
Introduction
With an estimated 22,530 cases reported and 13,980 estimated deaths in the year 2019, ovarian cancer is the deadliest gynecological disease accounting for more deaths than any other cancer in the female reproductive tract. The disease is often diagnosed at an advanced stage, which contributes to a low five-year survival rate of only 47.6% in the United States [1].
Our laboratory studies epithelial ovarian cancer (EOC) in laying hens, the only known natural animal model that spontaneously develops the disease over its lifespan. The disease in hens is very similar to the human form in expression of similar molecular markers such as CA-125 and e-cadherin, and symptoms such as accumulation of ascitic fluid and peritoneal metastasis in the advanced stage. Onset of EOC in laying hens is also positively correlated with the number of lifetime ovulation. Suppressing ovulation reduces ovarian cancer incidence in laying hens, similar to the preventative effects of reduced ovulation observed in women.
Our research has shown that dietary ingestion of flaxseed reduces the onset and severity of ovarian cancer in laying hens [2, 3]. Flaxseed is one of the richest plant sources of omega-3 polyunsaturated fatty acids (OM3FA), mostly α-linoleic acid (ALA), phytoestrogen lignans, namely secoisolariciresinol diglucoside (SDG) and both soluble and insoluble fibers. ALA is converted to docosahexaenoic acid (DHA) by action of desaturase and elongase enzymes. DHA has potent anti-inflammatory actions by regulating Nuclear Factor kappa B (NFκB) activation and cyclooxygenase 2 (COX-2) expression (manuscript in preparation).
Estradiol is metabolized by three cytochrome P450 (CYP) enzymes in the liver. We have also shown that the flaxseed diet in hens induces CYP1A1 expression in the liver while suppressing both CYP1B1 and CYP3A4 expressions. The upregulation of CYP1A1 parallels the increase in 2hydroxyestradiol and the 2MeOE2 level in the serum of the chickens [4]. 2MeOE2 has established anti-proliferative and pro-apoptotic properties [5, 6] that have been tested on various cancer cells [7–13], although its molecular mechanisms are yet to be fully understood.
Recently we have shown that 2MeOE2 induces apoptosis in human ovarian cancer cells. The pro-apoptotic and anti-angiogenic effects of 2MeOE2 are dependent on the p38 MAPK pathway [14]. In a time-course study, caspase-3 cleavage was first detected at 24 h and TUNEL positive staining followed [14, 15]. This prompted us to probe the 2MeOE2 -induced apoptosis, especially the apoptotic histone modifications. Among the multiple upstream activators which triggers these histone marks during apoptosis, protein kinase Cδ (PKCδ) is one common factor. Therefore, we investigated whether the pro-apoptotic actions of 2MeOE2 involve PKCδ signaling.
The objective of this study was to investigate the epigenetic modifications exerted by 2MeOE2 and assess the role of PKCδ in 2MeOE2 actions in order to gain an in-depth mechanistic understanding of its molecular and cellular pro-apoptotic actions.
Results
2MeOE2 increases γH2Ax and phH3ser10 expression in human ovarian cancer cells
We hypothesized that 2MeOE2 treatment results in apoptotic histone modifications. Therefore, we investigated phospho histone H2A at ser139 (γH2Ax) and phospho histone H3 at ser10 (phH3ser10) expressions in human ovarian cancer cells after 10 μM 2MeOE2 treatment for 24 hours. 2MeOE2 treatment increased the number of cells expressing γH2Ax, a marker for DNA damage, and phH3ser10 in all three cell lines. Co-immunostained sections suggested that γH2Ax and phH3ser10 are co-expressed by a number of cells following 2MeOE2 treatment (Figure 1A). In order to verify our observation and to confirm that this upregulation is not because of a reduction in total number of viable cells, we quantified γH2Ax and phH3ser10 expression by western blot on total protein isolated from cells following 2MeOE2 treatment. The expression of γH2Ax and phH3ser10 significantly increases following 2MeOE2 treatment (Figure 1B).
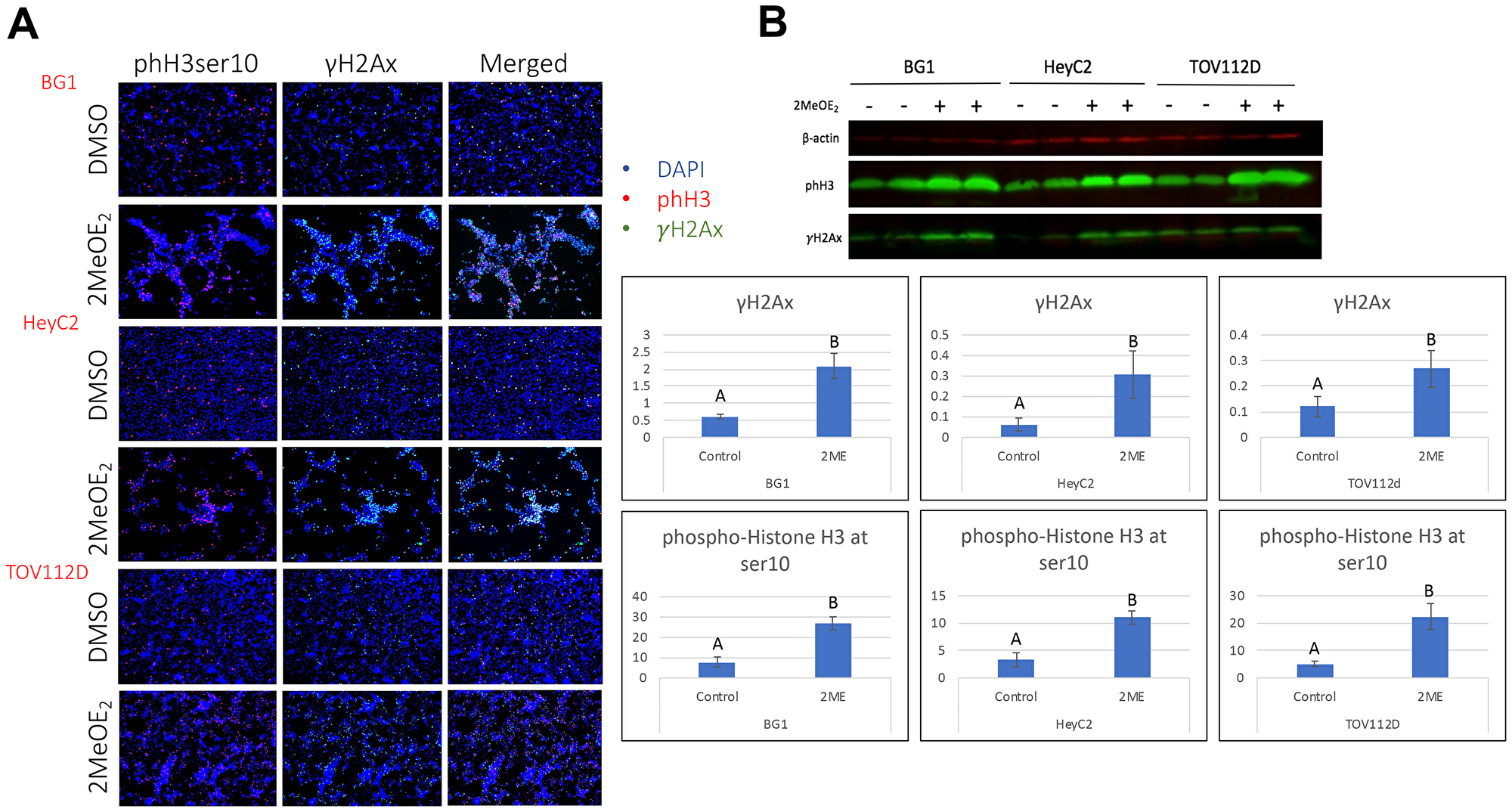
Figure 1: 2MeOE2 induced histone modifications in human ovarian cancer cells. BG1, HeyC2 and TOV112D cells were treated with 10 μM 2MeOE2 for 24 h. (A) Control and 2MeOE2-treated cells seeded on coverslip were then immunostained with anti-γH2Ax co-stained with anti-phospho histone H3 (ser10). (B) Western blot for γH2Ax and phospho histone H3 (ser10) from whole cell lysates (n = 4). Student’s unpaired t-test, error bars: SEM, p < 0.05.
2MeOE2 increases phH2Bser14 expression in human ovarian cancer cells
To examine whether 2MeOE2 treatment induces apoptotic histone modification, we investigated phospho histone H2B at ser14 (phH2Bser14) expression after 2MeOE2 treatment. Cells were seeded on coverslips and harvested after a 24 h treatment with 10 μM 2MeOE2. 2MeOE2 treatment increased number of phH2Bser14 expressing cells in all three cell lines. Co-immunostaining against phH2Bser14 and γH2Ax revealed that similar to our earlier observation with phH3ser10, a number of cells co-expressed phH2Bser14 and γH2Ax, the marker for DNA damage (Figure 2A). Western blot data on total protein lysates confirmed significant upregulation of phH2Bser14 following 2MeOE2 treatment (Figure 2B).
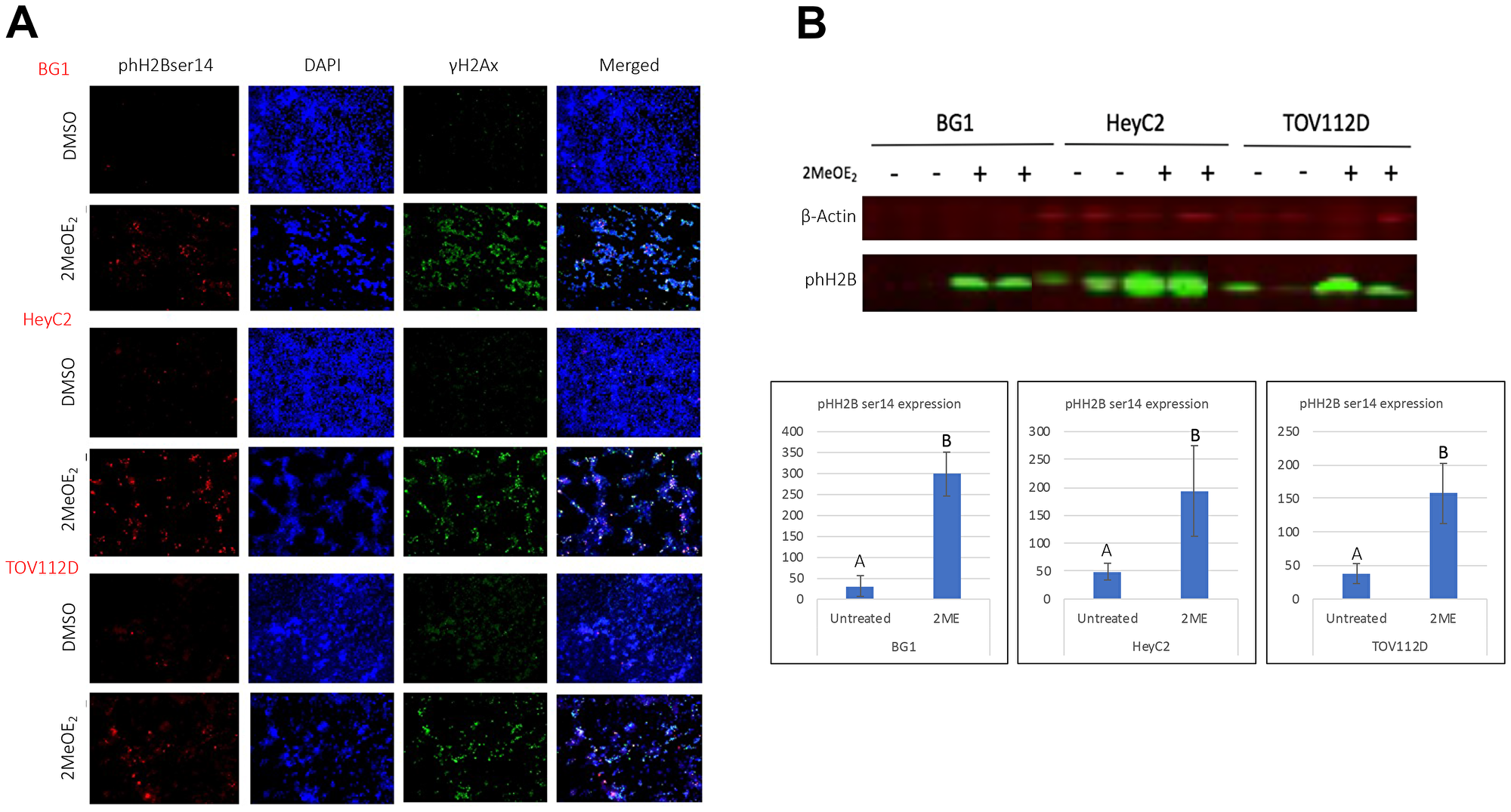
Figure 2: 2MeOE2 induces phosphorylation on ser 14 of histone H2B in human ovarian cancer cells. BG1, HeyC2 and TOV112D cells were seeded on coverslips and treated with 10 μM 2MeOE2 for 24 h. (A) Control and 2MeOE2-treated cells were immunostained with anti-phospho histone H2B (ser14) co-stained with anti-γH2Ax. (B) Western blot from whole cell lysates for phospho histone H2B (ser14) (n = 4). Student’s unpaired t-test, error bars: SEM, p < 0.05.
PKCδWT increases pro-apoptotic actions of 2MeOE2
Histone modifications such as phH3ser10 and phH2Bser14, have been reported to be mediated by PKCδ during apoptosis. Therefore, we hypothesized that 2MeOE2-mediated apoptosis in the ovarian cancer cells may involve PKCδ signaling. To address this question, BG1 cells were transfected with a wild type PKCδ (PKCδWT) or a kinase-negative PKCδ (PKCδDN). BG1 cells transfected with a pGFP expression vector was taken as a transfection control (mock). Cells showed no apparent changes in total numbers or gross morphology following the transfections (Figure 3A). Western blot on total cellular lysates confirmed that the amount of PδWT and PKCδDN were increased after the transfection. PKCδWT or PKCδDN did not induce cleavage of caspase-3 or phosphorylation of p38 MAPK. Also, neither PKCδWT nor PKCδDN altered phH2Bser14 expression in the BG1 cells (Figure 3A).
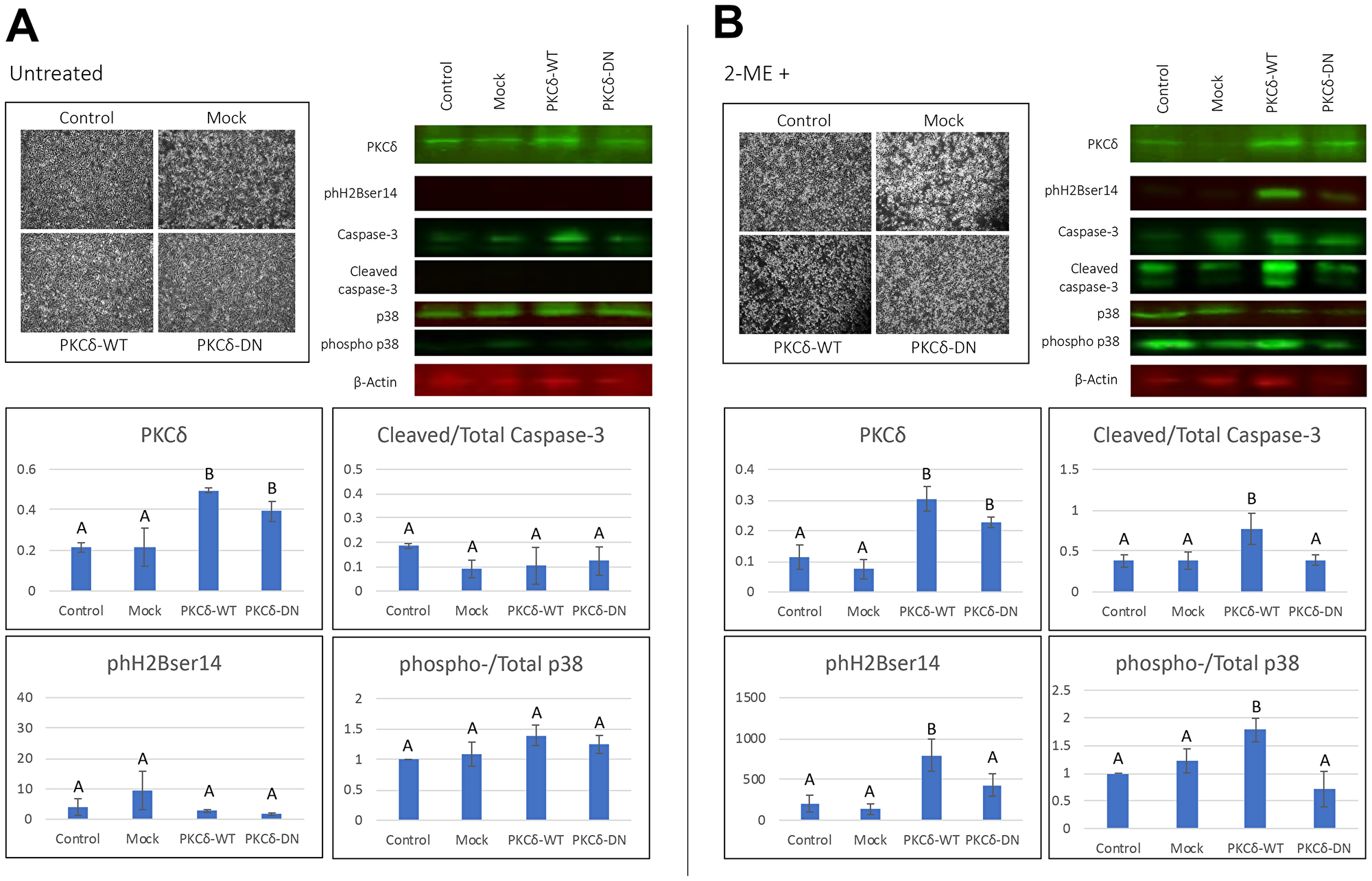
Figure 3: Effect of transient transfection of PKCδWT and PKCδDN to human ovarian cancer cells. (A) BG1 cells were either untransfected (control) or transfected with a pGFP expression vector (mock), a wild type PKCδ expression vector (PKCδWT) or a C3-domain mutated PKCδ expression vector (PKCδDN). (B) 10 μM 2MeOE2 was treated to BG1 cells that were either untransfected (control) or transfected with a GFP (mock control), PKCδWT or PKCδDN expression vector for 24 h. Cells were photographed and harvested after 24 h from transfection. Western blot analysis performed on total protein lysates from untransfected, mock, PKCδWT and PKCδDN transfected BG1 cells against PKCδ, phospho histone H2B (ser 14), caspase-3, cleaved caspase-3, total p38 and phospho-p38 (n = at least 3 for each dataset). Ratio of normalized phospho-p38 to normalized total p38 was quantified to estimate p38 phosphorylation resulting from individual transfection. Similarly, ratio of normalized cleaved caspase-3 to normalized pan caspase-3 was quantified to estimate amount of activated caspase-3. One-way ANOVA, error bars: SEM, p < 0.05.
Although PKCδWT or PKCδDN did not induce or prevent apoptosis alone, PKCδWT significantly potentiated the pro-apoptotic actions of 2MeOE2. BG1 cells transfected with PKCδWT, PKCδDN or pGFP (mock) were treated with 10 μM 2MeOE2 for 24 h. Western blots demonstrated that 2MeOE2 treatment of BG1 cells transfected with PKCδWT resulted in a significantly higher amount of cleaved caspase-3, phosphorylated p38 MAPK and phH2Bser14 expression compared to untransfected and mock transfected BG1 cells. Notably, 2MeOE2 treatment of the PKCδDN -transfected cells did not alter cleavage of caspase-3, phosphorylation of p38 MAPK or phH2Bser14 expression compared to the untransfected and mock (Figure 3B).
Knock down of PKCδ decreases 2MeOE2 mediated apoptosis in BG1 cells
To investigate the role of PKCδ in 2MeOE2 -mediated apoptosis in the ovarian cancer cells, we performed a siRNA-mediated knockdown of PKCδ. BG1 cells were transfected with a PKCδRNAi expression vector to knock down endogenous PKCδ. BG1 cells transfected with a plasmid expressing scrambled shRNA was used as a silencing control. PKCδRNAi -transfected cells were visibly more resistant to 2MeOE2 treatment compared to the untransfected and scrambled shRNA transfected cells (Figure 4). Western blots confirmed the decrease of PKCδ in BG1 cells. In the PKCδRNAi transfected cells, 2MeOE2 treatment had significantly decreased cleavage of caspase-3 and phosphorylation of p38 MAPK and phH2Bser14 expression, compared to the untransfected and scrambled shRNA transfected cells (Figure 4).
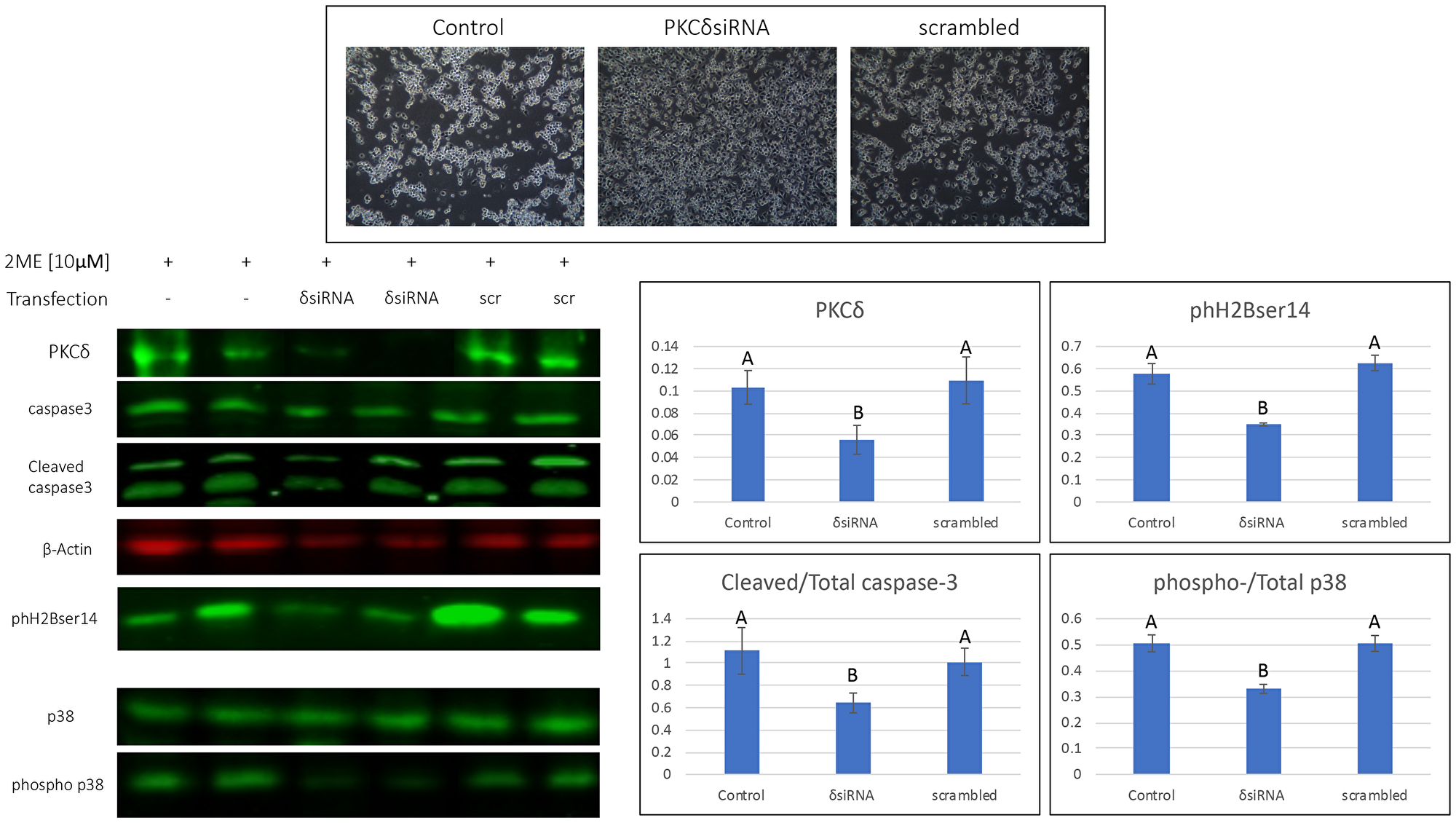
Figure 4: 2MeOE2 effects on human ovarian cancer cells after silencing PKCδ. BG1 cells were either untransfected or transfected with a PKCδ siRNA or scrambled shRNA expression vectors, before treating them with 10 μM 2MeOE2. Cells were photographed after a 24 h incubation in 2MeOE2. Western blot analysis performed on total protein lysates against PKCδ, cleaved and total caspase-3, phospho histone H2B (ser 14), total and phosphorylated p38 (n = 4 for each dataset). One-way ANOVA, error bars: SEM, p < 0.05.
PKCδCAT parallels some 2MeOE2 induced effects, induces apoptosis in a p38-dependent manner
PKCδ can be activated by either phosphorylation leading to structural changes in the full length molecule or catalytic cleavage by caspases which liberates its catalytic fragment (PKCδCAT). To investigate the role of PKCδCAT in the pro-apoptotic effects, we tested whether overexpression of PKCδCAT alone can exert the 2MeOE2 -mediated pro-apoptotic effects. BG1 cells were transfected with the PKCδCAT expression vector and pGFP transfected cells were used as transfection controls. Following 24 h of transfection, PKCδCAT transfected cells were visibly fewer in number and had more floating cells compared to the untransfected and the mock control (Figure 5A). Western blots demonstrated the cleavage of caspase-3, phosphorylation of p38 MAPK and phH2Bser14 expression in the PKCδCAT transfected cells were significantly higher than the controls (Figure 5A). In summary, PKCδCAT significantly induced apoptosis in the BG1 cells and the effects were similar to that exerted by 2MeOE2.
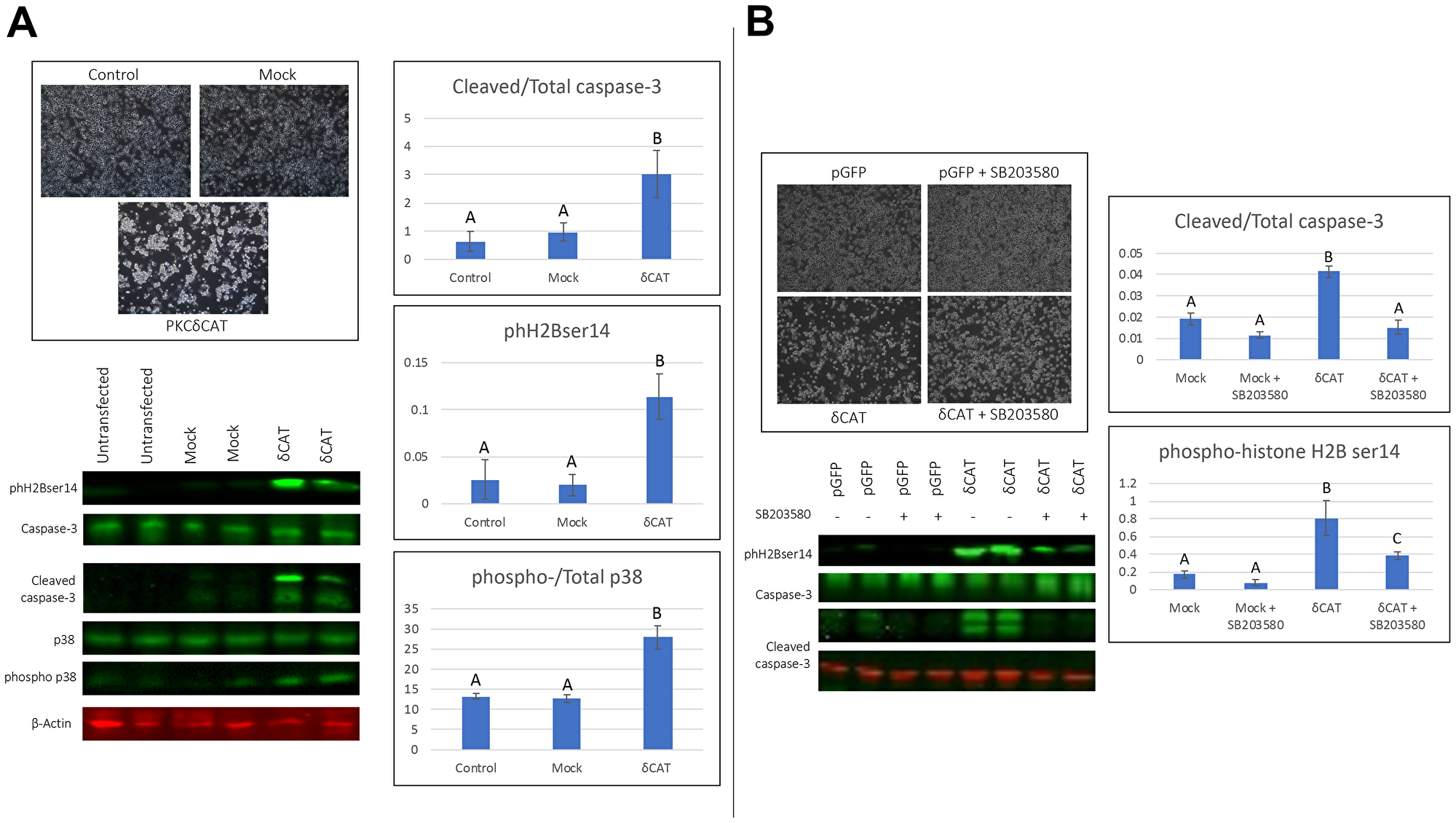
Figure 5: Expression of the catalytically active fragment of PKCδ to human ovarian cancer cells. (A) BG1 cells were either untransfected or transfected with a GFP expression vector (mock) or an expression vector with the catalytically active fragment of PKCδ (PKCδCAT). Cells were photographed and harvested after 24 h of transfection. Western blot analysis on total cell lysates against cleaved and total caspase-3, phospho histone H2B (ser 14), total and phosphorylated p38 (n = 4 for each dataset). One-way ANOVA, error bars: SEM, p < 0.05. (B) BG1 cells were transfected with either a GFP expression vector (mock) or an expression vector with catalytically active fragment of PKCδ (PKCδCAT) with or without 10 μM SB203580, a p38 MAPK inhibitor. Cells were photographed after 24 h of transfection. Western blot analysis on total cell lysates against phospho histone H2B (ser 14), cleaved and total caspase-3 (n = 3 for each dataset). Two-way ANOVA, error bars: SEM, p < 0.05.
To test whether the activation of p38 MAPK by PKCδCAT is associated with its pro-apoptotic effects, BG1 cells were transfected with a pGFP or PKCδCAT and treated with +/− 10 μM SB203580, a selective p38 MAPK inhibitor. SB203580 had no visible effect on pGFP transfected cells, but it reduced the number of rounded up and floating cells that were transfected with PKCδCAT (Figure 5B). Western blots demonstrated that SB203580 reduces phH2Bser14 expression and cleavage of caspase-3 in the PKCδCAT transfected BG1 cells, while having no significant effect in the pGFP transfected cells. Notably, SB203580 treated PKCδCAT cells had a significantly higher phH2Bser14 expression compared to its pGFP control (Figure 5B).
PKCδDN partially reverses 2MeOE2 induced pro-apoptotic effects
To test whether the pro-apoptotic effects of 2MeOE2 are dependent on PKCδ, BG1 cells were transfected with an increasing amount of PKCδDN (2 μg, 3 μg and 4 μg of plasmid DNA) with same amount of respective pGFP controls. After 24 h of transfection, 10 μM 2MeOE2 was added to the cells for another 24 h. Western blot on total protein lysates indicated that higher amounts of PKCδDN (3 μg and 4 μg) significantly reduces 2MeOE2 -induced γH2Ax expression and cleavage of caspase-3 compared to their respective pGFP controls. phH3ser10 expression was decreased in 2 μg PKCδDN transfected cells, however there was no change observed in 3 μg and 4 μg PKCDN transfected groups (Figure 6A).
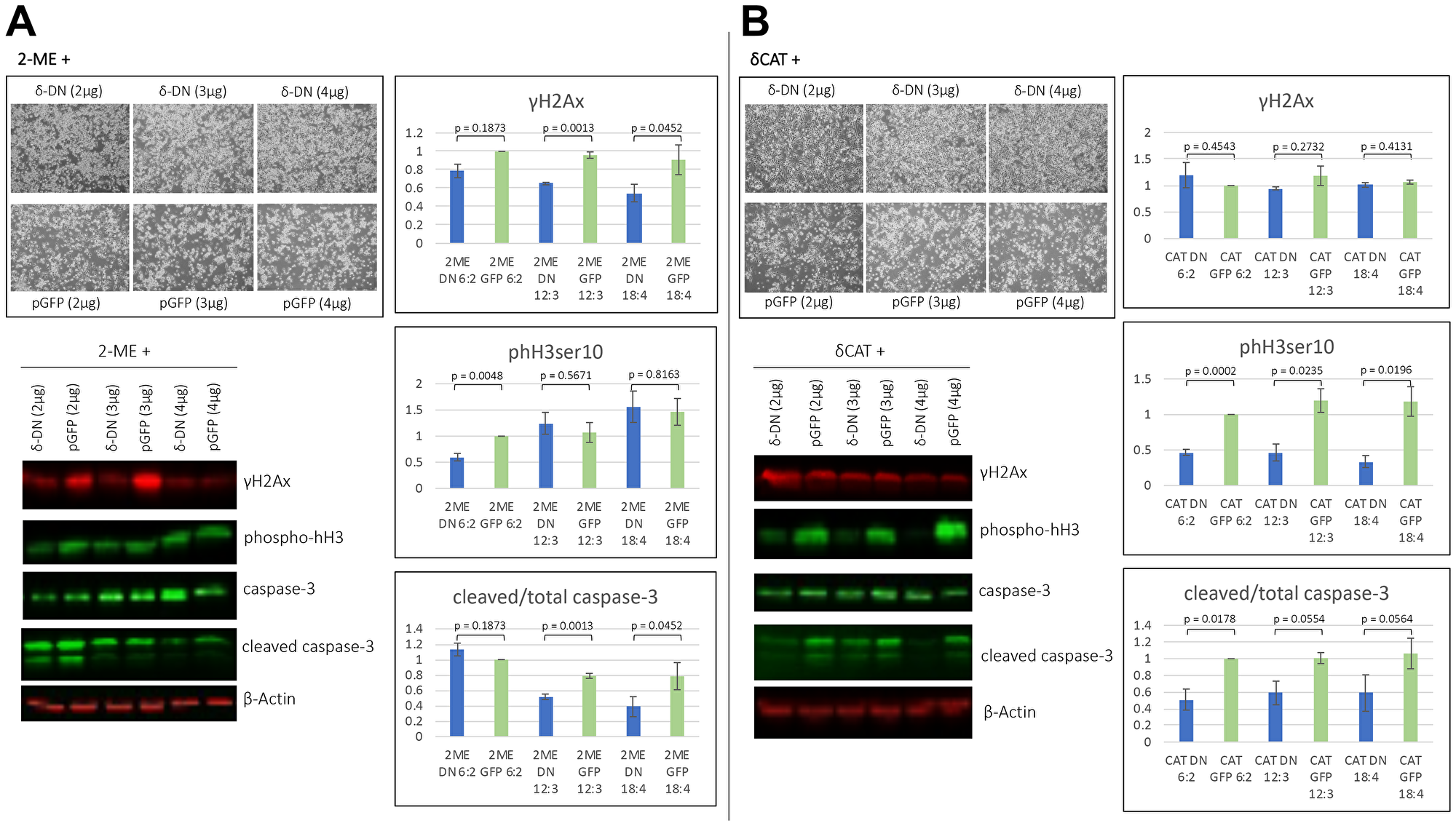
Figure 6: 2MeOE2 effects are in part dependent on PKCδ. (A) BG1 cells were transfected with a fixed amount (2 μg) of PKCδCAT and increasing amount (2 μg, 3 μg, and 4 μg) of either PKCδDN or pGFP (mock). Cells harvested and photographed after 24 h of transfection. Western blot analysis on cell lysates against γH2Ax, phospho histone H3 (ser10), total and cleaved caspase-3 (n = 3 for each dataset). Student’s unpaired t-test, error bars: SEM, p < 0.05. (B) BG1 cells were transfected with increasing amount (2 μg, 3 μg, and 4 μg) of PKCδDN or pGFP (mock). After 24 h of transfection, cells were treated with 10 μM 2MeOE2 for next 24 h, photographed and harvested. Western blot on total cell lysates against γH2Ax, phospho histone H3 (ser10), total and cleaved caspase-3 (n = 3 for each dataset). Student’s unpaired t-test, error bars: SEM, p < 0.05.
To examine the PKCδ-specific effects, we transfected BG1 cells with PKCδCAT and co-transfected with an increasing amount (2 μg, 3 μg and 4 μg) of PKCδDN or pGFP as respective controls. Western blot on the total protein lysates indicated that PKCδDN has no effect on γH2Ax expression, but phH3ser10 and cleaved caspase-3 expressions were found to be significantly decreased in all PKCδDN transfected groups compared to their respective pGFP controls (Figure 6B). These data suggest PKCδ augments 2MeOE2 -induced DNA damage response, resulting in apoptosis. However, no consistent trend was observed in phH3ser10 expression upon PKCδDN treatment possibly because phH3ser10 is also expressed in multiple other events such as mitosis, oxidative stress, etc.
DISCUSSION
2MeOE2 is a natural metabolite of estradiol (E2) that has known anti-angiogenic and anti-tumor activity. Anti-proliferative effects of 2MeOE2 have been extensively studied in ovarian cancer [16, 17], melanoma [18, 19], lung cancer [20–23], breast cancer [24–28], prostate cancer [29–31] and several other cancers [7, 9, 32, 33]. According to the current understanding, the anti-tumor actions of 2MeOE2 are accomplished by binding at/near the colchicine binding sites of the growing microtubules [34]. This suppresses the microtubule dynamics resulting in a G2/M phase arrest, and subsequent phosphorylation of Bcl2 [31, 35, 36] and Bcl-xL [37], preventing their anti-apoptotic activity. 2MeOE2 has also been reported to inhibit the expression, nuclear retention and transcriptional activity of hypoxia-inducible factor 1α (HIF-1α) and therefore preventing hypoxia-driven angiogenesis [38]. However, the cellular and molecular mechanism of action of 2MeOE2 is not thoroughly understood. In addition to disrupting microtubule dynamics, 2MeOE2 has been reported to activate a number of cellular kinases and several pro-apoptotic factors. Recently, we have reported that 2MeOE2 induces apoptosis in human ovarian cancer cells, which is partially dependent on activation of p38 MAPK. The current study was designed to gain a comprehensive understanding of the molecular actions of 2MeOE2 in ovarian cancer cells.
In the current study, we have extended our investigation to the epigenetic histone modifications induced by 2MeOE2. Histones were originally thought to be simple static scaffolds that package the DNA of a cell. However, it is well established now that the histones are dynamic proteins that undergo critical post-translational modifications for proper chromatin function [39]. The fate of a cell is dependent on distinct combination of histone modifications [40–42]. The Histone Code hypothesis proposed by Allison and Turner [43] states that not a single histone modification mark, but a collective combination of different histone modifications code for the fate of the cell. The apoptotic histone code includes severe dephosphorylation in the H1 linker region [44–46] and a number of phosphorylation, acetylation and methylation events on different histone monomers [47], as illustrated in Supplementary Figure 1A.
2MeOE2 treatment of human ovarian cancer cells causes increased phospho histone H3 at ser10 (phH3ser10) expression. Phosphorylation at this site has been traditionally regarded as a mitotic marker which correlates with chromosome condensation during mitosis and meiosis [48]. Therefore, phH3ser10 is classically associated with cell division and proliferation. On the other hand, growing evidence suggests that cells undergoing apoptosis after being exposed to various death stimuli also express phH3ser10. Therefore, this epigenetic mark is associated with chromosome compaction but the specific cellular effects of this phosphorylation is unclear. When we have co-immunostained 2MeOE2 treated human ovarian cancer cells with phH3ser10 and phospho histone H2A at ser139 (γH2Ax), a known DNA damage marker, we have observed that some cells expressing phH3ser10 are also undergoing DNA damage. A growing body of evidence suggests that distinctly different upstream kinases phosphorylate histone H3 at ser 10 under different conditions. During mitosis, this phosphorylation is mediated by mitotic kinases such as Aurora B and Vaccinia-related kinase 1 (VRK1) [49], whereas during apoptosis, the same phosphorylation is mediated by protein kinase Cδ (PKCδ) [50].
Similar to the phH3ser10 mark, phosphorylation of histone H2B at ser 14 (phH2Bser14) was also upregulated following 2MeOE2 in human ovarian cancer cells. Most of the phH2Bser14 positive cells were also positive for γH2Ax, indicative of DNA damage. The phH2Bser14 mark is associated with apoptosis whereas acetylation of the adjacent lysine residue (K15) is a property of surviving cells [51]. Acetylated K15 mark in surviving cells prevents phosphorylation in the ser 14 residue, which is deacetylated during apoptosis [52]. The ser 14 site of histone H2B is known to be phosphorylated by both Mammalian sterile 20 kinase (Mst-1) [53] and PKCδ [54, 55] during apoptosis. Interestingly, both of these enzymes are activated by caspase-3-mediated cleavage. Taken together, from the observations that 2MeOE2 induces both phH3ser10 and phH2Bser14 in the cells undergoing DNA damage, we hypothesized that PKCδ, being a common activator of both histone modifications, potentially plays a role in 2MeOE2 induced apoptosis.
The full-length human PKCδ has a catalytic domain which is essential for its enzymatic activity. The catalytic domain has a C3 (ATP binding domain) and a C4 (substrate binding) domain [56]. PKCδ also has a regulatory domain which contains two constant regions (C1 and C2-like) and a pseudo-substrate region, that keeps the full length PKCδ in a folded in an inactive conformation, preventing access to the substrate-binding pocket (C4 domain) [56, 57]. The catalytic and regulatory domains are connected by a hinge region (as described in Supplementary Figure 1B). Several apoptotic agents can induce a caspase-mediated cleavage of full-length PKCδ. This liberates the catalytic fragment (PKCδCAT) which is capable of inducing chromatin condensation and DNA damage leading to apoptosis [58–60]. The PKCδCAT can freely translocate to nucleus and/or mitochondria [61–63] and promote apoptosis [60, 64, 65]. PKCδ is also suggested to interact with c-Abl tyrosine kinase. Phosphorylation at the tyrosine residues in the C4 domain mediated by c-Abl have been found to induce the nuclear translocation of PKCδ [66], although the exact mechanism of nuclear transport of PKCδ is largely unknown.
In the current study, we have seen that overexpressing the full-length PKCδ (PKCδWT) and a C3 domain-mutated (K376R) PKCδ (PKCδDN) transfection in the BG1 cells had no effect on apoptosis, suggesting that the full-length PKCδ needs to be activated to trigger apoptosis. After cells were treated with 2MeOE2, the PKCδWT transfected cells evoked a greater apoptotic response compared to the mock control and the PKCδDN transfected cells. This suggested that the 2MeOE2 treatment activates full-length PKCδ which triggers the apoptotic cascade. Following 2MeOE2 treatment, only the PKCδWT transfected cells showed significantly higher levels of phH2Bser14 expression, suggesting that the PKCδ activation can induce this epigenetic histone modification. We have also observed that a siRNA mediated PKCδ depletion made the BG1 cells significantly more resistant towards 2MeOE2-mediated apoptosis. PKCδ knockdown also reduced the phH2Bser14 expression, paralleling the cleavage of caspase-3.
These findings indicate that the 2MeOE2-activated PKCδ is responsible for its pro-apoptotic actions. Transfection of the catalytic fragment of PKCδ (PKCδCAT) induced apoptosis in BG1 cells. PKCδCAT alone could induce cleavage of caspase-3 and also increase phH2Bser14 indicating that catalytic cleavage of PKCδ, leading to the liberation of PKCδCAT fragment is necessary for the pro-apoptotic effects.
p38 MAPK regulates a number of cellular processes in response to a variety of stress signals [67] such as UV radiation and inflammatory cytokines. In our previous study, we reported that 2MeOE2 treatment induces p38 phosphorylation and a selective p38 inhibitor, SB203580 [68], reduces its pro-apoptotic effect in the human ovarian cancer cells [14]. Evidence suggests that catalytically cleaved PKCδ can phosphorylate p38 MAPK in smooth muscle cells [69, 70], fibroblasts [71], prostate cancer cells [72] and hepatic stellate cells [73]. In our current study, we have also investigated whether 2MeOE2 mediated phosphorylation of p38 MAPK involves PKCδ signaling. Following 2MeOE2 treatment, PKCδWT transfection shows a significantly higher amount of phosphorylated p38 MAPK. 2MeOE2 treatment of PKCδ-depleted BG1 cells had decreased amount of phosphorylated p38 MAPK compared to scrambled siRNA control. These data suggest that 2MeOE2 mediated activation of PKCδ also phosphorylates p38 MAPK and the functional C3 domain of PKCδ is required for this activity. PKCδCAT alone successfully increased phosphorylation of p38 MAPK in BG1 cells. SB203580 reduced the pro-apoptotic effects of PKCδCAT. Previously, we reported that SB203580 reduces the amount of caspase-3 cleavage induced by 2MeOE2 in human ovarian cancer cells [14]. In contrast, SB203580 almost entirely inhibited downstream caspase-3 activation by PKCδ, indicating that p38 MAPK pathway is a principal pathway through which PKCδ-mediated apoptotic effects are elicited. Interestingly, SB203580 significantly reduced the phH2Bser14 expression induced by PKCδCAT, although it was still significantly higher than pGFP transfected or SB203580 treated pGFP cells. This suggests that p38 MAPK and subsequent activation of caspase-3 plays a major role in cleavage and activation of more PKCδ. The catalytic activation of PKCδ therefore results in a feedforward cycle amplifying the apoptotic signal.
Transfection of PKCδDN significantly reduced cleavage of caspase-3 and γH2Ax expression compared to their respective pGFP controls, suggesting that the pro-apoptotic actions of 2MeOE2 are partially dependent on PKCδ activation. The phH3ser10 expression was lower in 2 μg PKCδDN transfected cells however there was no significant alteration in the groups receiving higher amounts of PKCδDN. Therefore no discernible pattern was observed with phH3ser10. Interestingly, when we co-transfected PKCδDN in an increasing amount (2 μg, 3 μg and 4 μg) with the same amount of PKCδCAT (2 μg), we found that PKCδDN transfected cells had lower cleavage of caspase-3 and phH3ser10 expression compared to their respective pGFP controls. Notably, γH2Ax expression was found unaltered in these groups suggesting that γH2Ax is not directly affected by PKCδ. This also corroborates the role of PKCδ in the apoptotic phH3ser10 expression.
Our findings indicate that 2MeOE2 -mediated anti-tumor actions involve the catalytic activation of PKCδ in the pro-apoptotic pathway. The catalytic fragment of PKCδ is responsible for the apoptotic histone modifications and acceleration of the apoptotic cascade through p38 MAPK pathway. Dietary flaxseed supplementation activates PKCδ secondary to increasing endogenous production of 2MeOE2 which drives apoptosis in ovarian cancer cells (Figure 7). This study offers new insight into the molecular underpinnings of dietary flaxseed’s chemopreventative actions in ovarian cancer.
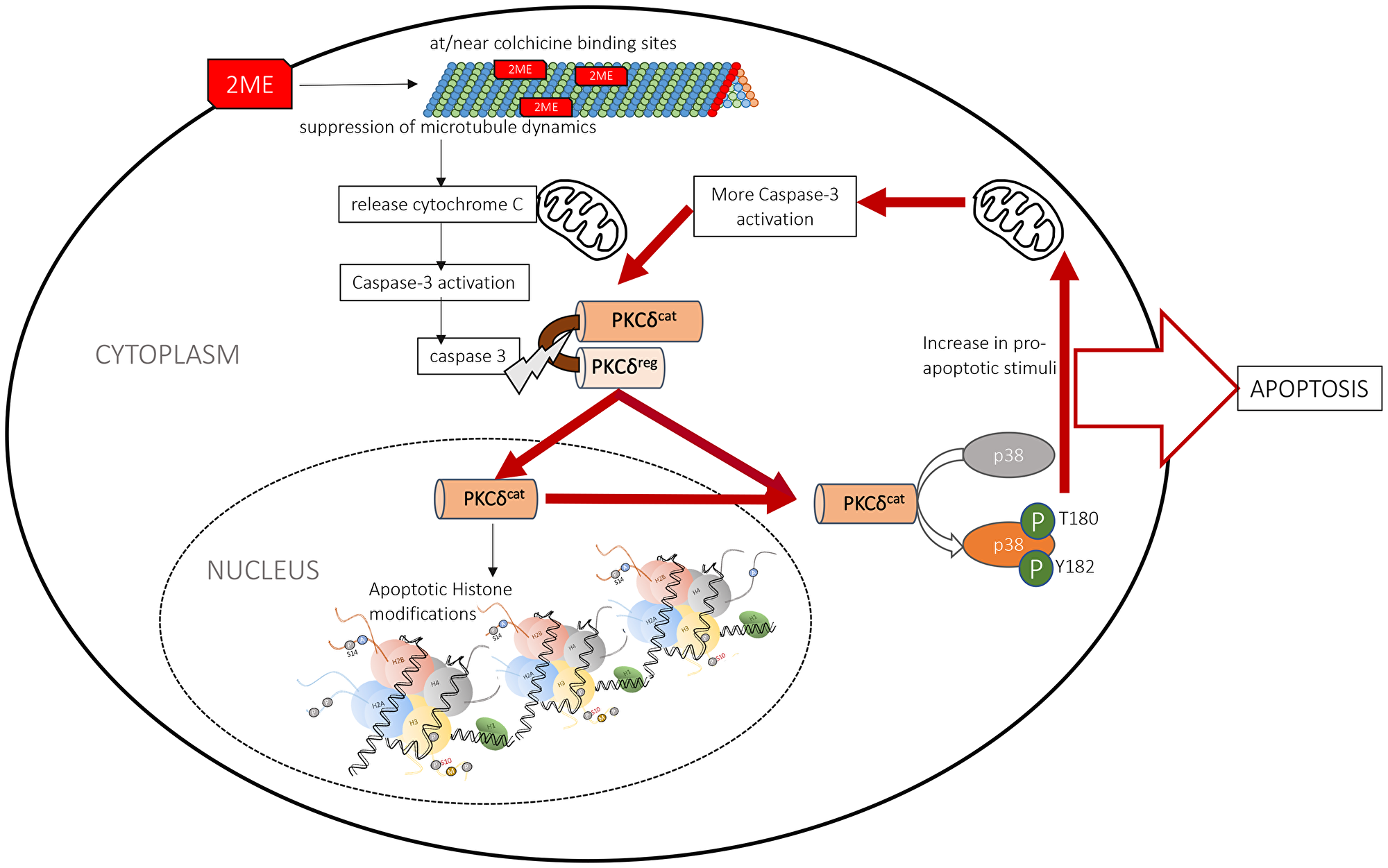
Figure 7: Schematic diagram of the molecular action of 2MeOE2. 2MeOE2 -mediated pro-apoptotic actions involve the catalytic activation of PKCδ by cellular caspases. The catalytic fragment of PKCδ is responsible for the apoptotic histone modifications in the nucleus and activates p38 MAPK pathway in the cytosol, which induces more activation of caspases that consequently cleave and activate more PKCδ. Therefore this cycle accelerates and amplifies the 2MeOE2-mediated apoptotic signal.
Materials and Methods
Materials
BG1 cells were obtained from the laboratory of Dr. Ken Korach at NIEHS and Hey C2 cell line was obtained from Dr. Jean Hurteau at Northshore University Health, Evanston Hospital. TOV112D (CRL11731) cell line was purchased from ATCC. HyClone DMEM culture media (with and without phenol red) from ThermoFisher (SH30604.02); 2-methoxyestradiol from Sigma-Aldrich (M6383); 100× HALT protease and phosphatase inhibitor cocktail from ThermoFisher (78440); DyLight™800 conjugated goat anti-rabbit IgG antibody (H&L) (35571) and DyLight™680 conjugated goat anti-rabbit IgG antibody (H&L) (35518) from Thermofisher. Alexa-594 donkey anti-rabbit secondary (133200) from Jackson Immuno Research. jet-PEI® purchased from Polyplus transfection (catalog no.101-10). Expression plasmids encoding PKCδWT (Addgene plasmid # 16386), PKCδDN (Addgene plasmid # 16389), and PKCδCAT (Addgene plasmid # 16388) were gifts from Bernard Weinstein [74], pSUPER-PKCδRNAi (Addgene plasmid # 10819) was a gift from Alex Toker [75] and pSUPER-scramble (Addgene plasmid # 118349) was a gift from Lea Sistonen [76]. E. Z. N. A.® plasmid maxi kit (D6922-02) was purchased from Omega Bio-Tek. SB203580 p38 MAPK inhibitor was purchased from Cayman Chemical (13067).
Cell culture and treatments
BG1, HeyC2 and TOV112D cells were cultured in DMEM (with phenol red) media supplemented with 10% fetal bovine serum and 7500 IU penicillin, 7500 μg streptomycin, incubated at 5% CO2 and 37°C. Cells were seeded with a density of 4 × 105 cells per well in 6-well tissue culture plates. Media was changed after 24 hours to DMEM (phenol red-free) supplemented with 10% charcoal stripped newborn calf serum and 0.75% of 10,000 μg/ml penicillin-streptomycin. 2MeOE2 (stock in DMSO) dilutions were prepared in phenol red-free DMEM before adding to the cells. Following a 24 h incubation, cells were photographed harvested and total proteins were extracted.
Protein isolation from cells
Cells were scraped off after adding 200 μl ice-cold PBS and collected in Eppendorf tubes. Tubes were centrifuged at 2000 g at 4°C for 3 min, supernatant discarded and the pellets were resuspended in 30 μl of protein lysis buffer (1× HALT protease and phosphatase inhibitor cocktail in 0.1% SDS/1xPBS). Following a short burst of sonication, protein quantities were estimated by BCA method and stored at −20°C.
Immunocytochemistry
Cells were cultured on cover glass (2 × 104 cells per cover glass) and treated after 24 h of initial seeding. Following treatment, cells were fixed with 1:1 methanol: acetone (v/v) for 20 mins. Samples were then blocked with 5% bovine serum albumin for 30 mins and incubated with primary antibodies (1:100 dilution in blocking solution) overnight at 4°C (Table 1). Following 1x PBS rinse, secondary antibodies (Alexa Fluor® 594 nm, red, 1:200 and Alexa Fluor® 488 nm, green, 1:200, diluted in blocking solution) were applied for a 1 h incubation at room temperature. Samples were then washed with 1× PBS and mounted with DAPI Fluoromount G (Southern Biotech). Slides were then visualized with a Leica DM5500Q microscope, images were captured with a Leica DFC365FX camera, taken from A4 (DAPI), Alexa 488 nm (green) and Alexa 594 nm (red) channels. Images from multiple channels were superimposed using the Leica Application Suite-Advanced Fluorescence version 2.6.0.7266 software.
Table 1: List of primary antibodies
| Target protein | Manufacturer | Raised in | Dilutions |
|---|---|---|---|
| Caspase-3 | Cell signaling technology, 9665S | Rabbit | 1:500 |
| Cleaved caspase-3 | Cell signaling technology, 9664S | Rabbit | 1:500 |
| p38 MAPK | Cell signaling technology, 9212S | Rabbit | 1:700 |
| Phospho p38 MAPK | Cell signaling technology, 4511S | Rabbit | 1:700 |
| Phospho histone H3 (ser10) | Cell signaling technology, 9701S | Rabbit | 1:1000 |
| Phospho histone H2B (ser14) | Cell signaling technology, 6959S | Rabbit | 1:1000 |
| γH2Ax | Novus Bio, NBP1-19255 | Mouse | 1:1000 |
| Protein kinase Cδ | Cell signaling technology, 2058S | Rabbit | 1:1000 |
| β-actin | Cell signaling technology, 3700S | Mouse | 1:1000 |
Bacterial culture and plasmid DNA preparation
DH5α cells transfected with respective plasmids were grown in standard LB broth at 37°C with 100 μg/ml ampicillin. Plasmid DNA was prepared from 200 ml. overnight grown bacterial culture with E.Z.N.A. plasmid maxi kit following manufacturer’s protocol. Extracted DNA was purified by re-precipitation with 0.1 volume of 3M Na-acetate (pH 5.2) and 0.7 volume of isopropanol at −20°C overnight. Final DNA pellet was resuspended in 1× TE buffer, concentration determined by Nanodrop 2000 (Thermo Scientific).
Transient transfection
BG1 cells were seeded in 6-well tissue culture plates (2 × 104 cells per well). Medium was replaced on the next day with DMEM (with phenol red) without antibiotics and transfection was performed with jet-PEI. Per well, 2 μg of plasmid DNA was added to 200 μl of Opti-MEM media; then 6 μl of jet-PEI (1 μg/μl stock) was added; the mastermix was vortexed every 5 mins for 20 mins and added to respective wells. Transfected cells were cultured for 24 h at 5% CO2 and 37°C. At this point, cells were either harvested or treated for further analysis.
Western blot analysis
Western blot was performed as described previously. 30 μg of total protein was resolved using an SDS-PAGE gel and transferred to a PVDF membrane. Membranes were blocked by Sea Block blocking buffer (Pierce) for an hour at RT, followed by overnight incubation at 4°C with the primary antibodies diluted in the blocking buffer (Table 1). Membranes were washed with 1×TBS with 0.01% Tween-20 followed by an hour incubation at room temperature with an anti-mouse Dylight 680 and anti-rabbit Dylight 800 secondary antibodies (1:2000 dilution in 1×TBST with 0.01% Tween-20). After washing the membranes with 1x TBST with 0.01% Tween-20, the membranes were imaged in Odyssey CLx imaging system (Li-COR Biosciences). All target proteins were normalized to β-actin expression.
Statistical analysis
Statistical analysis was performed by GraphPad Prism v5.0. Unpaired student’s t-test were performed for analysis of two groups. One-way or Two-way analysis of variance (ANOVA) was performed for multiple groups followed by Tukey’s range test. Statistically significant change was considered for a p value of < 0.05.
Abbreviations
EOC: Epithelial ovarian cancer; 2MeOE2: 2-methoxyestradiol; PKCδ: Protein kinase C delta; CYP: Cytochrome P450; MAPK: Mitogen-activated protein kinase; GFP: Green fluorescent protein; DMEM: Dulbecco’s modified Eagle’s medium; DMSO: Dimethyl sulfoxide; PBS: Phosphate buffered saline; DAPI: 4′,6-diamidino phenylindole; PEI: Polyethylenimine; SDS-PAGE: Sodium dodecyl sulphate polyacrylamide gel electrophoresis; PVDF: Polyvinylidene difluoride.
ACKNOWLEDGMENTS
We sincerely thank Dr. Brent Bany, Department of Physiology, Southern Illinois University School of Medicine, for his invaluable input in the development of this project. We also thank Ms. Kara Starkweather for her help with the cell culture studies and immunocytochemical staining.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
NIH R01AT005295.
References
1. Cramer DW, Elias KM. Perspectives on Ovarian Cancer From SEER: Today and Tomorrow. J Natl Cancer Inst. 2019; 111:5–6. https://doi.org/10.1093/jnci/djy074. [PubMed].
2. Eilati E, Bahr JM, Hales DB. Long term consumption of flaxseed enriched diet decreased ovarian cancer incidence and prostaglandin E(2)in hens. Gynecol Oncol. 2013; 130:620–628. https://doi.org/10.1016/j.ygyno.2013.05.018. [PubMed].
3. Ansenberger K, Richards C, Zhuge Y, Barua A, Bahr JM, Luborsky JL, Hales DB. Decreased severity of ovarian cancer and increased survival in hens fed a flaxseed-enriched diet for 1 year. Gynecol Oncol. 2010; 117:341–347. https://doi.org/10.1016/j.ygyno.2010.01.021. [PubMed].
4. Dikshit A, Gomes Filho MA, Eilati E, McGee S, Small C, Gao C, Klug T, Hales DB. Flaxseed reduces the pro-carcinogenic micro-environment in the ovaries of normal hens by altering the PG and oestrogen pathways in a dose-dependent manner. Br J Nutr. 2015; 113:1384–1395. https://doi.org/10.1017/S000711451500029X. [PubMed].
5. Nakagawa-Yagi Y, Ogane N, Inoki Y, Kitoh N. The endogenous estrogen metabolite 2-methoxyestradiol induces apoptotic neuronal cell death in vitro. Life Sci. 1996; 58:1461–1467. https://doi.org/10.1016/0024-3205(96)00116-6. [PubMed].
6. Klauber N, Parangi S, Flynn E, Hamel E, D’Amato RJ. Inhibition of angiogenesis and breast cancer in mice by the microtubule inhibitors 2-methoxyestradiol and taxol. Cancer Res. 1997; 57:81–86. [PubMed].
7. Fotsis T, Zhang Y, Pepper MS, Adlercreutz H, Montesano R, Nawroth PP, Schweigerer L. The endogenous oestrogen metabolite 2-methoxyoestradiol inhibits angiogenesis and suppresses tumour growth. Nature. 1994; 368:237–239. https://doi.org/10.1038/368237a0. [PubMed].
8. Zoubine MN, Weston AP, Johnson DC, Campbell DR, Banerjee SK. 2-methoxyestradiol-induced growth suppression and lethality in estrogen-responsive MCF-7 cells may be mediated by down regulation of p34cdc2 and cyclin B1 expression. Int J Oncol. 1999; 15:639–646. https://doi.org/10.3892/ijo.15.4.639. [PubMed].
9. Seegers JC, Aveling ML, Van Aswegen CH, Cross M, Koch F, Joubert WS. The cytotoxic effects of estradiol-17 beta, catecholestradiols and methoxyestradiols on dividing MCF-7 and HeLa cells. J Steroid Biochem. 1989; 32:797–809. https://doi.org/10.1016/0022-4731(89)90455-X. [PubMed].
10. Kuo KL, Lin WC, Ho IL, Chang HC, Lee PY, Chung YT, Hsieh JT, Pu YS, Shi CS, Huang KH. 2-methoxyestradiol induces mitotic arrest, apoptosis, and synergistic cytotoxicity with arsenic trioxide in human urothelial carcinoma cells. PLoS One. 2013; 8:e68703. https://doi.org/10.1371/journal.pone.0068703. [PubMed].
11. Gorska-Ponikowska M, Kuban-Jankowska A, Daca A, Nussberger S. 2-Methoxyestradiol Reverses the Pro-Carcinogenic Effect of L-Lactate in Osteosarcoma 143B Cells. Cancer Genomics Proteomics. 2017; 14:483–493. https://doi.org/10.21873/cgp.20058. [PubMed].
12. Massaro RR, Faiao-Flores F, Rebecca VW, Sandri S, Alves-Fernandes DK, Pennacchi PC, Smalley KSM, Maria-Engler SS. Inhibition of proliferation and invasion in 2D and 3D models by 2-methoxyestradiol in human melanoma cells. Pharmacol Res. 2017; 119:242–250. https://doi.org/10.1016/j.phrs.2017.02.013. [PubMed].
13. Xiong XX, Qiu XY, Hu DX, Chen XQ. Advances in Hypoxia-Mediated Mechanisms in Hepatocellular Carcinoma. Mol Pharmacol. 2017; 92:246–255. https://doi.org/10.1124/mol.116.107706. [PubMed].
14. Pal P, Hales K, Petrik J, Hales DB. Pro-apoptotic and anti-angiogenic actions of 2-methoxyestradiol and docosahexaenoic acid, the biologically derived active compounds from flaxseed diet, in preventing ovarian cancer. J Ovarian Res. 2019; 12:49. https://doi.org/10.1186/s13048-019-0523-3. [PubMed].
15. Dikshit A, Hales K, Hales DB. Whole flaxseed diet alters estrogen metabolism to promote 2-methoxtestradiol-induced apoptosis in hen ovarian cancer. J Nutr Biochem. 2017; 42:117–125. https://doi.org/10.1016/j.jnutbio.2017.01.002. [PubMed].
16. Cushman M, He HM, Katzenellenbogen JA, Lin CM, Hamel E. Synthesis, antitubulin and antimitotic activity, and cytotoxicity of analogs of 2-methoxyestradiol, an endogenous mammalian metabolite of estradiol that inhibits tubulin polymerization by binding to the colchicine binding site. J Med Chem. 1995; 38:2041–2049. https://doi.org/10.1021/jm00012a003. [PubMed].
17. Day JM, Newman SP, Comninos A, Solomon C, Purohit A, Leese MP, Potter BV, Reed MJ. The effects of 2-substituted oestrogen sulphamates on the growth of prostate and ovarian cancer cells. J Steroid Biochem Mol Biol. 2003; 84:317–325. https://doi.org/10.1016/S0960-0760(03)00045-1. [PubMed].
18. Dobos J, Timar J, Bocsi J, Burian Z, Nagy K, Barna G, Petak I, Ladanyi A. In vitro and in vivo antitumor effect of 2-methoxyestradiol on human melanoma. Int J Cancer. 2004; 112:771–776. https://doi.org/10.1002/ijc.20473. [PubMed].
19. Ghosh R, Ott AM, Seetharam D, Slaga TJ, Kumar AP. Cell cycle block and apoptosis induction in a human melanoma cell line following treatment with 2-methoxyoestradiol: therapeutic implications? Melanoma Res. 2003; 13:119–127. https://doi.org/10.1097/00008390-200304000-00003. [PubMed].
20. Mukhopadhyay T, Roth JA. Superinduction of wild type p53 protein after 2-methoxyestradiol treatment of Ad5p53-transduced cells induces tumor cell apoptosis. Oncogene. 1998; 17:241–246. https://doi.org/10.1038/sj.onc.1201909. [PubMed].
21. Mukhopadhyay T, Roth JA. Induction of apoptosis in human lung cancer cells after wild type p53 activation by methoxyestradiol. Oncogene. 1997; 14:379–384. https://doi.org/10.1038/sj.onc.1200835. [PubMed].
22. Kataoka M, Schumacher G, Cristiano RJ, Atkinson EN, Roth JA, Mukhopadhyay T. An agent that increases tumor suppressor transgene product coupled with systemic transgene delivery inhibits growth of metastatic lung cancer in vivo. Cancer Res. 1998; 58:4761–4765. [PubMed].
23. Maxwell SA, Roth JA, Mukhopadhyay T. Analysis of phosphorylated isoforms of the p53 tumor suppressor protein in human lung carcinoma cells undergoing apoptosis. Electrophoresis. 1996; 17:1772–1775. https://doi.org/10.1002/elps.1150171115. [PubMed].
24. Amorino GP, Freeman ML, Choy H. Enhancement of radiation effects in vitro by the estrogen metabolite 2-methoxyestradiol. Radiat Res. 2000; 153:384–391. https://doi.org/10.1667/0033-7587(2000)153[0384:EOREIV]2.0.CO;2. [PubMed].
25. Liu ZJ, Lee WJ, Zhu BT. Selective insensitivity of ZR-75-1 human breast cancer cells to 2-methoxyestradiol: evidence for type II 17beta-hydroxysteroid dehydrogenase as the underlying cause. Cancer Res. 2005; 65:5802–5811. https://doi.org/10.1158/0008-5472.CAN-04-3714. [PubMed].
26. Sutherland TE, Schuliga M, Harris T, Eckhardt BL, Anderson RL, Quan L, Stewart AG. 2-methoxyestradiol is an estrogen receptor agonist that supports tumor growth in murine xenograft models of breast cancer. Clin Cancer Res. 2005; 11:1722–1732. https://doi.org/10.1158/1078-0432.CCR-04-1789. [PubMed].
27. Han GZ, Liu ZJ, Shimoi K, Zhu BT. Synergism between the anticancer actions of 2-methoxyestradiol and microtubule-disrupting agents in human breast cancer. Cancer Res. 2005; 65:387–393. [PubMed].
28. Lewis JS, Thomas TJ, Pestell RG, Albanese C, Gallo MA, Thomas T. Differential effects of 16alpha-hydroxyestrone and 2-methoxyestradiol on cyclin D1 involving the transcription factor ATF-2 in MCF-7 breast cancer cells. J Mol Endocrinol. 2005; 34:91–105. https://doi.org/10.1677/jme.1.01599. [PubMed].
29. Qadan LR, Perez-Stable CM, Anderson C, D’Ippolito G, Herron A, Howard GA, Roos BA. 2-Methoxyestradiol induces G2/M arrest and apoptosis in prostate cancer. Biochem Biophys Res Commun. 2001; 285:1259–1266. https://doi.org/10.1006/bbrc.2001.5320. [PubMed].
30. Kumar AP, Garcia GE, Slaga TJ. 2-methoxyestradiol blocks cell-cycle progression at G(2)/M phase and inhibits growth of human prostate cancer cells. Mol Carcinog. 2001; 31:111–124. https://doi.org/10.1002/mc.1046. [PubMed].
31. Bu S, Blaukat A, Fu X, Heldin NE, Landstrom M. Mechanisms for 2-methoxyestradiol-induced apoptosis of prostate cancer cells. FEBS Lett. 2002; 531:141–151. https://doi.org/10.1016/S0014-5793(02)03478-6. [PubMed].
32. Kinuya S, Kawashima A, Yokoyama K, Kudo M, Kasahara Y, Watanabe N, Shuke N, Bunko H, Michigishi T, Tonami N. Anti-angiogenic therapy and radioimmunotherapy in colon cancer xenografts. Eur J Nucl Med. 2001; 28:1306–1312. https://doi.org/10.1007/s002590100585. [PubMed].
33. Carothers AM, Hughes SA, Ortega D, Bertagnolli MM. 2-Methoxyestradiol induces p53-associated apoptosis of colorectal cancer cells. Cancer Lett. 2002; 187:77–86. https://doi.org/10.1016/S0304-3835(02)00409-3. [PubMed].
34. Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003; 6:355–361. https://doi.org/10.1016/j.drup.2003.10.001. [PubMed].
35. Tinley TL, Leal RM, Randall-Hlubek DA, Cessac JW, Wilkens LR, Rao PN, Mooberry SL. Novel 2-methoxyestradiol analogues with antitumor activity. Cancer Res. 2003; 63:1538–1549. [PubMed].
36. Shimada K, Nakamura M, Ishida E, Kishi M, Konishi N. Roles of p38- and c-jun NH2-terminal kinase-mediated pathways in 2-methoxyestradiol-induced p53 induction and apoptosis. Carcinogenesis. 2003; 24:1067–1075. https://doi.org/10.1093/carcin/bgg058. [PubMed].
37. Basu A, Haldar S. Identification of a novel Bcl-xL phosphorylation site regulating the sensitivity of taxol- or 2-methoxyestradiol-induced apoptosis. FEBS Lett. 2003; 538:41–47. https://doi.org/10.1016/S0014-5793(03)00131-5. [PubMed].
38. Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, Willard MT, Zhong H, Simons JW, Giannakakou P. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003; 3:363–375. https://doi.org/10.1016/S1535-6108(03)00077-1. [PubMed].
39. Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011; 12:7–18. https://doi.org/10.1038/nrg2905. [PubMed].
40. Turner BM. Defining an epigenetic code. Nat Cell Biol. 2007; 9:2–6. https://doi.org/10.1038/ncb0107-2. [PubMed].
41. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000; 403:41–45. https://doi.org/10.1038/47412. [PubMed].
42. Jenuwein T, Allis CD. Translating the histone code. Science. 2001; 293:1074–1080. https://doi.org/10.1126/science.1063127. [PubMed].
43. Turner BM. Histone acetylation and an epigenetic code. BioEssays. 2000; 22:836–845. https://doi.org/10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [PubMed].
44. Th’ng JP. Histone modifications and apoptosis: cause or consequence? Biochem Cell Biol. 2001; 79:305–311. https://doi.org/10.1139/o01-031. [PubMed].
45. Kratzmeier M, Albig W, Hanecke K, Doenecke D. Rapid dephosphorylation of H1 histones after apoptosis induction. J Biol Chem. 2000; 275:30478–30486. https://doi.org/10.1074/jbc.M003956200. [PubMed].
46. Talasz H, Helliger W, Sarg B, Debbage PL, Puschendorf B, Lindner H. Hyperphosphorylation of histone H2A.X and dephosphorylation of histone H1 subtypes in the course of apoptosis. Cell Death Differ. 2002; 9:27–39. https://doi.org/10.1038/sj.cdd.4400925. [PubMed].
47. Fullgrabe J, Hajji N, Joseph B. Cracking the death code: apoptosis-related histone modifications. Cell Death Differ. 2010; 17:1238–1243. https://doi.org/10.1038/cdd.2010.58. [PubMed].
48. Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999; 274:25543–25549. https://doi.org/10.1074/jbc.274.36.25543. [PubMed].
49. Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, Sen S, Allis CD, Sassone-Corsi P. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002; 22:874–885. https://doi.org/10.1128/MCB.22.3.874-885.2002. [PubMed].
50. Park CH, Kim KT. Apoptotic phosphorylation of histone H3 on Ser-10 by protein kinase Cdelta. PLoS One. 2012; 7:e44307. https://doi.org/10.1371/journal.pone.0044307. [PubMed].
51. Thorne AW, Kmiciek D, Mitchelson K, Sautiere P, Crane-Robinson C. Patterns of histone acetylation. Eur J Biochem. 1990; 193:701–713. https://doi.org/10.1111/j.1432-1033.1990.tb19390.x. [PubMed].
52. Ajiro K, Scoltock AB, Smith LK, Ashasima M, Cidlowski JA. Reciprocal epigenetic modification of histone H2B occurs in chromatin during apoptosis in vitro and in vivo. Cell Death Differ. 2010; 17:984–993. https://doi.org/10.1038/cdd.2009.199. [PubMed].
53. Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, Beeser A, Etkin LD, Chernoff J, Earnshaw WC, Allis CD. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003; 113:507–517. https://doi.org/10.1016/S0092-8674(03)00355-6. [PubMed].
54. Ajiro K. Histone H2B phosphorylation in mammalian apoptotic cells. An association with DNA fragmentation. J Biol Chem. 2000; 275:439–443. https://doi.org/10.1074/jbc.275.1.439. [PubMed].
55. Hu Y, Liu Z, Yang SJ, Ye K. Acinus-provoked protein kinase C delta isoform activation is essential for apoptotic chromatin condensation. Cell Death Differ. 2007; 14:2035–2046. https://doi.org/10.1038/sj.cdd.4402214. [PubMed].
56. Duquesnes N, Lezoualc’h F, Crozatier B. PKC-delta and PKC-epsilon: foes of the same family or strangers? J Mol Cell Cardiol. 2011; 51:665–673. https://doi.org/10.1016/j.yjmcc.2011.07.013. [PubMed].
57. Rosse C, Linch M, Kermorgant S, Cameron AJ, Boeckeler K, Parker PJ. PKC and the control of localized signal dynamics. Nat Rev Mol Cell Biol. 2010; 11:103–112. https://doi.org/10.1038/nrm2847. [PubMed].
58. Sun X, Wu F, Datta R, Kharbanda S, Kufe D. Interaction between protein kinase C delta and the c-Abl tyrosine kinase in the cellular response to oxidative stress. J Biol Chem. 2000; 275:7470–7473. https://doi.org/10.1074/jbc.275.11.7470. [PubMed].
59. Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem. 2005; 280:28721–28730. https://doi.org/10.1074/jbc.M501092200. [PubMed].
60. Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996; 184:2399–2404. https://doi.org/10.1084/jem.184.6.2399. [PubMed].
61. Cho W. Membrane targeting by C1 and C2 domains. J Biol Chem. 2001; 276:32407–32410. https://doi.org/10.1074/jbc.R100007200. [PubMed].
62. Steinberg SF. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem J. 2004; 384:449–459. https://doi.org/10.1042/BJ20040704. [PubMed].
63. Hurley JH, Misra S. Signaling and subcellular targeting by membrane-binding domains. Annu Rev Biophys Biomol Struct. 2000; 29:49–79. https://doi.org/10.1146/annurev.biophys.29.1.49. [PubMed].
64. Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med. 2003; 7:341–350. https://doi.org/10.1111/j.1582-4934.2003.tb00237.x. [PubMed].
65. Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 1995; 14:6148–6156. https://doi.org/10.1002/j.1460-2075.1995.tb00305.x. [PubMed].
66. Yuan ZM, Utsugisawa T, Ishiko T, Nakada S, Huang Y, Kharbanda S, Weichselbaum R, Kufe D. Activation of protein kinase C delta by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene. 1998; 16:1643–1648. https://doi.org/10.1038/sj.onc.1201698. [PubMed].
67. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994; 265:808–811. https://doi.org/10.1126/science.7914033. [PubMed].
68. Wrobleski ST, Doweyko AM. Structural comparison of p38 inhibitor-protein complexes: a review of recent p38 inhibitors having unique binding interactions. Curr Top Med Chem. 2005; 5:1005–1016. https://doi.org/10.2174/1568026054985894. [PubMed].
69. Ryer EJ, Sakakibara K, Wang C, Sarkar D, Fisher PB, Faries PL, Kent KC, Liu B. Protein kinase C delta induces apoptosis of vascular smooth muscle cells through induction of the tumor suppressor p53 by both p38-dependent and p38-independent mechanisms. J Biol Chem. 2005; 280:35310–35317. https://doi.org/10.1074/jbc.M507187200. [PubMed].
70. Larroque-Cardoso P, Swiader A, Ingueneau C, Negre-Salvayre A, Elbaz M, Reyland ME, Salvayre R, Vindis C. Role of protein kinase C delta in ER stress and apoptosis induced by oxidized LDL in human vascular smooth muscle cells. Cell Death Dis. 2013; 4:e520. https://doi.org/10.1038/cddis.2013.47. [PubMed].
71. Kim DS, Kim JH, Lee GH, Kim HT, Lim JM, Chae SW, Chae HJ, Kim HR. p38 Mitogen-activated protein kinase is involved in endoplasmic reticulum stress-induced cell death and autophagy in human gingival fibroblasts. Biol Pharm Bull. 2010; 33:545–549. https://doi.org/10.1248/bpb.33.545. [PubMed].
72. Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J Biol Chem. 2003; 278:33753–33762. https://doi.org/10.1074/jbc.M303313200. [PubMed].
73. de Galarreta MR, Navarro A, Ansorena E, Garzon AG, Modol T, Lopez-Zabalza MJ, Martinez-Irujo JJ, Iraburu MJ. Unfolded protein response induced by Brefeldin A increases collagen type I levels in hepatic stellate cells through an IRE1alpha, p38 MAPK and Smad-dependent pathway. Biochim Biophys Acta. 2016; 1863:2115–2123. https://doi.org/10.1016/j.bbamcr.2016.05.002. [PubMed].
74. Soh JW, Weinstein IB. Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem. 2003; 278:34709–34716. https://doi.org/10.1074/jbc.M302016200. [PubMed].
75. Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol. 2004; 24:2614–2626. https://doi.org/10.1128/MCB.24.7.2614-2626.2004. [PubMed].
76. Ostling P, Bjork JK, Roos-Mattjus P, Mezger V, Sistonen L. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J Biol Chem. 2007; 282:7077–7086. https://doi.org/10.1074/jbc.M607556200. [PubMed].