
Introduction
Basal cell carcinoma (BCC) is the most common skin malignancy, affecting more than 10 million new patients annually worldwide and with an incidence that increases by approximately 1% each year [1–3]. Most BCCs are treated with surgery, with a favorable prognosis [4]. In cases of locally advanced BCC (laBCC) where surgery is contraindicated, inhibition of the Hedgehog pathway is one of the few approved and recommended treatment options [4, 5].
Most BCCs exhibit constitutive activation of the Hedgehog pathway due to mutations in pathway members, most often Patched 1 (PTCH1) and Smoothened (SMO) [6–8]. Expression of the transcription factor Glioma-associated oncogene homolog 1 (GLI1) is a biomarker for Hedgehog pathway activation [9].
Sonidegib, a Hedgehog pathway inhibitor (HHI), blocks pathway signaling by selective inhibition of SMO [10]. It is approved in the US, EU, Switzerland, and Australia for the treatment of adult patients with laBCC not amenable to curative surgery or radiotherapy [11–14]. In Switzerland and Australia, sonidegib is also approved for the treatment of metastatic BCC (mBCC) [13, 14].
The efficacy and safety of 2 doses of sonidegib (200 and 800 mg once daily [QD]) were assessed through 42 months of treatment in patients with advanced BCC in the Basal cell carcinoma Outcomes with LDE225 (sonidegib) Treatment (BOLT) study (NCT01327053) [15–18]. The primary analysis at 6 months showed objective response rate (ORR) (95% confidence interval [CI]) of 43% (28%–59%) and 38% (28%–48%) in laBCC and 15% (2%–45%) and 17% (5%–39%) in mBCC for the 200 and 800 mg QD doses, respectively [18]. The final analysis at 42 months was the longest clinical trial follow-up to date with an HHI and reported ORR (95% CI) of 56% (43%–68%) and 46% (37%–55%) in laBCC and 8% (< 1%–36%) and 17% (5%–39%) in mBCC for 200 and 800 mg QD, respectively [15]. At 42 months, adverse events (AEs) with the approved 200 mg QD dose were mostly Grade 1 or 2, manageable, and reversible with dose interruptions [15]. While some results regarding GLI1 expression associations with clinical outcomes were reported in the BOLT primary analysis [18], here we report the complete secondary biomarker analysis of Hedgehog pathway inhibition with sonidegib from the BOLT study.
Results
Patient disposition, demographics, and clinical characteristics
Overall, 230 patients were enrolled in the study, of whom 79 and 151 were randomized to sonidegib 200 and 800 mg QD, respectively (Supplementary Figure 1). At the time of data cutoff for biomarker analysis (6 months), 39 (49.4%) patients in the 200 mg group and 46 (30.5%) patients in the 800 mg group were still receiving treatment. The biomarker population included 67 and 83 patients randomized to sonidegib 200 and 800 mg QD, respectively. AEs were the most common reason for discontinuation, reported for 16 (20.3%) and 48 (31.8%) discontinuing patients in the 200 and 800 mg group, respectively.
Patient demographics and clinical characteristics were similar between the biomarker and intent-to-treat (ITT) populations (Supplementary Table 1). The biomarker population was 62.7% and 65.1% male with mean (standard deviation [SD]) age of 65.6 (15.6) and 64.0 (14.9) years for the 200 and 800 mg groups, respectively. The ITT population was 60.8% and 63.6% male with mean (SD) age of 65.6 (15.7) and 63.6 (14.6) years for the 200 and 800 mg groups, respectively. Patients with laBCC comprised 91.0% and 91.6% of the biomarker population, and 83.5% and 84.8% of the ITT population for the 200 and 800 mg groups, respectively.
Reduction of GLI1 expression levels
Median (95% CI) GLI1 expression at baseline was −2.64 (−3.22, −2.27) and −3.04 (−3.21, −2.56) for 200 and 800 mg groups, respectively. Longitudinal analyses showed substantial reductions in GLI1 expression from baseline. Expression was reduced by a median (95% CI) of 87.4% (77.0%–96.1%) and 96.2% (94.1%–98.4%) at week 9, 92.7% (78.4%–96.8%) and 95.8% (91.8%–98.2%) at week 17, and 93.0% (33.1%–97.6%) and 97.1% (87.8%–99.5%) at the end of treatment (EOT) for the 200 and 800 mg groups, respectively (Supplementary Figure 2).
When patients in the 2 dose groups were stratified by the type of BCC, median (95% CI) reduction in GLI1 expression at week 9 was 87.4% (55.6%–96.9%) and 94.2% (87.8%–98.4%) for patients with aggressive laBCC, 86.7% (80.8%–96.1%) and 97.2% (95.0%–98.9%) for patients with nonaggressive laBCC, and 98.2% (85.1%–98.5%) and 99.2% (94.3%–99.6%) for patients with mBCC receiving sonidegib 200 and 800 mg QD, respectively (Figure 1). At week 17, median (95% CI) percent reduction was 88.7% (54.6%–93.0%) and 97.0% (77.5%–98.9%) for aggressive laBCC, 97.5% (80.3%–98.8%) and 95.0% (80.7%–97.5%) for nonaggressive laBCC, and 99.1% (96.4%–99.6%) and 99.3% (95.9%–99.9%) for mBCC in the 200 and 800 mg groups, respectively. Median (95% CI) reduction in GLI1 expression at EOT was 96.0% (67.9%–98.9%) and 98.0% (90.6%–99.7%) for patients with aggressive laBCC, 73.0% (49.5%–98.9%) and 92.5% (40.5%–99.8%) for patients with nonaggressive laBCC, and 77.0% (56.8%–97.3%) and 96.1% (22.6%–99.7%) for patients with mBCC receiving sonidegib 200 and 800 mg QD, respectively.
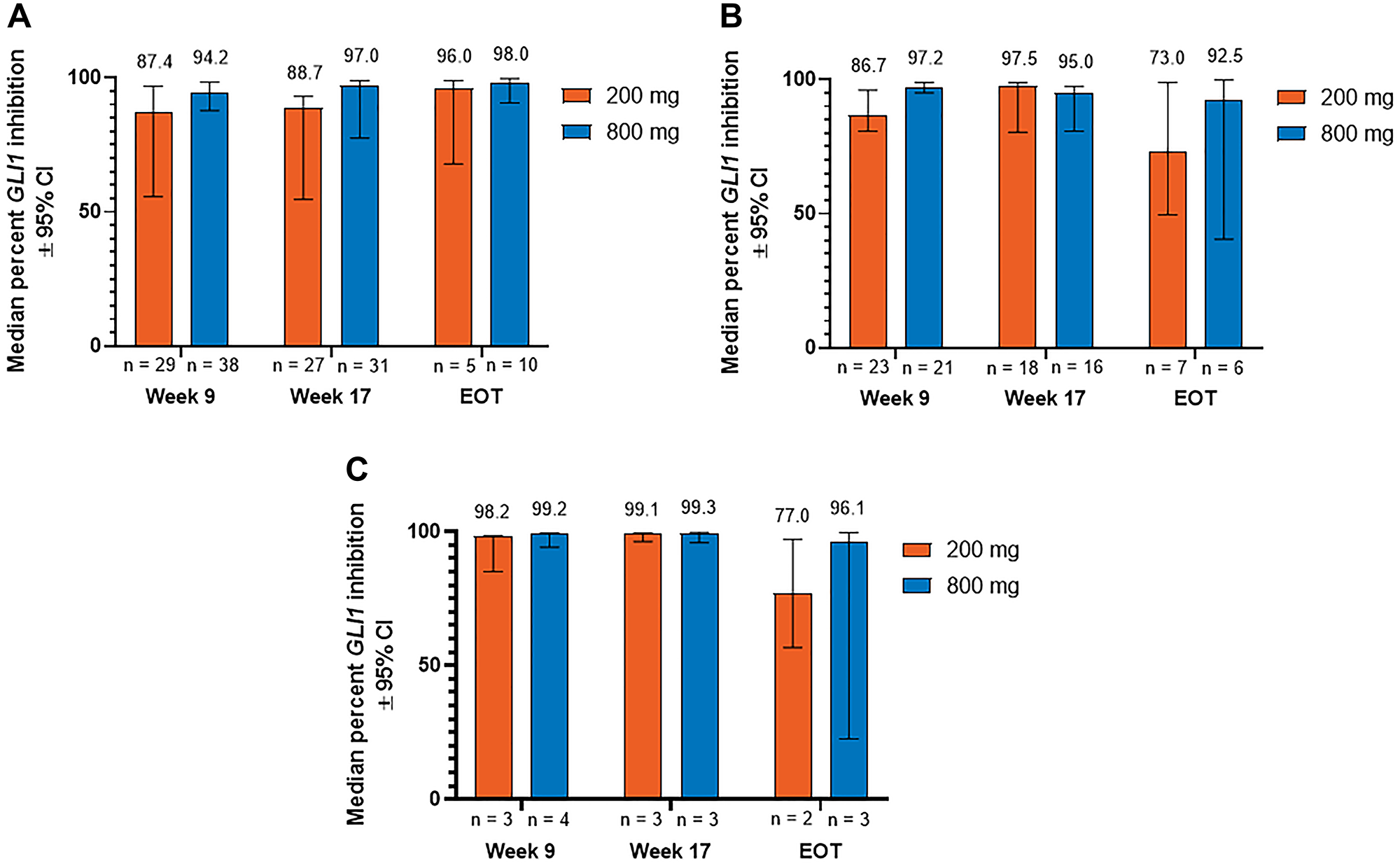
Figure 1: Percent inhibition of GLI1 expression relative to baseline in patients with (A) aggressive laBCC, (B) nonaggressive laBCC, and (C) mBCC. BCC, basal cell carcinoma; CI, confidence interval; EOT, end of treatment; GLI1, Glioma associated oncogene homolog 1; laBCC, locally advanced BCC; mBCC, metastatic BCC.
Subgroup analyses of GLI1 expression
Marked reduction in GLI1 expression from baseline was overall consistent between subgroups of patients with laBCC and mBCC stratified by demographic (Figure 2) and baseline clinical characteristics (Figures 3 and 4). Median percent reduction for patients with aggressive laBCC ranged longitudinally (week 9 to EOT; lowest and highest value shown) 75.1%–98.9% and 90.6%–97.5% for those < 65 years, and 64.4%–94.8% and 92.9%–98.9% for those ≥ 65 years receiving sonidegib 200 and 800 mg QD, respectively. For patients with nonaggressive laBCC, median percent reduction ranged longitudinally 73.0%–96.6% and 62.6%–96.4% in patients younger than 65 years, and 48.1%–98.1% and 87.9%–98.9% in patients ≥ 65 years receiving sonidegib 200 and 800 mg QD, respectively. Patients with mBCC achieved median percent reduction ranging longitudinally 91.7%–97.8% and 96.1%–99.3% for those younger than 65 years, and 56.8%–99.6% and 99.0% (data available for 1 patient for week 9 only) for patients ≥ 65 years receiving sonidegib 200 and 800 mg QD, respectively.
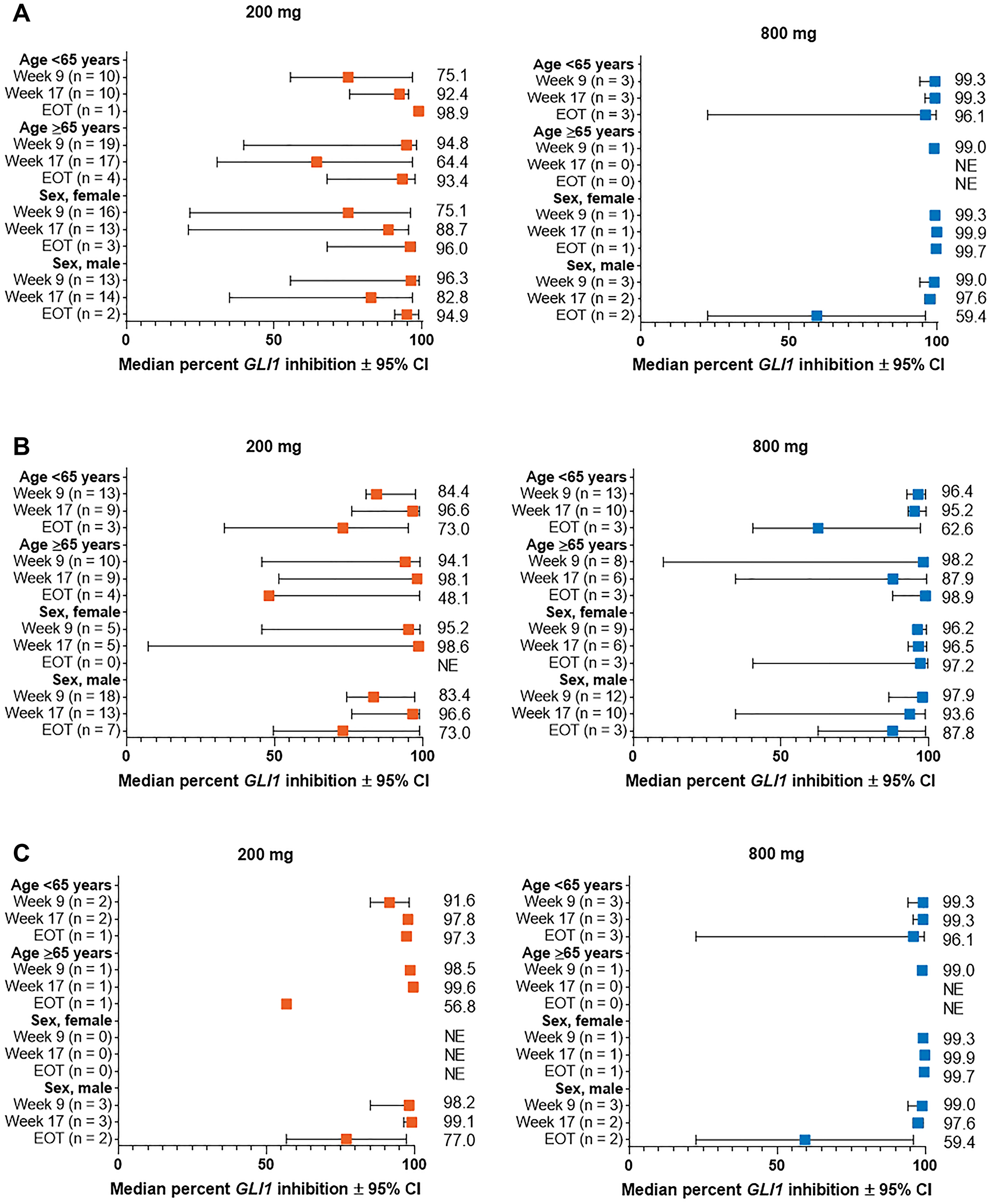
Figure 2: Percent change from baseline in GLI1 expression in subgroups by demographic characteristics for patients with (A) aggressive laBCC, (B) nonaggressive laBCC, and (C) mBCC. BCC, basal cell carcinoma; CI, confidence interval; EOT, end of treatment; GLI1, Glioma associated oncogene homolog 1; laBCC, locally advanced BCC; mBCC, metastatic BCC; NE, not estimable.
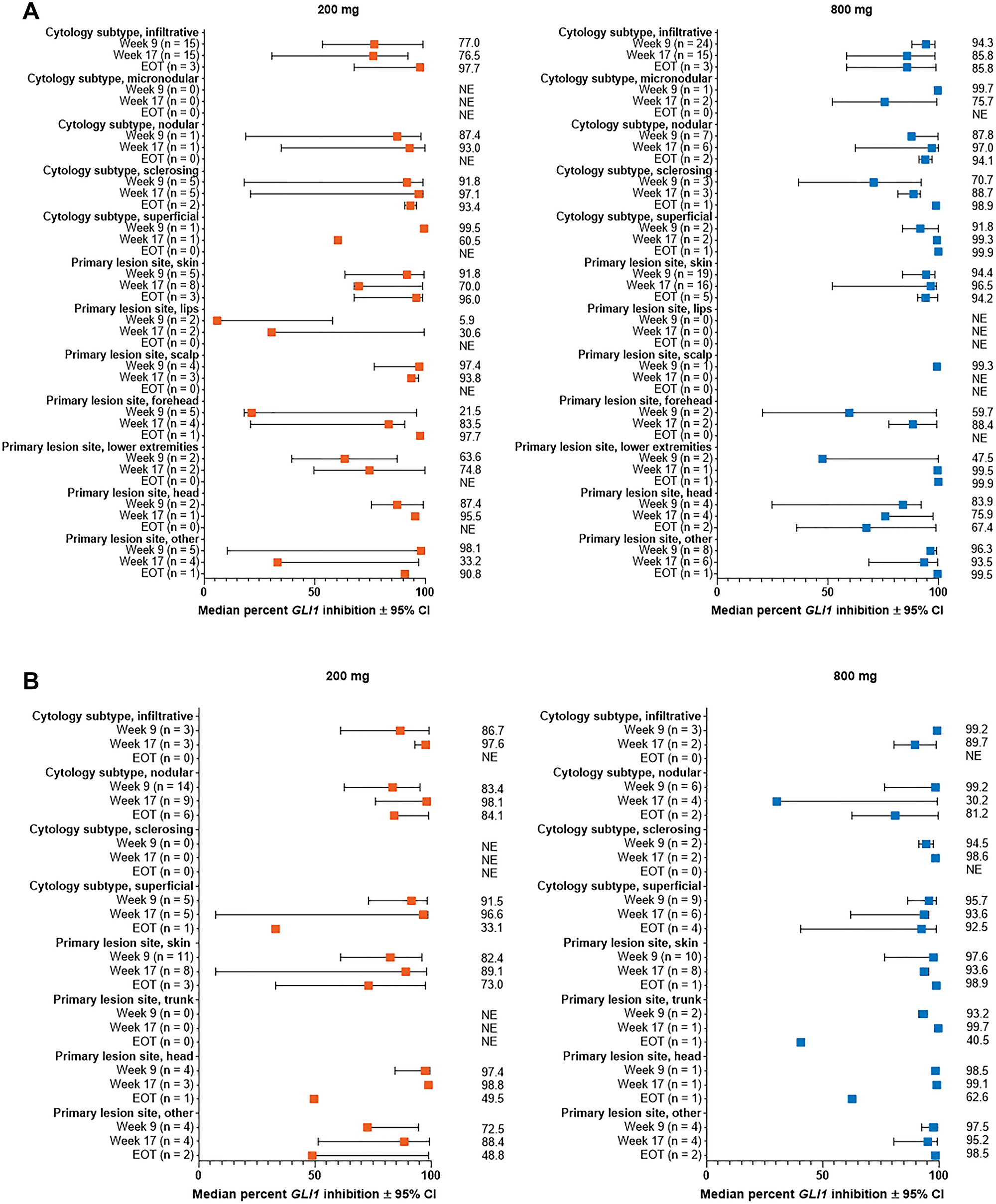
Figure 3: Percent change from baseline in GLI1 expression in subgroups by lesion cytology and site for patients with (A) aggressive laBCC, (B) nonaggressive laBCC. Results are shown for subgroups with > 1 patient in either dose group. All subgroups for patients with mBCC had ≤ 1 patient. BCC, basal cell carcinoma; CI, confidence interval; EOT, end of treatment; GLI1, Glioma associated oncogene homolog 1; laBCC, locally advanced BCC; mBCC, metastatic BCC; NE, not estimable.
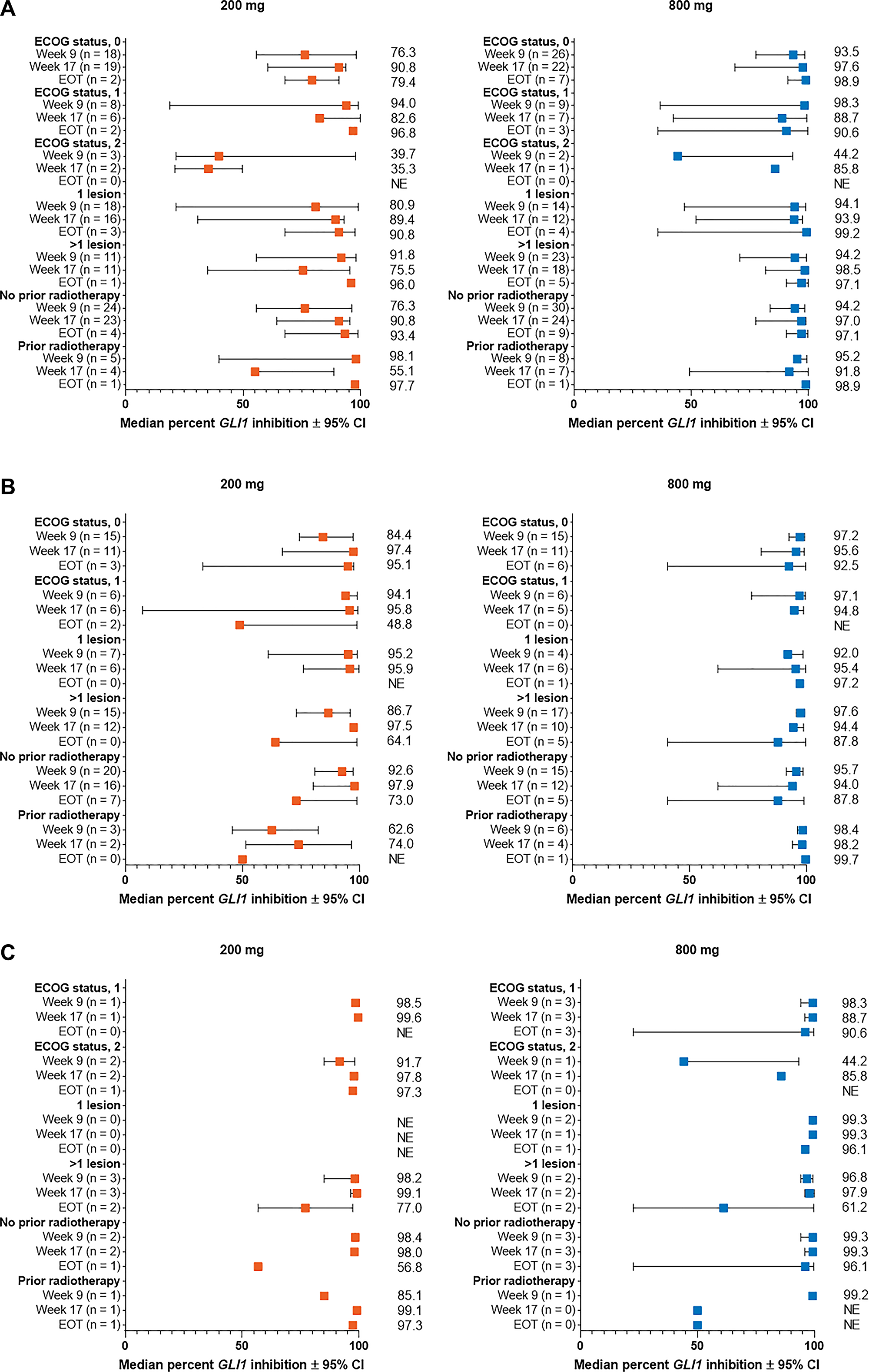
Figure 4: Percent change from baseline in GLI1 expression in subgroups by Eastern Cooperative Oncology Group status, number of lesions, and prior radiotherapy for patients with (A) aggressive laBCC, (B) nonaggressive laBCC, and (C) mBCC. BCC, basal cell carcinoma; CI, confidence interval; EOT, end of treatment; GLI1, Glioma associated oncogene homolog 1; laBCC, locally advanced BCC; mBCC, metastatic BCC; NE, not estimable.
Patient subgroups stratified by BCC cytological subtype demonstrated relatively consistent reduction in GLI1 expression from baseline across subtypes (Table 1).
Table 1: Percent inhibition of GLI1 expression relative to baseline by cytological subtype
| 200 mg QD | 800 mg QD | |||||
|---|---|---|---|---|---|---|
| laBCC | mBCC | laBCC | mBCC | |||
| Aggressive | Nonaggressive | Aggressive | Nonaggressive | |||
| Basosquamous | n = 0 | n = 1 | n = 2 | n = 1 | n = 0 | n = 0 |
| Week 9 | — | 99.1 (NE) | — | 98.3 (NE) | — | — |
| Week 17 | — | 85.2 (NE) | — | 99.3 (NE) | — | — |
| EOT | — | — | — | — | — | — |
| Infiltrative | n = 21 | n = 3 | n = 3 | n = 28 | n = 6 | n = 3 |
| Week 9 | 77.0 (53.7–99.1) | 86.7 (61.0–99.1) | 91.7 (85.1–98.2) | 94.3 (88.0–98.4) | 99.2 (98.0–99.5) | 99.2 (99.0–99.3) |
| Week 17 | 76.5 (30.7–92.3) | 97.6 (93.0–99.1) | 97.8 (96.4–99.1) | 85.8 (58.5–98.9) | 89.7 (80.7–98.8) | 99.9 (NE) |
| EOT | 97.7 (67.9–98.9) | — | 97.3 (NE) | 96.9 (35.8–99.7) | — | 99.7 (NE) |
| Micronodular | n = 0 | n = 0 | n = 0 | n = 2 | n = 0 | n = 0 |
| Week 9 | — | — | — | 99.7 (NE) | — | — |
| Week 17 | — | — | — | 75.7 (52.0–99.3) | — | — |
| EOT | — | — | — | — | — | — |
| Multifocal | n = 1 | n = 0 | n = 0 | n = 0 | n = 2 | n = 0 |
| Week 9 | 63.7 (NE) | — | — | — | 99.1 (NE) | — |
| Week 17 | 75.5 (NE) | — | — | — | — | — |
| EOT | — | — | — | — | — | — |
| Nodular | n = 8 | n = 14 | n = 1 | n = 9 | n = 8 | n = 1 |
| Week 9 | 87.4 (18.8–98.2) | 83.4 (62.6–95.2) | 98.5 (NA) | 87.8 (5.0–99.8) | 98.5 (76.5–99.4) | 94.2 (NE) |
| Week 17 | 93.0 (34.9–99.9) | 98.1 (76.0–99.5) | 99.6 (NA) | 97.0 (62.4–99.9) | 30.2 (93.2–99.3) | 95.9 (NE) |
| EOT | — | 84.1 (49.5–98.9) | — | 94.1 (91.2–97.1) | 81.2 (62.6–99.7) | 22.6 (NE) |
| Morpheaform | n = 5 | n = 0 | n = 1 | n = 3 | n = 2 | n = 0 |
| Week 9 | 91.8 (18.2–99.1) | — | — | 70.7 (36.7–92.2) | 94.5 (91.4–97.6) | — |
| Week 17 | 97.1 (21.0–99.1) | — | — | 88.7 (81.7–91.8) | 98.6 (97.5–99.7) | — |
| EOT | 93.4 (90.8–96.0) | — | 56.8 (NE) | 98.9 (NE) | — | — |
| Superficial | n = 1 | n = 7 | n = 1 | n = 2 | n = 11 | n = 0 |
| Week 9 | 99.5 (NE) | 91.5 (73.0–98.3) | — | 91.8 (83.7–99.9) | 95.7 (86.5–98.9) | — |
| Week 17 | 60.5 (NE) | 96.6 (7.3–98.6) | — | 99.3 (99.2–99.5) | 93.6 (62.1–95.6) | — |
| EOT | — | 33.1 (NE) | — | 99.9 (NE) | 92.5 (40.5–98.9) | — |
| Other | n = 0 | n = 0 | n = 0 | n = 1 | n = 1 | n = 1 |
| Week 9 | — | — | — | — | 97.2 (NE) | 99.6 (NE) |
| Week 17 | — | — | — | — | 95.6 (NE) | 99.3 (NE) |
| EOT | — | — | — | — | — | 96.1 (NE) |
Association between GLI1 levels and efficacy outcomes
Substantial reductions in GLI1 levels from baseline were observed in patients with disease control (complete response [CR], partial response [PR], or stable disease [StDis]), with median percent reduction ranging 74.5%–97.9% and 95.7%–98.0% at week 9, and 90.8%–99.5% and 96.1%–97.0% at week 17, for the 200 and 800 mg groups, respectively (Supplementary Table 2). However, a significant association was not observed between strength of GLI1 repression and odds of tumor response (CR+PR), with hazard ratio (HR; 95% CI, P-value) for low vs high tumor inhibition of 1.4 (0.5–3.8, P = 0.4838) and 0.8 (0.3–2.0, P = 0.6627), for the 200 and 800 mg groups, respectively (Supplementary Table 3).
Overall, no significant association was observed between extent of GLI1 inhibition and time to tumor response (TTR), with HR (95% CI, P-value) for low vs high expression of 0.9 (0.4–1.9, P = 0.4932) and 1.4 (0.7–2.8, P = 0.3148) for the 200 and 800 mg groups, respectively (Supplementary Table 4).
Association between GLI1 levels and time to onset of grade ≥ 2 creatine kinase elevation
There was no overall significant association between extent of GLI1 inhibition and time to onset of grade ≥ 2 creatine kinase (CK) elevation, with HR (95% CI, P-value) for low vs high expression of 0.6 (0.2–2.5, P = 0.3348) and 1.2 (0.5–2.6, P = 0.3348) for the 200 and 800 mg groups, respectively (Supplementary Table 5). Among patients with greater GLI1 inhibition from baseline, those in the 800 mg dose group had an increased risk of grade ≥ 2 CK elevation, with HR (95% CI, P-value) vs the 200 mg dose of 2.3 (0.8–6.2, P = 0.0406).
DISCUSSION
Results from this secondary analysis suggest that sonidegib treatment led to substantial reductions in GLI1 levels from baseline across doses, BCC subtypes, examined time points, and demographic and baseline disease characteristics in patients with advanced BCC. Marked reductions in GLI1 levels from baseline were observed in patients who achieved disease control with sonidegib, consistent with Hedgehog pathway inhibition.
The Hedgehog pathway plays a key role in regulating cell proliferation and differentiation during development and is involved in the maintenance and repair of many adult tissues including the skin, hair, muscles, and nervous system [19]. Upon activation of the pathway, a signaling ligand of the Hedgehog family binds the transmembrane receptor PTCH1 [20]. This causes the G-protein coupled receptor SMO—sonidegib’s target in the pathway—to dissociate from PTCH1 and migrate to the primary cilium, where it releases the cytoplasmic sequestration of the GLI family of transcription factors by Suppressor of Fused through a multistep signaling cascade [20]. Noncanonical pathways of GLI1 activation have also been reported, including by the Ras and p53 families of tumor suppressors [21]. Aberrant GLI1 activation promotes tumor growth, migration, and angiogenesis [21].
Despite the overall durable efficacy of sonidegib treatment observed in the BOLT study, a subset of patients develop resistance to HHIs that can be either primary or acquired after initial response to treatment [22]. Moreover, although GLI1 suppression is dose- and exposure-dependent, a reduction of GLI1 expression did not always correlate with tumor response in the phase 1 efficacy and safety study for sonidegib, most likely due to limited sample size, indicating resistance may develop despite GLI1 inhibition [10]. The strong—but not complete—inhibition of GLI1 expression in this biomarker analysis is consistent with the possibility that patients with low GLI1 inhibition are resistant to sonidegib. However, there was no significant correlation between sonidegib efficacy and the strength of GLI1 inhibition. This is possibly due to the study not being sufficiently powered to confirm this correlation. Additionally, inhibition of GLI1 in the different patient subgroups examined was uniformly strong, and none of the examined demographic and baseline clinical characteristics showed strong correlation with sonidegib resistance.
Animal and cell-line cancer models studied the impact of sonidegib on Hedgehog pathway activity. In human primary glioblastoma initiating cells, sonidegib reduces levels of GLI1, GLI2, PTCH1, and PTCH2 messenger ribonucleic acid (RNA) and protein, as well as GLI1 and GLI2 translocation into the nucleus [23]. Epistasis testing using sonidegib in combination with direct GLI1 and GLI2 inhibition by RNA interference revealed the effect of sonidegib on Hedgehog pathway downstream targets, including upregulation of the cell death mediators, Fas, Death receptor (DR)4, and DR5, and downregulation of the cell proliferation mediators B-cell lymphoma 2 and Platelet-derived growth factor receptor A [23]. In human renal cell carcinoma lines, sonidegib inhibited GLI1 and GLI2 and reduced cell proliferation in combination with everolimus or sunitinib, whereas direct inhibition of GLI1 and GLI2 in combination with everolimus or sunitinib had no impact on proliferation, suggesting that other targets downstream of SMO may play a role in tumor response [24].
This analysis found no significant association between the magnitude of GLI1 inhibition and time to onset of grade ≥ 2 CK elevation—suggesting that CK elevation is not influenced by the extent of GLI1 inhibition. CK elevation is a common treatment-emergent AE observed in patients treated with Hedgehog inhibitors. While the exact mechanism responsible for muscle spasms and increased CK levels in patients receiving HHIs is not completely understood, muscle spasms are considered to be linked with paradoxical activation of the SMO/calcium/AMP-activated protein kinase axis, and the inhibition of SMO signaling leads to an influx of calcium into muscle cells [25]. The pivotal clinical studies of vismodegib (Erivedge®, Genentech, South San Francisco, CA), an HHI indicated for the treatment of advanced BCC, did not include CK monitoring; however, muscle spasms were reported in 71.2% of patients [26]. All currently approved HHIs have the same mechanism of action and similar safety profiles; it is thought that CK elevation and muscle spasms are HHI class-effect AEs.
Limitations of the current study include the lack of statistical power to assess specific biomarker-related hypotheses. All biomarker analysis should be considered exploratory and hypothesis-generating, and all P-values were nominal or were adjusted for multiplicity only at the biomarker level within a specific model or analysis. GLI1 was the only examined biomarker, and data on the expression of other prominent members of the pathway were not collected. Additionally, GLI1 expression as related to reduction in tumor mass was not directly evaluated in this analysis. Subsequently, decreases in tumor size may have contributed to reductions in GLI1 levels measured following treatment.
In summary, the reduction of GLI1 expression is consistent with potent inhibition of the Hedgehog pathway by sonidegib in patients with laBCC and mBCC. These results support further clinical studies on the impact of sonidegib on Hedgehog pathway biomarkers.
Materials and Methods
BOLT study design
This randomized, double-blind, adaptive phase 2 multicenter study adhered to the ethical principles of the Declaration of Helsinki and received approval for its protocol and all amendments from an Independent Ethics Committee or Institutional Review Board at each study site. Written informed consent was obtained from all patients before any study-specific procedures.
Study design is described in detail elsewhere and is briefly summarized here and in Supplementary Figure 2. The study enrolled men and women age ≥ 18 years with a histologically confirmed diagnosis of mBCC or laBCC (not amenable to radiation therapy, curative surgery, or other local therapies) and a World Health Organization (WHO) performance status ≤ 2. Patients were randomized 1:2 to receive sonidegib 200 or 800 mg QD until disease progression, unacceptable toxicity, withdrawal of consent, study termination, or death.
BOLT efficacy assessments
Tumor response was evaluated with Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 for patients with mBCC. For laBCC, the standard RECIST 1.1 criteria are inadequate, since posttreatment morphological changes such as ulceration, cyst formation, and scarring may confound tumor response evaluation. A modified (m)RECIST protocol was developed to assess tumor response in laBCC, integrating central histological review, one-dimensional localized soft-tissue magnetic resonance imaging (MRI) along the lesion’s longest diameter per RECIST 1.1, and bidimensional color photography measurements per WHO guidelines.
The primary efficacy endpoint was ORR per central review. Secondary efficacy assessments included TTR and best overall response (either CR, PR, StDis, progressive disease, or unknown). Best overall response was assessed by central review according to mRECIST in patients with laBCC and RECIST 1.1 in patients with mBCC. mRECIST criteria includes a combination of MRI scans, color photography, and baseline and follow-up histopathology data for patients to assess tumor response.
BOLT safety assessments
Safety assessments included AEs monitored throughout the study, coded using the medical Dictionary for Regulatory Activities version 19.0, and assessed for toxicity using the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. CK levels were assessed prior to starting treatment or within 72 hours of the first dose, then weekly during the first 2 months, and every 4 weeks thereafter while on study treatment.
Biomarker assessments
Biopsies were collected from accessible lesions at Screening, weeks 9 and 17 predose, and within 21 days after the last dose of study drug. For patients with multiple lesions, no specific lesion was designated for biopsy, and any lesion could be used. Presence of tumor tissue in biopsy samples was histologically confirmed prior to biomarker analysis.
GLI1 expression at Screening was used as baseline measurement. Gene expression at all examined time points was assessed using reverse transcriptase polymerase chain reaction, in terms of the number of reaction cycles needed to reach the threshold amount of product. Expression of GLI1 at each time point was normalized to the housekeeping gene ubiquitin C, and fold and percent change from baseline of normalized GLI1 expression were computed.
Percent change from baseline in GLI1 expression was stratified by type of BCC (aggressive and nonaggressive laBCC and mBCC), age, sex, number of lesions at baseline, cytology subtype, primary site of cancer, prior radiotherapy, and Eastern Cooperative Oncology Group performance status. Association between GLI1 expression and select efficacy and safety assessments was evaluated, including best overall response, TTR, and time to onset of grade ≥ 2 CK elevation.
Statistical analyses
The ITT population included all randomized patients. The subset of the ITT population with valid biomarker samples comprised the biomarker population used for all biomarker analyses. Data collected up to 6 months after the last patient randomization date were included in the analysis. All statistical evaluations were exploratory since the study was not powered to assess specific hypotheses regarding change in GLI1 expression. Since inhibition of the Hedgehog pathway with sonidegib should result in GLI1 inhibition, postbaseline records with > 100% change in GLI1 levels from baseline were considered outliers and were excluded from analyses and models.
Visual assessments of strip plots determined the relationship between baseline GLI1 levels or changes from baseline in GLI1 levels and clinical efficacy outcomes. Percent change in GLI1 levels was summarized using descriptive statistics (mean, 95% CI, median, 95% distribution-free CI, SD, quartiles, and range). Longitudinal analyses of GLI1 expression were performed using a linear mixed model, including fold change in GLI1 expression from baseline as a response variable and visit, treatment group, and visit-by-treatment interaction as covariates. Median percent change from baseline and 95% CI for each dose group and time point were computed based on model-based least squares mean fold change estimates.
Cox proportional hazards models determined associations between baseline GLI1 levels or changes from baseline in GLI1 levels, and TTR and time to onset of grade ≥ 2 CK elevation. Kaplan-Meier curves and log-rank test–based P-values for the difference in time-to-onset curves were produced for groups defined by dose level and categorized GLI1 levels. Normalized baseline GLI1 levels were categorized as low or high based on the median (both doses combined). Percent change in GLI1 from baseline at week 9 or 17 was categorized as low (greater inhibition) or high (lesser inhibition) based on the third quartile (both doses combined).
Abbreviations
AE: adverse event; BCC: basal cell carcinoma; BOLT: Basal cell carcinoma Outcomes with LDE225 (sonidegib) Treatment; CI: confidence interval; CK: creatine kinase; CR: complete response; DR: death receptor; EOT: end of treatment; GLI1: Glioma-associated oncogene homolog 1; HHI: Hedgehog pathway inhibitor; HR: hazard ratio; ITT: intent-to-treat; laBCC: locally advanced BCC; mBCC: metastatic BCC; mRECIST: modified RECIST; MRI: magnetic resonance imaging; ORR: objective response rate; PR: partial response; PTCH1: Patched 1; QD: once daily; RECIST: Response Evaluation Criteria in Solid Tumors; RNA: ribonucleic acid; SMO: Smoothened; SD: standard deviation; StDis: stable disease; TTR: time to tumor response; WHO: World Health Organization.
ACKNOWLEDGMENTS
This study was sponsored and funded by Novartis and Sun Pharmaceutical Industries, Inc. Writing and editorial support for manuscript preparation were provided by Ginny Feltzin, PhD, and Zehra Gundogan, VMD, of AlphaBioCom, LLC, and funded by Sun Pharmaceutical Industries, Inc. All authors met the International Council of Medical Journal Editors criteria and received neither honoraria nor payment for authorship.
CONFLICTS OF INTEREST
R. Dummer has participated on advisory boards and consulted for Amgen; Bristol-Myers Squibb; CatalYm; Merck Sharp & Dohme; Novartis Pharmaceutical Corporation; Pierre-Fabre; Roche; Sanofi; Second Genome; Sun Pharmaceutical Industries, Inc.; and Takeda. L. Liu and N. Squittieri are employees of Sun Pharmaceutical Industries, Inc. R. Gutzmer serves as consultant to Almirall; Amgen; Bristol-Myers Squibb; Merck Serono; Merck Sharp & Dohme; Novartis; Pfizer; Pierre-Fabre; Roche; Sanofi Genzyme; Sun Pharmaceutical Industries, Inc.; and 4SC; has received travel grants and honoraria for lectures from Almirall; Amgen; Bristol-Myers Squibb; Merck-Serono; Merck Sharp & Dohme; Novartis; Pierre-Fabre; Roche; Sun Pharmaceutical Industries, Inc.; and received research funding from Amgen, Johnson & Johnson, Merck-Serono, Novartis. J. Lear receives personal fees from Novartis and Sun Pharmaceutical Industries, Inc.
FUNDING
This work was supported by Sun Pharmaceutical Industries, Inc.
References
1. Asgari MM, Moffet HH, Ray GT, Quesenberry CP. Trends in Basal Cell Carcinoma Incidence and Identification of High-Risk Subgroups, 1998–2012. JAMA Dermatol. 2015; 151:976–981. https://doi.org/10.1001/jamadermatol.2015.1188. [PubMed].
2. Lucas RM, McMichael AJ, Armstrong BK, Smith WT. Estimating the global disease burden due to ultraviolet radiation exposure. Int J Epidemiol. 2008; 37:654–667. https://doi.org/10.1093/ije/dyn017. [PubMed].
3. Xiang F, Lucas R, Hales S, Neale R. Incidence of nonmelanoma skin cancer in relation to ambient UV radiation in white populations, 1978–2012: empirical relationships. JAMA Dermatol. 2014; 150:1063–1071. https://doi.org/10.1001/jamadermatol.2014.762. [PubMed].
4. Work G, Invited R, Kim JYS, Kozlow JH, Mittal B, Moyer J, Olencki T, Rodgers P. Guidelines of care for the management of basal cell carcinoma. J Am Acad Dermatol. 2018; 78:540–559. https://doi.org/10.1016/j.jaad.2017.10.006. [PubMed].
5. Dreier J, Cheng PF, Bogdan Alleman I, Gugger A, Hafner J, Tschopp A, Goldinger SM, Levesque MP, Dummer R. Basal cell carcinomas in a tertiary referral centre: a systematic analysis. Br J Dermatol. 2014; 171:1066–1072. https://doi.org/10.1111/bjd.13217. [PubMed].
6. Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008; 8:743–754. https://doi.org/10.1038/nrc2503. [PubMed].
7. Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, Glynn M, Zaphiropoulos PG, Pressman C, Undén AB, Dean M, Brash DE, Bale AE, Toftgård R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996; 14:78–81. https://doi.org/10.1038/ng0996-78. [PubMed].
8. Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, Rosenthal A, Epstein EH Jr, de Sauvage FJ. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998; 391:90–92. https://doi.org/10.1038/34201. [PubMed].
9. Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009; 30:303–312. https://doi.org/10.1016/j.tips.2009.03.007. [PubMed].
10. Rodon J, Tawbi HA, Thomas AL, Stoller RG, Turtschi CP, Baselga J, Sarantopoulos J, Mahalingam D, Shou Y, Moles MA, Yang L, Granvil C, Hurh E, et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin Cancer Res. 2014; 20:1900–1909. https://doi.org/10.1158/1078-0432.CCR-13-1710. [PubMed].
11. Lu T, Wang B, Gao Y, Dresser M, Graham RA, Jin JY. Semi-mechanism-based population pharmacokinetic modeling of the hedgehog pathway inhibitor vismodegib. CPT Pharmacometrics Syst Pharmacol. 2015; 4:680–689. https://doi.org/10.1002/psp4.12039. [PubMed].
12. Lorusso PM, Jimeno A, Dy G, Adjei A, Berlin J, Leichman L, Low JA, Colburn D, Chang I, Cheeti S, Jin JY, Graham RA. Pharmacokinetic dose-scheduling study of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors. Clin Cancer Res. 2011; 17:5774–5782. https://doi.org/10.1158/1078-0432.CCR-11-0972. [PubMed].
13. LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, Graham RA, Zerivitz KL, Low JA, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011; 17:2502–2511. https://doi.org/10.1158/1078-0432.CCR-10-2745. [PubMed].
14. Graham RA, Lum BL, Cheeti S, Jin JY, Jorga K, Von Hoff DD, Rudin CM, Reddy JC, Low JA, Lorusso PM. Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin Cancer Res. 2011; 17:2512–2520. https://doi.org/10.1158/1078-0432.CCR-10-2736. [PubMed].
15. Dummer R, Guminksi A, Gutzmer R, Lear JT, Lewis KD, Chang ALS, Combemale P, Dirix L, Kaatz M, Kudchadkar R, Loquai C, Plummer R, Schulze HJ, et al. Long-term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42-month analysis of the phase 2 randomised, double-blind BOLT study. Br J Dermatol. 2020; 182:1369–1378. https://doi.org/10.1111/bjd.18552. [PubMed].
16. Dummer R, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, Herd RM, Kaatz M, Loquai C, Stratigos AJ, Schulze HJ, Plummer R, Gogov S, et al. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J Am Acad Dermatol. 2016; 75:113–25.e5. https://doi.org/10.1016/j.jaad.2016.02.1226. [PubMed].
17. Lear JT, Migden MR, Lewis KD, Chang ALS, Guminski A, Gutzmer R, Dirix L, Combemale P, Stratigos A, Plummer R, Castro H, Yi T, Mone M, et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J Eur Acad Dermatol Venereol. 2018; 32:372–381. https://doi.org/10.1111/jdv.14542. [PubMed].
18. Migden MR, Guminski A, Gutzmer R, Dirix L, Lewis KD, Combemale P, Herd RM, Kudchadkar R, Trefzer U, Gogov S, Pallaud C, Yi T, Mone M, et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): a multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015; 16:716–728. https://doi.org/10.1016/S1470-2045(15)70100-2. [PubMed].
19. Petrova R, Joyner AL. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development. 2014; 141:3445–3457. https://doi.org/10.1242/dev.083691. [PubMed].
20. Athar M, Li C, Kim AL, Spiegelman VS, Bickers DR. Sonic hedgehog signaling in Basal cell nevus syndrome. Cancer Res. 2014; 74:4967–4975. https://doi.org/10.1158/0008-5472.CAN-14-1666. [PubMed].
21. Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012; 13:105–113. [PubMed].
22. Migden MR, Chang ALS, Dirix L, Stratigos AJ, Lear JT. Emerging trends in the treatment of advanced basal cell carcinoma. Cancer Treat Rev. 2018; 64:1–10. https://doi.org/10.1016/j.ctrv.2017.12.009. [PubMed].
23. Fu J, Rodova M, Nanta R, Meeker D, Van Veldhuizen PJ, Srivastava RK, Shankar S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro Oncol. 2013; 15:691–706. https://doi.org/10.1093/neuonc/not011. [PubMed].
24. D’Amato C, Rosa R, Marciano R, D’Amato V, Formisano L, Nappi L, Raimondo L, Di Mauro C, Servetto A, Fulciniti F, Cipolletta A, Bianco C, Ciardiello F, et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br J Cancer. 2014; 111:1168–1179. https://doi.org/10.1038/bjc.2014.421. [PubMed].
25. Dinehart MS, McMurray S, Dinehart SM, Lebwohl M. L-Carnitine reduces muscle cramps in patients taking vismodegib. SKIN The Journal of Cutaneous Medicine. 2018; 2:90–95.
26. Sekulic A, Migden MR, Basset-Seguin N, Garbe C, Gesierich A, Lao CD, Miller C, Mortier L, Murrell DF, Hamid O, Quevedo JF, Hou J, McKenna E, et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: final update of the pivotal ERIVANCE BCC study. BMC Cancer. 2017; 17:332. https://doi.org/10.1186/s12885-017-3286-5. [PubMed].