
Introduction
Hepatocellular carcinoma (HCC)—the most common type of primary liver cancer—is an aggressive cancer that spans more than 850,000 new yearly diagnoses and causes 800,000 annual deaths, representing the fifth most common cancer globally and the second most common cause of cancer-related death worldwide [1]. The incidence of HCC in the United States has tripled over the past three decades, and is projected to increase for the foreseeable future given the growing prevalence of HCC risk factors, including hepatitis B or C virus infection, obesity, diabetes mellitus, and excessive alcohol consumption [2]. HCC results from chronic liver disease, termed cirrhosis, with cancer developing at a 5-year incidence up to 30% in at-risk cirrhotic populations [3]. The prognosis for HCC patients is dismal, with an overall 5-year survival rate of 18.4% [4], and the increasing prevalence of liver cirrhosis ensures that HCC will continue to represent an important public health concern in the future [5].
A large number of rodent HCC models have been developed and utilized for preclinical research [6]. Despite their benefits, current HCC animal models have significant disadvantages that limit the testing of novel therapies and their translation to clinical practice. First, rodents are poor preclinical models of drug toxicity, sensitivity, and efficacy due to significant differences in xenobiotic receptors and drug metabolism [7]. This factor is of immense importance as less than 8% of drugs translate successfully from animal testing into Phase 1 clinical cancer trials [8]. Furthermore, the small size of rodents prohibits the testing of device-based tools and techniques widely employed in clinical practice. This is of significant consequence given the central role of locoregional therapies (LRTs) in HCC clinical management. The rabbit VX2 model has been considered the most relevant and widely used model to test HCC LRTs to date [9]. However, the VX2 model also has significant drawbacks, such as squamous cell origin, unknown tumor biology, internal necrosis, only peripheral vascularization, and varying tumor kinetics [10]. As such, there is a crucial need for more clinically relevant large animal models that faithfully recapitulates human HCC to address unmet clinical needs and serve as a bridge between murine studies and clinical practice.
This study describes utilization of the Oncopig Cancer Model for development of a clinically relevant, translational porcine HCC model. The Oncopig Cancer Model is a transgenic pig model that develops site and cell specific tumors following Cre recombinase induced expression of heterozygous KRASG12D and TP53R167H transgenes [11]. The large size of the pig and its similarities with humans in terms of anatomy, physiology, metabolism, immunity, and genetics make it an ideal model species for development of a large animal cancer model. Development of Oncopig HCC cell lines has been previously described [12], however, prior work was limited to characterization of HCC cell lines derived from three Oncopigs, minimal in vitro and in vivo profiling, and no description of intrahepatic tumors. As such, this study was undertaken to test the hypothesis that phenotypically consistent Oncopig HCC cells that faithfully recapitulate the in vitro features of human HCC can be developed across a large Oncopig cohort, and that these cells can be utilized to develop clinically relevant intrahepatic HCC tumors in Oncopigs.
Results
Oncopig HCC cells recapitulate in vitro features of human HCC cells
HCC cell lines were successfully developed from 15 Oncopigs by exposure of isolated hepatocytes to Cre recombinase. As we have previously demonstrated Oncopig primary hepatocytes do not express Oncopig transgenes and become apoptotic within 14 days of culturing [12], primary hepatocytes were not included in any downstream analyses. Characterization of Oncopig HCC cells confirmed expression of KRASG12D and TP53R167H, sustained propagation in vitro, positive arginase-1 staining (median 100%, range 88–100% purity), and positive KRASG12D staining (Figure 1A, 1B). Oncopig HCC cell lines displayed cell cycle lengths that mirrored human HCC cell lines (HepG2, Hep3B, Huh7, SNU-387, and SNU-475; Figure 1C and Supplementary Figure 1). In a wound healing assay, Oncopig HCC cell lines exhibited comparable time to half gap closure as Hep3B cell line (Figure 1D). Oncopig HCC cell lines produced alpha fetoprotein (AFP) at similar levels as HepG2 cells (Figure 1E), demonstrating the potential utility of serum AFP as a biomarker for tumor growth in the Oncopig HCC model, similar to clinical practice [13]. Together, these data demonstrate the generation of 15 distinct Oncopig HCC cell lines displaying consistent in vitro features similar to human HCC cells.
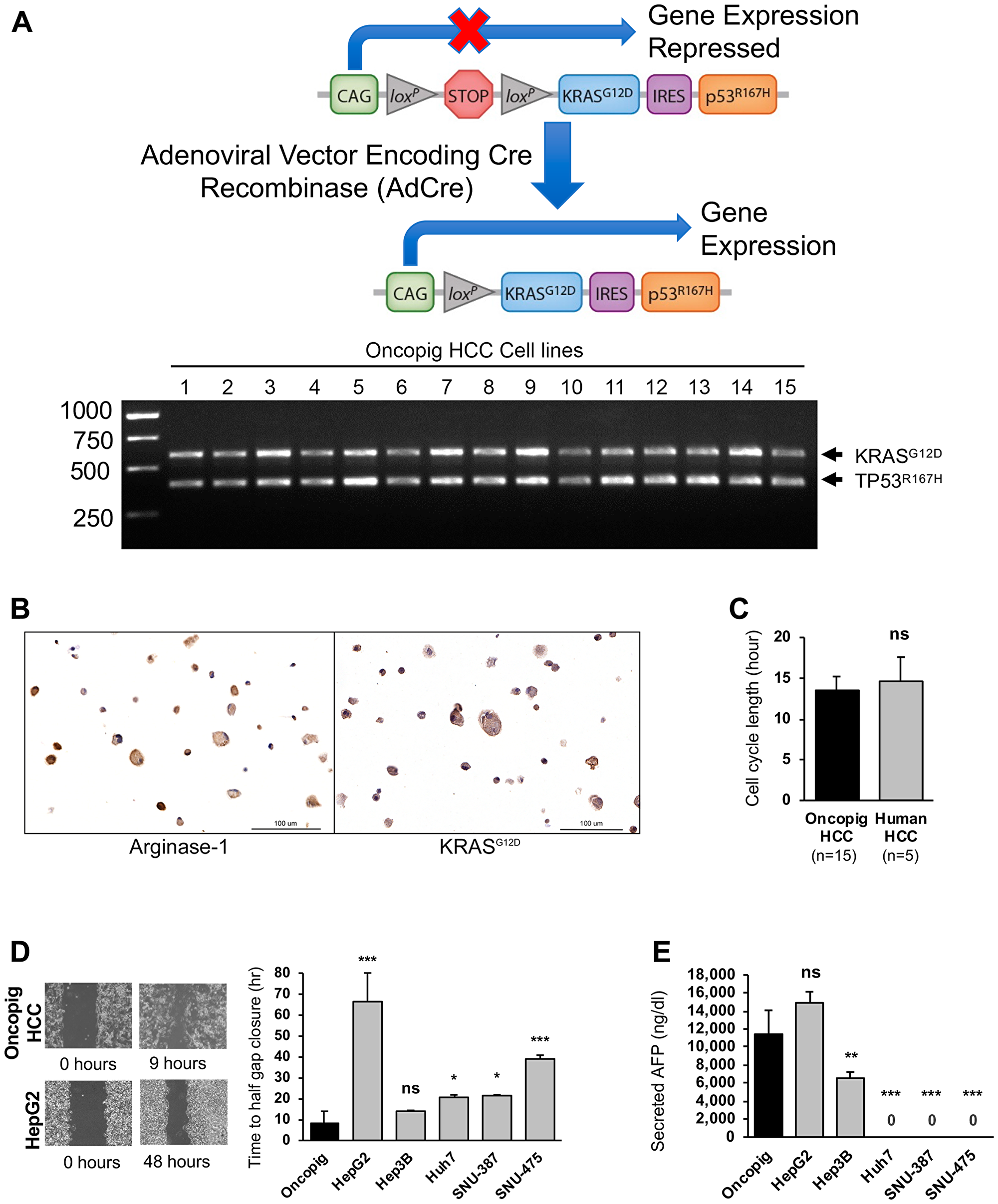
Figure 1: Oncopig and human HCC in vitro phenotypes. (A) Schematic of Oncopig transgene construct and agarose gel electrophoresis of RT-PCR products confirming Oncopig transgene (KRASG12D and TP53R167H) expression following exposure to AdCre. (B) Positive arginase-1 and KRASG12D staining (brown) of cultured Oncopig HCC cell lines (20×). (C) Oncopig and human HCC cell cycle lengths. (D) Representative cell migration images depicting faster gap closure in Oncopig compared to HepG2 and half gap closure rates for Oncopig (n = 15 cell lines) and human HCC cells. (E) AFP secretion from Oncopig (n = 15 cell lines) and human HCC cells. Huh7, SNU-387, and SNU475 are known non-AFP producing cell lines. ns = non-significant, *denotes p-value < 0.05, **denotes p-value ≤ 0.01, ***denotes p-value ≤ 0.0001.
Oncopig HCC is predictive of human HCC chemotherapeutic susceptibility
As comparative expression of genes involved in drug metabolism and transport between animal models and humans can predict similarities in treatment responses [14], expression levels of key drug metabolizing enzymes and transporters were compared in human (HepG2, Huh7, and Hep3B) and Oncopig HCC cells. Similar expression of the uptake transporter SLC22A1, the efflux pump ABCB1, as well as the drug metabolizing enzyme UGT1A1 [15–17], were observed between Oncopig and human HCC cell lines (Figure 2A). In contrast, reduced expression of the phase 1 sorafenib metabolizing enzyme CYP3A4 (porcine homologue CYP3A39) [18, 19] and increased expression of CRB1, which is involved in doxorubicin metabolism [20] were observed in Oncopig cell lines compared to human HCC cell lines (Figure 2A).
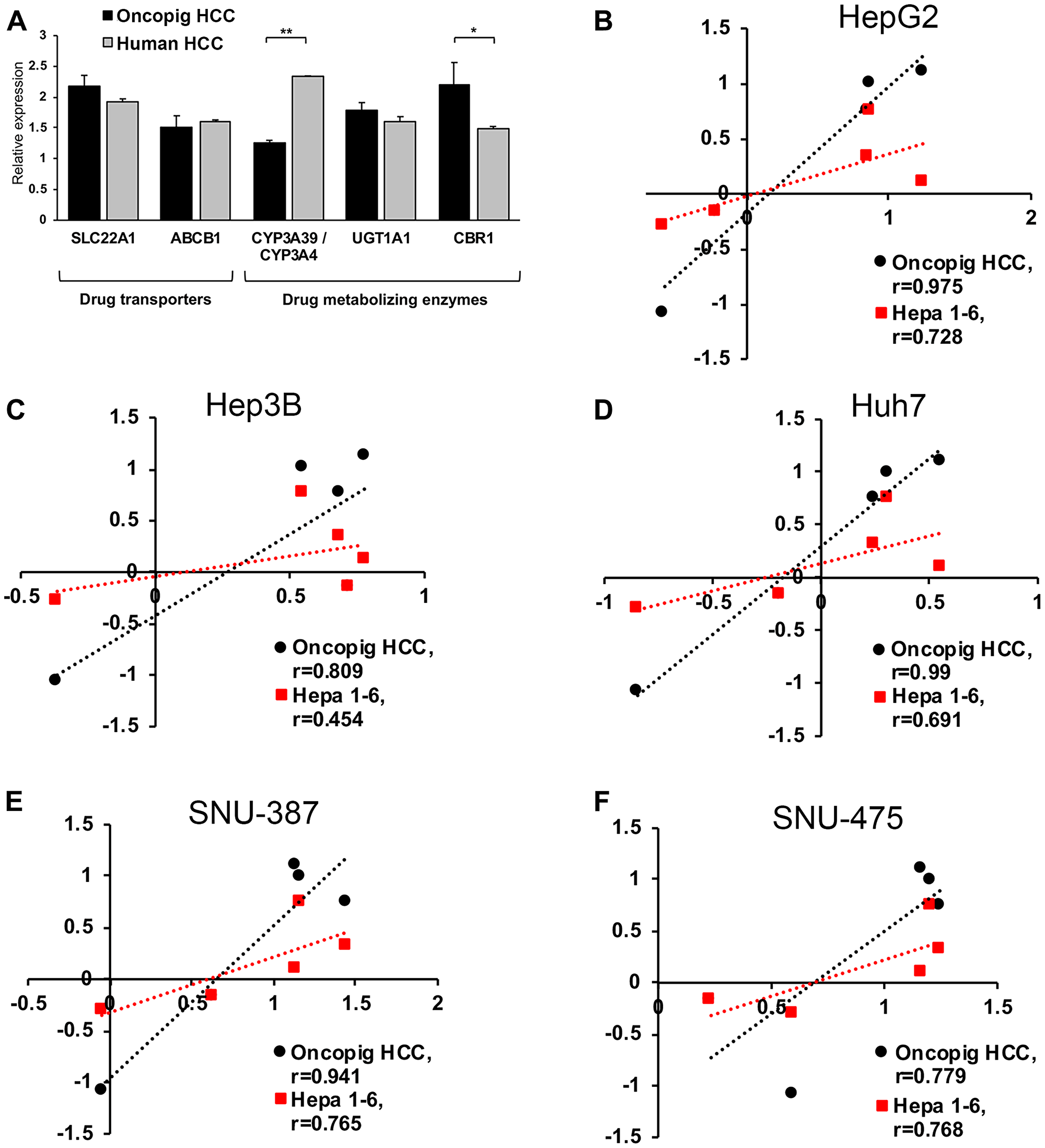
Figure 2: Oncopig, human, and murine HCC in vitro chemotherapeutic susceptibility. (A) Gene expression levels in Oncopig (n = 3 cell lines) and human HCC cells (HepG2, Huh7, and Hep3B). (B–F) Correlation analysis of logIC50 values demonstrating more similar in vitro chemotherapeutic responses between Oncopig and human compared to murine Hepa1-6 and human HCC cells. Chemotherapeutic response of each HCC cell line towards sorafenib, doxorubicin, cisplatin, mitomycin C, and 5-FU was determined. Pearson correlation between logIC50 in Oncopig HCC cells or murine Hepa1-6 cells and the following human HCC cells was analyzed: (B) HepG2, (C) Hep3B, (D) Huh7, (E) SNU-387, and (F) SNU-475. *denotes P < 0.05, **denotes P ≤ 0.001.
To evaluate the ability of the Oncopig HCC model to predict human HCC therapeutic responses, Oncopig, human, and murine HCC cell line chemotherapeutic susceptibility was tested for agents clinically employed for locoregional (doxorubicin, cisplatin, and mitomycin C) and systemic (sorafenib) HCC treatment [21–23], in addition to 5-fluorouracil (5-FU), which is relatively ineffective for HCC treatment [24]. Oncopig HCC cell lines (n = 6) displayed consistent susceptibility to both sorafenib and doxorubicin (Supplementary Figure 2A, 2B). In addition, Oncopig HCC cell line log half maximal inhibitory concentrations (logIC50) values were highly correlated with logIC50 values for all five human HCC cell lines (Pearson’s r = 0.779–0.990; Figure 2B–2F and Supplementary Table 1), suggesting Oncopig HCC chemotherapeutic responses are highly predictive of human responses. In addition, Oncopig HCC responses were more predictive than the murine HCC line Hepa1-6, as evidenced by higher correlation coefficients for Oncopig compared to murine HCC cells for all 5 human comparisons (Figure 2B–2F). Importantly, while Oncopig HCC cells correctly predicted resistance of human HCC to 5-FU treatment, murine HCC cells displayed markedly increased susceptibility to 5-FU (Supplementary Figure 2C). Together, these results demonstrate similar expression of key genes involved in drug metabolism and transport between Oncopig and human HCC, and the ability of Oncopig HCC cells to predict human HCC chemotherapeutic susceptibility in vitro, providing a rationale for utilization of the Oncopig HCC model as a translational model to bridge the gap between murine and human studies.
Oncopig HCC cells produce viable SCID mouse xenografts
To confirm in vivo tumorigenicity, Oncopig HCC cell lines (n = 15) were injected subcutaneously (SQ) into severe combined immunodeficiency (SCID) mice. In total, 68 SQ Oncopig HCC tumors were successfully developed (median 4, range 3–8 tumors/cell line; Figure 3A, 3B). Tumors measured median 6.7 × 5.4 mm in size at 21-days post injection. Histologic evaluation confirmed neoplastic masses comprised of spindle shaped malignant cells displaying arginase-1 positivity (Figure 3C, 3D). In addition, whereas control untransformed Oncopig tissues did not produce AFP, Oncopig xenograft HCC tumors produced AFP (Figure 3E), further confirming their identity as HCC tumors.
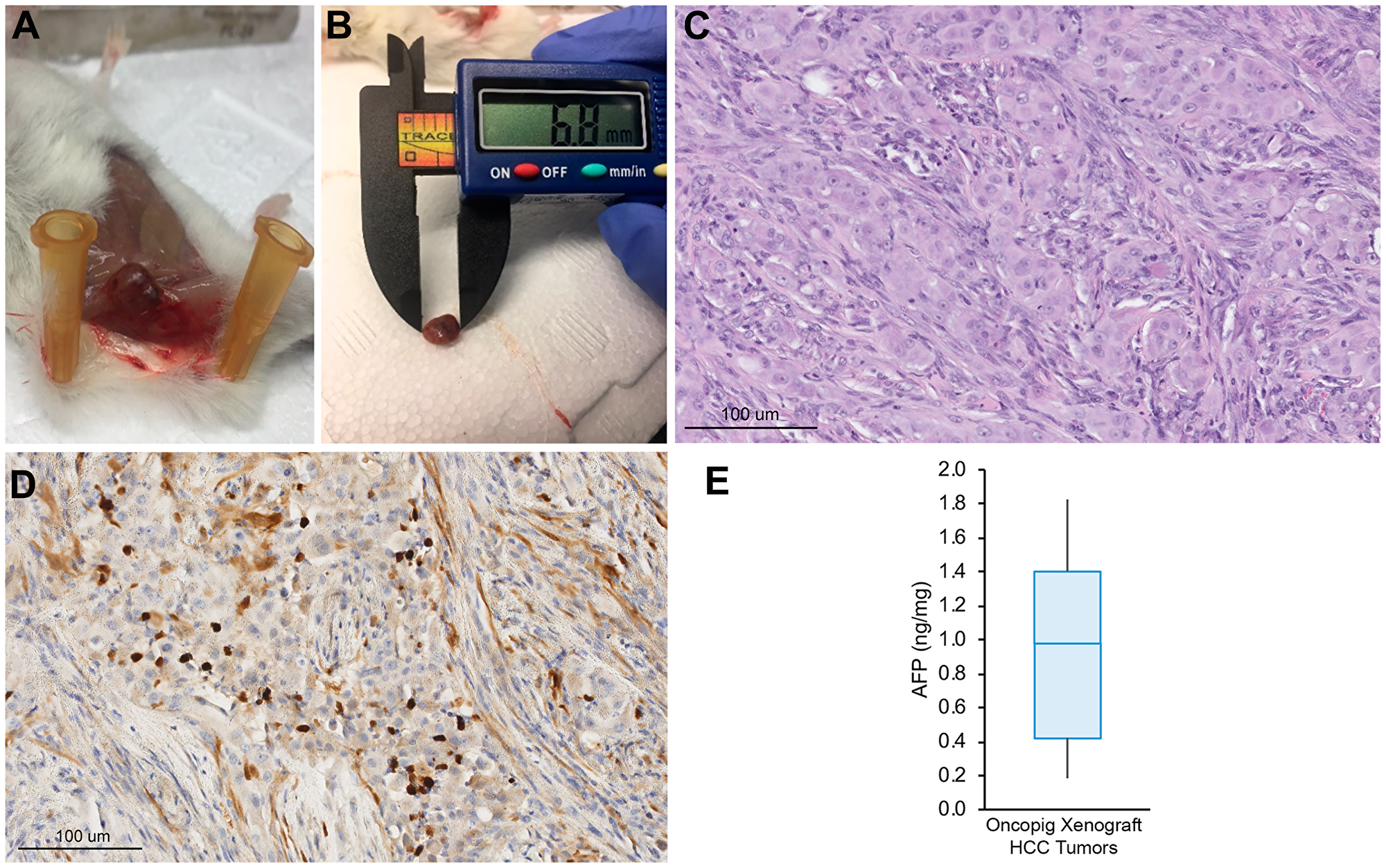
Figure 3: Oncopig HCC xenograft tumor development. (A) Representative SQ Oncopig HCC xenograft tumor. (B) Excised Oncopig HCC xenograft tumor. (C) H & E (20×) of Oncopig HCC xenograft tumor reveals densely cellular subcutaneous nodule with interspersed fat cells. Intervening fibrous vascular septae noted. (D) On arginase-1 IHC (20×), epithelial cells show focal arginase-1 expression (brown) consistent with hepatocellular differentiation. (E) AFP expression across Oncopig HCC xenograft tumors (n = 10).
Oncopig HCC cell lines reproducibly generate autograft SQ tumors
In order to confirm the reproducibility of our previous report demonstrating development of a single SQ Oncopig HCC tumor [12], autologous SQ injection of Oncopig HCC cells was performed across 13 Oncopigs, which resulted in successful tumor formation at a rate of 68% (34/50) per injection and 85% (11/13) per pig (Figure 4A–4C), reaching a median size of 16 × 13 mm within 2-weeks post injection. Histologic evaluation confirmed neoplasm in all cases, characterized by malignant epithelial cells with invasion into adjacent skeletal muscle, regions of inflammation, and positive arginase-1 and KRASG12D staining (Figure 4D). Oncopig SQ HCC tumors also produced AFP (Figure 4E), further confirming their identity as HCC tumors. Control Oncopig tissues were negative for AFP production.
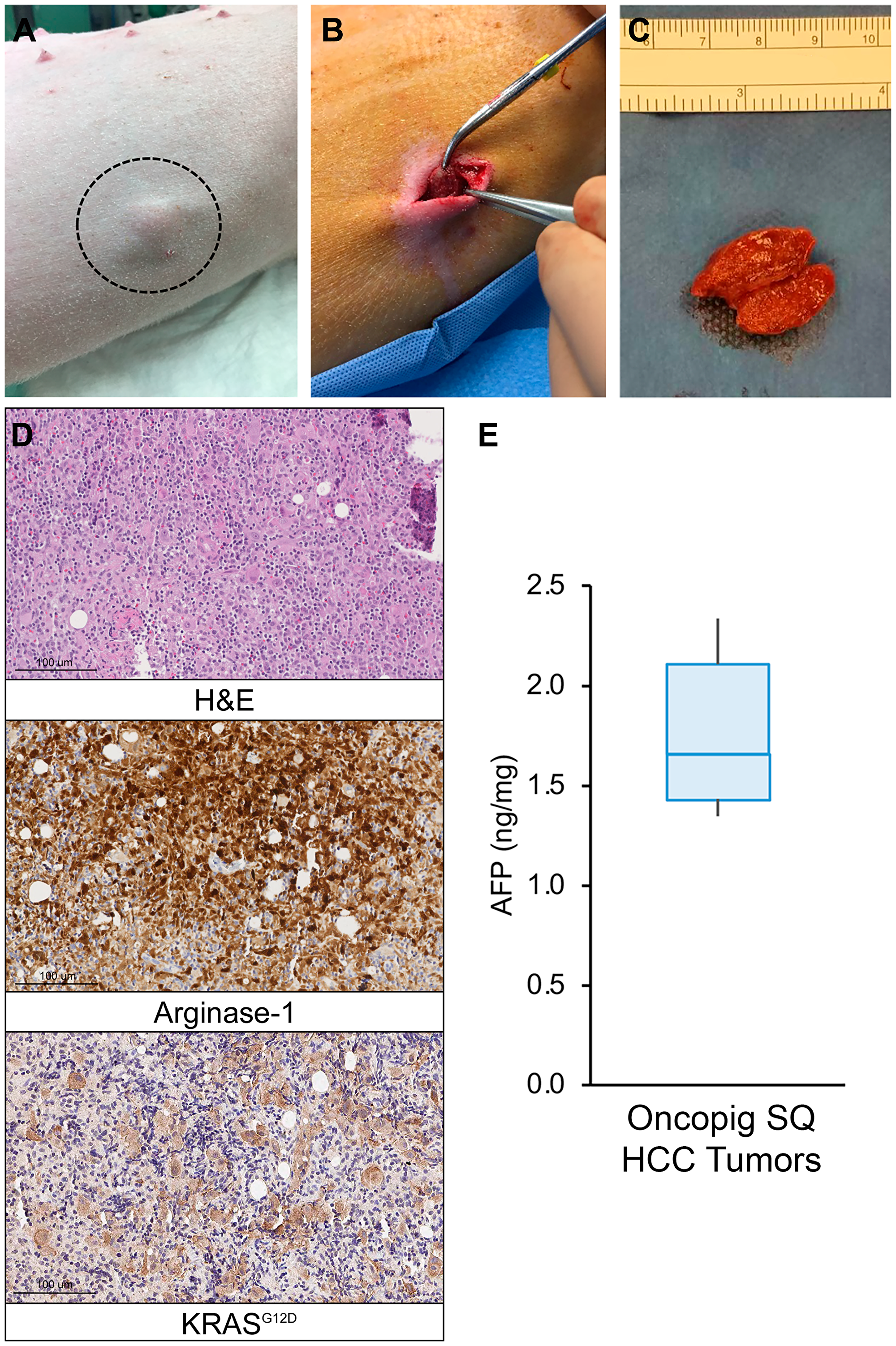
Figure 4: Oncopig SQ HCC autograft formation. (A) Photograph of visible SQ HCC tumor (circled) in Oncopig flank. (B) Excision of 2.0 cm SQ HCC tumor. (C) Excised and transected SQ HCC tumor. (D) H & E (20×) of Oncopig SQ HCC tumor demonstrates prominent, dispersed, pleomorphic large atypical cells, 5–10× the size of lymphocytes, flanking fibrous vascular septae, surrounded by dense mixed immune cell infiltrates. Arginase-1 IHC (20×) shows that these atypical cells show patchy arginase-1 expression (brown) consistent with hepatocellular differentiation. KRASG12D IHC (20×) confirms KRASG12D expression (brown) consistent with malignancy. (E) AFP expression across Oncopig SQ HCC tumors (n = 6).
Reproducible development of Oncopig intrahepatic HCC tumors
Following confirmation of successful, reproducible Oncopig SQ HCC tumor formation, Oncopig intrahepatic HCC tumor development was performed via autologous engraftment of SQ tumor fragments into Oncopig livers following 15 days of SQ tumor growth. Consistent with findings in murine HCC models demonstrating improved tumor growth when implanting HCC cells into cirrhotic compared to non-cirrhotic livers [25], attempts to develop intrahepatic tumors in healthy Oncopig livers were unsuccessful (n = 3). Therefore, in order to allow for development in a clinically relevant liver microenvironment, alcoholic liver fibrosis was induced immediately prior to engraftment in a single Oncopig. The Oncopig underwent biweekly ultrasound surveillance for intrahepatic tumor formation, which resulted in identification of a 1.0 cm mass 4-weeks post engraftment (Figure 5A). Intrahepatic HCC tumor formation was confirmed 5 days later via computed tomography (CT) scan (Figure 5B). The Oncopig was then euthanized and tumor samples were collected for histological and genomic analyses (Figure 5C). Histological evaluation confirmed an HCC tumor showing architectural distortion characterized by expansion of liver cords, nuclear pleomorphism, anisonucleosis, and nodular fibrosis in a background of dense collagen bands within adjacent non-tumorous liver consistent with METAVIR grade 2–3 fibrosis (Figure 5D), as well as arginase-1 and KRASG12D positivity (Figure 5E).
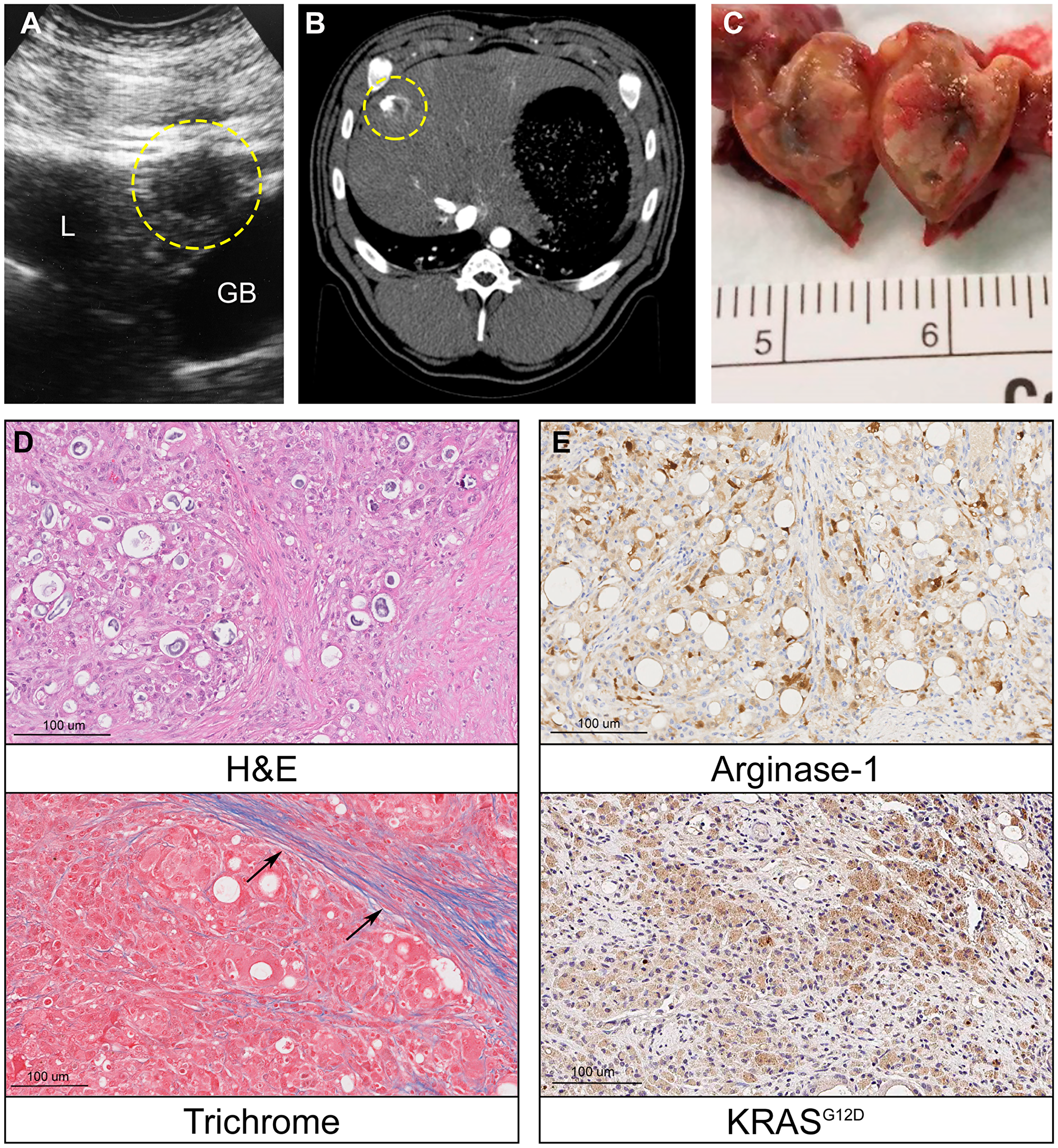
Figure 5: Oncopig intrahepatic HCC tumor formation. (A) Liver ultrasound depicting a hypoechoic 1 cm round intrahepatic HCC tumor (circled, L = liver, GB = gallbladder). (B) Contrast enhanced liver CT depicts same HCC tumor (circled). (C) Photograph of transected intrahepatic HCC tumor. (D) H & E (20×) of Oncopig intrahepatic HCC tumor reveals architectural distortion characterized by expansion of liver cords, nuclear pleomorphism, anisonucleosis, and nodular fibrosis. Masson’s trichrome of adjacent non-tumorous liver demonstrates dense collagen bands (arrows) consistent with METAVIR grade 2-3 fibrosis. (E) Arginase-1 IHC (20×) shows patchy arginase-1 expression (brown) consistent with hepatocellular differentiation. KRASG12D IHC (20×) confirms KRASG12D expression (brown) consistent with malignancy.
Following confirmation of successful Oncopig intrahepatic HCC tumor formation, the ability to reproducibly develop Oncopig intrahepatic HCC tumors was confirmed by performing HCC cell line development, SQ injection, and concurrent liver fibrosis induction and engraftment of SQ tumor fragments into the liver of 2 additional Oncopigs following 11 days of SQ tumor growth. Consistent with the first experiment, imaging assessment with ultrasound and CT resulted in identification of liver tumors. These included a hypoechoic intrahepatic mass measuring 1.0 cm 4-weeks post engraftment in the first Oncopig (Supplementary Figure 3A) that was not visible on subsequent follow-up, and a hypoechoic liver mass measuring 0.6 cm 2-weeks post engraftment that grew to 1.4 cm 4-weeks post engraftment in the second Oncopig (Supplementary Figure 3B, 3C). Finally, a hypervascular liver tumor measuring 1.0 cm was observed 10-weeks post tumor engraftment in the second Oncopig (Supplementary Figure 3D). Together, these results demonstrate reproducible development of Oncopig intrahepatic HCC tumors of clinically relevant sizes, in addition to the ability to image and characterize using clinically relevant modalities.
Genomic signatures of Oncopig intrahepatic HCC
In order to assess the ability of Oncopig HCC to mimic human HCC genomic signatures observed clinically, five spatially distinct tumor biopsies from the intrahepatic HCC tumor depicted in Figure 5C underwent whole-genome sequencing. Consistent with the relatively young age of the tumor (5 weeks old), copy number calling showed a mostly copy-neutral tumor (Figure 6A). In total 5,257 single nucleotide variants (SNVs) were identified, with 337 variants common between each tumor sample, and between 249 to 385 SNVs unique to each sample (Figure 6B and Supplementary Figure 4), indicating the presence of intratumor heterogeneity resulting from the accumulation of somatic mutations in distinct tumor cells as commonly observed in human HCC. To assess whether the SNVs resulted from mutational processes observed in human HCC, Oncopig mutational profiles were decomposed into mutational signatures, resulting in identification of 11 COSMIC v2 signatures (Figure 6C). Of these, signature 1 (33.7%) is observed in almost all human tumors, while signatures 12 and 17 (4.7% and 3.5%, respectively) are associated with human HCC. Finally, 133 variants were found to occur in 89 driver genes, all of which are known to be mutated in human HCC (Supplementary Table 2). Although these variants did not occur in coding regions, studies have suggested that noncoding mutations can affect gene regulation and may be important to elucidate mechanisms of tumorigenesis [26]. In summary, intratumor heterogeneity and SNV signatures previously identified in human HCC were observed in Oncopig intrahepatic HCC, indicating Oncopig HCC tumors resemble human HCC at the genomic level.
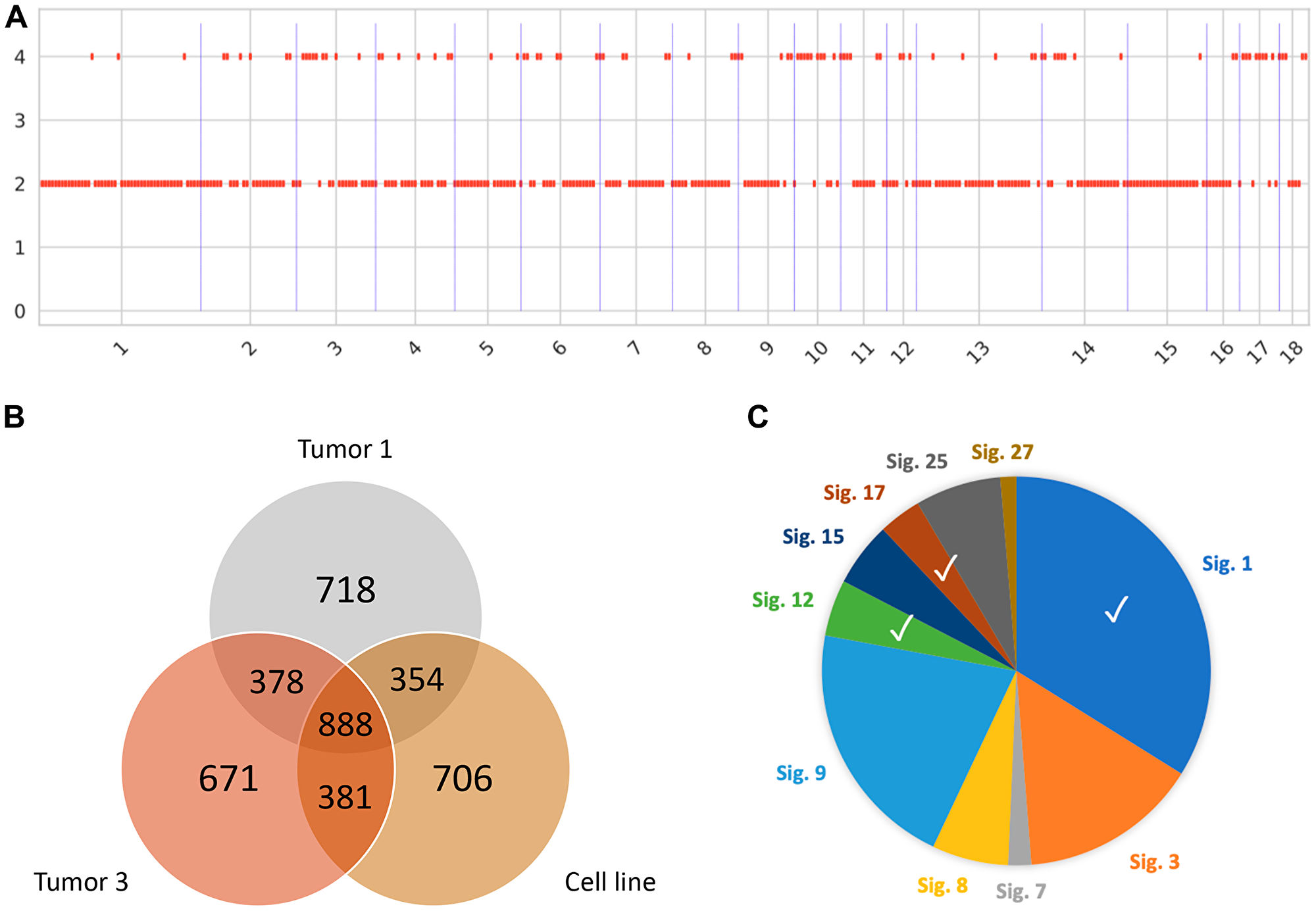
Figure 6: Genomic signatures of Oncopig HCC. (A) Somatic copy-number calling reveals a largely copy-neutral tumor in line with the young age of the tumor. (B) Representative venn diagram showing distribution of SNVs in the cell line and 2 out of 5 tumor samples. (C) Mutational signatures identified resemble signatures observed in human HCC tumors (Signatures 1, 12, and 17).
Oncopig HCC cell lines are genetically manipulatable
Advances in animal modeling and gene editing provide an opportunity to develop genetically tailored tumors. This enables investigation of the contribution of clinically relevant driver mutations on tumor progression and treatment susceptibility, as well as preclinical testing of novel precision medicine approaches. As a first step towards generation of genetically tailored Oncopig HCC tumors, we tested our ability to knockout (KO) the Oncopig TP53R167H and KRASG12D driver mutations using CRISPR-Cas9 (Figure 7A). Oncopig HCC cells were successfully edited at a rate of over 80% (Figure 7B) with insertions or deletions (INDELs) occurring around the predicted cleavage sites (Supplementary Figure 5A, 5B). In addition, simultaneous targeting of TP53R167H and KRASG12D resulted in deletion of the region between the two gRNAs (Figure 7A, 7B) which was confirmed via Sanger sequencing (Supplementary Figure 5C). While the percentage of TP53R167H edited cells was maintained in culture for up to two weeks, the proportion of cells harboring KRASG12D edits decreased over time (Figure 7B) suggesting KRASG12D is required for Oncopig HCC cell survival. Isolation and screening of 5 single cell clones from the TP53R167H edited cell pool resulted in development of two TP53R167H KO HCC cell lines harboring frameshift mutations (17 and 4 bp deletions) leading to protein truncation (Figure 7C). The parental and TP53R167H KO cell lines stained positive for arginase-1 (Figure 7D), confirming their identity as HCC cells. As expected, TP53R167H KO resulted in reduced cell proliferation compared to the parental line (Figure 7E) further demonstrating the ability to introduce genetic alterations with significant effects on malignant potential. These results suggest combining the Oncopig orthotopic HCC model with in vitro gene editing could lead to development of genetically tailored HCC tumors for investigating the contribution of driver mutations on clinically relevant cancer phenotypes and testing of novel precision medicine approaches.
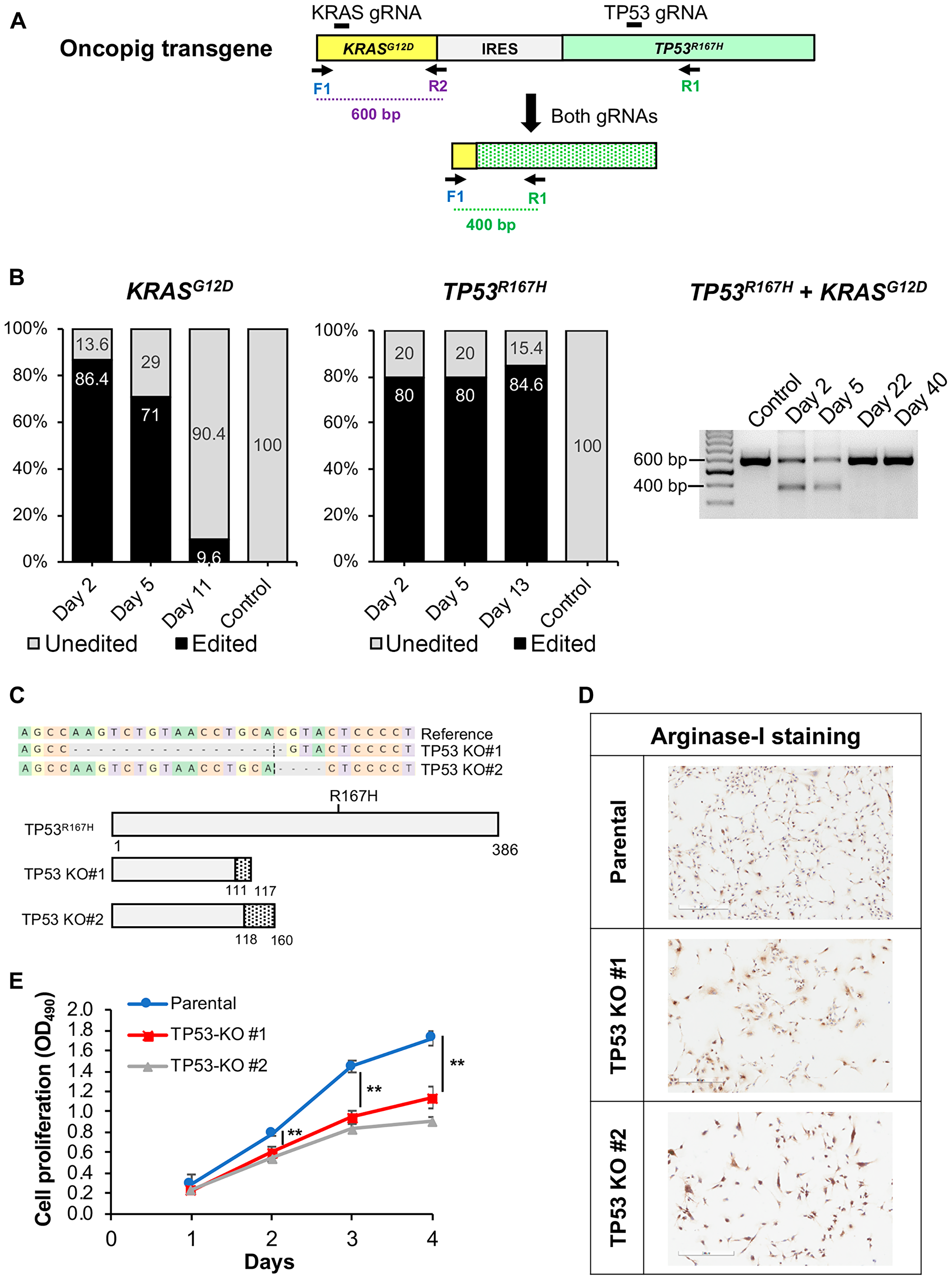
Figure 7: CRISPR/Cas9-mediated disruption of Oncopig KRASG12D and TP53R167H transgenes. (A) Schematic representation of the Oncopig transgene showing gRNA target sites and primers used for PCR. IRES, Internal ribosome entry site. (B) KRASG12D and TP53R167H editing efficiencies at multiple time points post transfection with Cas9 and gRNAs. (C) Frameshift mutations resulting in protein truncation for 2 Oncopig TP53R167H KO HCC cell lines developed via single cell clone isolation and screening. Dashed line marks the cleavage position, and dashed grey boxes represent nucleotide deletions. Dotted regions represent frameshifts in predicted protein sequences. (D) Positive arginase-1 staining (brown) of parental and TP53R167H KO cell lines (scale bar, 300 μm). (E) Cellular proliferation of Oncopig parental and TP53R167H KO HCC cell lines. Values represent mean ± S. D. (n ≥ 3). **indicates P < 0.001.
DISCUSSION
Advances in cancer care are dependent upon the use of preclinical in vivo model systems to test new diagnostic and imaging modalities, therapeutic strategies, and improve treatment outcomes. Due to the limitations of current HCC animal models, more clinically relevant large animal HCC models are required to more effectively test novel LRTs and other therapies for translation to clinical practice. This study presents a genetically adjustable, reproducible Oncopig HCC model that closely predicts human HCC chemotherapeutic responses. Expanding on previous reports [12], reproducible generation of Oncopig SQ HCC tumors via autologous injection of Oncopig HCC cells is demonstrated, in addition to engraftment of SQ tumor fragments into the liver resulting in reproducible development of intrahepatic HCC tumors that reach clinically relevant sizes within 1–2 months and are trackable using clinically employed imaging modalities. Although alcohol-induced liver fibrosis was observed in liver adjacent to intrahepatic tumors, it did not reach an irreversible cirrhotic state (METAVIR grade 4), indicating further work is required to increase the severity of liver disease in this model. This result is further confirmed by our previous publication demonstrating alcohol-induced liver fibrosis disease severity peaks at 8-weeks post induction, with fibrosis levels reducing to METAVIR grade F1–F2 by 20-weeks post induction [27]. Liver fibrosis recovery during tumor development could also explain the lack of sustained intrahepatic tumor growth observed over the 10-week observation period, although further studies in a larger cohort are required to confirm this hypothesis.
The successful development and implementation of novel therapeutic strategies for HCC is critically dependent on the capability to screen, test, and apply such approaches across the in vitro to in vivo continuum. The presented in vitro chemotherapy results support the concept that the Oncopig HCC model can be used to screen and test promising drugs with results translatable to clinical trials, and subsequently clinical practice. The Oncopig HCC model therefore represents an ideal platform that permits in vitro therapeutic screening using HCC cell lines with the ability to translate promising strategies to in vivo testing in a clinically relevant large animal model. In addition, as pigs are commonly used for toxicology studies [28], the Oncopig HCC model provides the opportunity to simultaneously test safety and efficacy in the same animal. However, as in vivo chemotherapeutic responses were not assessed in this study, it is unclear whether Oncopig HCC in vitro chemotherapeutic responses are representative of in vivo responses. This represents a limitation of the current study, highlighting the need for future studies assessing Oncopig intrahepatic HCC tumor chemotherapeutic susceptibility.
While the Oncopig HCC model is not the only porcine HCC model reported to date, it does provide significant advantages over previously published models. Autochthonous HCC has been developed in chemically induced porcine models [29–31]; however, such models take 1–2 years to develop clinically relevant tumors and do not allow for control of tumor number, location, underlying genetics, or comorbidities, rendering them potentially less suitable for preclinical and co-clinical trials. On the other hand, the Oncopig HCC model presented allows for development of genetically defined intrahepatic HCC tumors of clinically relevant size in animals as young as 4–5 months of age. In addition, Oncopig HCC tumors recapitulate human histologic and molecular features, including arginase-1 staining and AFP production. Another potentially significant advantage of the Oncopig HCC model is highlighted by our successful development of genetically defined Oncopig HCC cell lines. This work provides proof-of-concept validation for combining the Oncopig orthotopic HCC model with in vitro gene editing to develop genetically tailored HCC tumors that can be used for investigating the contribution of driver mutations on cancer phenotypes and testing of novel precision medicine approaches. In addition, the large size and segmental nature of the porcine liver enables development of several distinct spatially separated tumors in the same animal. Hence, one animal can be used to investigate therapeutic effectiveness on tumors bearing differential mutational profiles. Further studies are required to assess the ability to develop genetically tailored HCC tumors through knockout of tumor suppressor genes or introduction of activating oncogenic mutations.
In summary, the Oncopig HCC model offers a novel, physiologically and anatomically relevant cancer model for which a multitude of innovative therapeutic modalities can be applied and tested while significantly reducing the costs, confounding variables seen in human subjects, and lengthy conduct of human clinical trials. Importantly, the Oncopig can be utilized to conduct correlative studies for more efficient and consistent investigation of new therapies. Its size allows for utilization of the same methods and instruments used in human clinical practice, including CT and magnetic resonance imaging technologies. This model is thus amenable to developing and establishing medical imaging standards related to diagnosing HCC tumors and tracking treatment response using accepted radiologic criteria, a critical facet of therapeutic discovery and validation. Importantly, the Oncopig is also immunocompetent, lending itself to investigation of immunotherapies [32]. Therefore, the Oncopig fulfills the currently unmet clinical modeling needs for HCC, particularly for pilot investigations of experimental therapies or experimental therapeutic combinations not feasible in human subjects.
Materials and Methods
Animal subjects
This study was completed at the University of Illinois at Urbana-Champaign (UIUC) and the University of Illinois at Chicago (UIC). Institutional Animal Care and Use Committee approval was obtained at both sites (UIUC: protocol #16-065 approved 6/23/2016, UIC: protocol #16-090 approved 7/13/2016). All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals. Seventeen female Oncopigs were utilized for this study.
Oncopig HCC cell line development
Oncopig HCC cell lines were developed as previously described [12]. KRASG12D and TP53R167H expression was confirmed by RT-PCR using primers listed in Supplementary Table 3 as previously described [12].
Cell culture
Human (HepG2, Hep3B, Huh7, SNU-387, and SNU-475; ATCC, Manassas VA), murine (Hepa1-6; ATCC), and Oncopig HCC cell lines were maintained in DMEM (or DMEM/F12 for HepG2) supplemented with 10% FBS and 1% Penicillin-Streptomycin.
Proliferation assays
Oncopig and human HCC cell cycle lengths were analyzed using the Cell Trace CFSE Cell Proliferation Kit (Thermo Fisher Scientific, Waltham MA, USA) following the manufacturer’s instructions. Cell cycle length was calculated using linear regression estimation from a percent fluorescence versus time plot. The proliferation of Oncopig HCC TP53R167H knockout cells was determined using the CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (#G3580; Promega, Madison WI, USA) following the manufacturer’s instructions.
Cell migration
Cell migration was analyzed by plating 3 × 104 HCC cells into each chamber of a cell culture insert (Ibidi #81176, Munich, Germany). The cell culture insert was removed 24-hours post seeding, leaving a defined cell-free gap of 500 μm between confluent cell monolayers. Images were taken at 10× magnification at multiple time points until gap closure. Image analysis was performed using the ImageJ [33] MRI Wound Healing Tool plugin (NIH, Rockville MD, USA). Gap area was quantified at each time point, and time to half gap closure was calculated using linear regression estimation from a gap area versus time plot.
In vitro chemotherapeutic susceptibility
The sensitivity of Oncopig, human, and murine HCC cell lines was tested for five chemotherapeutic agents: sorafenib (Bayer, Leverkusen Germany), doxorubicin (Pfizer Inc., New York NY, USA), cisplatin (Sigma-Aldrich, St. Louis MO, USA), mitomycin C (Accord Healthcare Inc., Durham NC, USA), and 5-FU (Acros Organics, Geel Belgium). Briefly, 1 × 104 cells/well (2 × 104 for HepG2) were seeded in 96-well plates. The following day, culture medium was replaced with fresh medium supplemented with each chemotherapeutic agent at 8-point serial dilutions. The following drug concentrations were used to assay clinically relevant administered dosages [34, 35], and to allow calculation of the IC50: sorafenib and mitomycin C: 0.5–100 μM, doxorubicin: 0.1–20 μM, cisplatin: 1–200 μM, and 5-FU: 1–500 μM. Cell viability was assessed after 72 hours using a MTT assay (#V13154; Invitrogen, Carlsbad, CA, USA) following the manufacturer’s recommendations using a BioTek 800 TS Absorbance Reader (BioTek, Winooski, VT, USA). IC50 values were determined by nonlinear regression analysis using GraphPad Prism 8 (GraphPad, San Diego, CA, USA) from plots of relative percent viability versus log10 drug concentration.
Gene expression analysis
qPCR was performed to assess the expression of genes involved in drug uptake (human: SLC22A1; porcine homolog: SLC22A1), metabolism (human: CYP3A4, CBR1, UGT1A1; porcine homologs: CYP3A39, CBR1, UGT1A1) and export (human: ABCB1; porcine homolog: ABCB1) [15–20]. Total RNA was extracted from Oncopig and human HCC cell lines (HepG2, Huh7, Hep3B) using the All Prep DNA/RNA extraction kit (Qiagen, Germantown MD, USA) according to the manufacturer’s protocol. cDNA was synthesized from RNA using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific). qPCR was performed using primers listed in Supplementary Table 3 on the ABI Real Time PCR system (Thermo Fisher Scientific) using the Power SYBR Green Gene PCR Master Mix (Thermo Fisher Scientific). The relative expression level for each gene of interest was determined by normalizing to GAPDH.
Oncopig HCC xenografts
Ten- to 12-week-old female SCID mice (#001303 NOD. CB17-Prkdcscid/J; Jackson Laboratory, Bar Harbor ME, USA) were used for xenograft tumor generation. Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (5–10 mg/kg). The flanks were shaved and sterilized with alcohol. 1 × 107 Oncopig HCC cells suspended in 50 μL serum free DMEM were SQ injected into each flank using a 21-gauge needle. Mice were sacrificed in a carbon dioxide (CO2) chamber followed by cervical dislocation at 21-days post-inoculation. Tumors were harvested by incising the overlying skin, and carefully freeing tumors from SQ tissues using blunt dissection. Tumors were formalin fixed for histologic processing, or homogenized in PBS for AFP analysis.
Oncopig HCC SQ autografts
Oncopigs were anesthetized by intramuscular administration of telazol (2.6–4.4 mg/kg), ketamine (1.3–2.2 mg/kg), and xylazine (1.3–2.2 mg/kg). The flanks were shaved and sterilized with betadine and alcohol scrub. 1 × 107 Oncopig HCC cells were suspended in 100 μL PBS and autologously injected SQ into 1–6 flank sites per pig (median age 83, range 60–126, days). Oncopigs were euthanized at median 28 (range 21–77) days post SQ tumor inoculation. Tumors were extracted and formalin fixed for histologic processing, or homogenized in PBS for AFP analysis.
Oncopig intrahepatic HCC tumor development
SQ Oncopig HCC tumor fragments were autografted into the liver following alcohol-induced fibrosis induction in three Oncopigs 11–15 days post SQ injection. Fibrosis was induced by hepatic transarterial infusion of 0.75 mL/kg ethanol-ethiodized oil (1:3 v/v) as described previously [27]. Intrahepatic tumor induction was performed 30 minutes after fibrosis induction. A flank mass was surgically excised, and three 3- to 4-mm3 tumor fragments were harvested. Next, the accessible right medial hepatic lobe was identified using ultrasound guidance. A 15-gauge needle with stylet (Echogenic Co-axial Introducer Needle; Argon Medical Devices, Wheeling IL, USA) was advanced percutaneously into the liver, and the Oncopig HCC fragments were pushed into the hepatic parenchyma using the stylet. Ultrasound imaging was performed to monitor for tumor growth and collect tumor biopsies, followed by multiphase contrast-enhanced CT to confirm tumor development (CT protocol: non-contrast, arterial phase at 30–35 seconds, portal venous phase at 60–70 seconds, and delayed phase at 180 seconds after 120–150 mL of iodinated contrast injected intravenously at a rate of 4–5 mL/s). Following CT confirmation, Oncopig subjects were euthanized and tumor masses were harvested, transected, and measured. Tumor samples were either stored in formalin for histologic processing, or flash frozen in liquid nitrogen and stored at –80°C for genomic analysis.
AFP quantification
AFP levels were determined using a porcine AFP ELISA kit (#MBS945039; MyBioSource Inc., San Diego CA, USA). 3 × 106 HCC cells in 2 ml medium were seeded into 6-well plates and incubated for 72 hours followed by withdrawal of 100 μl of supernatant for analysis. The assay was performed in triplicate for each cell line. For tumor AFP levels, 100 mg was homogenized in 1 ml PBS, followed by 2 freeze-thaw cycles to break the cell membranes. Homogenates were then centrifuged for 5 minutes at 5,000 × g at 4°C and the supernatants were assayed in triplicate immediately following the manufacturer’s protocol. Untransformed Oncopig brain, lung, kidney, and bladder tissues were used as negative controls.
Immunohistochemistry (IHC)
Oncopig HCC cells in a nearly confluent T75 cm2 flask were gently scraped in ice-cold PBS and centrifuged at 100 g at 4°C for 5 minutes. The pellets were re-suspended in 10% formalin and the cells were fixed for 4 hours at room temperature. Cells were then centrifuged at 100× g and the cell pellet was re-suspended in 2% agarose and stored at 4°C for 30 minutes, followed by the addition of 70% ethanol. Cell pellets and formalin fixed tumor samples were provided to the Research Histology and Tissue Imaging Core at the UIC for processing, embedding, sectioning, and staining. Oncopig HCC TP53R167H knockout cells were seeded in a chamber slide (#PEZGS0416; Millipore-Sigma, Burlington MA, USA) and incubated overnight. Cells were then washed with PBS, fixed with 10% formalin and provided for staining. Tissue sections were stained using hematoxylin and eosin (H&E). IHC was performed to detect arginase-1 (#ab91279; Abcam; Cambridge, UK) and KRASG12D (#ab221163; Abcam). Whole slides were scanned using a Hamamatsu Nanozoomer scanner (Hamamatsu Photonics, Hamamatsu City Japan), and digital images were visualized with NDP. view2 software (Hamamatsu Photonics). Cell line purity was quantified by determining the number of arginase-1 positive cells divided by total number of cells across three 40× microscopic regions. Blinded histopathological analyses were performed by a board-certified veterinary pathologist, and board-certified human pathologist with subspecialty training in Liver and Transplantation Pathology.
CRISPR-Cas9 gene editing
As the Oncopig transgenes are inserted as cDNA, gRNAs targeting an exon-exon junction in each transgene (TP53R167H or KRASG12D) were designed using CRISPOR web tool [36]. Each gRNA was synthesized by incubating equimolar ratios of Alt-R™ crRNA (Supplementary Table 3) and tracrRNA (#1072532; IDT Corporation, Chicago, IL, USA) at 95°C for 5 minutes and cooling to room temperature. Each gRNA was combined with purified S. pyogenes Cas9 nuclease (#1081058; IDT Corporation) to form a ribonucleoprotein (RNP) complex. Oncopig HCC cells were reverse transfected with 25 nM RNP complexes using the Lipofectamine CRISPRMAX kit (#CMAX00003; Invitrogen) following the manufacturer’s instructions.
Confirmation of gene editing
Genomic DNA was extracted from Oncopig HCC cells using QuickExtract DNA Extraction Solution (#QE09050; Lucigen, Middleton WI, USA) following the manufacturer’s instructions. The genomic locus that flanks the Cas9 target site was amplified by PCR using primers listed in Supplementary Table 3. PCR products were provided to the Genome Editing Core at UIC, where a second PCR was performed to attach Fluidigm adaptor and barcode sequences. Targeted sequencing was performed using a MiSeq (Illumina, San Diego, CA, USA) following the manufacturer’s instructions. Sequencing reads were analyzed using CRISPResso2 with default parameters [37].
Whole genome sequencing
DNA was extracted from Oncopig tissues and cell lines using the DNeasy Blood & Tissue Kit (Qiagen) following the manufacturer’s instructions. DNA (300 ng) was used to develop shotgun genomic libraries using the Hyper Library Construction Kit from Kapa Biosystems (Roche, Indianapolis, IN, USA) by the Roy J. Carver Biotechnology Center (University of Illinois, Urbana IL, USA) following standard protocols. Paired-end, 150 bp reads were generated by sequencing libraries on a NovaSeq 6000. All datasets are available in the NCBI Short Read Archive under accession number PRJNA599402.
Intratumor heterogeneity
Sequencing reads were aligned to the Sscrofa11.1 reference genome using BWA MEM v0.7.17 [38]. Duplicate reads were marked using the GATK 4.1 MarkDuplicates function [39]. Following alignment, Strelka v2.9.10 [40] was used for somatic SNV calling using a multi-sample workaround described by Strelka’s developers (https://github.com/Illumina/strelka/issues/59), retaining variants flagged as PASS in at least one sample. HATCHet was used for somatic copy number alteration (CNA) calling [41]. Mutational signature exposure analysis was performed on the union of SNVs across all tumor samples using SignaturesEstimation [42]. To investigate the presence of SNVs in driver genes, homologous porcine genes were identified for a subset of 723 human driver genes from the COSMIC v90 database [43]. SNVs in driver genes were identified using bedtools v2.28.0 [44] and annotated using SnpEff [45].
Statistical analysis
Comparison of Oncopig and human cell cycle length and gene expression was done using the Student’s t-test. Time to half gap closure, AFP secretion and cell proliferation at each time point were compared using 1-way analysis of variance (ANOVA). Pearson’s correlations were used to assess similarities of IC50 values across cell lines. Statistical analyses were performed using SPSS Statistics version 22 (SPSS Inc., Chicago IL, USA) and GraphPad Prism 8 (GraphPad, San Diego, CA, USA). P-values < 0.05 were considered statistically significant.
Abbreviations
HCC: hepatocellular carcinoma; LRT: locoregional therapy; UIC: University of Illinois at Chicago; RT-PCR: reverse transcription polymerase chain reaction; DMEM: Dulbecco’s Modified Eagle Medium; FBS: fetal bovine serum; CFSE: carboxyfluorescein succinimidyl ester; NIH: National Institutes of Health; 5-FU: 5-fluorouracil; DMSO: dimethylsulfoxide; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; IC50: half maximal inhibitory concentration; qPCR: quantitative polymerase chain reaction; RNA: ribonucleic acid; DNA: deoxyribonucleic acid; cDNA: complementary deoxyribonucleic acid; SCID: severe combined immunodeficiency; SQ: subcutaneous; CO2: carbon dioxide; PBS: phosphate buffered saline; AFP: alpha fetoprotein; CT: computed tomography; ELISA: enzyme-linked immunosorbent assay; IHC: immunohistochemistry; H&E: hematoxylin and eosin; CRISPR: clustered regularly interspaced short palindromic repeats; gRNA: guide ribonucleic acid; crRNA: CRISPR ribonucleic acid; tracrRNA: trans-activating CRISPR ribonucleic acid; RNP: ribonucleoprotein; bp: base pair; CNA: copy number alteration; SNV: single nucleotide variant; ANOVA: analysis of variance; KO: knockout; INDEL: insertion or deletion; AdCre: adenoviral vector encoding Cre recombinase.
ACKNOWLEDGMENTS
Histological services were provided by the Research Resources Center Research Histology and Tissue Imaging Core at the University of Illinois at Chicago, established with the support of the Vice Chancellor of Research. We acknowledge the UICentre for Drug Discovery at the University of Illinois at Chicago for consultation.
CONFLICTS OF INTEREST
Ron C. Gaba receives research support from Guerbet USA LLC, Janssen Research & Development LLC, the United States Department of Defense, and the United States National Institutes of Health. F. Edward Boas is a co-founder of Claripacs, LLC. He received research funding (investigator-initiated) from Guerbet. He received research support (investigator-initiated) from GE. He received research supplies (investigator-initiated) from Bayer. He received a research grant and speaker fees from Society of Interventional Oncology, which were sponsored by Guerbet. He attended research meetings sponsored by Guerbet. He is an investor in Labdoor, Qventus, CloudMedx, Notable Labs, and Xgenomes. He is the inventor and assignee on US patent 8233586, and is an inventor on US provisional patent applications 62/754,139 and 62/817,116. Regina M. Schwind receives research support from Guerbet USA LLC and Janssen Research & Development LLC. Lawrence B. Schook research support from Guerbet USA LLC, Janssen Research & Development LLC, the United States Department of Defense, and the United States National Institutes of Health. Kyle M. Schachtschneider receives research support from Guerbet USA LLC, Janssen Research & Development LLC, and the United States National Institutes of Health. The remaining authors—Lobna Elkhadragy, Sulalita Chaki, Hanna H. Chen, Mohammed El-Kebir, Kelly D. Garcia, Eileena F. Giurini, Grace Guzman, Francesca V. LoBianco, Mario F. Neto, Jordan L. Newson, Aisha Qazi, Maureen Regan, Lauretta A. Rund, Matthew C. Stewart, Faith M. Thomas, Herbert E. Whiteley, and Jiaqi Wu—have no reported conflicts of interest.
FUNDING
This work was supported by the United States Department of Defense (Translational Team Science Award CA150590), Guerbet USA LLC (Healthcare Professional Grant for ethiodized oil material transfer), and the University of Illinois Department of Radiology.
References
1. Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, Dandona L, Fleming T, Forouzanfar MH, et al; Global Burden of Disease Cancer Collaboration. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017; 3:524–548. https://doi.org/10.1001/jamaoncol.2016.5688. [PubMed].
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913–2921. https://doi.org/10.1158/0008-5472.CAN-14-0155. [PubMed].
3. Singal AG, El-Serag HB. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015; 13:2140–2151. https://doi.org/10.1016/j.cgh.2015.08.014. [PubMed].
4. National Cancer Institute Surveillance, Epidemiology, and End Results Program (NCI SEER). Cancer Stat Facts: Liver and Intrahepatic Bile Duct Cancer. 2019. Available from: https://seer.cancer.gov/statfacts/html/livibd.html.
5. Moon AM, Singal AG, Tapper EB. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin Gastroenterol Hepatol. 2019; S1542–3565:30849–3. https://doi.org/10.1016/j.cgh.2019.07.060. [PubMed].
6. Santos NP, Colaco AA, Oliveira PA. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumour Biol. 2017; 39:1010428317695923. https://doi.org/10.1177/1010428317695923. [PubMed].
7. Swanson KS, Mazur MJ, Vashisht K, Rund LA, Beever JE, Counter CM, Schook LB. Genomics and clinical medicine: rationale for creating and effectively evaluating animal models. Exp Biol Med (Maywood). 2004; 229:866–875. https://doi.org/10.1177/153537020422900902. [PubMed].
8. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014; 6:114–118. [PubMed].
9. Parvinian A, Casadaban LC, Gaba RC. Development, growth, propagation, and angiographic utilization of the rabbit VX2 model of liver cancer: a pictorial primer and “how to” guide. Diagn Interv Radiol. 2014; 20:335–340. https://doi.org/10.5152/dir.2014.13415. [PubMed].
10. Aravalli RN, Golzarian J, Cressman EN. Animal models of cancer in interventional radiology. Eur Radiol. 2009; 19:1049–1053. https://doi.org/10.1007/s00330-008-1263-8. [PubMed].
11. Schook LB, Collares TV, Hu W, Liang Y, Rodrigues FM, Rund LA, Schachtschneider KM, Seixas FK, Singh K, Wells KD, Walters EM, Prather RS, Counter CM. A Genetic Porcine Model of Cancer. PLoS One. 2015; 10:e0128864. https://doi.org/10.1371/journal.pone.0128864. [PubMed].
12. Schachtschneider KM, Schwind RM, Darfour-Oduro KA, De AK, Rund LA, Singh K, Principe DR, Guzman G, Ray CE Jr, Ozer H, Gaba RC, Schook LB. A validated, transitional and translational porcine model of hepatocellular carcinoma. Oncotarget. 2017; 8:63620–63634. https://doi.org/10.18632/oncotarget.18872. [PubMed].
13. Lou J, Zhang L, Lv S, Zhang C, Jiang S. Biomarkers for Hepatocellular Carcinoma. Biomark Cancer. 2017; 9:1–9. https://doi.org/10.1177/1179299X16684640. [PubMed].
14. Chu X, Bleasby K, Evers R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin Drug Metab Toxicol. 2013; 9:237–252. https://doi.org/10.1517/17425255.2013.741589. [PubMed].
15. Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005; 106:97–132. https://doi.org/10.1016/j.pharmthera.2004.10.013. [PubMed].
16. Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, Altman RB. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharmacogenet Genomics. 2011; 21:152–161. https://doi.org/10.1097/FPC.0b013e3283385a1c. [PubMed].
17. Lozano E, Herraez E, Briz O, Robledo VS, Hernandez-Iglesias J, Gonzalez-Hernandez A, Marin JJ. Role of the plasma membrane transporter of organic cations OCT1 and its genetic variants in modern liver pharmacology. Biomed Res Int. 2013; 2013:692071. https://doi.org/10.1155/2013/692071. [PubMed].
18. Achour B, Barber J, Rostami-Hodjegan A. Cytochrome P450 Pig liver pie: determination of individual cytochrome P450 isoform contents in microsomes from two pig livers using liquid chromatography in conjunction with mass spectrometry. Drug Metab Dispos. 2011; 39:2130–2134. https://doi.org/10.1124/dmd.111.040618. [PubMed].
19. Ghassabian S, Rawling T, Zhou F, Doddareddy MR, Tattam BN, Hibbs DE, Edwards RJ, Cui PH, Murray M. Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites. Biochem Pharmacol. 2012; 84:215–223. https://doi.org/10.1016/j.bcp.2012.04.001. [PubMed].
20. Kassner N, Huse K, Martin HJ, Godtel-Armbrust U, Metzger A, Meineke I, Brockmoller J, Klein K, Zanger UM, Maser E, Wojnowski L. Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab Dispos. 2008; 36:2113–2120. https://doi.org/10.1124/dmd.108.022251. [PubMed].
21. Solomon B, Soulen MC, Baum RA, Haskal ZJ, Shlansky-Goldberg RD, Cope C. Chemoembolization of hepatocellular carcinoma with cisplatin, doxorubicin, mitomycin-C, ethiodol, and polyvinyl alcohol: prospective evaluation of response and survival in a U.S. population. J Vasc Interv Radiol. 1999; 10:793–798. https://doi.org/10.1016/S1051-0443(99)70117-X. [PubMed].
22. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–390. https://doi.org/10.1056/NEJMoa0708857. [PubMed].
23. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10:25–34. https://doi.org/10.1016/S1470-2045(08)70285-7. [PubMed].
24. Furukawa S. In vitro chemosensitivity of hepatocellular carcinoma for hepatic arterial infusion chemotherapy using the MTT assay with the combinations of antitumor drugs. Kurume Med J. 2004; 51:25–33. https://doi.org/10.2739/kurumemedj.51.25. [PubMed].
25. Reiberger T, Chen Y, Ramjiawan RR, Hato T, Fan C, Samuel R, Roberge S, Huang P, Lauwers GY, Zhu AX, Bardeesy N, Jain RK, Duda DG. An orthotopic mouse model of hepatocellular carcinoma with underlying liver cirrhosis. Nat Protoc. 2015; 10:1264–1274. https://doi.org/10.1038/nprot.2015.080. [PubMed].
26. Diederichs S, Bartsch L, Berkmann JC, Frose K, Heitmann J, Hoppe C, Iggena D, Jazmati D, Karschnia P, Linsenmeier M, Maulhardt T, Mohrmann L, Morstein J, et al. The dark matter of the cancer genome: aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol Med. 2016; 8:442–457. https://doi.org/10.15252/emmm.201506055. [PubMed].
27. Gaba RC, Mendoza-Elias N, Regan DP, Garcia KD, Lokken RP, Schwind RM, Eichner M, Thomas FM, Rund LA, Schook LB, Schachtschneider KM. Characterization of an Inducible Alcoholic Liver Fibrosis Model for Hepatocellular Carcinoma Investigation in a Transgenic Porcine Tumorigenic Platform. J Vasc Interv Radiol. 2018; 29:1194–202.e1. https://doi.org/10.1016/j.jvir.2018.03.007. [PubMed].
28. Singh VK, Thrall KD, Hauer-Jensen M. Minipigs as models in drug discovery. Expert Opin Drug Discov. 2016; 11:1131–1134. https://doi.org/10.1080/17460441.2016.1223039. [PubMed].
29. Li X, Zhou X, Guan Y, Wang YX, Scutt D, Gong QY. N-nitrosodiethylamine-induced pig liver hepatocellular carcinoma model: radiological and histopathological studies. Cardiovasc Intervent Radiol. 2006; 29:420–428. https://doi.org/10.1007/s00270-005-0099-8. [PubMed].
30. Mitchell J, Tinkey PT, Avritscher R, Van Pelt C, Eskandari G, Konnath George S, Xiao L, Cressman E, Morris JS, Rashid A, Kaseb AO, Amin HM, Uthamanthil R. Validation of a Preclinical Model of Diethylnitrosamine-Induced Hepatic Neoplasia in Yucatan Miniature Pigs. Oncology. 2016; 91:90–100. https://doi.org/10.1159/000446074. [PubMed].
31. Ho J, Ware M, Law J, Nagaraj A, Jain S, Rios J, Calderon R, Toombs B, Anderson A, Bray C, Curley S, Corr SJ. Improved, Shorter-Latency Carcinogen-Induced Hepatocellular Carcinoma Model in Pigs. Oncology. 2018; 95:360–369. https://doi.org/10.1159/000491092. [PubMed].
32. Schachtschneider KM, Schwind RM, Newson J, Kinachtchouk N, Rizko M, Mendoza-Elias N, Grippo P, Principe DR, Park A, Overgaard NH, Jungersen G, Garcia KD, Maker AV, et al. The Oncopig Cancer Model: An Innovative Large Animal Translational Oncology Platform. Front Oncol. 2017; 7:190. https://doi.org/10.3389/fonc.2017.00190. [PubMed].
33. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–675. https://doi.org/10.1038/nmeth.2089. [PubMed].
34. Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist. 2007; 12:426–437. https://doi.org/10.1634/theoncologist.12-4-426. [PubMed].
35. Namur J, Citron SJ, Sellers MT, Dupuis MH, Wassef M, Manfait M, Laurent A. Embolization of hepatocellular carcinoma with drug-eluting beads: doxorubicin tissue concentration and distribution in patient liver explants. J Hepatol. 2011; 55:1332–1338. https://doi.org/10.1016/j.jhep.2011.03.024. [PubMed].
36. Concordet JP, Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. https://doi.org/10.1093/nar/gky354. [PubMed].
37. Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol. 2016; 34:695–697. https://doi.org/10.1038/nbt.3583. [PubMed].
38. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013; 1303.3997. https://arxiv.org/abs/1303.3997.
39. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. https://doi.org/10.1101/gr.107524.110. [PubMed].
40. Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, Saunders CT. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018; 15:591–594. https://doi.org/10.1038/s41592-018-0051-x. [PubMed].
41. Zaccaria S, Raphael BJ. Accurate quantification of copy-number aberrations and whole-genome duplications in multi-sample tumor sequencing data. BioRxiv. 2018; 496174. https://doi.org/10.1101/496174.
42. Huang X, Wojtowicz D, Przytycka TM. Detecting presence of mutational signatures in cancer with confidence. Bioinformatics. 2018; 34:330–337. https://doi.org/10.1093/bioinformatics/btx604. [PubMed].
43. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, Fish P, Harsha B, Hathaway C, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019; 47:D941–D947. https://doi.org/10.1093/nar/gky1015. [PubMed].
44. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. https://doi.org/10.1093/bioinformatics/btq033. [PubMed].
45. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012; 6:80–92. https://doi.org/10.4161/fly.19695. [PubMed].