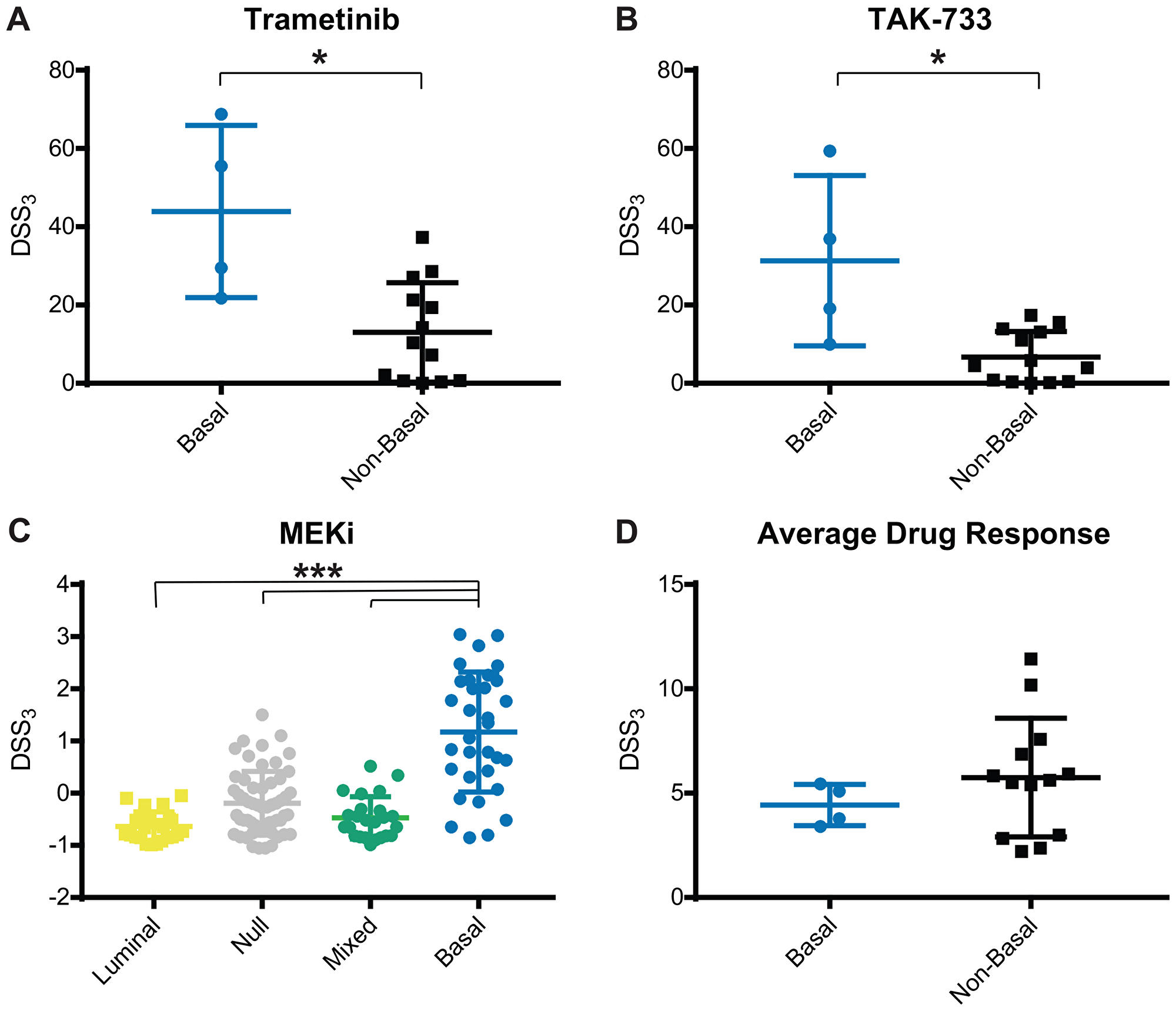
Figure 4: MEK inhibitor response correlates with basal subtype. Average and standard deviation for DSS3 response to (A) Trametinib, (B) TAK-733, (C) Normalized MEK inhibitors, and (D) Average drug response, grouped by cell line subtype. Each point represents an individual cell line. Center line is average and brackets are standard deviation. Significance determined using Mann-Whitney test, *p < 0.05, or Kruskal-Wallis with Dunn test for multiple comparisons, ***p < 0.001.
DISCUSSION
There is an increasing amount of literature that shows growing cells in 3D culture offers advantages over 2D, most importantly in that 3D cell culture signaling is more faithful of in vivo signaling [21–26]. This distinction is critical because we found many key drugs have differential responses when comparing 2D and 3D screening results, even when all other components of the assay are identical (Supplementary Figure 7). These changes in signaling in 3D can lead to differential drug response in key cancer pathways [33]. Screens in 2D can both identify false lead compounds and miss identifying compounds that have a profound effects. There are a number of existing resources for drug screening bladder cell lines in 2D [15–17]; thus, the goal of this work is to contribute to the field by providing a large screening resource in 3D culture. While there have been many advances in screening bladder cancer patient-derived organoids (PDOs) [28, 29, 34], PDOs are a limited resource, and a gap remains in how to identify the best compounds from approved or IND-stage, drugs to test in screens.
In this work, we screened 652 investigational compounds and 3 clinically relevant combinations using 17 bladder cancer cell lines. The 652 compounds mostly comprise investigational new drugs or drugs that are approved for other cancer indications. The combinations tested were those most commonly used in the clinic, namely MVAC, cisplatin with gemcitabine, and carboplatin with paclitaxel [35–37]. It was notable that the DSS3 of the carboplatin and paclitaxel combination was superior to both MVAC and cisplatin with gemcitabine, but the maximum efficacy was lower for nearly all cell lines. These data are confirmation of what is seen in the literature, that carboplatin with paclitaxel is a preferred therapy following failure of standard of care [37].
Nearly 17% of the drugs tested showed some level of efficacy, as determined by the DSS3, Figure 1 [30]. Many of the compounds identified in this screen as the most efficacious have been previously implicated in bladder cancer. Romidepsin has been extensively studied in bladder cancer preclinical models, showing promise [38–41]. A Phase II clinical trial of romidepsin in bladder cancer was withdrawn (NCT00087295), tempering expectations of this drug in bladder cancer; however, recent work has shown that romidepsin spares normal cells and acts as a radiosensitizer in bladder cancer, opening the door for future studies with this drug [42]. Bortezomib has been studied in bladder cancer and failed a Stage II clinical trial, determined to be safe but not efficacious as a second line therapy [43]. Triptolide has been studied within a combination therapy in bladder pre-clinical studies [44, 45]. Though there are no current clinical trials with this drug, our results confirm previous literature that it is a promising investigational compound in bladder cancer and may warrant further studies. Many of the 30 “active” compounds (such as gemcitabine, paclitaxel, vinblastine, and doxorubicin) are currently used in the standard treatment of bladder cancer, further supporting the validity of our approach to be empirically concordant with the clinical setting. We believe that many of the remaining compounds may be worthy of further study, including screening in patient-derived organoids.
Utilizing previous sequencing efforts, we wanted to determine the impact of mutations and CNA on drug response. In particular, DNA alterations are utilized in the clinic in many basket clinical trials, such as the MATCH trial (NCT02465060) [46]. We find that bladder cell lines have a very heterogeneous mutational spectrum, with most prioritized mutations only present in 1–2 cell lines, Figure 2, consistent with current literature [47]. For mutations found in 3+ cell lines, we identify correlates with TP53 and PIK3CA mutation status. We identify onalespib and clofarabine as significantly more efficacious in TP53 mutant cell lines compared to wildtype, Supplementary Figure 1, a novel finding. Paradoxically, in the PIK3CA mutants, we identify that AZD-8186 responds best in PIK3CA WT cells, Supplementary Figure 2. This is the opposite of what we would predict clinically [48, 49]; however, it is important to note that AZD-8186 is one of many PI3K inhibitors in our drug screen, and other PI3K inhibitors with better efficacy (e.g., gedatolisib) show no significant difference in DSS3 between groups, supporting that PI3K mutation status alone is unlikely to be a predictor of which PI3K pathway modulating drug to use. When we incorporate PTEN mutation and CNA data, the correlation is no longer significant and we instead see a correlation with the Aurora inhibitor, MLN228, where the drug is most effective in wildtype cells, Supplementary Figure 3. It is notable that in neither the mutational nor mutational and CNA combination do we see correlations with PI3K inhibitors. Similarly, we fail to observe significant correlations of FGFR3 mutation with FGFR inhibition response, ERBB2 mutation with ERBB2 inhibition response, or HRAS and KRAS mutation and PI3K/AKT/mTOR inhibitor response. Taken together, our findings emphasize the great need for biomarkers of drug response in bladder cancer.
Integration of mutational and CNA data greatly improved our ability to identify correlations of drug response. CDKN2A loss correlated with relatively poor response to purine analogs as well as HDAC inhibitors. The purine analog result is counter to what one may expect, because CDKN2A deletion is known to increase the cell cycle, and the two drugs would be expected to have a more profound effect in highly proliferative cells [50, 51]. It is known however, that these drugs are effective in lowly proliferating lymphocytes [52, 53], and thus it is possible that they are effective in CDKN2A WT cells through a mechanism unrelated to proliferation. Previous literature has shown that some HDAC inhibitors are less efficacious in cells with TP53 loss, suggesting that different HDAC inhibitors have a differential dependence on TP53 status [54]. RB1 loss significantly correlated with average drug response across the 17 cell lines, Supplementary Figure 5. It has been established that RB1 status predicts response to cisplatin-based chemotherapy in bladder cancer [55, 56]. Our data suggest that this trend may hold for additional mechanisms of cell death.
MEK inhibition has been proposed as a promising treatment option in NMIBC by a mechanism related to enhancing the efficacy of BCG therapy [57]. Additionally, MEK inhibition has been proposed as a promising strategy in bladder cancers with high expression of KIF15, which upregulates the MEK pathway [58]. Pre-clinical work in bladder cancer [49, 59] and extensive studies in other cancers [60–63] suggest that MEK inhibition could be a potential combination approach to PI3K/AKT/mTOR inhibition, by targeting a parallel compensatory pathway. Despite these reports, the only clinical trial that utilizes MEK inhibition in bladder cancer is the ongoing MATCH screening trial in advanced solid tumors (NCT02465060), using mutation status of BRAF, GNA11, and NF1 to identify potential responders to the MEK inhibitor, Trametinib. These results suggest an opportunity to identify a population where MEK inhibition could be a particularly promising treatment strategy in bladder cancer.
Our panel of 17 cell lines have no prioritized mutations in BRAF, GNA11, or NF1, yet a subgroup responds very well to MEK inhibition. We identify the strongest responding cell lines as basal bladder cancer subtype, Figures 3 and 4. Basal bladder cancer is frequently clinically aggressive and has the worst overall prognosis [3], and thus, additional therapeutic options for these tumors would be highly clinically relevant. One cell line has a high-level EGFR amplification and another has an activating KRAS mutation, but the remaining two cell lines do not have molecular changes that would a priori indicate MEK sensitivity, including SCaBER, which is frequently the most sensitive cell line to MEK inhibitors. Only one other drug, atuveciclib (PTEFb/CDK9 inhibitor) (Supplementary Figure 3), was identified as significantly more effective in basal cell lines of drugs with and average DSS3 > 10 for basal bladder cancer, while 4 individual MEK inhibitors are significantly more effective in basal bladder cell lines, using the same criteria. Further, normalizing all MEK inhibitors with an average DSS3 > 0 and plotting them together, the basal subtype is significantly more sensitive to MEK inhibition than any other subtype, Figure 4C. Previous work in breast cancer has identified basal breast cancer as particularly susceptible to MEK inhibition, and importantly notes a PI3K feedback loop that requires combination inhibition to overcome the compensatory signaling [64]. Overall, this work strongly supports that MEK inhibitors should be explored as a potential therapeutic for bladder tumors with a basal signature that are refractory to standard of care therapy, particularly in combination with an inhibitor of parallel signaling.
In summary, this work is a valuable resource for the identification of experimental therapeutics in bladder cancer, having screened 652 investigational therapeutics and 3 drug combinations in 17 bladder cancer cell lines, using a 3D cell culture format. As next steps, we pose that this work be used to further test additional therapeutic options for patients with bladder cancer. Moreover, this work highlights a need for biomarkers of drug response, beyond mutational data. Lastly, using these methods, we identify MEK inhibitors as a promising therapeutic in the basal bladder cancer subtype. Important future work will investigate the specific molecular features of the basal subtype that make these cells more sensitive to MEK inhibition, and if this MEK sensitivity signature is applicable to other cancer subtypes.
Materials and Methods
Cell culture
Cells used in this manuscript have all been previously published and include: 5637, UM PDX BC8149, J82, SCaBER, SW780, T-24, TCCSUP, UM-UC1, UM-UC-3, UM-UC-5, UM-UC-6, UM-UC-9, UM-UC-12, UM-UC-14, UM-UC-15, UM-UC-17, and UM-UC-18. 5637, J82, SCaBER, SW780, and TCCSUP were purchased from ATCC and expanded under ATCC recommended conditions. Remaining cell lines were expanded in HyClone DMEM w/ glutamine (Thermo Fisher Scientific), 10% HyClone FBS (Thermo Fisher Scientific), Penicillin-Streptomycin (Thermo Fisher Scientific), amphotericin b (Thermo Fisher Scientific), and gentamicin (Thermo Fisher Scientific) at 5% CO2. All cell lines were drug tested in 3D growth media: DMEM (Thermo Fisher Scientific), 10% FBS (Thermo Fisher Scientific), 1% matrigel (Corning), B-27 (Thermo Fisher Scientific), anti-anti (Thermo Fisher Scientific), gentamicin (Thermo Fisher Scientific), Human EGF (25 ug/500 ml) (Sigma Aldrich), Human Heregulin β-1 (25 ug/500 ml) (Stemcell Technologies), Human KGF (FGF-7)(5 ug/500 ml) (Stemcell Technologies), Human FGF-10 (5 ug/500 ml) (Stemcell Technologies), Human Noggin (50 ug/500 ml) (Stemcell Technologies), Human RSPO1 (250 ug/500 ml) (Stemcell Technologies) at 10% CO2.
Chemicals and reagents
Chemicals were purchased from Selleckchem (Houston, TX, USA), Sigma-Aldrich (St. Louis, MO, USA), Cayman Chemical (Ann Arbor, MI, USA), and Med Chem Express (Monmouth Junction, NJ, USA). Included is the Selleckchem L-3500 screening library, the highly selective inhibitor library of 339 inhibitors covering 123 targets. Compounds were diluted in DMSO (Sigma-Aldrich, D2650) or water, depending on solubility, except for copanlisib which was diluted in 10% Trifluoroacetic acid in DMSO due (Sigma-Aldrich, T6508). Compounds were prepared as a 10 mM solution unless solubility constraints required lower concentrations.
Drug screening
Cells were screened in 384-well ultra-low attachment plates (Corning, 4516 or S-Bio, MS-9384WZ) in singlicate or duplicate, 7-point dose-response format. Cells were plated on day 0 at 3000 cells per well. On day 0, drugs were added at 1:1000 using a 50 nl pin tool, resulting in 0.1% final DMSO concentration per well. On day 5, viability was measured using CellTiter-Glo 3D (Promega, G9683) on an Envision plate reader (Perkin Elmer).
Drug sensitivity score calculation
Drug dose response data were fit to the equation Y = bottom + (top − bottom)/(1 + 10(Log10IC50-X) × HillSlope) where X = Log10 (concentration, M) and Y = % inhibition (vs. vehicle) using CDD Vault. Constraints used were bottom = 0 and top ≤ 100. DSS3 values were calculated as described by Yadav et al. [30]. IC50 value, hillslope, maximum inhibition, and drug range were entered into the DSS package for Rstudio and DSS3 values (ranging 0 to 100) were calculated.
Prioritized somatic variants and copy number alterations
For all cell lines except SCaBER and TCCSUP, next-generation DNA sequencing (DNAseq) data using a targeted pan-cancer assay (Oncomine Comprehensive Panel) was previously published by our group [7, 65]. Briefly, OCP targets hotspot regions in recurrent mutated oncogenes (e.g., BRAF exon 15) and the entire coding region of tumor suppressor genes (e.g., TP53, etc.); well-supported variants with established or presumptive oncogenic roles were considered prioritized mutations. For a subset of genes with recurrent CNA, OCP targets genomic regions across the gene body to detect copy number gains and losses [65]; for the purposes of this study, only high-level amplifications (≥ 2 copy gain) or deep deletions (2 copy loss) were considered prioritized. For the SCaBER and TCCSUP cell lines, genome-wide somatic mutation and CNA data was obtained from the CCLE via cBioPortal for Cancer Genomics (http://www.cbioportal.org/) [66] and reviewed by an experienced molecular pathologist (A.M.U.); only prioritized molecular alterations (as described above) occurring within the targeted regions of OCP were included for subsequent analyses.
RNA-based molecular subtyping
All transcriptomic data utilized in this study was generated from 2-D cultures using standard growth conditions. For all cell lines except SCaBER and TCCSUP, gene-level FPKM values were generated from whole-transcriptome next-generation RNA sequencing (RNAseq) data as described previously [31]; for the SCaBER and TCCSUP cell lines, gene-level RPKM values derived from whole-transcriptome RNAseq data were obtained directly from the CCLE database (portals. broadinstitute. org/ccle) [67]. All FPKM and RPKM values were log2-transformed, and sample-level gene expression Z-scores were generated for inter-sample standardization and comparison. For each sample, the Basal-Luminal (B-L) score was calculated as the average of basal gene expression less the average of luminal gene expression, as described previously [7, 32]. Basal genes included CD44, CDH3, EGFR, TP63, KRT14, KRT16, KRT5, KRT6A, and KRT6B, while luminal genes included ERBB2, FGFR3, FOXA1, GATA3, KRT19, KRT20, PPARG, UPK1B, UPK2, and UPK3A. The median B-L score across all samples was -0.15 (inter-quartile range = −0.39 to 0.55). All samples were assigned a molecular subtype based on their B-L score, as well as the average expression across all basal and luminal genes: B-L score > 0.55 was classified as “basal” subtype; B-L score < −0.39 was classified as “luminal” subtype; −0.39 ≤ B-L score ≤ 0.55 was classified as “mixed” subtype; and, average expression < 0 across all basal and luminal genes was classified as “null” subtype.
Correlations and statistics
Significant differences in groups were first identified by using the kruskalwallis function in R to prioritize compounds. Results were then filtered to eliminate compounds that had an average DSS3 < 10 for all groups. Remaining significant differences were validated in GraphPad Prism 7 using either a Mann-Whitney test (2 groups) or a Dunn test (3+ groups). MEK inhibitor normalization was carried out using the scale function in R. Normalized heat maps were generated using Morpheus (Broad Institute).
Abbreviations
NMIBC: non-muscle invasive bladder cancer; MIBC: muscle invasive bladder cancer; BCG: bacille Calmette-Guerin; CCLE: Cancer Cell Line Encyclopedia; GDSC: Genomics of Drug Sensitivity in Cancer; PDO: patient-derived organoid; MVAC: methotrexate, vinblastine, adriamycin and cisplatin; DSS3: drug sensitivity score 3; HDAC: histone deacetylase; MATCH: molecular analysis for therapy choice; B-L: Basal-Luminal.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This research was supported by the National Institutes of Health (1R21CA218498 to M.B.S. and S.D.M.). This research was also supported in part by a University of Michigan Rogel Cancer Center grant to A.M.U.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020; 70:7–30. https://doi.org/10.3322/caac.21590. [PubMed].
2. Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Kim WY. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A. 2014; 111:3110–3115. https://doi.org/10.1073/pnas.1318376111. [PubMed].
3. Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014; 25:152–165. https://doi.org/10.1016/j.ccr.2014.01.009. [PubMed].
4. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315–322. https://doi.org/10.1038/nature12965. [PubMed].
5. Rebouissou S, Bernard-Pierrot I, de Reynies A, Lepage ML, Krucker C, Chapeaublanc E, Herault A, Kamoun A, Caillault A, Letouze E, Elarouci N, Neuzillet Y, Denoux Y, et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med. 2014; 6:244ra91. https://doi.org/10.1126/scitranslmed.3008970. [PubMed].
6. Ho PL, Kurtova A, Chan KS. Normal and neoplastic urothelial stem cells: getting to the root of the problem. Nat Rev Urol. 2012; 9:583–594. https://doi.org/10.1038/nrurol.2012.142. [PubMed].
7. Hovelson DH, Udager AM, McDaniel AS, Grivas P, Palmbos P, Tamura S, Lazo de la Vega L, Palapattu G, Veeneman B, El-Sawy L, Sadis SE, Morgan TM, Montgomery JS, et al. Targeted DNA and RNA Sequencing of Paired Urothelial and Squamous Bladder Cancers Reveals Discordant Genomic and Transcriptomic Events and Unique Therapeutic Implications. Eur Urol. 2018; 74:741–753. https://doi.org/10.1016/j.eururo.2018.06.047. [PubMed].
8. McConkey DJ, Choi W. Molecular Subtypes of Bladder Cancer. Curr Oncol Rep. 2018; 20:77. https://doi.org/10.1007/s11912-018-0727-5. [PubMed].
9. Ashton A, Murthy Kusuma VR, Crew J, Mostafid AH. BCG therapy in NMIBC: How much and for how long? Arch Esp Urol. 2018; 71:342–348. [PubMed].
10. Gontero P, Bohle A, Malmstrom PU, O’Donnell MA, Oderda M, Sylvester R, Witjes F. The role of bacillus Calmette-Guerin in the treatment of non-muscle-invasive bladder cancer. Eur Urol. 2010; 57:410–429. https://doi.org/10.1016/j.eururo.2009.11.023. [PubMed].
11. Boegemann M, Krabbe LM. Prognostic Implications of Immunohistochemical Biomarkers in Non-Muscle-Invasive Bladder Cancer and Muscle-Invasive Bladder Cancer. Mini Rev Med Chem. 2020; 20:1133–1152. https://doi.org/10.2174/1389557516666160512151202. [PubMed].
12. Jayaratna IS, Navai N, Dinney CP. Risk based neoadjuvant chemotherapy in muscle invasive bladder cancer. Transl Androl Urol. 2015; 4:273–282. https://doi.org/10.3978/j.issn.2223-4683.2015.06.07. [PubMed].
13. Li S, Wu J, Chen Y, Tang W, Peng Q, Deng Y, Xie L, Wang J, Huang S, Li R, Qin X, Zhao J. ERCC1 expression levels predict the outcome of platinum-based chemotherapies in advanced bladder cancer: a meta-analysis. Anticancer Drugs. 2014; 25:106–114. https://doi.org/10.1097/CAD.0000000000000021. [PubMed].
14. Mucaki EJ, Zhao JZL, Lizotte DJ, Rogan PK. Predicting responses to platin chemotherapy agents with biochemically-inspired machine learning. Signal Transduct Target Ther. 2019; 4:1. https://doi.org/10.1038/s41392-018-0034-5. [PubMed].
15. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. Addendum: The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2019; 565:E5–E6. https://doi.org/10.1038/s41586-018-0722-x. [PubMed].
16. Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015; 528:84–87. https://doi.org/10.1038/nature15736. [PubMed].
17. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013; 41:D955–D961. https://doi.org/10.1093/nar/gks1111. [PubMed].
18. Goodspeed A, Jean A, Costello JC. A Whole-genome CRISPR Screen Identifies a Role of MSH2 in Cisplatin-mediated Cell Death in Muscle-invasive Bladder Cancer. Eur Urol. 2019; 75:242–250. https://doi.org/10.1016/j.eururo.2018.10.040. [PubMed].
19. Tong Z, Sathe A, Ebner B, Qi P, Veltkamp C, Gschwend JE, Holm PS, Nawroth R. Functional genomics identifies predictive markers and clinically actionable resistance mechanisms to CDK4/6 inhibition in bladder cancer. J Exp Clin Cancer Res. 2019; 38:322. https://doi.org/10.1186/s13046-019-1322-9. [PubMed].
20. Xylinas E, Hassler MR, Zhuang D, Krzywinski M, Erdem Z, Robinson BD, Elemento O, Clozel T, Shariat SF. An Epigenomic Approach to Improving Response to Neoadjuvant Cisplatin Chemotherapy in Bladder Cancer. Biomolecules. 2016; 6:37. https://doi.org/10.3390/biom6030037. [PubMed].
21. Shield K, Ackland ML, Ahmed N, Rice GE. Multicellular spheroids in ovarian cancer metastases: Biology and pathology. Gynecol Oncol. 2009; 113:143–148. https://doi.org/10.1016/j.ygyno.2008.11.032. [PubMed].
22. Zietarska M, Maugard CM, Filali-Mouhim A, Alam-Fahmy M, Tonin PN, Provencher DM, Mes-Masson AM. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol Carcinog. 2007; 46:872–885. https://doi.org/10.1002/mc.20315. [PubMed].
23. Lee J, Cuddihy MJ, Kotov NA. Three-dimensional cell culture matrices: state of the art. Tissue Eng Part B Rev. 2008; 14:61–86. https://doi.org/10.1089/teb.2007.0150. [PubMed].
24. Edmondson R, Broglie JJ, Adcock AF, Yang L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev Technol. 2014; 12:207–218. https://doi.org/10.1089/adt.2014.573. [PubMed].
25. Antoni D, Burckel H, Josset E, Noel G. Three-dimensional cell culture: a breakthrough in vivo. Int J Mol Sci. 2015; 16:5517–5527. https://doi.org/10.3390/ijms16035517. [PubMed].
26. Smith SJ, Wilson M, Ward JH, Rahman CV, Peet AC, Macarthur DC, Rose FR, Grundy RG, Rahman R. Recapitulation of tumor heterogeneity and molecular signatures in a 3D brain cancer model with decreased sensitivity to histone deacetylase inhibition. PLoS One. 2012; 7:e52335. https://doi.org/10.1371/journal.pone.0052335. [PubMed].
27. Amaral RLF, Miranda M, Marcato PD, Swiech K. Comparative Analysis of 3D Bladder Tumor Spheroids Obtained by Forced Floating and Hanging Drop Methods for Drug Screening. Front Physiol. 2017; 8:605. https://doi.org/10.3389/fphys.2017.00605. [PubMed].
28. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LJ, Kandoth C, Williams AB, Bergren SK, Pietzak EJ, Anderson CB, et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell. 2018; 173:515–28.e17. https://doi.org/10.1016/j.cell.2018.03.017. [PubMed].
29. Mullenders J, de Jongh E, Brousali A, Roosen M, Blom JPA, Begthel H, Korving J, Jonges T, Kranenburg O, Meijer R, Clevers HC. Mouse and human urothelial cancer organoids: A tool for bladder cancer research. Proc Natl Acad Sci U S A. 2019; 116:4567–4574. https://doi.org/10.1073/pnas.1803595116. [PubMed].
30. Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, Majumder MM, Malani D, Murumagi A, Knowles J, Porkka K, Heckman C, Kallioniemi O, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep. 2014; 4:5193. https://doi.org/10.1038/srep05193. [PubMed].
31. Tamura S, Wang Y, Veeneman B, Hovelson D, Bankhead A 3rd, Broses LJ, Lorenzatti Hiles G, Liebert M, Rubin JR, Day KC, Hussain M, Neamati N, Tomlins S, et al. Molecular Correlates of In Vitro Responses to Dacomitinib and Afatinib in Bladder Cancer. Bladder Cancer. 2018; 4:77–90. https://doi.org/10.3233/BLC-170144. [PubMed].
32. Udager AM, McDaniel AS, Hovelson DH, Fields K, Salami SS, Kaffenberger SD, Spratt DE, Montgomery JS, Weizer AZ, Reichert ZR, Alva AS, Chinnaiyan AM, Tomlins SA, et al. Frequent PD-L1 Protein Expression and Molecular Correlates in Urinary Bladder Squamous Cell Carcinoma. Eur Urol. 2018; 74:529–531. https://doi.org/10.1016/j.eururo.2018.06.019. [PubMed].
33. Riedl A, Schlederer M, Pudelko K, Stadler M, Walter S, Unterleuthner D, Unger C, Kramer N, Hengstschlager M, Kenner L, Pfeiffer D, Krupitza G, Dolznig H. Comparison of cancer cells in 2D vs 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J Cell Sci. 2017; 130:203–218. https://doi.org/10.1242/jcs.188102. [PubMed].
34. Yoshida T, Singh AK, Bishai WR, McConkey DJ, Bivalacqua TJ. Organoid culture of bladder cancer cells. Investig Clin Urol. 2018; 59:149–151. https://doi.org/10.4111/icu.2018.59.3.149. [PubMed].
35. Sagaster P, Flamm J, Flamm M, Mayer A, Donner G, Oberleitner S, Havelec L, Lepsinger L, Ludwig H. Neoadjuvant chemotherapy (MVAC) in locally invasive bladder cancer. Eur J Cancer. 1996; 32A:1320–1324. https://doi.org/10.1016/0959-8049(96)00114-1. [PubMed].
36. Dash A, Pettus JA 4th, Herr HW, Bochner BH, Dalbagni G, Donat SM, Russo P, Boyle MG, Milowsky MI, Bajorin DF. A role for neoadjuvant gemcitabine plus cisplatin in muscle-invasive urothelial carcinoma of the bladder: a retrospective experience. Cancer. 2008; 113:2471–2477. https://doi.org/10.1002/cncr.23848. [PubMed].
37. Furubayashi N, Negishi T, Yamashita T, Kusano S, Taguchi K, Shimokawa M, Nakamura M. The combination of paclitaxel and carboplatin as second-line chemotherapy can be a preferred regimen for patients with urothelial carcinoma after the failure of gemcitabine and cisplatin chemotherapy. Mol Clin Oncol. 2017; 7:1112–1118. https://doi.org/10.3892/mco.2017.1452. [PubMed].
38. Fan J, Stanfield J, Guo Y, Karam JA, Frenkel E, Sun X, Hsieh JT. Effect of trans-2,3-dimethoxycinnamoyl azide on enhancing antitumor activity of romidepsin on human bladder cancer. Clin Cancer Res. 2008; 14:1200–1207. https://doi.org/10.1158/1078-0432.CCR-07-1656. [PubMed].
39. Li QQ, Hao JJ, Zhang Z, Hsu I, Liu Y, Tao Z, Lewi K, Metwalli AR, Agarwal PK. Histone deacetylase inhibitor-induced cell death in bladder cancer is associated with chromatin modification and modifying protein expression: A proteomic approach. Int J Oncol. 2016; 48:2591–2607. https://doi.org/10.3892/ijo.2016.3478. [PubMed].
40. Karam JA, Fan J, Stanfield J, Richer E, Benaim EA, Frenkel E, Antich P, Sagalowsky AI, Mason RP, Hsieh JT. The use of histone deacetylase inhibitor FK228 and DNA hypomethylation agent 5-azacytidine in human bladder cancer therapy. Int J Cancer. 2007; 120:1795–1802. https://doi.org/10.1002/ijc.22405. [PubMed].
41. Pinkerneil M, Hoffmann MJ, Deenen R, Kohrer K, Arent T, Schulz WA, Niegisch G. Inhibition of Class I Histone Deacetylases 1 and 2 Promotes Urothelial Carcinoma Cell Death by Various Mechanisms. Mol Cancer Ther. 2016; 15:299–312. https://doi.org/10.1158/1535-7163.MCT-15-0618. [PubMed].
42. Paillas S, Then CK, Kilgas S, Ruan JL, Thompson J, Elliott A, Smart S, Kiltie AE. The Histone Deacetylase Inhibitor Romidepsin Spares Normal Tissues While Acting as an Effective Radiosensitizer in Bladder Tumors in Vivo. Int J Radiat Oncol Biol Phys. 2020; 107:212–221. https://doi.org/10.1016/j.ijrobp.2020.01.015. [PubMed].
43. Rosenberg JE, Halabi S, Sanford BL, Himelstein AL, Atkins JN, Hohl RJ, Millard F, Bajorin DF, Small EJ, Cancer and Leukemia Group B. Phase II study of bortezomib in patients with previously treated advanced urothelial tract transitional cell carcinoma: CALGB 90207. Ann Oncol. 2008; 19:946–950. https://doi.org/10.1093/annonc/mdm600. [PubMed].
44. Yang Y, Zhang LJ, Bai XG, Xu HJ, Jin ZL, Ding M. Synergistic antitumour effects of triptolide plus gemcitabine in bladder cancer. Biomed Pharmacother. 2018; 106:1307–1316. https://doi.org/10.1016/j.biopha.2018.07.083. [PubMed].
45. Ho JN, Byun SS, Lee S, Oh JJ, Hong SK, Lee SE, Yeon JS. Synergistic antitumor effect of triptolide and cisplatin in cisplatin resistant human bladder cancer cells. J Urol. 2015; 193:1016–1022. https://doi.org/10.1016/j.juro.2014.09.007. [PubMed].
46. Park JJH, Siden E, Zoratti MJ, Dron L, Harari O, Singer J, Lester RT, Thorlund K, Mills EJ. Systematic review of basket trials, umbrella trials, and platform trials: a landscape analysis of master protocols. Trials. 2019; 20:572. https://doi.org/10.1186/s13063-019-3664-1. [PubMed].
47. Meeks JJ, Al-Ahmadie H, Faltas BM, Taylor JA 3rd, Flaig TW, DeGraff DJ, Christensen E, Woolbright BL, McConkey DJ, Dyrskjot L. Genomic heterogeneity in bladder cancer: challenges and possible solutions to improve outcomes. Nat Rev Urol. 2020; 17:259–270. https://doi.org/10.1038/s41585-020-0304-1. [PubMed].
48. Ross RL, McPherson HR, Kettlewell L, Shnyder SD, Hurst CD, Alder O, Knowles MA. PIK3CA dependence and sensitivity to therapeutic targeting in urothelial carcinoma. BMC Cancer. 2016; 16:553. https://doi.org/10.1186/s12885-016-2570-0. [PubMed].
49. Zeng SX, Zhu Y, Ma AH, Yu W, Zhang H, Lin TY, Shi W, Tepper CG, Henderson PT, Airhart S, Guo JM, Xu CL, deVere White RW, et al. The Phosphatidylinositol 3-Kinase Pathway as a Potential Therapeutic Target in Bladder Cancer. Clin Cancer Res. 2017; 23:6580–6591. https://doi.org/10.1158/1078-0432.CCR-17-0033. [PubMed].
50. Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine. 2016; 8:30–39. https://doi.org/10.1016/j.ebiom.2016.04.017. [PubMed].
51. Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993; 366:704–707. https://doi.org/10.1038/366704a0. [PubMed].
52. Seto S, Carrera CJ, Kubota M, Wasson DB, Carson DA. Mechanism of deoxyadenosine and 2-chlorodeoxyadenosine toxicity to nondividing human lymphocytes. J Clin Invest. 1985; 75:377–383. https://doi.org/10.1172/JCI111710. [PubMed].
53. Johnston JB. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk Lymphoma. 2011; 52:43–45. https://doi.org/10.3109/10428194.2011.570394. [PubMed].
54. Sonnemann J, Marx C, Becker S, Wittig S, Palani CD, Kramer OH, Beck JF. p53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. Br J Cancer. 2014; 110:656–667. https://doi.org/10.1038/bjc.2013.742. [PubMed].
55. Plimack ER, Dunbrack RL, Brennan TA, Andrake MD, Zhou Y, Serebriiskii IG, Slifker M, Alpaugh K, Dulaimi E, Palma N, Hoffman-Censits J, Bilusic M, Wong YN, et al. Defects in DNA Repair Genes Predict Response to Neoadjuvant Cisplatin-based Chemotherapy in Muscle-invasive Bladder Cancer. Eur Urol. 2015; 68:959–967. https://doi.org/10.1016/j.eururo.2015.07.009. [PubMed].
56. Yin M, Grivas P, Emamekhoo H, Mendiratta P, Ali S, Hsu J, Vasekar M, Drabick JJ, Pal S, Joshi M. ATM/RB1 mutations predict shorter overall survival in urothelial cancer. Oncotarget. 2018; 9:16891–16898. https://doi.org/10.18632/oncotarget.24738. [PubMed].
57. Whang YM, Jin SB, Park SI, Chang IH. MEK inhibition enhances efficacy of bacillus Calmette-Guerin on bladder cancer cells by reducing release of Toll-like receptor 2-activated antimicrobial peptides. Oncotarget. 2017; 8:53168–53179. https://doi.org/10.18632/oncotarget.18230. [PubMed].
58. Zhao H, Bo Q, Wu Z, Liu Q, Li Y, Zhang N, Guo H, Shi B. KIF15 promotes bladder cancer proliferation via the MEK-ERK signaling pathway. Cancer Manag Res. 2019; 11:1857–1868. https://doi.org/10.2147/CMAR.S191681. [PubMed].
59. Cirone P, Andresen CJ, Eswaraka JR, Lappin PB, Bagi CM. Patient-derived xenografts reveal limits to PI3K/mTOR- and MEK-mediated inhibition of bladder cancer. Cancer Chemother Pharmacol. 2014; 73:525–538. https://doi.org/10.1007/s00280-014-2376-1. [PubMed].
60. Temraz S, Mukherji D, Shamseddine A. Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers. Int J Mol Sci. 2015; 16:22976–22988. https://doi.org/10.3390/ijms160922976. [PubMed].
61. Blaser B, Waselle L, Dormond-Meuwly A, Dufour M, Roulin D, Demartines N, Dormond O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer. 2012; 12:86. https://doi.org/10.1186/1471-2407-12-86. [PubMed].
62. Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M, Rogler G. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget. 2015; 6:42183–42196. https://doi.org/10.18632/oncotarget.5619. [PubMed].
63. Milella M, Falcone I, Conciatori F, Matteoni S, Sacconi A, De Luca T, Bazzichetto C, Corbo V, Simbolo M, Sperduti I, Benfante A, Del Curatolo A, Cesta Incani U, et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci Rep. 2017; 7:43013. https://doi.org/10.1038/srep43013. [PubMed].
64. Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y, Hu Z, Knight Z, Feiler HS, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009; 69:565–572. https://doi.org/10.1158/0008-5472.CAN-08-3389. [PubMed].
65. Hovelson DH, McDaniel AS, Cani AK, Johnson B, Rhodes K, Williams PD, Bandla S, Bien G, Choppa P, Hyland F, Gottimukkala R, Liu G, Manivannan M, et al. Development and validation of a scalable next-generation sequencing system for assessing relevant somatic variants in solid tumors. Neoplasia. 2015; 17:385–399. https://doi.org/10.1016/j.neo.2015.03.004. [PubMed].
66. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
67. Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019; 569:503–508. https://doi.org/10.1038/s41586-019-1186-3. [PubMed].