
CURRENT THERAPEUTIC STRATEGIES FOR ADVANCED OVARIAN CANCER
Epithelial ovarian cancer (OVCA) originates from a layer of cells covering the surface of the ovaries or fallopian tubes and accounts for ~90% of the primary ovarian tumors [1]. Throughout the world, OVCA is the most lethal gynecologic cancer, with 46% survival five years after diagnosis [2]. NCI Seer data (https://seer.cancer.gov/) predict that approximately 21,750 American women will be diagnosed with OVCA in 2020, which will lead to the death of 13,940 American women. The diagnosis of OVCA is challenging due to the vague and non-specific symptoms at the initial stage. Thus, OVCA is often misdiagnosed as other common ailments. Moreover, due to the deficiency of early-stage OVCA screening strategies, the correct diagnosis of OVCA usually occurs at advanced stages, resulting in poor prognosis and low survival rate [3, 4]. For most low-grade OVCA confined in ovaries and pelvis, a debulking surgery is curative. For high-grade OVCA, standard therapy involves surgical debulking of the tumors followed by combination chemotherapies with carboplatin and paclitaxel [5]. Most patients initially respond favorably to this combined treatment and achieve remission [6]. However, in many cases, tumors will eventually recur, and recurrent tumors will become resistant to chemotherapies, which were effective for primary tumors. Therefore, angiogenesis inhibitors, PARP inhibitors, and immunotherapies are employed to treat recurrent OVCA [7, 8].
Angiogenesis inhibitors aim to inhibit the growth of new blood vessels in tumors by blocking the vascular endothelial growth factor (VEGF), VEGF receptors, or its downstream signaling pathway [9]. In most solid tumors, including OVCA, the uncontrolled tumor growth, combined with inadequate blood perfusion, leads to low tumor pO2, tumor hypoxia, and other tumor microenvironmental stresses [10–13]. Hypoxia triggers the HIF-mediated hypoxia gene expression program that leads to the invasion, migration, and metastasis of tumor cells [14–16]. Tumor hypoxia also induces abnormal angiogenesis, creates dysregulated blood vessel networks defective in drug delivery and contributes to chemo-resistance [14–16]. Therefore, angiogenesis inhibitors, such as bevacizumab, may normalize tumor blood vessels, mitigate tumor hypoxia, and restore response to chemotherapeutics [17].
Poly (ADP-ribose) polymerases (PARPs) are a family of proteins that catalyze the transfer of ADP-ribose to target proteins (poly ADP-ribosylation). PARPs mediate many biological processes, including the repair of single-strand break (SSBs) through base excision repair [18]. PARP inhibitors, such as Olaparib and Rucaparib, have emerged as effective treatments for a subset of OVCA bearing mutations in BRCA1 and BRCA2 [18]. Both BRCA1 and BRCA2 proteins promote homology-directed repair (HDR) of DNA double-strand break (DSB) [19]. Therefore, BRCA1 and BRCA2 mutations lead to defects in DNA DSB repair, rendering these BRCA1 and BRCA2 mutated cells rely on a PARP-mediated DNA repair pathway. Hence, they are highly sensitive to the death caused by PARP inhibitors [20, 21]. In contrast, healthy cells with intact BRCA1/BRCA2 are not susceptible to PARP inhibitors, creating the synthetic lethal relationship and significant therapeutic window [20, 21]. Olaparib was initially approved for maintenance for BRCA-mutated recurrent OVCA [18]. Recently, the FDA expanded the approval of Olaparib and bevacizumab as the front-line treatment for women with advanced ovarian cancer [22].
Immune checkpoint blockade is a powerful new therapeutic option for many cancers [23]. The most common immune checkpoint blockage refers to blocking immune inhibitory receptors (CTLA4, PD1 on T cells, or PDL1 on tumor cells and tumor-infiltrating immune cells) using antagonistic antibodies. Programmed death 1 (PD1) and its ligands PDL1 and PDL2 play a key role in dampening T cell responses in the tumor [24–26]. Blocking the PD1/PDL1 inhibitory axis allow the CD8+ CTL to attack tumor cells, leading to a sustained anti-tumor response. However, the clinical responses of advanced OVCA to immunotherapy are unsatisfactory, with response seen in only 10–25% patients [27–31]. Therefore, there are increasing interests in combining other novel therapeutic approaches with the immune checkpoint blockade to improve response rate and efficacy.
FERROPTOSIS–A NOVEL FORM OF REGULATED CELL DEATH WITH SIGNIFICANT THERAPEUTIC POTENTIAL
Even with all these advancements, clinical outcomes of advanced OVCA are still unsatisfactory [7]. Therefore, new therapeutic options are urgently needed. One new strategy to eliminate tumor cells is to identify and target their metabolic Achilles’ heel and specific nutrient preference [32]. Cystine deprivation of cancer cells with specific cellular origins and somatic mutations triggers ferroptosis, a novel form of regulated cell death characterized by lipid peroxidation [33, 34]. Ferroptosis was first uncovered during the investigation of the death mechanisms induced by erastin, an agent that was selected based on its ability to selectively eradicate RAS-mutated cancer cells [33]. Since then, significant progress has been made in understanding the biological processes and genetic determinants of ferroptosis, as summarized in some excellent reviews [35–38]. Here we review some key players relevant to the ferroptosis of OVCA.
GPX4 and FSP1 mediate two ferroptosis protection pathways
There are two known ferroptosis protection mechanisms mediated by glutathione peroxidase 4 (GPX4) and ferroptosis suppressor protein 1 (FSP1). Both proteins neutralize ROS and prevent lipid peroxidation. GPX4 is a phospholipid hydroperoxidase that protects cells against membrane lipid peroxidation using glutathione (GSH) as its cofactor. Therefore, ferroptosis can be triggered by either the depletion of GSH or direct inhibition of GPX4. RSL3 and several other ferroptosis-inducing agents (FINs) [39] induce ferroptosis by blocking the function of GPX4 downstream of the NADPH-GSH that supply the cofactors for GPX4.
Many ferroptosis-inducing agents work by the depletion of GSH or cysteine. For example, erastin is an xCT inhibitor that induces ferroptosis by preventing cystine import and depleting GSH. Similarly, the cystine deprivation also leads to GSH depletion and death in a subset of cystine-addicted cancer cells [40–42]. In contrast, enhanced GSH synthesis upon the activation of NRF2 by various mechanisms would protect cells from ferroptosis [43, 44].
Several pathways can also compensate for the cystine deprivation and rescue ferroptosis. Upon cystine deprivation or xCT inhibitors, the cysteine can be generated by the transsulfuration pathway to prevent ferroptosis. For example, a forward genetic screen revealed that the removal of cysteinyl-tRNA synthetase (CARS) protected ferroptosis [45]. This protection occurred through the induction of the transsulfuration pathway to replenish cysteine [45]. Furthermore, the addition of coenzyme A (CoA), from the de novo CoA synthesis pathway [46], also replenishes cysteine and rescued ferroptosis [47]. In addition, pharmacogenomic analyses identified NAPDH as a robust determinant of ferroptosis [48], probably by regenerating GSH. Consistently, we have found that MESH1, the metazoa homolog of SpoT, is the first cytosolic NADPH phosphatase [49] whose induction is responsible for the NAPDH depletion during ferroptosis [50].
FSP1 and Coenzyme Q10 (CoQ10) axis have been identified as a new ferroptosis protection mechanism [51, 52]. FSP1 is an NADH-dependent CoQ10 oxidoreductase that reduces CoQ10. When FSP1 is myristoylated, it moves to the plasma membrane to limit lipid peroxidation and suppress ferroptosis. Therefore, the removal of FSP1 also leads to lipid peroxidation, membrane damage, and ferroptosis.
Promotion of ferroptosis by NOXs and iron
During ferroptosis, the oxidative radicals are generated by NOXs (nicotinamide adenine dinucleotide phosphate (NADPH) oxidases), a family of oxidases that use NADPH as cofactors. Therefore, NOX inhibitors consistently inhibit ferroptosis. Interestingly, each member of NOXs expresses differently in a tissue-specific manner [53]. Therefore, the specific members of the NOXs mediate ferroptosis may vary in distinct cell and tissue types.
Iron metabolism and labile iron pools are also critical for ferroptosis [33]. “Ferroptosis” indicates that iron is indispensable. Iron is postulated to drive the Fenton reaction that amplifies the free oxidative radicals, generated by NOXs and other sources, to trigger ferroptosis [54]. Therefore, iron chelator blocks ferroptosis by limiting cellular iron levels. For example, enhanced ferroptosis susceptibility is noted in erythrocyte-ingested macrophages [55] and hepatocytes in patients with hemochromatosis [56].
Similarly, NRF2 activation also limits ferroptosis by inducing the transcription of Ferritin Heavy Chain 1 (FTH1, involved in iron storage) to reduce labile iron [57]. Chen et al. found that the serine/threonine kinase ATM involved in the DNA damage pathway also regulated ferroptosis. Inhibition of ATM by genetic and chemical means prevents ferroptosis by reducing cellular iron through the induction of both iron storage (FTH1, FTL – Ferritin Heavy, and Light Chain), and export (FPN1-Ferroportin) [58].
FERROPTOSIS SUSCEPTIBILITY OF OVARIAN CANCER CELLS AND RELEVANT GENETIC DETERMINANTS
Inducing ferroptosis has been shown to have potent anti-tumor potential for many tumor types [35–37]. However, relatively little is known about the determinants and therapeutic potential of ferroptosis in OVCA. Several recent studies have investigated the genetic determinants of ferroptosis and demonstrated the potential role in OVCA therapy. We summarize these studies and their findings in Figure 1. Torti group first described that the high-grade OVCA has a lower level of the iron exporter (ferroportin) and a higher level of the iron importer (transferrin receptor), resulting in the accumulation of intracellular labile iron. High intracellular labile iron enhances the invasion and metastasis of OVCA by inducing matrix metalloproteases and interleukin 6 [59]. Since the iron chelators can eradicate these OVCA, they termed such observations “iron addiction” [59]. As expected, such high intracellular iron also promotes the ferroptosis of these OVCA [59]. In the follow-up studies, the Torti group elucidated the role of stearoyl-CoA desaturase (SCD1) in ferroptosis. SCD1 catalyzes the rate-limiting step in the monounsaturated fatty acid synthesis. Inhibition of SCD1 depletes CoQ10, an endogenous membrane antioxidant used by FSP1 to protect cells from ferroptosis [60] (Figure 1).
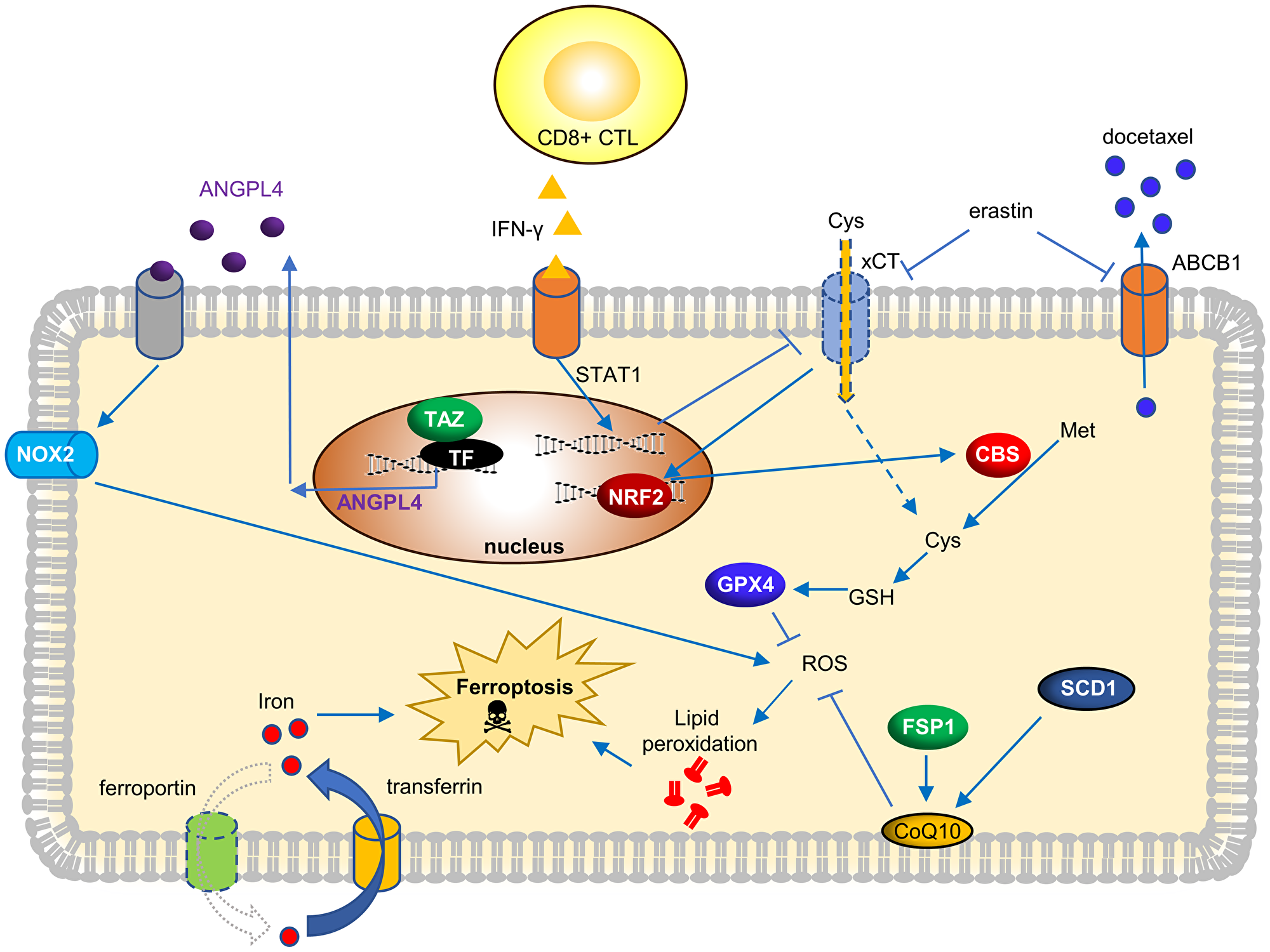
Figure 1: Current genetic determinants and its molecular mechanisms to trigger ferroptosis in OVCA.
The altered metabolism of cancer cells may render specific nutrients indispensable. Such nutrient addiction can be identified in a nutrigenetic screen by dropping off individual nutrient and analyze its transcriptome responses and cell viability [61]. Such a nutrigenetic approach revealed glutamine [62] and cystine addiction of renal cell carcinoma [40], breast cancer [41] and non-small cell lung cancers (NSCLC) [42]. When a similar nutrigenetic screen was used to analyze a panel of serous and clear-cell type OVCA cells, these OVCA cells were highly sensitive to cystine-deprived death [63]. Interestingly, cell density seemed to be a critical factor. As the Hippo effectors YAP/TAZ are the sensors of cell density [64], we identified that TAZ regulated ferroptosis in OVCA by affecting the expression of ANGPTL4 and NOX2 [63]. Therefore, ferroptosis-inducing agents may have significant therapeutic potential for OVCA with activated TAZ [63].
The immunotherapy depends on the ability of the CD8+ CTL to eradicate target tumor cells. Li’s group recently showed that CD8+ CTL and γ-interferon (IFN-γ) killed ID8, a murine OVCA cell, through the ferroptosis mechanism [65]. IFN-γ suppressed the cystine import by repressing the expression of SLC3A2 and SLC7A11, both subunits of the xCT that mediate the cystine import (Figure 1). Importantly, ferroptosis-inducing agents can enhance the efficacy of immunotherapy. This landmark study draws the unexpected connection between ferroptosis and immunotherapy.
One major clinical challenge of OVCA is the chemo-resistance of recurrent OVCA [7, 8]. One mechanism of docetaxel resistance is the overexpression ATP Binding Cassette Subfamily B Member 1 (ABCB1), which pumps out the docetaxel [66]. A recent study on the docetaxel-resistant OVCA [67] has shown that erastin mitigates the overexpression of ABCB1. Thus, when erastin was combined with docetaxel, erastin significantly increased the intracellular level of docetaxel. Therefore, erastin reverses the ABCB1-mediated chemo-resistance in OVCA, showing the therapeutic value of combining erastin and docetaxel [67] (Figure 1).
Another interesting paper focuses on ferroptosis resistance in OVCA induced by long-term erastin exposure [67]. Prolonged erastin eventually leads to ferroptosis resistance by activation of the transsulfuration pathway. Ferroptosis resistance is caused by the NRF2-mediated upregulation of cystathionine β-synthase (CBS) and transsulfuration. Therefore, genetically repression of NRF2 enhanced ferroptosis susceptibility of these ferroptosis-resistant cells, consistent with the anti-ferroptosis role of NRF2 [43, 44, 68].
THERAPEUTIC IMPLICATION AND FUTURE DIRECTION
These studies have provided compelling evidence that OVCA is highly sensitive to ferroptosis. However, much remained unknown about the genetic determinants of ferroptosis in OVCA to enable the selection of ovarian tumors, which may best respond to ferroptosis-inducing therapies. First, OVCA is an extremely heterogeneous disease based on the histopathology, somatic mutations, cellular origins, and various clinical parameters. For example, OVCA is classified into different histological subtypes, including serous, mucinous, endometrioid, clear cell, transitional cell, carcinosarcoma, mixed epithelial tumor, and undifferentiated carcinoma [69]. It is not clear whether these histological types guide the use of ferroptosis for OVCA. While most of the current studies of ferroptosis focus on the serous OVCA, future efforts will expand the investigations to other histological types. It is interesting to note that clear-cell type OVCA, characterized by the clear cytoplasm due to lipid and glycogen accumulation, is highly addicted to cystine and sensitive to the GPX4-removal ferroptosis [70]. However, the ferroptosis phenotypes of other OVCA remain largely unknown.
Another critical source of OVCA heterogeneity is the somatic mutations. TGCA analysis of OVCA has revealed the landscapes of somatic mutations [71]. As expected, TP53 was found to be mutated in > 90% of tumors. The next most common mutations are in BRCA1 or BRCA2 in 11–12% of OVCA. Other statistically recurrently mutated genes include RB1, NF1, FAT3, CSMD3, GABRA6, and CDK12 [71]. TP53 is one of the most important tumor suppressor genes, and different mutations of TP53 have been reported to either promote or limit ferroptosis in a highly context-dependent manner [72–75]. Therefore, it will be fascinating to elucidate further whether and how p53 mutations affect the ferroptosis in OVCA. Other than p53, retinoblastoma protein (RB1) also limited the sorafenib-induced ferroptosis [76]. Therefore, these somatic mutations may alter the metabolic states of the OVCA to enhance or limit ferroptosis sensitivity. However, much remains unknown about how to incorporate these histological subtypes and somatic mutations into reliable and robust predictors of ferroptosis sensitivity of OVCA.
Even many studies have identified genetic determinants of ferroptosis in other cancer types, it will still be important to validate and identify the specific determinants in OVCA. For example, the Hippo pathway has been shown to regulate ferroptosis in multiple tumor cell types [63, 77–80]. However, different Hippo effectors are employed in different cancer cells. In breast cancer and mesothelioma, YAP regulates ferroptosis in response to cellular contacts [79]. In contrast, in renal and ovarian cancer, TAZ is the relevant Hippo effector [63, 78] due to the predominant expression pattern. Similarly, while NOXs are essential for ferroptosis, distinct NOX members execute ferroptosis in different tumors. In OVCA, NOX2 was highly expressed to mediate ferroptosis [63]. In contrast, the ferroptosis of renal cell carcinoma is mediated by renal-specific NOX4 [78]. Therefore, Identifying the particular genetic determinants and relevant mediators of ferroptosis in OVCA may help to predict the response to ferroptosis-inducing therapies and potential resistant mechanisms.
Employing ferroptosis in combination therapeutics may have the opportunities to enhance the efficacy of existing therapeutic approaches. Ferroptosis is found to enhance the efficacy of immunotherapies [65], chemotherapies [81], and ionization radiations [82–84]. DNA damage and ATM/ATR activation have been found to promote ferroptosis [58]. PARP inhibitors may also trigger DNA damage, ATM/ATR activation [85], thus sensitizing OVCA to ferroptosis. Therefore, future efforts on optimizing the best strategies combining ferroptosis with standard cancer therapeutics would greatly improve outcomes and survival of patients with advanced OVCA.
While xCT inhibitors and cystine deprivation are established means to induce ferroptosis in vitro, it is not clear how best to induce ferroptosis in vivo for therapeutic purposes. Recently, imidazole ketone erastin (IKE) has been developed for in vivo application because of its potency, solubulity and metabolic stability [86]. Another promising agent with significant translational potential is the engineered human cyst(e)inase modified from CBS [87]. Cyst(e)inase suppresses tumor growth in multiple syngeneic and xenograft tumor models without apparent weight loss or other adverse effects. Cyst(e)inase can also synergize with immunotherapy [65] and is effective in pancreatic cancers [47]. Therefore, these reagents will be further optimized for the future clinical application of triggering ferroptosis to improve the outcomes of women with advanced OVCA. Studies have demonstrated a different angle on targeting anti-ferroptosis components, GPX4, or FSP1. However, it remains unknown whether the inhibitors of GPX4 or FSP1 can safely induce ferroptosis in vivo without severe side effects. The genetic removal of GPX4 leads to acute renal injuries [88] and hepatocyte death that can be preventable by vitamin E [89]. Therefore, we would not be surprised if GPX4 inhibitors have significant liver and renal toxicities. In contrast, the genetic removal of FSP1 in mice results in modest phenotypes [90, 91]. Therefore, targeting FSP1 may have fewer side effects and better tolerated than GPX4 inhibitors.
We expect that ferroptosis will emerge as a promising therapy to enhance the efficacy of immunotherapy, chemotherapeutics, and PARP inhibitors for advanced OVCA. However, much work remains to be accomplished toward that goal. Especially, the identification of robust predictive biomarkers of ferroptosis sensitivity to select tumors that are most likely to respond. Additionally, it is critical to identify the best means of inducing in vivo ferroptosis as well as optimize the combination strategies. In the long-term, we expect that the similar targeting of the altered metabolisms in OVCA may present an entirely new avenue of therapeutic opportunity for OVCA, which can be incorporated with current treatments.
ACKNOWLEDGMENTS
We are grateful for the critical feedback and technical support from the members of Chi lab. We acknowledge the financial support in part by DOD grants (W81XWH-17-1-0143, W81XWH-15-1-0486, W81XWH-19-1-0842), Emerson Collective, NIH grants (GM124062, 1R01NS111588-01A1), the Duke Bridge Fund and Duke Cancer Institute (DCI) pilot fund.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
References
1. Gadducci A, Guarneri V, Peccatori FA, Ronzino G, Scandurra G, Zamagni C, Zola P, Salutari V. Current strategies for the targeted treatment of high-grade serous epithelial ovarian cancer and relevance of BRCA mutational status. J Ovarian Res. 2019; 12:9. https://doi.org/10.1186/s13048-019-0484-6. [PubMed].
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30. https://doi.org/10.3322/caac.21332. [PubMed].
3. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004; 351:2519–29. https://doi.org/10.1056/NEJMra041842. [PubMed].
4. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008; 58:71–96. https://doi.org/10.3322/CA.2007.0010. [PubMed].
5. Aletti GD, Gallenberg MM, Cliby WA, Jatoi A, Hartmann LC. Current management strategies for ovarian cancer. Mayo Clin Proc. 2007; 82:751–70. https://doi.org/10.4065/82.6.751. [PubMed].
6. Ozols RF. Systemic therapy for ovarian cancer: current status and new treatments. Semin Oncol. 2006; 33:S3–11. https://doi.org/10.1053/j.seminoncol.2006.03.011. [PubMed].
7. Lheureux S, Gourley C, Vergote I, Oza AM. Epithelial ovarian cancer. The Lancet. 2019; 393:1240–53. https://doi.org/10.1016/S0140-6736(18)32552-2. [PubMed].
8. Cortez AJ, Tudrej P, Kujawa KA, Lisowska KM. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol. 2018; 81:17–38. https://doi.org/10.1007/s00280-017-3501-8. [PubMed].
9. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014; 26:605–22. https://doi.org/10.1016/j.ccell.2014.10.006. [PubMed].
10. Chen JL, Lucas JE, Schroeder T, Mori S, Wu J, Nevins J, Dewhirst M, West M, Chi JT. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008; 4:e1000293. https://doi.org/10.1371/journal.pgen.1000293. [PubMed].
11. Chen JL, Merl D, Peterson CW, Wu J, Liu PY, Yin H, Muoio DM, Ayer DE, West M, Chi JT. Lactic acidosis triggers starvation response with paradoxical induction of TXNIP through MondoA. PLoS Genet. 2010; 6:e1001093. https://doi.org/10.1371/journal.pgen.1001093. [PubMed].
12. Gatza ML, Kung HN, Blackwell KL, Dewhirst MW, Marks JR, Chi JT. Analysis of tumor environmental response and oncogenic pathway activation identifies distinct basal and luminal features in HER2-related breast tumor subtypes. Breast Cancer Res. 2011; 13:R62. https://doi.org/10.1186/bcr2899. [PubMed].
13. Lucas JE, Kung HN, Chi JT. Latent factor analysis to discover pathway-associated putative segmental aneuploidies in human cancers. PLoS Comput Biol. 2010; 6:e1000920. https://doi.org/10.1371/journal.pcbi.1000920. [PubMed].
14. Keenan MM, Liu B, Tang X, Wu J, Cyr D, Stevens RD, Ilkayeva O, Huang Z, Tollini LA, Murphy SK, Lucas J, Muoio DM, Kim SY, et al. ACLY and ACC1 Regulate Hypoxia-Induced Apoptosis by Modulating ETV4 via alpha-ketoglutarate. PLoS Genet. 2015; 11:e1005599. https://doi.org/10.1371/journal.pgen.1005599. [PubMed].
15. Chi JT, Wang Z, Nuyten DS, Rodriguez EH, Schaner ME, Salim A, Wang Y, Kristensen GB, Helland A, Borresen-Dale AL, Giaccia A, Longaker MT, Hastie T, et al. Gene expression programs in response to hypoxia: cell type specificity and prognostic significance in human cancers. PLoS Med. 2006; 3:e47. https://doi.org/10.1371/journal.pmed.0030047. [PubMed].
16. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002; 2:38–47. https://doi.org/10.1038/nrc704. [PubMed].
17. Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol Oncol. 2011; 121:230–8. https://doi.org/10.1016/j.ygyno.2010.11.035. [PubMed].
18. Slade D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020; 34:360–94. https://doi.org/10.1101/gad.334516.119. [PubMed].
19. Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol. 2018; 2:313–36. https://doi.org/10.1146/annurev-cancerbio-030617-050502. [PubMed].
20. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–21. https://doi.org/10.1038/nature03445. [PubMed].
21. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–7. https://doi.org/10.1038/nature03443. [PubMed].
22. Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, Fujiwara K, Vergote I, Colombo N, Mäenpää J, Selle F, Sehouli J, Lorusso D, et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N Engl J Med. 2019; 381:2416–28. https://doi.org/10.1056/NEJMoa1911361. [PubMed].
23. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015; 348:56–61. https://doi.org/10.1126/science.aaa8172. [PubMed].
24. Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015; 112:1421–7. https://doi.org/10.1038/bjc.2015.124. [PubMed].
25. Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015; 23:32–8. https://doi.org/10.1016/j.coph.2015.05.011. [PubMed].
26. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015; 27:450–61. https://doi.org/10.1016/j.ccell.2015.03.001. [PubMed].
27. Normann MC, Türzer M, Diep LM, Oldenburg J, Gajdzik B, Solheim O, Rud E. Early experiences with PD-1 inhibitor treatment of platinum resistant epithelial ovarian cancer. J Gynecol Oncol. 2019; 30:e56. https://doi.org/10.3802/jgo.2019.30.e56. [PubMed].
28. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, Kanai M, Mori Y, Matsumoto S, Chikuma S, Matsumura N, Abiko K, Baba T, et al. Safety and Antitumor Activity of Anti–PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015; 33:4015–22. https://doi.org/10.1200/jco.2015.62.3397. [PubMed].
29. Infante JR, Braiteh F, Emens LA, Balmanoukian AS, Oaknin A, Wang Y, Liu B, Molinero L, Fasso M, O’Hear C, Gordon M. Safety, clinical activity and biomarkers of atezolizumab (atezo) in advanced ovarian cancer (OC). Annals of Oncology. 2016; 27. https://doi.org/10.1093/annonc/mdw374.18.
30. Varga A, Piha-Paul SA, Ott PA, Mehnert JM, Berton-Rigaud D, Johnson EA, Cheng JD, Yuan S, Rubin EH, Matei DE. Antitumor activity and safety of pembrolizumab in patients (pts) with PD-L1 positive advanced ovarian cancer: Interim results from a phase Ib study. Journal of Clinical Oncology. 2015; 33:5510. https://doi.org/10.1200/jco.2015.33.15_suppl.5510.
31. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Matsumura N, Abiko K, Baba T, Yamaguchi K, Ueda A, Kanai M, Mori Y, Matsumoto S, et al. Efficacy and safety of anti-PD-1 antibody (Nivolumab: BMS-936558, ONO-4538) in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2015; 33:4015–22. https://doi.org/10.1200/jco.2015.62.3397. [PubMed].
32. Keenan MM, Chi JT. Alternative fuels for cancer cells. Cancer J. 2015; 21:49–55. https://doi.org/10.1097/PPO.0000000000000104. [PubMed].
33. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012; 149:1060–72. https://doi.org/10.1016/j.cell.2012.03.042. [PubMed].
34. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol. 2008; 15:234–45. https://doi.org/10.1016/j.chembiol.2008.02.010. [PubMed].
35. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017; 171:273–85. https://doi.org/10.1016/j.cell.2017.09.021. [PubMed].
36. Dixon SJ, Stockwell BR. The Hallmarks of Ferroptosis. Annual Review of Cancer Biology. 2019; 3:35–54. https://doi.org/10.1146/annurev-cancerbio-030518-055844.
37. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016; 23:369–79. https://doi.org/10.1038/cdd.2015.158. [PubMed].
38. Sun T, Chi JT. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes & Diseases. 2020. [Epub ahead of print]. https://doi.org/10.1016/j.gendis.2020.05.004.
39. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014; 156:317–31. https://doi.org/10.1016/j.cell.2013.12.010. [PubMed].
40. Tang X, Wu J, Ding CK, Lu M, Keenan MM, Lin CC, Lin CA, Wang CC, George D, Hsu DS, Chi JT. Cystine Deprivation Triggers Programmed Necrosis in VHL-Deficient Renal Cell Carcinomas. Cancer Res. 2016; 76:1892–903. https://doi.org/10.1158/0008-5472.CAN-15-2328. [PubMed].
41. Tang X, Ding CK, Wu J, Sjol J, Wardell S, Spasojevic I, George D, McDonnell DP, Hsu DS, Chang JT, Chi JT. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene. 2017; 36:4379. https://doi.org/10.1038/onc.2017.192. [PubMed].
42. Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, Triplett K, Roy R, Pardo OE, Seckl MJ, Rowlinson SW, Stone E, Lamb RF. Oncogene-Selective Sensitivity to Synchronous Cell Death following Modulation of the Amino Acid Nutrient Cystine. Cell Reports. 2017; 18:2547–56. https://doi.org/10.1016/j.celrep.2017.02.054. [PubMed].
43. Chen PH, Chi JT, Boyce M. KEAP1 has a sweet spot: A new connection between intracellular glycosylation and redox stress signaling in cancer cells. Mol Cell Oncol. 2017; 4:e1361501. https://doi.org/10.1080/23723556.2017.1361501. [PubMed].
44. Chen PH, Smith TJ, Wu J, Siesser PF, Bisnett BJ, Khan F, Hogue M, Soderblom E, Tang F, Marks JR, Major MB, Swarts BM, Boyce M, et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017; 36:2233–50. https://doi.org/10.15252/embj.201696113. [PubMed].
45. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016; 23:270–8. https://doi.org/10.1038/cdd.2015.93. [PubMed].
46. Lin CC, Kitagawa M, Tang X, Hou MH, Wu J, Qu DC, Srinivas V, Liu X, Thompson JW, Mathey-Prevot B, Yao TP, Lee SH, Chi JT. CoA synthase regulates mitotic fidelity via CBP-mediated acetylation. Nat Commun. 2018; 9:1039. https://doi.org/10.1038/s41467-018-03422-6. [PubMed].
47. Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, Decker AR, Sastra SA, Palermo CF, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020; 368:85–9. https://doi.org/10.1126/science.aaw9872. [PubMed].
48. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol. 2016; 23:225–35. https://doi.org/10.1016/j.chembiol.2015.11.016. [PubMed].
49. Ding CK, Rose J, Wu J, Sun T, Chen KY, Chen PH, Xu E, Tian S, Akinwuntan J, Guan Z, Zhou P, Chi JT. Mammalian stringent-like response mediated by the cytosolic NADPH phosphatase MESH1. bioRxiv. 2018; 325266. https://doi.org/10.1101/325266.
50. Ding CK, Rose J, Sun T, Wu J, Chen PH, Lin CC, Yang WH, Chen KY, Lee H, Xu E, Tian S, Akinwuntan J, Zhao J, et al. MESH1 is a cytosolic NADPH phosphatase that regulates ferroptosis. Nat Metab. 2020; 2:270–277. https://doi.org/10.1038/s42255-020-0181-1. [PubMed].
51. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH, Mourão A, Buday K, Sato M, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019; 575:693–8. https://doi.org/10.1038/s41586-019-1707-0. [PubMed].
52. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019; 575:688–92. https://doi.org/10.1038/s41586-019-1705-2. [PubMed].
53. Panday A, Sahoo MK, Osorio D, Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015; 12:5–23. https://doi.org/10.1038/cmi.2014.89. [PubMed].
54. Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018; 16:e2006203. https://doi.org/10.1371/journal.pbio.2006203. [PubMed].
55. Youssef LA, Rebbaa A, Pampou S, Weisberg SP, Stockwell BR, Hod EA, Spitalnik SL. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood. 2018; 131:2581–93. https://doi.org/10.1182/blood-2017-12-822619. [PubMed].
56. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, Zhang Z, Li Y, Wang X, Zhang J, Li G, Yang L, Liu W, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017; 66:449–65. https://doi.org/10.1002/hep.29117. [PubMed].
57. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology. 2016; 63:173–84. https://doi.org/10.1002/hep.28251. [PubMed].
58. Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, Xu Y, Yang WH, Mathey-Prevot B, Chi JT. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020; 27:1008–1022. https://doi.org/10.1038/s41418-019-0393-7. [PubMed].
59. Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, Xian W, McKeon F, Lynch M, Crum CP, Hegde P, Brewer M, Wang X, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017; 36:4089–99. https://doi.org/10.1038/onc.2017.11. [PubMed].
60. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM, Torti SV. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019; 79:5355–66. https://doi.org/10.1158/0008-5472.Can-19-0369. [PubMed].
61. Tang X, Keenan MM, Wu J, Lin CA, Dubois L, Thompson JW, Freedland SJ, Murphy SK, Chi JT. Comprehensive profiling of amino acid response uncovers unique methionine-deprived response dependent on intact creatine biosynthesis. PLoS Genet. 2015; 11:e1005158. https://doi.org/10.1371/journal.pgen.1005158. [PubMed].
62. Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011; 7:e1002229. https://doi.org/10.1371/journal.pgen.1002229. [PubMed].
63. Yang WH, Huang Z, Wu J, Ding CK, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. 2020; 18:79–90. https://doi.org/10.1158/1541-7786.Mcr-19-0691. [PubMed].
64. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016; 29:783–803. https://doi.org/10.1016/j.ccell.2016.05.005. [PubMed].
65. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019; 569:270–4. https://doi.org/10.1038/s41586-019-1170-y. [PubMed].
66. Borst P, Evers R, Kool M, Wijnholds J. The multidrug resistance protein family. Biochim Biophys Acta. 1999; 1461:347–57. https://doi.org/10.1016/s0005-2736(99)00167-4. [PubMed].
67. Liu N, Lin X, Huang C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br J Cancer. 2020; 122:279–92. https://doi.org/10.1038/s41416-019-0660-x. [PubMed].
68. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem Biol. 2020; 27:436–47. https://doi.org/10.1016/j.chembiol.2020.03.011. [PubMed].
69. Kaku T, Ogawa S, Kawano Y, Ohishi Y, Kobayashi H, Hirakawa T, Nakano H. Histological classification of ovarian cancer. Med Electron Microsc. 2003; 36:9–17. https://doi.org/10.1007/s007950300002. [PubMed].
70. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng YY, Deasy R, Kost-Alimova M, Dančík V, Leshchiner ES, Viswanathan VS, Signoretti S, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nature Communications. 2019; 10:1617. https://doi.org/10.1038/s41467-019-09277-9. [PubMed].
71. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166. [PubMed].
72. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, Gu W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019; 21:579–91. https://doi.org/10.1038/s41556-019-0305-6. [PubMed].
73. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, Lotze MT, Zeh HJ 3rd, Kang R, et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017; 20:1692–704. https://doi.org/10.1016/j.celrep.2017.07.055. [PubMed].
74. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015; 520:57–62. https://doi.org/10.1038/nature14344. [PubMed].
75. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, Dixon SJ. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018; 22:569–75. https://doi.org/10.1016/j.celrep.2017.12.077. [PubMed].
76. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, François C, Chatelain D, Debuysscher V, Barbare JC, Chauffert B, Galmiche A. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015; 356:971–7. https://doi.org/10.1016/j.canlet.2014.11.014. [PubMed].
77. Yang WH, Chi JT. Hippo pathway effectors YAP/TAZ as novel determinants of ferroptosis. Mol Cell Oncol. 2019:1699375. https://doi.org/10.1080/23723556.2019.1699375. [PubMed].
78. Yang WH, Ding CC, Sun T, Hsu DS, Chi JT. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep. 2019; 28:2501–8.e4. https://doi.org/10.1016/j.celrep.2019.07.107. [PubMed].
79. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, Jiang X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019; 572:402–406. https://doi.org/10.1038/s41586-019-1426-6. [PubMed].
80. Lin CC, Mabe NW, Lin YT, Yang WH, Tang X, Hong L, Sun T, Force J, Marks JR, Yao TP, Alvarez JV, Chi JT. RIPK3 upregulation confers robust proliferation and collateral cystine-dependence on breast cancer recurrence. Cell Death Differ. 2020; 27:2234–47. https://doi.org/10.1038/s41418-020-0499-y. [PubMed].
81. Zhou HH, Chen X, Cai LY, Nan XW, Chen JH, Chen XX, Yang Y, Xing ZH, Wei MN, Li Y, Wang ST, Liu K, Shi Z, et al. Erastin Reverses ABCB1-Mediated Docetaxel Resistance in Ovarian Cancer. Front Oncol. 2019; 9:1398. https://doi.org/10.3389/fonc.2019.01398. [PubMed].
82. Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, Dovas A, Higgins DM, Tan H, Zhang Y, Buonanno M, Wang TJC, Hei TK, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol. 2020; 15:469–84. https://doi.org/10.1021/acschembio.9b00939. [PubMed].
83. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, Ajani JA, Xiao Q, Liao Z, Wang H, Gan B. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020; 30:146–62. https://doi.org/10.1038/s41422-019-0263-3. [PubMed].
84. Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, Liao P, Zhou J, Zhang Q, Dow A, Saripalli AL, Kryczek I, Wei S, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019; 9:1673–1685. https://doi.org/10.1158/2159-8290.Cd-19-0338. [PubMed].
85. Lloyd RL, Wijnhoven PWG, Ramos-Montoya A, Wilson Z, Illuzzi G, Falenta K, Jones GN, James N, Chabbert CD, Stott J, Dean E, Lau A, Young LA. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene. 2020; 39:4869–83. https://doi.org/10.1038/s41388-020-1328-y. [PubMed].
86. Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, Stockwell BR. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol. 2019; 26:623–33.e9. https://doi.org/10.1016/j.chembiol.2019.01.008. [PubMed].
87. Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, Triplett K, Lamb C, Alters SE, Rowlinson S, Zhang YJ, Keating MJ, Huang P, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017; 23:120–7. https://doi.org/10.1038/nm.4232. [PubMed].
88. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014; 16:1180–91. https://doi.org/10.1038/ncb3064. [PubMed].
89. Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL, Conrad M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016; 9:22–31. https://doi.org/10.1016/j.redox.2016.05.003. [PubMed].
90. Mei J, Webb S, Zhang B, Shu HB. The p53-inducible apoptotic protein AMID is not required for normal development and tumor suppression. Oncogene. 2006; 25:849–56. https://doi.org/10.1038/sj.onc.1209121. [PubMed].
91. Nguyen HP, Yi D, Lin F, Viscarra JA, Tabuchi C, Ngo K, Shin G, Lee AY, Wang Y, Sul HS. Aifm2, a NADH Oxidase, Supports Robust Glycolysis and Is Required for Cold- and Diet-Induced Thermogenesis. Mol Cell. 2020; 77:600–17.e4. https://doi.org/10.1016/j.molcel.2019.12.002. [PubMed].