
Introduction
Chimeric antigen receptor (CAR) T cell therapies, and adoptive cell therapy (ACT) in general, represent one of the most promising anti-cancer strategies [1]. To date, two autologous CAR-T cell therapies directed against CD19 have been approved for use in the treatment of B-cell lymphomas including relapsed or refractory acute lymphoblastic lymphoma (ALL) and diffuse large cell B-cell lymphoma (DLBCL) [2]. Initial response rates in these heavily pretreated patients have been extraordinary, in the range of 80%, although durable responses have been considerably lower at 40–50% [2]. There are now nearly 1000 clinical trials involving CAR-T cells in the US (https://clinicaltrials.gov/), including expanding to new targets such as BCMA and CD123 in heme malignancies and various targets in solid tumors. Further, recombinant T cell receptor (TCR) engineered T cells, TCR-T that are directed against MHC-complexed peptides, are also being evaluated primarily in solid tumors [3].
It is unclear why some patients respond to treatment with adoptive cell therapies such as CAR-T, and others do not, though the tumor immune microenvironment is a likely contributor to variable effect of cell therapy in both hematologic and solid cancers. To this end, preclinical and clinical studies have shown that regulatory T cells (Tregs) can have an impact on the effect of ACT in mice and in patients with melanoma [4, 5]. In these studies, depletion of Tregs, whether by specific depletion or via conditioning with external beam radiation, had a favorable impact on the anti-tumor response to ACT. Interestingly, these and other studies suggest that Treg depletion is more sustained following treatment with radiation as opposed to chemotherapy-induced conditioning [6], where a rapid rebound of Tregs was seen with chemotherapy conditioning resulting in poorer outcomes. Other cell types that contribute to an immunosuppressive tumor microenvironment that may negatively impact CAR-T efficacy include myeloid derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) [7].
Another important area for improvement in CAR-T therapies involves the serious adverse events that have been reported, particularly with the drugs directed against CD19 such as tisagenlecleucel and axicabtagene ciloleucel. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) occur at very high rates following CD19 CAR-T administration, with CRS occurring in greater than 50% of patients and at least 10–30% patients experiencing high grade ICANS [2]. Recent preclinical studies have shown that cytokine release leading to CRS or neurotoxicity is due to activated macrophages following recruitment to the site of CAR-T and tumor cells. Mouse study results [8, 9] documented that macrophages secrete IL-1 or IL-6 following recruitment and activation by CAR-T cells at the tumor site.
Conditioning has been shown to improve the immune homeostatic environment to enable successful ACT or CAR-T engraftment and expansion in vivo following infusion, and represents a potential point of intervention to decrease serious toxicities following CAR-T treatment. Most CAR-T programs exploit the use of the combination of fludarabine and cyclophosphamide (flu/cy) as a lymphodepletive conditioning regimen prior to CAR-T. These drugs are often administered 2–7 days (2–5 day course of therapy) prior to ACT infusion. However, the commonly used flu/cy regimen is a non-specific and cytotoxic treatment that some patients may not be able to tolerate and may not offer tumor control. Additionally, flu/cy has been correlated with toxicities such as prolonged cytopenias and cytokine release syndrome (CRS) following CAR-T administration [10].
In contrast to relatively non-specific chemotherapy-derived lymphodepletion, targeted lymphodepletion with radioimmunotherapy (RIT) directed to CD45 may be a safer and more effective alternative to target and deplete immune cells. The CD45 antigen is found on all nucleated immune cells, with increased expression on mature lymphoid and myeloid lineages, leading to preferential depletion of mature immune cells compared to progenitor hematopoietic cells [11]. Importantly, immunomodulatory cells such as Tregs and MDSC express CD45 and are targets of lymphodepletion with a CD45-targeting antibody-radionuclide conjugate (ARC), potentially resulting in better engraftment, activation and persistence of the exogenously added CAR-T cells in patients. In addition, macrophages, implicated in CRS, are also sensitive to targeting with a CD45 ARC, and their transient reduction may result in mitigation of CRS risk. In addition, most hematologic malignancies such as leukemia and lymphoma abundantly overexpress CD45, at levels of 200 to 400,000 antigens per cell. Targeted lymphodepletion with a CD45 ARC is anticipated to result in a reduction in tumor burden, which may result in an improvement in overall response to the CAR-T therapy.
Anti-CD45 RIT with 131Iodine (131I)-apamistamab (Iomab-B), is in a Phase III clinical trial as a myeloablative targeted conditioning regimen prior to allogeneic stem cell transplant in patients with active relapsed/refractory acute myeloid leukemia (AML). Results from patients following dosimetry testing have shown that low non-myeloablative doses of 131I-apamistamab were able to safely induce transient lymphodepletion [12]. This data allowed us to hypothesize that low dose anti-CD45 RIT could be used as a targeted modality to effectively lymphodeplete prior to ACT. Here we describe the results of preclinical studies with an anti-mouse CD45 antibody 30F11, labeled with two different beta-emitters - 131I and 177Lutetium (177Lu), to investigate the effect of anti-CD45 RIT lymphodepletion on immune cell types and on tumor control in a model of adoptive cell therapy. Our results support CD45 targeted RIT lymphodepletion prior to adoptive cell therapy using a non-myeloablative dose of 131I-30F11 or 177Lu-30F11 antibody.
Results
131I-30F11 treatment transiently depleted CD45-expressing immune cell subsets in healthy mice
microSPECT/CT imaging of mice administered CD45-targeting antibody 30F11 radiolabeled with 111In (111In radiolabel was used in these experiments as imaging surrogate for therapeutic radionuclides 131I and 177Lu) showed that the antibody homed to immune system organs such as lymph nodes, spleen, and bone marrow as well as liver (Figure 1A). The imaging data was confirmed by the pharmacokinetics data which also demonstrated fast clearance of the antibody from the blood and kidneys, and low uptake in the pancreas and gonads (Figure 1B). Dose finding studies using 1.85–7.4 MBq 131I-30F11 antibody were performed next to determine the appropriate dose of 131I needed to define a non-myeloablative dose to safely lymphodeplete. Figure 2A shows that 3.7 MBq dose of 131I-30F11 transiently depleted lymphocytes, splenocytes, and myeloid derived cells (MDSC) but preserved bone marrow cells, platelets, and red blood cells. Experiments in which variable amounts of 30F11 were radiolabeled with 3.7 MBq 131I revealed no effect of the antibody amount on the efficacy of lymphodepletion (Figure 2B). Based on these results, 20 μg of antibody was labeled with 3.7 MBq 131I for targeted lymphodepletion in the follow-up experiments. Importantly, the detailed analyses of the depleted cells subpopulations showed that 131I-30F11 was able to deplete subsets such as spleen NK and B cells, neutrophils and spleen Tregs at a dose that did not impact bone marrow hematopoietic stem cells (HSCs) (Figure 3).
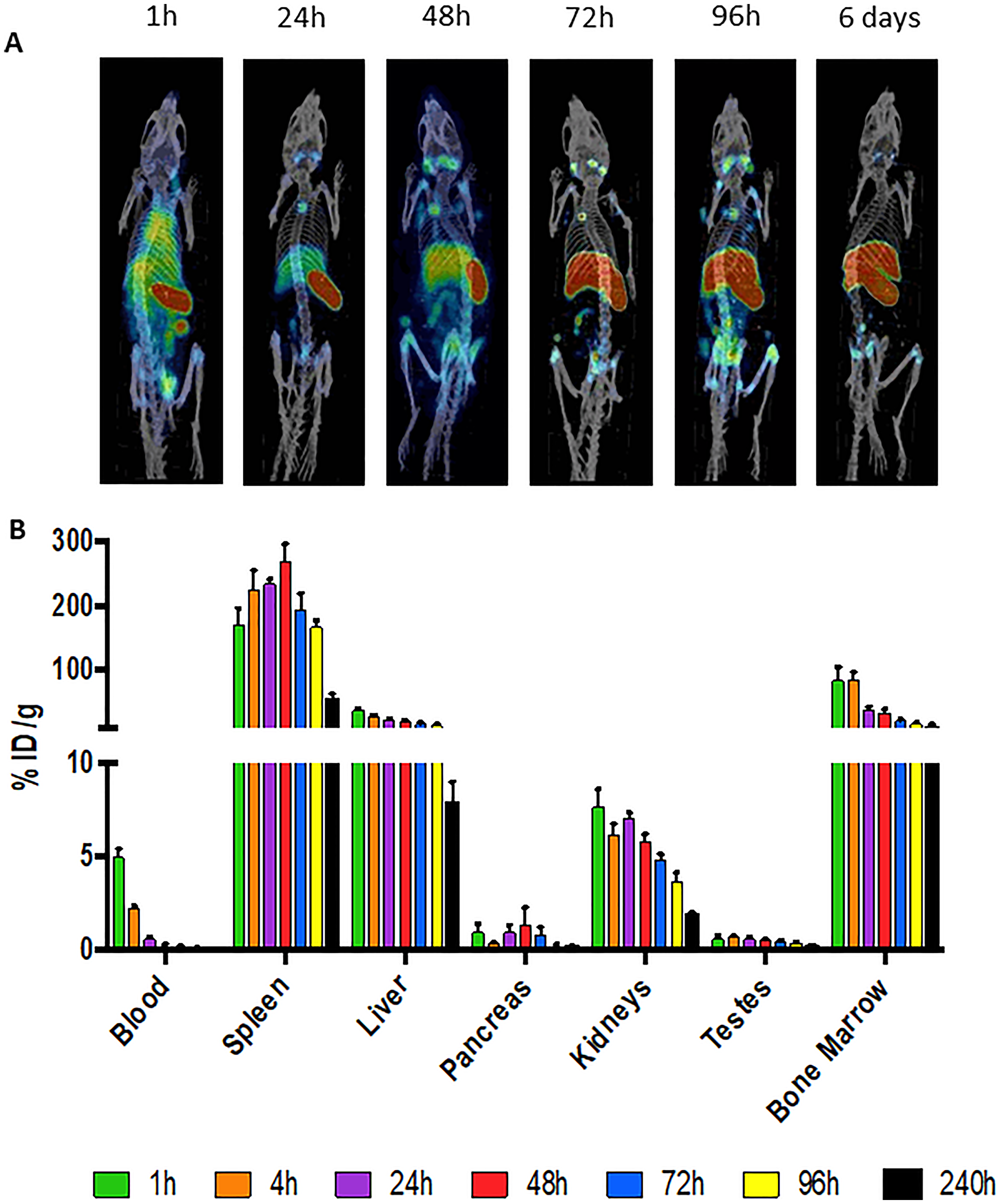
Figure 1: 111In-30F11 anti-CD45 antibody homes to immune organs. (A) C57Bl/6 mice were injected intraperitoneally (i.p.) with 60 μg 111In-30F11 antibody with a specific activity of 0.185 MBq/μg and antibody distribution was monitored by microSPECT/CT at 1, 24, 48, 72, 96 hours timepoints and then again at 6 days after antibody administration. 30F11 antibody homed to immune organs: lymph nodes, spleen, and bone marrow, it also homed to the liver. (B) pharmacokinetics of 111In-labeled 30F11 in male C57Bl/6 mice.
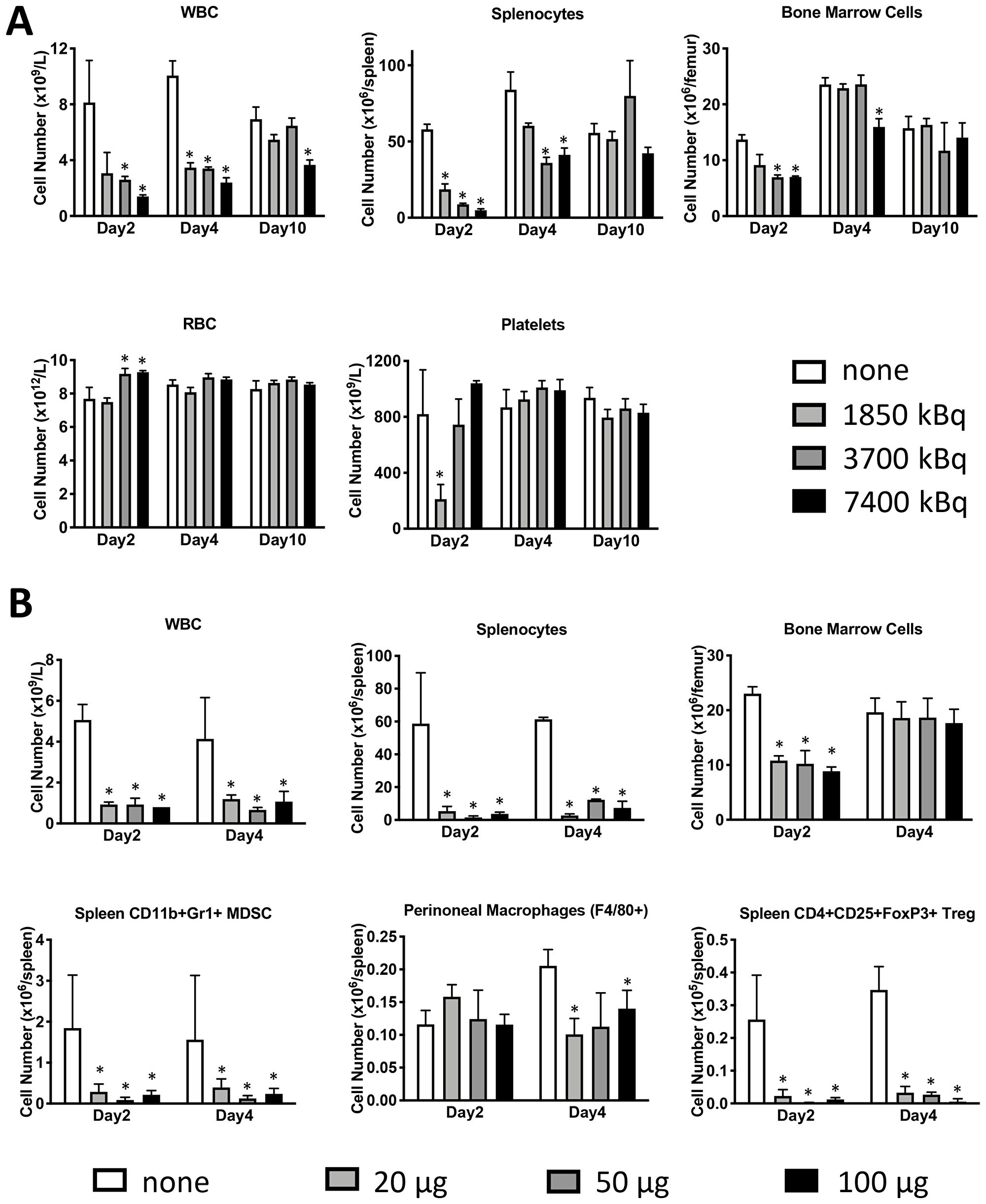
Figure 2: Lymphodepletion with 131I-labeled anti-CD45 30F11 antibody preserves bone marrow cells, platelets, and red blood cells but depletes splenocytes, Tregs and MDSC. (A) Dose finding studies were performed to determine the appropriate dose of 131I needed to define a non-myeloablative dose to safely lymphodeplete. Female C57Bl/6 mice were injected i.p. with 20 μg 30F11 labeled with 1.85-7.4 MBq 131I to determine the most effective and safe radioactivity dose required for transient lymphodepletion. Mice were euthanized at varying time points (2–21 days) and various immune cell subsets were quantified using flow cytometry and (B) To determine the appropriate amount of antibody for labeling - varying amounts of 30F11 antibody were administered to a mouse in 20–100 μg range, but with a constant activity of 3.7 MBq. Asterisks signify P values < 0.05.
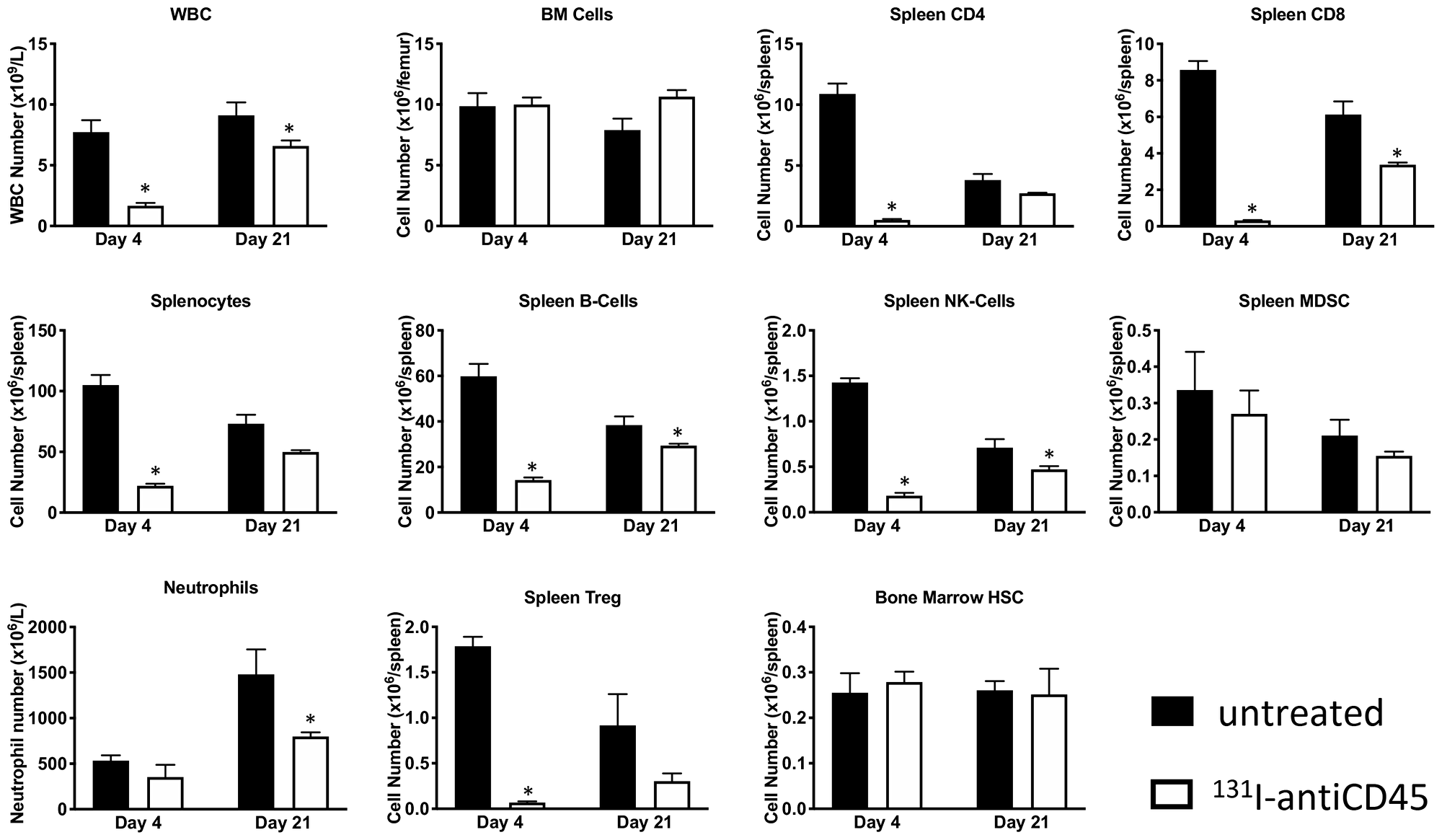
Figure 3: Treatment with 131I-labeled anti-CD45 30F11 antibody transiently depletes CD45-expressing immune cell subsets. Treatment of non-tumor bearing C57B/6 mice with 3.7 MBq 131I-30F11 antibody was effective in transiently lymphodepleting various lymphocyte populations such as WBC, CD4+ cells, CD8+ cells, splenocytes, B cells, MDSC, NK cells, neutrophils and Tregs at a dose that does not impact bone marrow HSCs. Asterisks signify P values < 0.05.
131I-30F11 treatment safely depletes immune cells in OT I tumor model and enables tumor control
Following E.G7 tumor engraftment, mice were conditioned with 3.7 MBq 131I-30F11 and received 106 OT I CD8 CD45.2 OVA reactive T cells on day 4. Figure 4A shows that targeted lymphodepletion with 131I-30F11 antibody enabled engraftment and persistence of adoptively transferred CD45.2 OT I CD8+ T cells in the spleen 17 days post injection. Similar to conditioning in non-tumor bearing mice, 131I-30F11 lymphodepletion mediated decreases in multiple lymphoid cell subsets such as spleen CD4+, CD8+, B and NK cells as well as Tregs. (Figure 4B). Importantly, while 131I-30F11 mediated targeted conditioning did not affect the tumor growth – the combination of 131I-30F11 mediated targeted conditioning and adoptively transferred OT I T cells enabled control of E.G7 tumor growth (Figure 4C). In the control group which received no conditioning or T cells and in 131I-30F11 alone groups, 0% of mice achieved complete response (CR), while in the treated group which received immunodepletion followed by OT I cells, 50% of mice (2/4) achieved CR (p = 0.02).
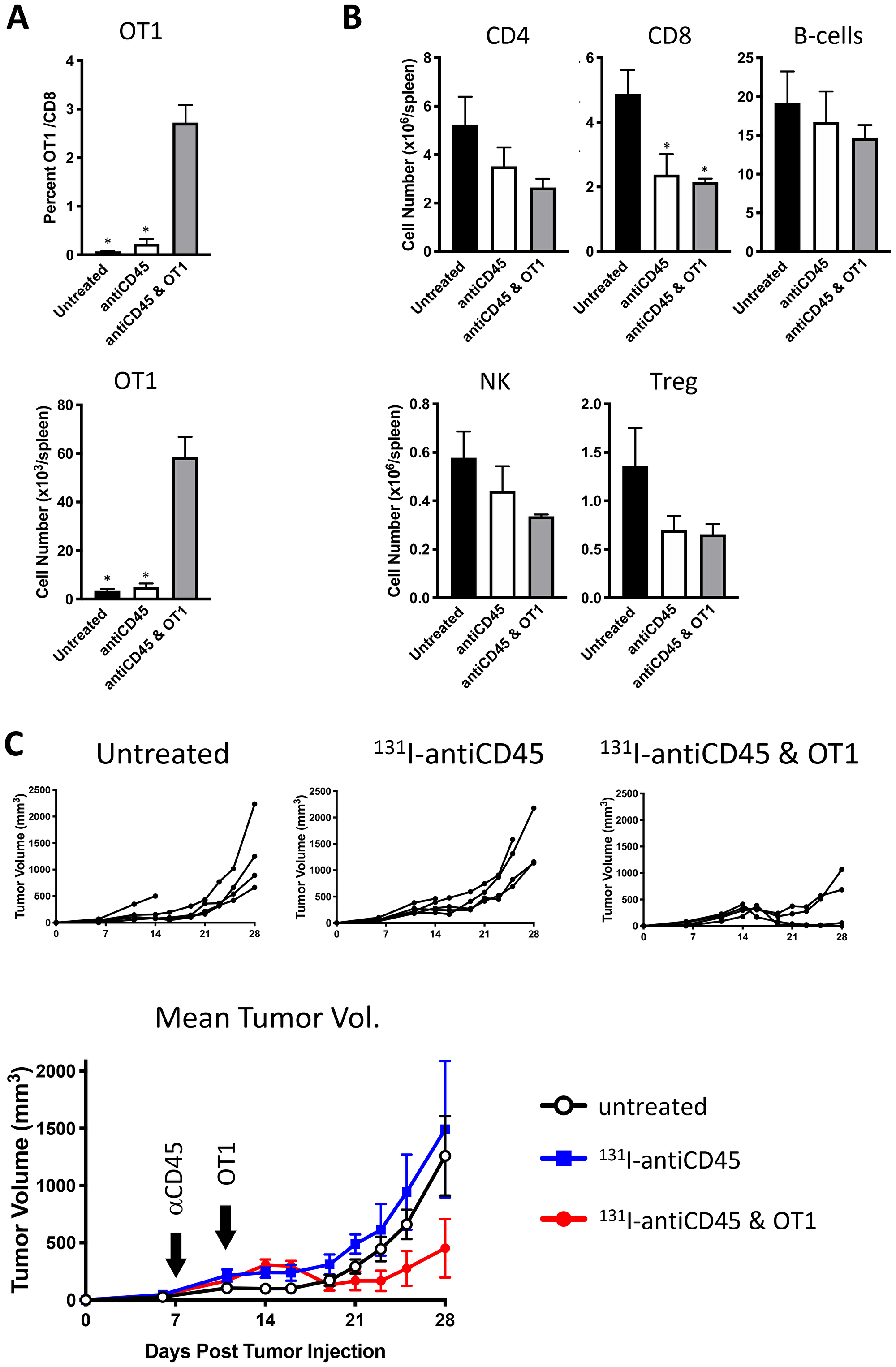
Figure 4: Lymphodepletion with 131I-labeled anti-CD45 30F11 antibody enables tumor control in OT I adoptive cell therapy model. Following E.G7 tumor engraftment, mice were conditioned with 3.7 MBq 131I-30F11; or received 3.7 MBq 131I-30F11 followed by 106 OT I CD8 CD45.2 OVA reactive T cells on day 4. Control mice received no conditioning or T cells. (A) Targeted lymphodepletion with 131I-30F11 antibody enables engraftment and persistence of adoptively transferred CD45.2 OT I CD8+ T cells in the spleen 17 days post injection. (B) Similar to conditioning in non-tumor bearing mice, lymphodepletion with 131I-30F11 mediated decreases in multiple lymphoid cell subsets. (C) 131I-30F11 mediated targeted conditioning and adoptively transferred OT I T cells enabled control of E.G7 tumor growth. In 3.7 MBq 131I-30F11 plus OT I T cells group 2/4 mice achieved CR; in 3.7 MBq 131I-30F11 alone group 0/5 mice achieved CR; in untreated control group, 0/5 mice achieved CR. N = 4–5 per group. Asterisks signify P values < 0.05.
131I-30F11 and 177Lu-30F11 treatment produced comparable effects on immune cells and tumor growth
Pilot treatment of non-tumor bearing C57B/6 mice with 0.74 or 1.48 MBq 177Lu-30F11 antibody revealed that 1.48 MBq 177Lu-30F11 was effective in transiently depleting various immune populations in the spleen including Tregs (Figure 5). Subsequently, we performed side by side comparison of lymphodepletion in non-tumor bearing C57B/6 mice with either 0.74 or 1.48 MBq 177Lu-30F11 or 1.85 or 3.7 MBq 131I-30F11. Both radiolabeled molecules were similarly effective in transiently lymphodepleting various immune cell populations without affecting bone marrow cells, red blood cells, or platelets (Figure 6). The experiment interrogating the ability of 177Lu- and 131I-30F11 mediated targeted conditioning prior to adoptively transferred OT I T cells to control E.G7 tumor growth showed comparable effect of both agents on the tumor size and overall survival with 20% better tumor control and higher overall survival in 131I-30F11 treated group (p = 0.03) (Figure 7).
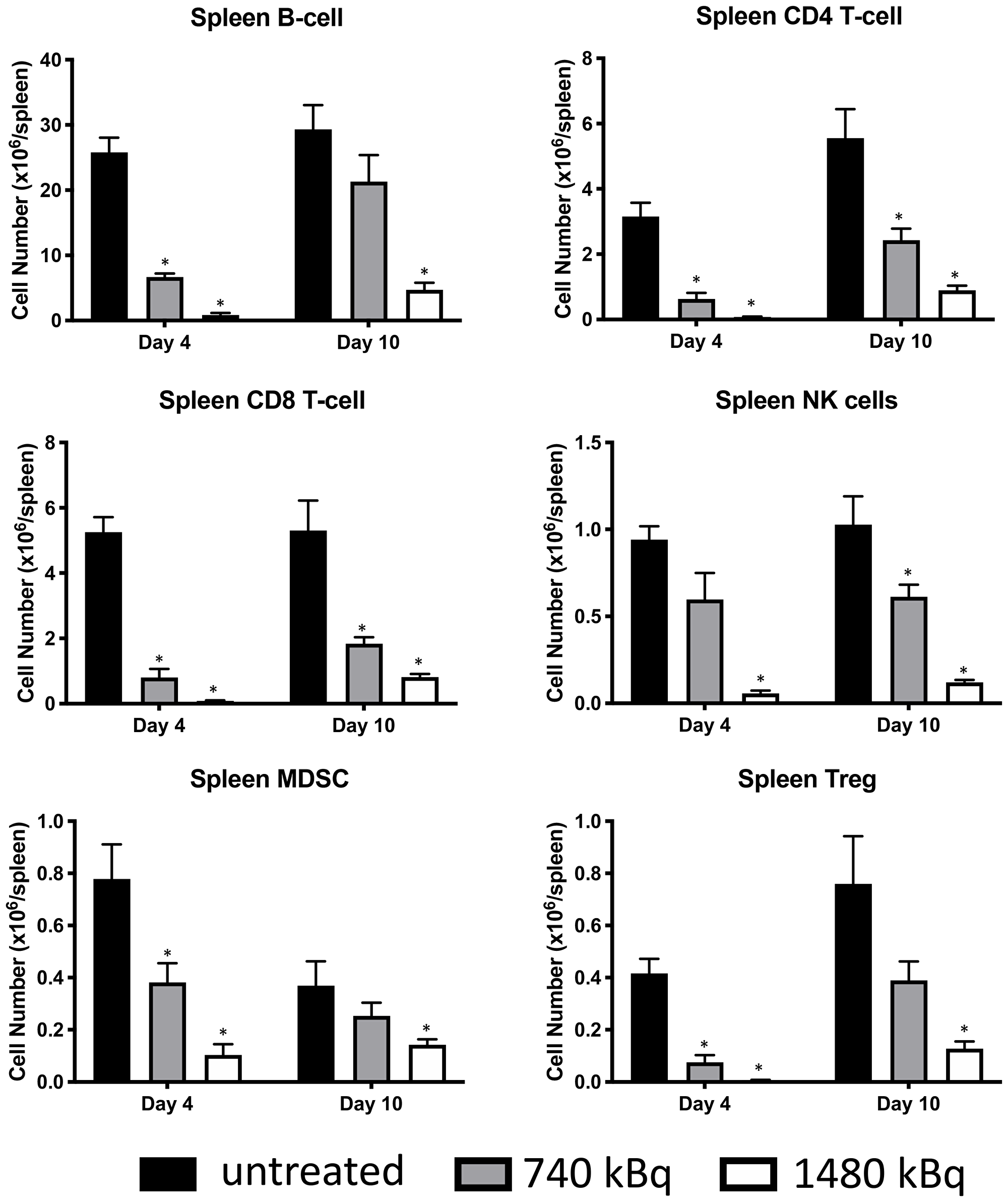
Figure 5: Treatment of non-tumor bearing C57B/6 mice with 1.48 MBq 177Lu-30F11 antibody was effective in transiently depleting various immune populations in the spleen such as CD4+ T cells, CD8+ T cells, B cells, MDSC, NK cells and Tregs. Asterisks signify P values < 0.05.
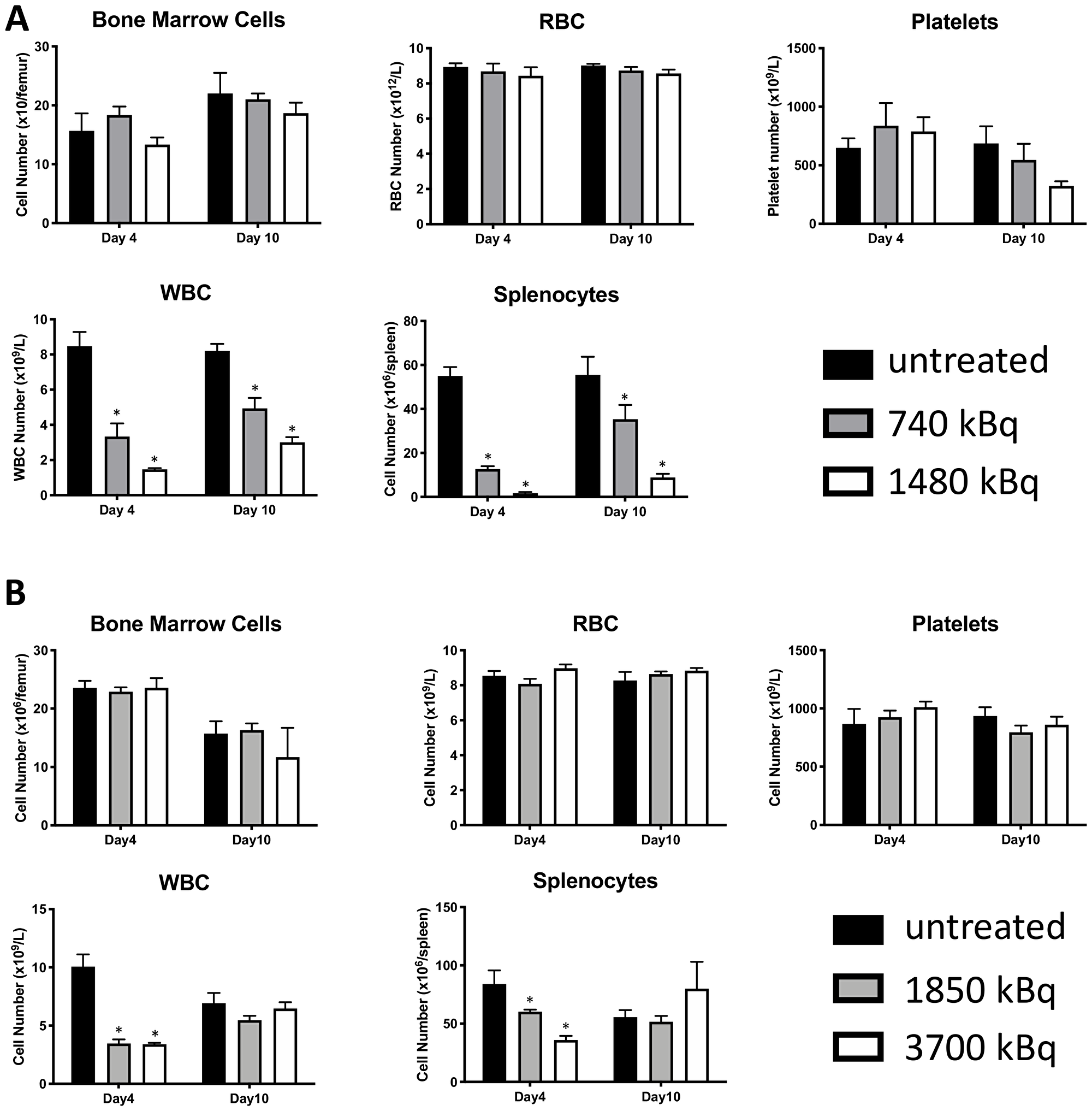
Figure 6: 177Lu-30F11 or 131I-30F11 antibody transiently depleted CD45+ immune cell subsets without affecting platelets, red blood cells, or bone marrow cells. Treatment of non-tumor bearing C57B/6 mice with (A) 0.74 or 1.48 MBq 177Lu-30F11; (B) 1.85 or 3.7 MBq 131I-30F11 antibody was similarly effective in transiently lymphodepleting various immune cell populations without affecting bone marrow cells, red blood cells, and platelets. Asterisks signify P values < 0.05.
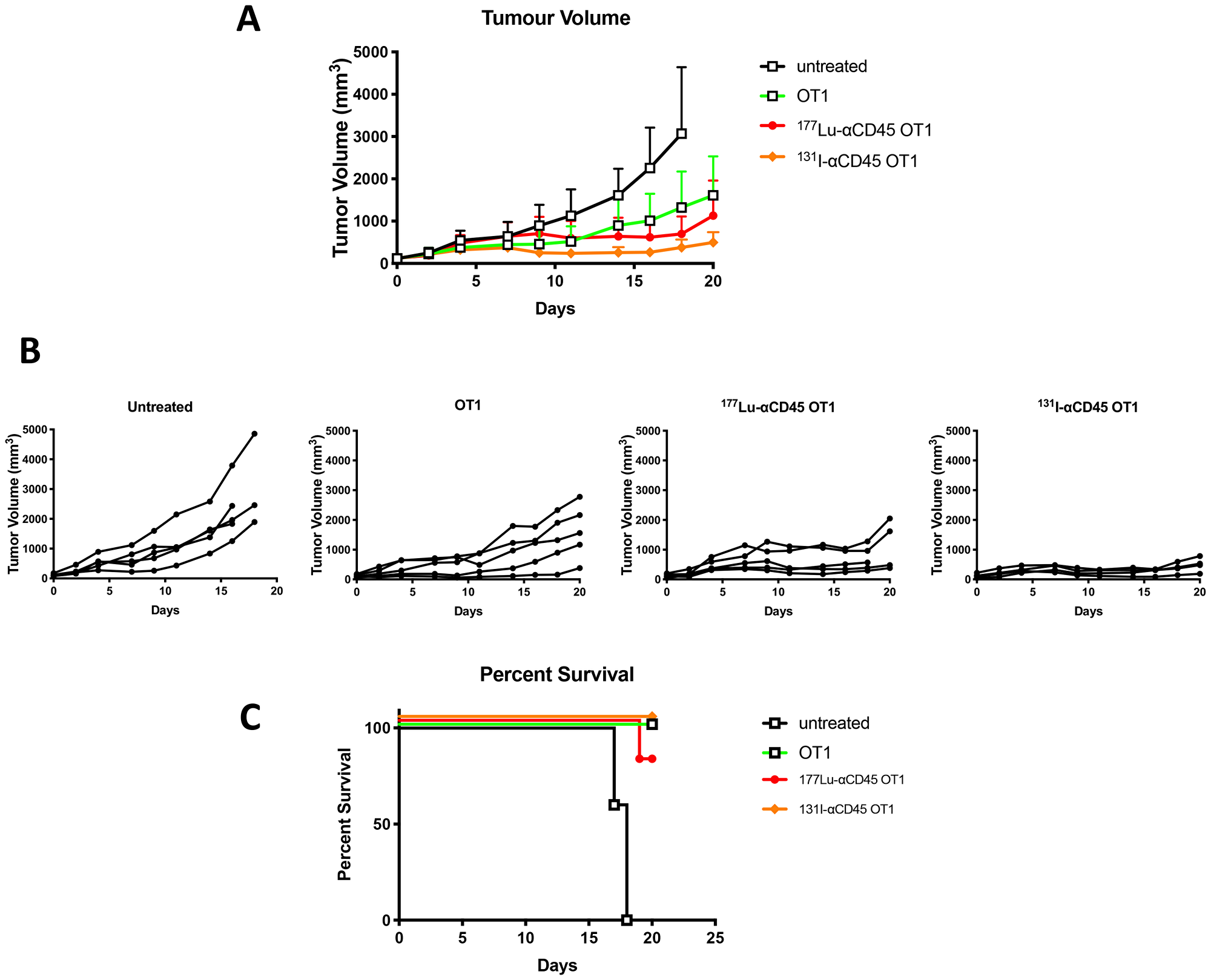
Figure 7: Comparative tumor control by 177Lu-30F11 or 131I-30F11 antibody. Following E. G7 tumor engraftment, mice either received no treatment or were conditioned with 1.48 MBq 177Lu-30F11 or 3.7 MBq 131I-30F11 on Day 0 and then received 106 OT I CD8+ CD45.2 OVA reactive T cells on day 4. (A) 177Lu-30F11 and 131I-CD45-mediated targeted conditioning prior to adoptively transferred OT I T cells enabled control of E.G7 tumor growth. (B) Tumor size for individual mice in each group is displayed. OT I T cell persistence and expansion was confirmed in mice at the time of sacrifice. (C) Survival of mice in treated and in untreated control groups.
DISCUSSION
Prior to a patient receiving a dose of an adoptive cell transfer such as engineered autologous or allogeneic CAR-T cells, it is common to perform a lymphodepletion step often using high dose chemotherapy [2, 6]. This process is considered important to create sufficient space in the immune microenvironment, e.g., bone marrow, to allow the transferred cells to engraft. Further, lymphodepletion appears to elicit a favorable cytokine profile for establishment and proliferation of the adoptively transferred lymphocytes [4]. In this study we demonstrate the feasibility and utility of radioimmunotherapy (RIT) using low non-myeloablative doses of 131I-radiolabeled anti-CD45 30F11 antibody to effectively lymphodeplete in a targeted manner in experimental models prior to administration of adoptive cell therapy. Significantly, targeted conditioning with pan-CD45 RIT, which selectively targets all nucleated immune cells, depletes not only lymphocytes, but also macrophages, as well as immune suppressive regulatory T cells and myeloid-derived suppressor cells in the immune microenvironment. It can potentially also exert a direct anti-tumor effect on CD45+ hematopoietic cancers.
Subsequently, we investigated use of an alternate payload selection, specifically 177Lu, for mediating lymphodepletion in mouse models. Both 131I and 177Lu are beta emitters with physical half lives of 8 and 6.6 days, respectively, and maximum beta energies of 0.6 and 0.5 MeV, respectively. The principle difference lies in the chemistry of these radionuclides, with 131I being a halogen and 177Lu – a trivalent radiometal. 177Lu-labeled somatostatin receptor targeting peptide (Lutathera) has been approved for treatment of neuroendocrine tumors [13], and a 177Lu-labeled small molecule binding to PSMA is currently in advanced stage clinical trials for treatment of metastatic prostate cancer [14]. We have selected 177Lu over another beta-emitting radiometal 90Y as it has been shown in long term follow up studies that patients treated with 177Lu-peptide therapy survive significantly longer than those treated with 90Y-peptide therapy due to more side effects from 90Y [15] which is a high energy beta emitter while 177Lu is a low energy beta emitter. We determined that 1.48 MBq 177Lu-30F11 could effectively deplete various immune cell subsets in mice but spare bone marrow cells, red blood cells, and platelets. In a model of adoptive cell therapy using CD45.1 OT1 mice bearing E.G7-OVA tumors, mice that received either 131I-30F11 or 177Lu-30F11 RIT-mediated lymphodepletion demonstrated enhanced tumor control over mice that did not receive lymphodepletion. Interestingly, lymphodepletion with 131I-30F11 resulted in somewhat greater tumor control than with 177Lu-30F11 (Figure 7) which might be explained by the residualizing nature of 177Lu which by persisting in the tumors due its residualization, can potentially kill some of the incoming adoptive T cells.
In conclusion, our data supports CD45 targeted RIT lymphodepletion with a non-myeloablative dose of 131I-30F11 or 177Lu-30F11 prior to adoptive cell therapy.
Materials and Methods
Antibodies and radiolabeling
The anti-mouse pan-CD45 binding antibody 30F11 (ThermoFisher Scientific, Catalog # 14-0451-82) was used in all experiments as a surrogate for pan-human CD45 131I-apamistamab (Iomab-ACT) to perform targeted lymphodepletion in mice. Radiolabeling of 30F11 antibody with 131I was carried out by adding 111 MBq Na131I to 15 μL PBS followed by 100 μg 30F11 antibody (1,111 MBq/mg) in a microcentrifuge tube. Chloramine-T (0.2 μL, 10 mg/mL concentration) was added and the solution was shaken at RT for 5 minutes. Sodium thiosulfate (22.8 μL, 0.3 mg/mL concentration) was added followed by 1 μL Sodium Iodide solution (10% w/v) and 7.5 μL of Ascorbic acid (500 mg/mL concentration). The antibody was then purified using spin filtration via an Amicon Ultra 0.5 mL centrifugal filter (30 K MW cut off, Fisher, Hampton, NH, USA). Radiochemical purity was then checked via instant thin layer chromatography (iTLC) and was greater than 99%. For radiolabeling with 177Lu and 111In (imaging surrogate of 177Lu) 30F11 antibody was conjugated to bifunctional chelating agent DOTA (Macrocyclics, USA) at a ratio 20:1 DOTA: Ab and then radiolabeled with 177Lu or 111In with the specific activity of 0.185 MBq /μg antibody as in [16, 17]. Immunoreactivity of the radiolabeled 30F11 antibody was evaluated by measuring binding to E.G7-OVA cells at concentrations ranging from 0.5 ng/mL to 10 μg/mL and detecting the bound antibody with flow cytometry and anti-rat IgG2bPE. The EC50 for naked and modified 30F11 was 394 ng/mL and 393 ng/mL, respectively (Supplementary Figure 1).
MicroSPECT/CT imaging of C57Bl/6 mice with 111In-labeled 30F11 antibody
Female 5 weeks old C57Bl/6 mice were injected intraperitoneally (i.p.) with 60 μg 111In-30F11 antibody with a specific activity of 0.185 MBq/μg and antibody distribution was monitored by microSPECT/CT at 1, 24, 48, 72, 96 hours timepoints and then again at 6 days after antibody administration. A MILabs VECTor4 (Netherlands) microSPECT/CT scanner was used to collect images. PMOD (version 3.9, PMOD Technologies, Inc, Zürich, Switzerland) was used for comprehensive image analysis. An Extra Ultra High Sensitivity Mouse (XUHS-M) collimator for 20–350 keV range was used to collect SPECT data using spiral trajectories. MILABS reconstruction software was used to reconstruct SPECT images using both 111In gamma emissions (245 keV and 171 keV) on a 0.4 mm voxel grid. For visual representation of accumulation, MIP images were utilized.
Biodistribution of 30F11 antibody
To determine the pharmacokinetics of 30F11 antibody in immunocompetent mice, a biodistribution was carried out with 111In-labeled 30F11. Immediately after labelling male C57bl/6 mice were injected i.v. with 1.11 MBq (30 μg) 111In-30F11. Five mice were euthanized at 1, 4, 24, 48, 72, 96 and 240 hours post injection, the organs were collected into pre-weighted tubes, and activity was assessed with a 2470 Wizard2 Gamma counter (Perkin Elmer, MA, USA). A standard that contained 10% of the injected dose was also read to perform decay correction. Percent injected dose per gram (%ID/g) was determined using the equation:
%ID/g = ((sample CPM)/(organ weight*(standard CPM*10)*radiolabeling yield)) × 100%, where CPM are counts per minute.
Lymphodepletion studies with 131I-30F11
In the first series of experiments, female 5 weeks old C57Bl/6 mice were injected i.p. with 20 μg of 131I-30F11 labeled with 1.85–7.4 MBq to determine the most effective and safe radioactivity dose required for transient lymphodepletion. Mice were euthanized at varying time points (2–21 days) and various immune cell subsets were quantified using flow cytometry as described below. The goal of the second series of experiments was to determine the optimal amount of antibody to be used for lymphodepletion. This was accomplished by varying the amount of 30F11 antibody administered to a mouse in 20–100 μg range, but with a constant specific activity of 3.7 MBq.
Flow cytometry
Single cell suspensions were made by passing spleens through a 70 μm cell strainer, washed three times with FACS buffer (0.01 M azide, 2% FBS, PBS), incubated with blocking antibody for 10 min at 4°C, then labeled with surface marker-specific fluorochrome-labeled antibodies (Supplementary Table 1) for an additional 20 min at 4°C and unbound Ab was washed away with FACS buffer. Cells that were probed for intracellular markers were permeabilized (Fix/Perm Buffer; ThermoFisher Scientific, Waltham, MA, USA) for 30 min, washed again with Perm buffer, incubated with blocking antibody in the same buffer for 10 min, and then labeled with marker-specific or isotype control fluorochrome-labeled antibodies for an additional 20 min. Stained cells were washed twice with Perm buffer and once with FACS buffer, and analyzed on a CytoFLEX flow cytometer (Beckman Coulter, Mississauga, ON). The data was processed using FlowJo software (Tree Star Inc., Ashland, OR). Supplementary Figure 2 shows gating scheme for the identifications of various immune cells.
Lymphodepletion and tumor control studies with 131I-30F11 in OT I mouse model
Three groups of female 5 weeks old C57Bl/6 CD45.1 mice, five animals per group, were injected subcutaneously with OVA expressing CD45+ E. G7-OVA lymphoma tumor cells. Seven days post-tumor cell injection, when tumor volume reached ~100 mm3, two groups of mice were treated with 3.7 MBq 131I-30F11 (20 μg), while the third group was left untreated. Four days post-lymphodepletion, OVA-specific CD8+ T cells were purified using anti-CD8 magnetic beads from CD45.2 OT I mice according to the manufacturer’s protocol (Miltenyi Biotec, Auburn, CA). One group of mice treated with 3.7 MBq 131I-30F11 was given a single i. v. injection of 106 CD45.2 OT I CD8+ cells. Tumor volume and body weight were monitored, and mice were sacrificed when tumor volume exceeded 4,000 mm3 or tumors became necrotic. Blood and spleen were assessed for immune cell subsets and presence of engrafted CD45.2 OT I cells by flow cytometry.
Comparative lymphodepletion studies with 131I-30F11 and 177Lu-30F11
Female adolescent C57Bl/6 mice were injected i.p. with 20 μg 30F11 labelled with 0.74 or 1.48 MBq of 177Lu or with 1.85 or 3.7 MBq of 131I to determine the ability to selectively deplete immune cell subsets. Immune cell subset quantitation was performed by flow cytometry as described above.
Comparative lymphodepletion and tumor control studies with 131I-30F11 and 177Lu-30F11 in OT I mouse model
Female 5 weeks old C57Bl/6 CD45.1 mice were injected subcutaneously with OVA expressing CD45+ E. G7-OVA lymphoma tumor cells until 100 mm3 tumor volume was reached. Seven days post-tumor cell injection, mice were treated with 177Lu-30F11 (1.48 MBq), 131I-30F11 (3.7 MBq), or received no lymphodepletion treatment. Four days post-lymphodepletion, OVA-reactive CD8+ T cells isolated from CD45.2 OT I mice were administered to tumor-bearing mice. The tumor volume, body weight, immune cell subsets and presence of engrafted cells were monitored as above.
Statistical analysis
Power analysis for the in vivo studies was estimated using PASS version 11 (NCSS, Inc.) using simulations of different cells depletions/tumor volumes based on pilot or literature data and conservative assumptions regarding the groups treated with the radiolabeled antibodies. All simulations showed power of at least 83% with only five animals per group because of the large differences between treated and untreated animals. Thus, 5 mice per group were utilized in the in vivo studies. GraphPad Prism 7 was used to analyze all the data (GraphPad Software, Inc., La Jolla, CA, USA). Differences among the groups were assessed using Student t-tests and one-way ANOVAs for multiple comparisons. Differences were considered significant at p < 0.05.
Abbreviations
CAR: chimeric antigen receptor; ACT: adoptive cell therapy; Tregs: regulatory T cells; MDSCs: myeloid derived suppressor cells; TAMs: tumor-associated macrophages; CRS: cytokine release syndrome; ICANS: immune effector cell-associated neurotoxicity syndrome; RIT: radioimmunotherapy; Ab: antibody; i.p.: intraperitoneally; OVA: ovalbumin; CPM: counts per minute.
ACKNOWLEDGMENTS
The authors would like to thank Mrs. Mackenzie Malo and Dr. Rubin Jiao for technical assistance.
CONFLICTS OF INTEREST
The research was funded by Actinium Pharmaceuticals and the funder had input into designing of the study and writing the manuscript. ED received the funding from Actinium Pharmaceuticals. DL and EG are employees of Actinium Pharmaceuticals. The rest of the authors declare no conflicts of interest.
FUNDING
This study was supported by research funding from Actinium Pharmaceutical Inc. and Fedoruk Center for Nuclear Innovation.
References
1. Makita S, Yoshimura K, Tobinai K. Clinical Development of anti-CD19 Chimeric Antigen Receptor T-cell Therapy for B-cell non-Hodgkin Lymphoma. Cancer Sci. 2017; 108:1109–1118. https://doi.org/10.1111/cas.13239. [PubMed].
2. Jacobson CA. CD19 Chimeric Antigen Receptor Therapy for Refractory Aggressive B-Cell Lymphoma. J Clin Oncol. 2019; 37:328–335. https://doi.org/10.1200/JCO.18.01457. [PubMed].
3. Zhang J, Wang L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat. 2019; 18:1533033819831068. https://doi.org/10.1177/1533033819831068. [PubMed].
4. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. Version 2. J Exp Med. 2005; 202:907–912. https://doi.org/10.1084/jem.20050732. [PubMed].
5. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, Schrump DS, Steinberg SM, Rosenberg SA, Robbins PF. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood. 2012; 119:5688–5696. https://doi.org/10.1182/blood-2011-10-386482. [PubMed].
6. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015; 348:62–68. https://doi.org/10.1126/science.aaa4967. [PubMed].
7. Porta C, Sica A, Riboldi E. Tumor-associated myeloid cells: new understandings on their metabolic regulation and their influence in cancer immunotherapy. FEBS J. 2018; 285:717–733. https://doi.org/10.1111/febs.14288. [PubMed].
8. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, Traversari C, Bordignon C, Ciceri F, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018; 24:739–748. https://doi.org/10.1038/s41591-018-0036-4. [PubMed].
9. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018; 24:731–738. https://doi.org/10.1038/s41591-018-0041-7. [PubMed].
10. Hay KA, Hanafi LA, Li D, Gust J, Liles WC, Wurfel MM, López JA, Chen J, Chung D, Harju-Baker S, Cherian S, Chen X, Riddell SR, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood. 2017; 130:2295–2306. https://doi.org/10.1182/blood-2017-06-793141. [PubMed].
11. Syrjälä M, Ruutu T, Jansson SE. A Flow Cytometric Assay of CD34-Positive Cell Populations in the Bone Marrow. Br J Haematol. 1994; 88:679–684. https://doi.org/10.1111/j.1365-2141.1994.tb05104.x. [PubMed].
12. Nath R, Geoghegan EM, Ulrickson ML, Spross JA, Lichtenstein RH, Konerth S, Fisher DR, Liang Q, Ludwig D, Reddy V, Berger MS, Gyurkocza B. Sierra Clinical Trial Dosimetry Results Support Low Dose Anti-CD45 Iodine (131I) Apamistamab [Iomab-B] for Targeted Lymphodepletion Prior to Adoptive Cell Therapy. Blood. 2019; 134:1958. https://doi.org/10.1182/blood-2019-128838.
13. Bushnell DL, Bodeker KL. Overview and Current Status of Peptide Receptor Radionuclide Therapy. Surg Oncol Clin N Am. 2020; 29:317–326. [PubMed].
14. Bögemann M, Herrmann K, Radtke JP, Rahbar K. [PSMA radioligand therapy in patients with advanced prostate cancer]. [Article in German]. Urologe A. 2020; 59:680–686. https://doi.org/10.1007/s00120-020-01205-w. [PubMed].
15. Gabriel M, Nilica B, Kaiser B, Virgolini IJ. Twelve-Year Follow-up After Peptide Receptor Radionuclide Therapy. J Nucl Med. 2019; 60:524–529. https://doi.org/10.2967/jnumed.118.215376. [PubMed].
16. Garg R, Mills K, Allen KJH, Causey P, Perron RW, Gendron D, Sanche S, Berman JW, Gorny MK, Dadachova E. Comparison of various radioactive payloads for a human monoclonal antibody to glycoprotein 41 for elimination of HIV-infected cells. Nucl Med Biol. 2020; 82–83:80–88. https://doi.org/10.1016/j.nucmedbio.2020.02.009. [PubMed].
17. Dawicki W, Allen KJH, Jiao R, Malo ME, Helal M, Berger MS, Ludwig DL, Dadachova E. Daratumumab-225Actinium conjugate demonstrates greatly enhanced antitumor activity against experimental multiple myeloma tumors. OncoImmunology. 2019; 8:1607673. https://doi.org/10.1080/2162402X.2019.1607673. [PubMed].