
Introduction
Breast cancer arises from a series of acquired mutations and epigenetic changes that disrupt normal mammary epithelial homeostasis and create multi-potent cells that can differentiate into biologically unique and clinically distinct subtypes [1–6]. Epithelial-to-mesenchymal transition (EMT)—a trans-differentiation process through which mammary epithelial cells acquire the aggressive mesenchymal phenotype—is a key driver of breast cancer progression, invasion and metastasis [7–12]. Transcription factors Snail, Slug, Twist, and Zeb1/2 contribute to EMT during early, normal development and have also been implicated in invasion [13–16]. Despite accumulating evidence that defines a broad understanding of EMT regulation and maintenance of the epithelial phenotype [7–12], the mechanism (s) by which mammary cells maintain their epithelial phenotype is unknown.
The runt-related RUNX family of proteins form heterodimeric transcription factor complexes containing one of the three DNA binding factors (i.e., RUNX1, RUNX2, and RUNX3) and a common non-DNA binding protein, CBFβ [17]. Although the three RUNX proteins can bind to DNA, interaction with CBFβ significantly enhances their DNA binding affinity and protects them from proteasome-mediated degradation [18–20]. Functionally, RUNX proteins are phenotypic transcription factors that dictate identity of specific lineages and are often tissue-restricted in expression [17]. Paradoxically, CBFβ is ubiquitous in expression, indicating non-canonical roles that are independent of DNA binding RUNX factors [21]. Conversely, several studies have shown that lineage restricted depletion of CBFβ only partially recapitulates phenotypes observed with deletion of the DNA binding RUNX factors [19, 22–25], suggesting that not all transcriptional regulatory activity of these proteins requires CBFβ. Furthermore, a recent study has shown that CBFβ functions post-transcriptionally to regulate gene expression in breast epithelial cells [26]. Together, these observations highlight knowledge gaps in the mechanistic understanding of gene regulation by the RUNX-CBFβ transcriptional complex.
In addition to the recognized role in hematopoiesis and hematological malignancies [22, 27–39], RUNX1 has been recently identified as a key player in breast cancer development and tumor progression [40–47]. Findings from our group [48], reinforced by studies from others [49, 50], have shown that RUNX1 plays a critical role in maintaining breast epithelial phenotype and prevents EMT. However, it is unclear whether CBFβ contributes to RUNX1-mediated regulation of epithelial cell identity and mechanisms by which RUNX1 prevents EMT have not been identified.
Mitotic gene bookmarking, i.e., transcription factor binding to target genes during mitosis for transcriptional regulation following cell division, is a key epigenetic mechanism to convey and sustain regulatory information for cell proliferation, growth, and cell identity from parent to progeny cells [51–58]. We have established that RUNX proteins, as well as other phenotypic transcription factors that include MYOD and CEBPα, are present during mitosis on RNA Pol I- and II-transcribed genes in osteoblasts and leukemia cells for coordinate control of cell growth, proliferation and phenotype [59–66]. It is increasingly evident that mitotic gene bookmarking by transcription factors is a key mechanism to determine and maintain cell fate across successive cell divisions [67–79].
In this study, we addressed the hypothesis that RUNX1 maintains the breast epithelial phenotype, at least in part through interaction with CBFβ, and that the RUNX1-CBFβ complex transcriptionally regulates genes that support mammary epithelial proliferation, growth, and phenotype. We find that compromising RUNX1 DNA binding using a pharmacological inhibitor that disrupts the interaction between RUNX1 and CBFβ results in loss of the epithelial phenotype and acquisition of mesenchymal properties that are accompanied by changes in expression of associated genes and pathways and represent early events in the onset of breast cancer. Fluorescence confocal microscopy of fixed and live mammary epithelial cells revealed that RUNX1 is localized in punctate subnuclear foci in interphase and dynamically redistributes during mitosis, where it is present on chromosomes throughout mitosis. To identify genes occupied by RUNX1, we performed chromatin immunoprecipitation coupled with high throughput sequencing (ChIP-Seq) using a RUNX1-specific antibody on mitotic, G1, and asynchronous normal mammary epithelial MCF10A cells. We found that ribosomal RNA genes, regulated by the RNA Pol I transcriptional machinery, were occupied by RUNX1. A fluorescence-based, global protein synthesis assay showed reduced protein synthesis when RUNX1 DNA binding was perturbed using a small molecule inhibitor. Consistent with dynamic redistribution during cell division, ChIP-Seq revealed that, in mitosis, RUNX1 remains associated with a subset of RNA Pol II regulated genes specifically involved in maintenance of the epithelial phenotype and EMT progression. These findings establish a critical transcriptional regulatory role for the RUNX1-CBFβ complex in preventing EMT and implicate RUNX1 occupancy of target genes at the mitosis into G1 transition as a potential mechanism to sustain the normal breast epithelial phenotype.
Results
Inhibition of RUNX1-CBFβ complex causes epithelial to mesenchymal transition and alters the associated transcriptome
To address experimentally whether functional disruption of the RUNX1-CBFβ transcriptional complex affects the mammary epithelial phenotype, we used a well-characterized small molecule inhibitor AI-14-91, which interferes with RUNX1-CBFβ interaction and disrupts RUNX1 DNA binding [80, 81]. We treated cells with the RUNX1-CBFβ inhibitor, as well as with the inactive control compound, and monitored changes in cell morphology over time. Consistent with previously reported role of RUNX1 in stabilizing the epithelial phenotype [48], disruption of the RUNX1-CBFβ complex for 48 hours resulted in mesenchymal morphology (Figure 1A). We next examined whether long-term inhibition of the RUNX1-CBFβ interaction caused a permanent change in cell phenotype. Longer term treatment (5 days) of actively proliferating normal mammary epithelial MCF10A cells showed significant apoptosis, although a small sub-population of cells survived and exhibited an altered phenotype; this phase was collectively denoted as the “crisis phase” (Figure 1B). The surviving sub-population at day 5 was recovered by culturing cells in media without the inhibitor, i.e., the “recovery phase”. By day 3–4 following media replacement, cells clearly showed a mesenchymal morphology (Figure 1B), indicating that interfering with the RUNX-CBFβ interaction and subsequent RUNX1 DNA binding causes loss of the normal mammary epithelial phenotype. Consistent with changes in cell morphology, we find alterations in expression and localization of the cytoskeletal F-actin protein (Figure 1C). These observations were confirmed by examining the expression of epithelial markers (e.g., E-cadherin, Figure 1D), as well as mesenchymal markers (e.g., Vimentin, Figure 1D). E-cadherin was largely unchanged; however, Vimentin expression was significantly increased, supporting initiation of an epithelial-to-mesenchymal transition upon inhibition of the RUNX1-CBFβ interaction.
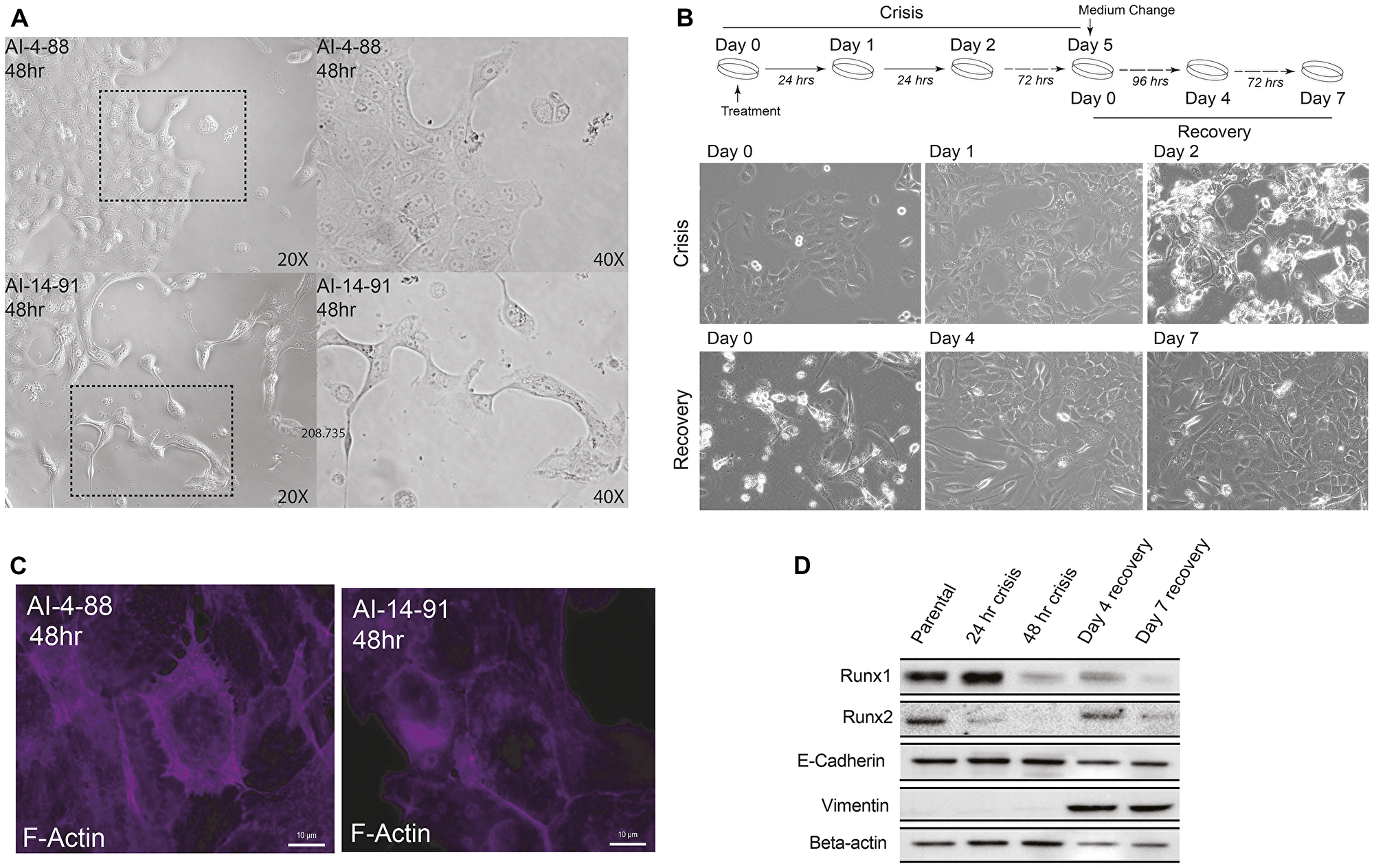
Figure 1: Disrupting transcriptional activity of the RUNX1-CBF Complex in MCF10A cells leads to a transformed cellular phenotype and EMT. (A) Phase contrast microscopy images of MCF10A cells treated with AI-4-88 or A-14-91 for 48 hours at 20 μM. Left panel: 20× magnification, right panel, 40× magnification. The outlines rectangle in the left panel is the resulting 40× magnification in the right panel. (B) Top panel: Experimental schematic depicting treatment schedule for the “crisis” and “recovery” stages. Bottom panel: Phase contrast microscopy images from Day 0, 1, and 2 or crisis where MCF10A cells were treated with AI-14-91 at 20 μM (top: left, middle, right, respectively). Phase contrast images from Day 0, 4, and 7 or recovery following a media replacement. (C) Morphological changes of the cytoskeletal protein F-actin upon inhibition of CBFβ-RUNX binding. When compared to inactive compound (left panel), cells treated with the active compound show substantial alteration in cytoarchitecture (right panel). (D) Western blot for RUNX1, RUNX2, epithelial marker E-cadherin, mesenchymal marker Vimentin, and loading control Beta-actin (top panel to bottom panel, respectively) in MCF10A whole cell lysate harvested from the crisis 24 hour and 48-hour timepoints and recovery day 4 and day 7 timepoints.
To identify transcriptome-wide changes associated with EMT upon inhibition of RUNX1 DNA binding activity, we performed RNA sequencing of three biological replicates at each of the indicated time points (Day 1 and 2 in Crisis Phase and Day 4 and 7 in Recovery Phase). Heatmap analysis of all time points identified substantial changes in gene expression as cells transitioned from an epithelial to mesenchymal phenotype (Figure 2A). A differential gene expression analysis between the crisis and recovery phases uncovered significant changes in expression of genes associated with EMT (e.g., IL32, SERPINB2, etc., Figure 2B). We performed pathway analysis on differentially expressed genes (Figure 2C and Supplementary Table 1). Consistent with phenotypic changes, we found that multiple signaling pathways that include TNF alpha, Interferon Gamma and estrogen responsiveness were altered during EMT caused by inhibition of the RUNX1-CBFβ complex.
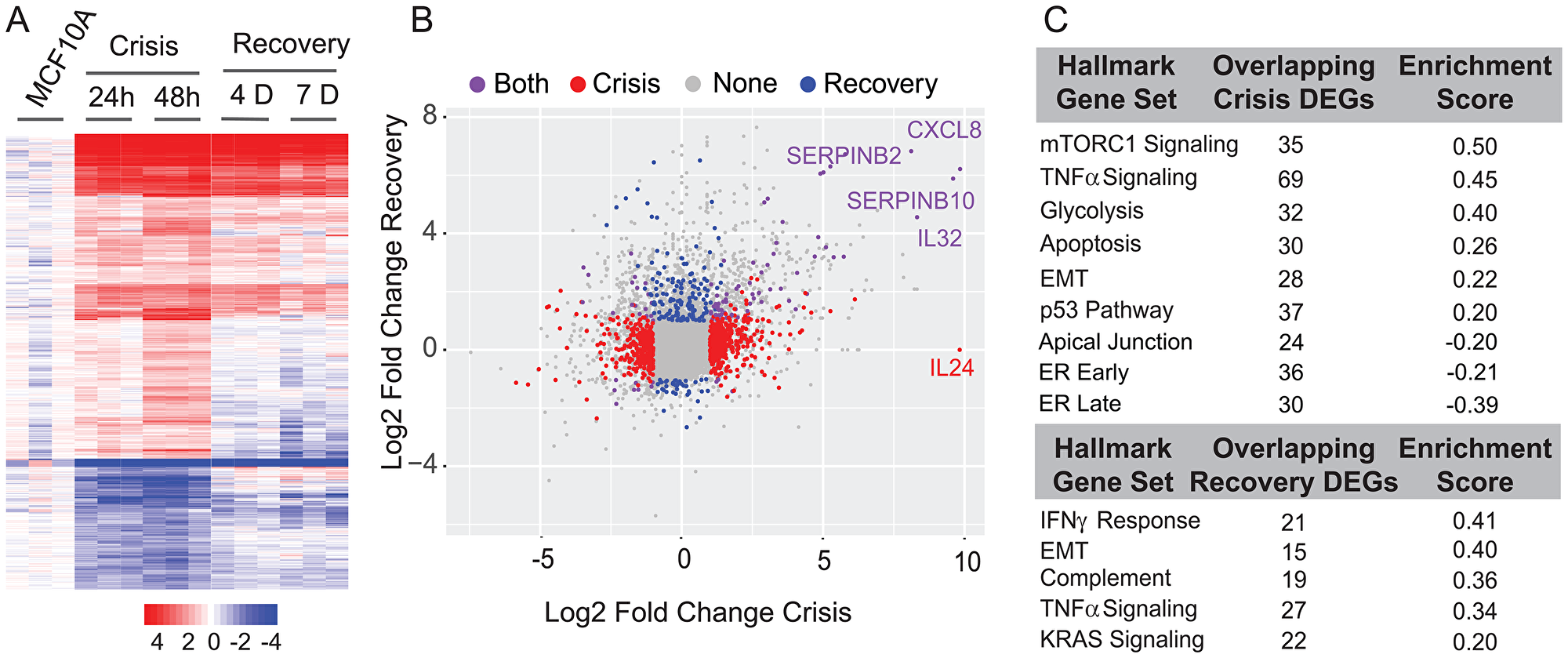
Figure 2: Differential expression and pathway analysis of RNA-Seq shows changes in key regulatory pathways involved in cell proliferation, metabolism, cell cycle control, ER (estrogen response), and EMT. (A) Expression heatmap of three biological replicates of 24-hour and 48-hour crisis timepoints and Day 4 and Day 7 recovery timepoints. (B) Scatterplot of log2 fold change between crisis and recovery phases. Most changed genes in each stage are indicated. (C) Table of overlapping pathways specific to crisis and recovery stages.
Together, these findings show that the interaction between RUNX1 and its heterodimeric partner CBFβ is a key determinant of the mammary epithelial cell identify and its inhibition leads to EMT and related transcriptomic changes.
RUNX1 organizes as punctate foci in the mammary epithelial cell nucleus during interphase and is dynamically redistributed during mitosis
We have shown that RUNX proteins reside in punctate subnuclear domains and their subnuclear localization is functionally linked with transcriptional activity [30, 82–94]. To investigate subcellular localization of RUNX1 in normal mammary epithelial cells, we performed immunofluorescence (IF) microscopy in actively proliferating MCF10A cells and imaged cells in interphase as well as those undergoing spontaneous mitoses. We observed that RUNX1 is distributed as punctate domains predominantly in the interphase nucleus; no cytoplasmic signal for the protein is detected above the background or in the antibody controls (Figure 3, interphase panel, and Supplementary Figure 1). Importantly, we found that a subset of RUNX1 foci is localized on mitotic chromatin at all topologically identified substages of mitosis (Figure 3, top panels). Two distinct types of foci are detectable on mitotic chromosomes: 2–8 large punctate foci that appear to be allelic as well as numerous smaller foci that are distributed across the chromosomes (Figure 3, bottom panels; white arrowheads). Importantly, the number of foci retained on mitotic chromosomes was reduced upon disruption of the RUNX1-CBFβ interaction with a pharmacological inhibitor (Supplementary Figure 2). Unlike the interphase nuclei, RUNX1 signal was also reproducibly detectable in cytosol of prophase and metaphase cells, likely due to the absence of nuclear membrane; this signal was much less apparent in anaphase and telophase cells, coinciding with reforming of the nuclear membrane in progeny cells (Figure 3). In all replicates, important secondary-antibody-only controls were included to confirm specificity of RUNX1 signal during interphase and on mitotic chromosomes (Supplementary Figure 1). To ensure reproducibility of our findings, the IF experiments were repeated at least 3 times and, at the minimum, 20 interphase and mitotic cells were imaged.
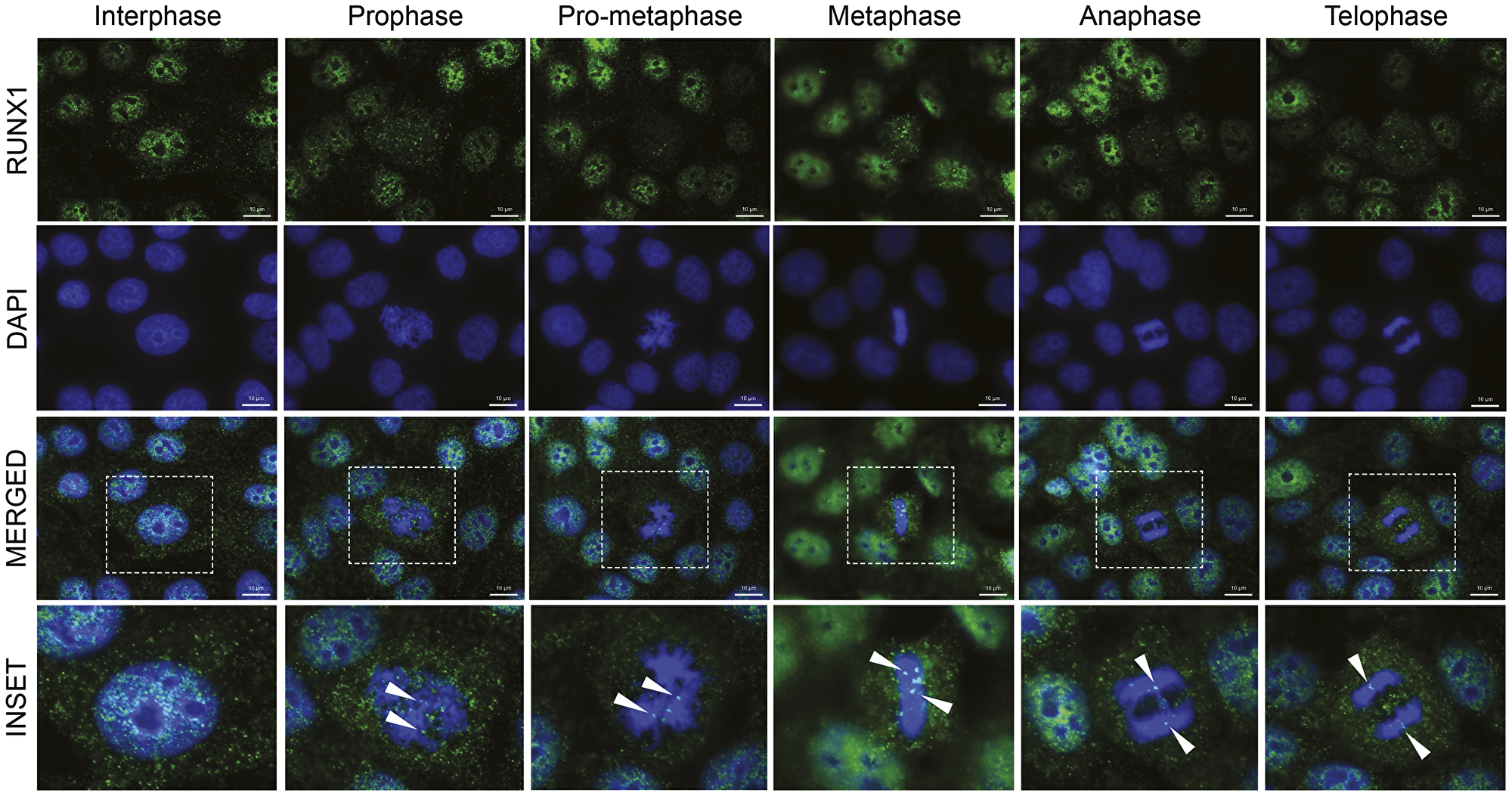
Figure 3: RUNX1 localizes to punctate subnuclear foci in interphase and dynamically redistributes throughout mitosis in the form of major and minor foci. Representative immunofluorescent images of interphase and mitotic MCF10A breast epithelial cells showing subcellular localization of RUNX1, identified using a specific antibody, throughout mitosis. Mitotic cells were further classified into substages of mitosis based on DAPI topology. RUNX1: green (top row); DAPI: blue (second row from top). Merged channel images (third row from top) contain an outlined region magnified in the bottom row labeled “inset”. White arrows highlight major Runx1 foci on mitotic chromatin. Three independent biological replicates were performed, and at least 20 cells for each mitotic substage were analyzed.
Because mitotic association of RUNX1 has potential functional ramifications—mitotic bookmarking is being studied as a key epigenetic mechanism to maintain cell identity [51–79]—we confirmed our findings in live cells. Multiple reports have indicated that formaldehyde fixation can prevent regulatory protein detection on mitotic chromosomes [67, 78]. To further confirm that association of RUNX1 with mitotic chromosomes is not under-represented because of formaldehyde fixation, we examined the localization of RUNX1-EGFP in actively proliferating, unfixed MCF10A cells. Consistent with our findings in fixed and synchronized cells, RUNX1-EGFP was associated with chromosomes in live MCF10A cells undergoing mitosis (Supplementary Figure 3). Together, these findings establish that RUNX1 is localized as punctate subnuclear foci in the interphase of mammary epithelial cells and is dynamically redistributed during mitosis, with a subset of foci retained on chromosomes at all stages of mitosis under physiological conditions in actively dividing, unfixed breast epithelial cells and, in agreement with our previous findings, equally distributed into resulting progeny cells [66].
RUNX1 occupies distinct gene sets associated with the mammary epithelial cell identity, growth, and proliferation during interphase and mitosis
To experimentally address whether RUNX1 presence on mitotic chromosomes reflects occupancy of target genes, MCF10A cells were synchronized in mitosis using nocodazole (50 ng/mL). Nocodazole dose and treatment time were empirically determined to minimize toxic effects of the drug, while maximizing mitotic enrichment (Supplementary Figure 4A). Mitotic cells were collected by mitotic shake-off and purity of harvested cells was confirmed by the presence of H3pS28 in > 70% of singlets. We chose the H3pS28 mark to identify mitotic cells because this histone mark is highly specific to condensed chromosomes during mitosis; the more commonly used H3pS10 mark is additionally observed in late G2 as well as in early G1 cells and has also been associated with replicating centers in S-phase [95, 96]. A parallel, nocodazole-treated cell population was released into early G1 by replacing nocodazole-containing growth medium with fresh, nocodazole-free, growth medium and was harvested 3 hours post-release (Supplementary Figure 4B). Western blot analysis of whole cell lysates from the three cell populations showed expected levels of expression for cell cycle-specific proteins Cyclin B and CDT1 (Supplementary Figure 4C). FACS profiles of the cell populations confirmed the characteristic enrichment of blocked cells in mitosis (Supplementary Figure 4D, Mitotic) and of G1 cells upon release into fresh media (Supplementary Figure 4D, G1) when compared to asynchronous cells (Supplementary Figure 4D, Asynch).
To determine whether RUNX1 remains bound to target genes during mitosis, ChIP-Seq was performed on Asynch, Mitotic, and G1 MCF10A cells using a RUNX1 specific antibody (Figure 4A). Enriched regions (peaks called by MACS2) were compared using k-means clustering (k = 4) of normalized enrichment profiles of the three cell populations. This analysis revealed subsets of genes that were either shared across the three groups or were specific for each, indicating dynamic binding of RUNX1 during and immediately after mitosis (Figure 4A and Supplementary Table 2). Peak calling identified RUNX1 occupancy of both protein coding and long non-coding RNA (lncRNA) genes. Specifically, RUNX1 occupied 1070 genes in cell population not in G1 or M phase (Figure 4A, green bar) and 1095 genes in G1-enriched cells (Figure 4A, light red bar). Importantly, RUNX1 occupied 551 genes (413 protein coding and 138 lncRNAs) in mitotically enriched MCF10A cells, of which 378 genes overlapped with those occupied in early G1 while 173 genes were only occupied during mitosis (Figure 4A, blue bar).
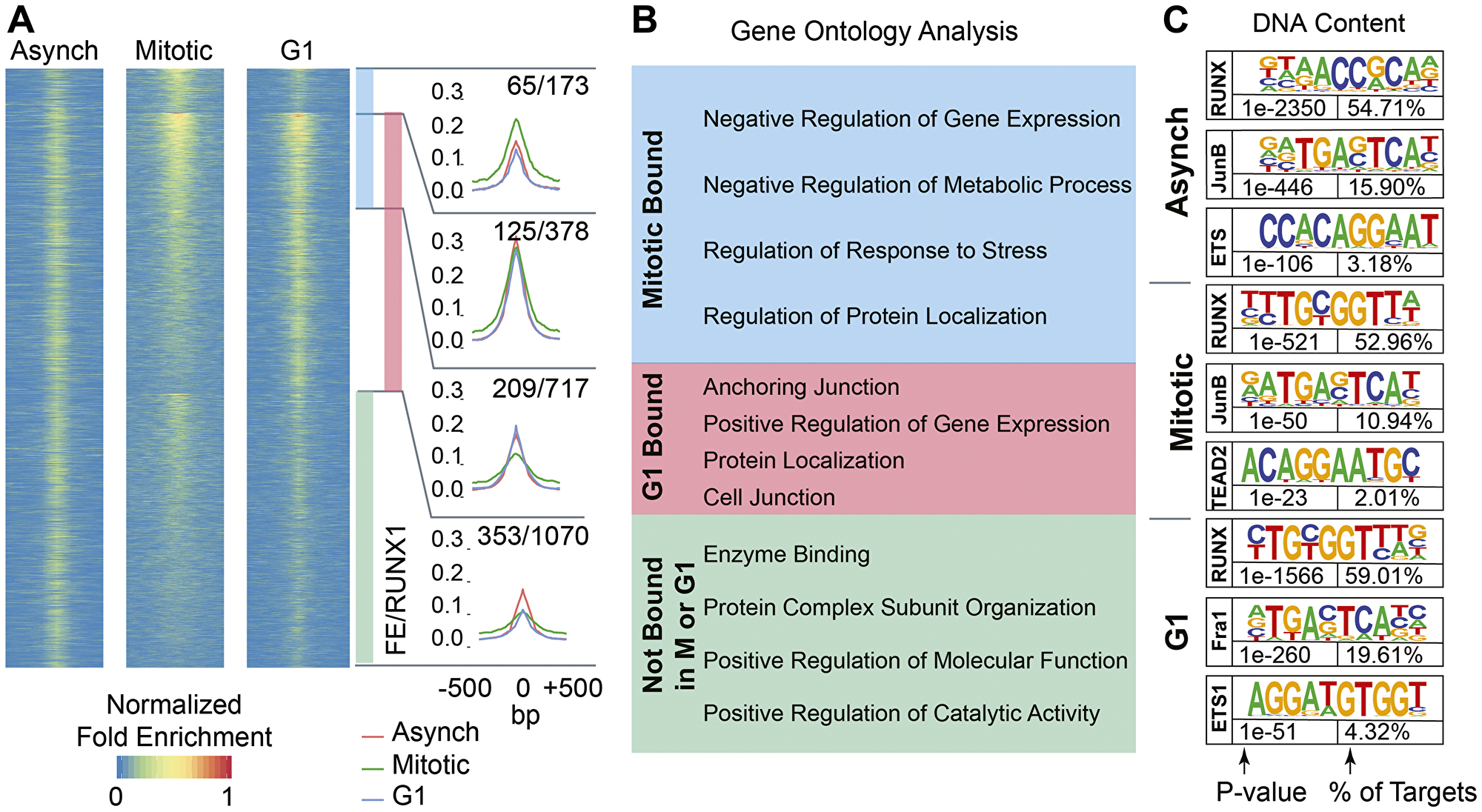
Figure 4: RUNX1 occupies protein coding genes and long non-coding RNAs across asynchronous, mitotic, and G1 populations of MCF10A breast epithelial cells. (A) Heatmaps showing peaks called between A, M, and G1 MCF10A cells (left, middle, and right, respectively). Cumulative occupancy of RUNX1 is shown as line graphs and genes that occupied by RUNX1 in each of the three cell populations are shown. Shown also are genes that are deregulated upon RUNX1 downregulation. For example, 1070 genes are bound by RUNX1 in cells that are neither in M nor G1 cells (green bar graph) and of those, 353 genes are deregulated upon RUNX1 downregulation. (B) Gene ontology analysis of RUNX1-bound genes identifies key regulatory pathways in the three cell populations. (C) Motif analysis of A, M, and G1 MCF10A cells reveals RUNX motif as one of the top motifs in all cell populations. Binding sites for key transcription factors that are known to cooperate with RUNX1 are also identified.
Functional relevance of RUNX1 occupancy in the three cell populations was determined by comparing RUNX1-occupied genes with those that are differentially regulated upon shRNA-mediated RUNX1 knockdown [48]. Of the 1070 genes occupied by RUNX1 in cell populations not in G1 or M phases, 353 genes were deregulated upon RUNX1 depletion (Figure 4A). Importantly, RUNX1 depletion deregulated 399 of 1268 RUNX1-bound genes in the M and early G1 populations, indicating a rapid and dynamic mode of transcriptional regulation as cells transition from mitosis into the next cell cycle. These findings, for the first time, reveal that several hundred target genes are bookmarked by RUNX1 during mitosis and transcriptionally regulated in normal mammary epithelial cells.
To identify cellular processes and pathways that are comprised of RUNX1-bookmarked genes, we performed gene set enrichment analysis (GSEA) on genes bound by RUNX1 during mitosis or G1, or not bound in either cell cycle stage (Figure 4B). Interestingly, most genes bookmarked by RUNX1 during mitosis were associated with negative regulation of gene expression and metabolic process (Figure 4B, blue box). Consistent with a cellular requirement to reattach and enter the next cell cycle and fully resume transcription, genes bound during early G1 were primarily enriched in biological processes involving cell anchorage, protein localization and positive regulation of gene expression (Figure 4B, red box). ChIP-seq results were further validated by motif analysis of RUNX1-bound peaks, which showed that the RUNX motif was the most enriched motif in all three cell populations (Figure 4C). Importantly, RUNX1-bound genomic regions were also enriched in motifs for transcription factors (e.g., Fra1, JunB, ETS) known to cooperate with RUNX1 for gene regulation [97] (Figure 4C). Together, these findings show that RUNX1 occupies genes involved in cell proliferation, growth, and phenotype during mitosis in normal mammary epithelial cells and suggest that mitotic gene bookmarking is a key mechanism by which RUNX1 sustains the mammary epithelial cell identity.
RUNX1-CBFβ complex transcriptionally regulates RNA Pol I-transcribed ribosomal RNA genes and impacts global protein synthesis
Our ChIP-Seq results revealed that RUNX1 occupies rDNA repeats in MCF10A mammary epithelial cells; all three MCF10A cell populations (Asynch, Mitotic, and G1) exhibited significant fold enrichment within the promoter region of hrDNA (Figure 5A), suggesting a potential role for RUNX1 in regulating rRNA genes in MCF10A cells. We confirmed this finding in actively proliferating MCF10A cells by immunofluorescence microscopy for antibodies specific against RUNX1 and upstream binding factor (UBF), a transcriptional activator that remains bound to rRNA genes during mitosis [98]. We observed that, in interphase, RUNX1 colocalization with UBF is primarily confined to the periphery of nucleoli. Interestingly, we found that large RUNX1 foci colocalized with UBF throughout each stage of mitosis (Figure 5B and Supplementary Figure 5). This colocalization between RUNX1 and UBF was validated by confocal microscopy; line scans of MCF10A cells show that although RUNX1 and UBF occupy distinct nuclear microenvironments in interphase (n = 15), both proteins substantially colocalize in metaphase (n = 15) (Supplementary Figure 5). Taken together, these findings establish RUNX1 binding to ribosomal DNA repeat regions by ChIP-Seq (Figure 5A) with confirmation at the cellular level by confocal microscopy (Figure 5B and Supplementary Figure 5).
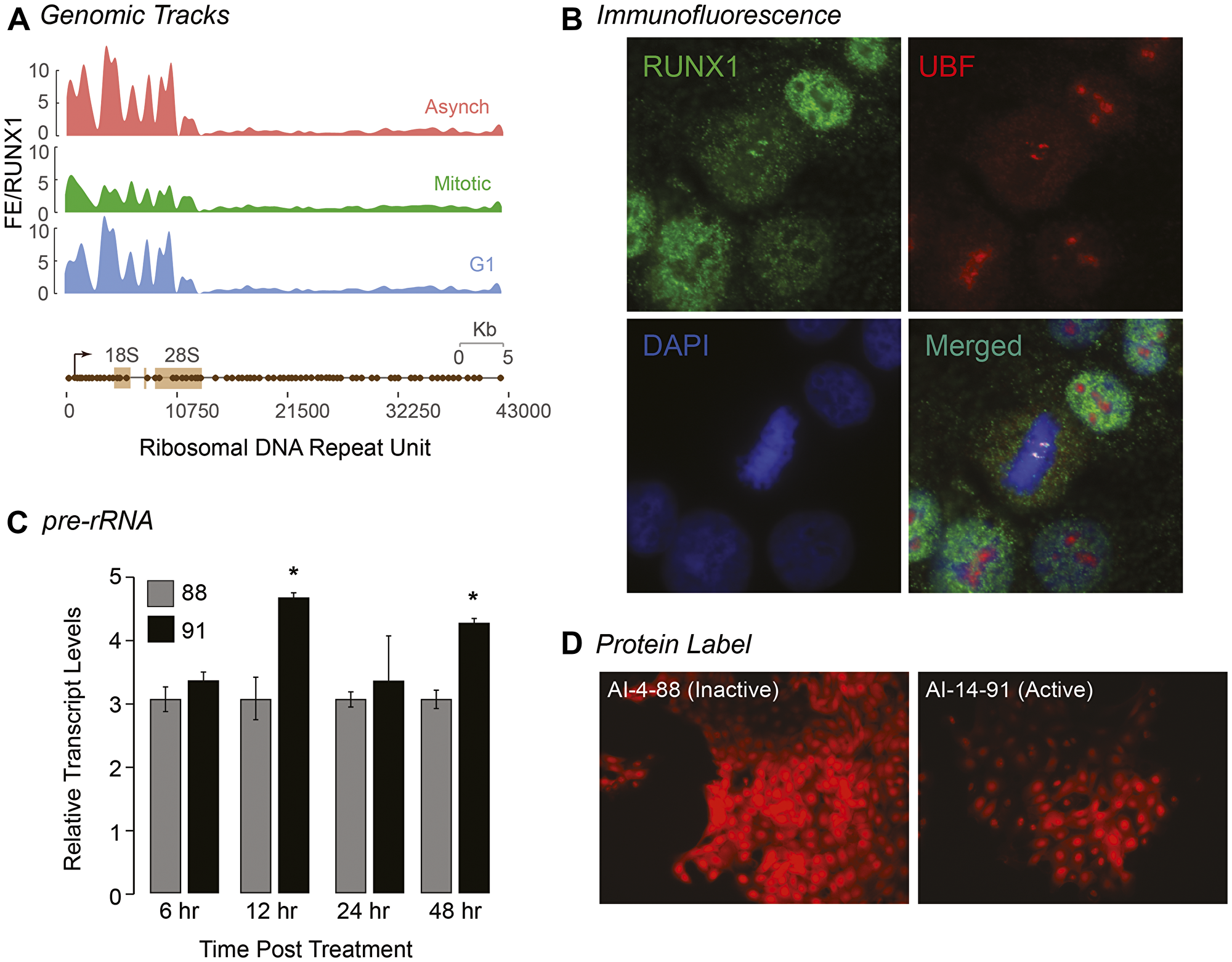
Figure 5: RUNX1 occupies rDNA promoter repeat regions in interphase and during mitosis and affects both pre-rRNA and global protein expression. (A) ChIP-Seq tracks of A, M, and G1 (top, middle, bottom, respectively) MCF10A cells mapped against rDNA repeat regions. (B) A representative metaphase MCF10A cell, stained for RUNX1 (green) and UBF1 (red) localization, is shown demonstrating that the two proteins colocalize during mitosis (merged). Cells are also counter stained with DAPI to visualize DNA (blue) and identify mitosis substages. (C) qRT-PCR data of pre-rRNA in actively proliferating MCF10A cells treated with either active (AI-14-91) or inactive (AI-4-88) compounds for 6, 12, 24, or 48 hrs. Expression of pre-rRNA was normalized relative to Beta Actin expression. Graph represents three independent biological replicates. Asterisks represents a p value of < 0.05. (D) Representative fluorescence microscopy images of global protein synthesis from MCF10A cells treated with either AI-4-88 (left) or AI-14-91 (right) for 24 hr at 20 μM (n = 3). Intensity of red fluorescence at 580 nm emission indicates nascent protein synthesis. All images were taken with 1000 ms exposures.
We have previously shown that CBFβ is associated with ribosomal RNA genes during mitosis in leukemia cells during mitosis [99]. We experimentally addressed the hypothesis that RUNX1-CBFβ regulates ribosomal RNA gene expression by using the AI-14-91 small molecule inhibitor. We examined the effect of RUNX1-CBFβ inhibitor on pre-rRNA expression and found that pre-rRNA expression was significantly increased at 12 hr and 48 hr time points after treatment of asynchronous cells with the AI-14-91 specific inhibitor but not the control inactive compound AI-4-88, indicating that RUNX1 suppresses rRNA gene expression in normal mammary epithelial cells (Figure 5C). Because levels of rRNA directly correlate with global protein synthesis, a fluorescent-based detection method was used to measure newly synthesized proteins. Cells treated with AI-14-91 for 24 hr or 48 hr showed a moderate change in levels of global protein synthesis in comparison to AI-4-88 control-treated cells under identical conditions (n = 3; Figure 5D). Together, our results demonstrate that the RUNX1-CBFβ interaction is critical for rRNA gene expression and global protein synthesis. Additionally, RUNX1 occupies RNA Pol I regulated rRNA genes during interphase and bookmarks them during mitosis which may act to transcriptionally repress them.
RUNX1-CBFβ complex is a key regulator of the epithelial transcriptome associated with hormone-responsiveness and mammary cell identity
Using RUNX1 occupied genes in mitosis and early G1, GSEA was performed to identify regulatory pathways (Figure 6A). In agreement with known roles of RUNX1 [100–104], the top 10 pathways identified included those involved in regulation of G2M Checkpoint, E2F targets, p53, and DNA repair (Figure 6A). Consistent with our finding that RUNX1 bookmarks and regulates rRNA genes, one of the pathways identified is mTOR signaling, a pathway that is required for cell growth and is a therapeutic target in breast cancers [105, 106]. Relevant to the normal mammary epithelial phenotype, both early and late estrogen responsive gene sets significantly overlap with RUNX1 mitotically bookmarked genes (Figure 6A). Because estrogen plays vital roles in promoting proliferative phenotypes of mammary epithelial cells [107–109], we interrogated RUNX1 bookmarked genes to identify those bound by RUNX1 and ERα in MCF7 cells, where RUNX1 contributes to higher order genome organization (Figure 6B) [110, 111]. Using publicly available datasets of ERα genome-wide occupancy and estradiol-regulated gene expression (GSE40129) [112], we discovered that a subset of genes mitotically bookmarked by RUNX1 is also bound by ERα, and either up or down regulated in response to estradiol. These findings indicate that RUNX1-occupied genes are involved in pathways that control hormone-responsiveness, proliferation and growth of normal mammary epithelial cells (Figure 6B).
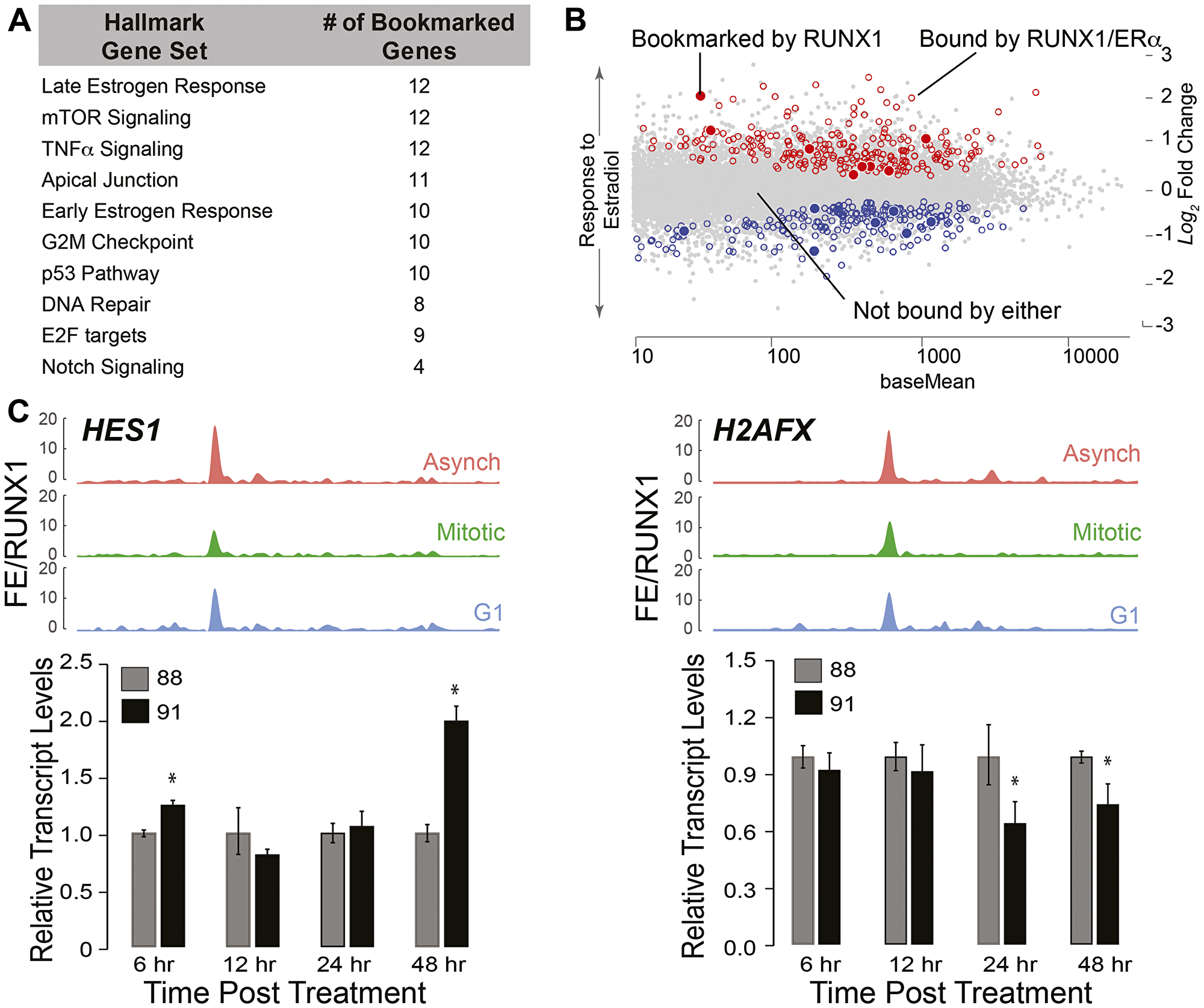
Figure 6: RUNX1-CBFβ complex is a key regulator of the epithelial transcriptome associated with hormone-responsiveness and mammary cell identity. (A) Gene Set Enrichment (GSE) analysis from interrogating mitotically bookmarked genes (i.e., RUNX1 mitotically occupied) against Hallmark Gene sets from Molecular Signatures Database (MSigDB). The top 10 most significantly overlapping gene sets are shown from top to bottom. (B) Scatter plot of genes identified to be up or down regulated in response to estradiol treatment, that are also bound by estrogen receptor α (ERα) and RUNX1 (empty circles, blue for downregulated and red for upregulated). Scatter plot also illustrates up or down regulated genes in response to estradiol treatment that are bound by ERα and mitotically bookmarked by RUNX1 (filled in circles, blue for downregulated and red for upregulated). (C) Top panel: ChIP-Seq tracks of HES1 (left) and H2AFX (right) from asynchronous (top-red), mitotic (middle-green), and G1 (bottom-blue). Bottom panel: qRT-PCR data of HES1 (left) and H2AFX (right) in asynchronous MCF10A cells treated with either active (AI-14-91) or inactive (AI-4-88) inhibitors for 6 hr, 12 hr, 24 hr, and 48 hr at 20 μM. Expression of target genes were normalized relative to beta actin.
We identified a novel subset of genes that are bookmarked by RUNX1 and relate to regulatory pathways involved in cellular phenotype including TNFα, Apical Junction and Notch signaling (Figure 6A). Furthermore, NEAT1 and MALAT1, lncRNAs often deregulated in breast cancer [113, 114], were also occupied by RUNX1 both in interphase and mitosis. Of the 413 RUNX1 bookmarked protein coding genes, TOP2A, MYC, HES1, RRAS, H2AFX, and CCND3 are representative of RNA Pol II-transcribed genes involved in phenotype maintenance and cell fate decisions (See Supplementary Table 2 for complete list). Recently, HES1 and H2AFX have been identified as regulators of breast epithelial phenotype [115–117]. In our ChIP-seq dataset, HES1 and H2AFX show significant fold enrichment of RUNX1 occupancy among the three populations of MCF10A cells (Figure 6C, top panels). Expression of HES1 increased upon inhibition of the RUNX1-CBFβ complex (Figure 6C: left panel, bar graph), indicating that RUNX1 represses HES1. In contrast, H2AFX expression at 24 hr and 48 hr of inhibitor treatment was decreased, suggesting RUNX1 activates H2AFX expression (Figure 6C: right panel, bar graph). These results show that RUNX1 occupies both protein coding and non-coding genes and establish that the activity of the RUNX1-CBFβ complex is a potential mechanism to stabilize the mammary epithelial phenotype.
DISCUSSION
This study establishes that pharmacological inhibition of the RUNX1-CBFβ complex causes transition of mammary epithelial cells to a mesenchymal phenotype, indicating that transcriptional regulation of target genes by the RUNX1-CBFβ complex is required to maintain the normal breast epithelial phenotype. Importantly, retention of RUNX1 on mitotic chromosomes and occupancy of target genes suggests that mitotic gene bookmarking is a potential epigenetic mechanism for coordinate regulation of RNA Pol I- and II-transcribed genes that are critical for mammary epithelial proliferation, growth, and phenotype maintenance.
Our findings are the first to examine RUNX1 occupancy of target genes during interphase and in mitosis in mammary epithelial cells and to report that RUNX1 coordinately controls cell growth-related ribosomal RNA (rRNA) genes and a large subset of cell proliferation/phenotype-related genes in these cells. One target gene of interest is hairy and enhancer of split-1 (HES1). Hes1 is a transcription factor that represses genes involved in cellular development and is regulated primarily by NOTCH signaling, one of our top ten overlapping hallmark gene sets occupied by RUNX1 in interphase and bookmarked in mitosis (Figure 6) [118, 119]. HES1 was recently shown to have a prominent role in proliferation and invasion of breast cancer cells, and its silencing led to downregulation of p-Akt signaling and ultimately prevented EMT [115]. Our findings indicate that the RUNX1-CBFβ complex stabilizes the normal mammary epithelial phenotype, in part, by RUNX1 bookmarking of the HES1 gene and suppression of its expression by the RUNX1-CBFβ complex.
Another important RNA Pol II-transcribed gene mitotically bookmarked by RUNX1 and critical for maintaining cellular phenotype is histone variant H2AFX (H2AFX). Silencing H2AFX in breast epithelial cells leads to induction of EMT through activation of SNAIL2/SLUG and TWIST1 [117]. Upon inhibition of the RUNX1-CBFβ interaction, we find a decrease in H2AFX expression and a concomitant, significant increase in SNAIL2/SLUG expression (preliminary data not shown). These data identify RUNX1 as a novel upstream regulator of H2AFX expression; RUNX1 bookmarking and activation of H2AFX and subsequent suppression of SNAIL2/SLUG prevents EMT in breast epithelial cells.
Several groups have shown that RUNX1 interacts with ERα at both enhancer regions and transcriptional start sites (TSSs) for regulation of specific genes [43, 110]. Our ChIP-Seq results, coupled with publicly available data sets, reveal a novel observation: In addition to previously reported RUNX1-ERα co-occupancy of genes critical for epithelial cell identity, RUNX1 bookmarks a subset of ERα-occupied, hormone-responsive genes during mitosis and this bookmarking may be critical for maintenance of the breast epithelial phenotype. It is important to note that MCF10A cells, used in this study, are considered ERα-low, limiting broader biological significance of our findings. Future studies, focused on mitosis specific RUNX1 and/or ERα depletion followed by nascent transcriptomic analyses in estrogen receptor positive mammary epithelial and breast cancer cells, will be required to investigate mechanistic significance of this observation.
Our study establishes that inhibition of RUNX1 DNA binding through pharmacological disruption of the RUNX1-CBFβ interaction specifically elicits an epithelial-to-mesenchymal transition that accompanies changes in critical genes and pathways involved in EMT. Recent findings have identified a non-canonical, post-transcriptional role for CBFβ in mammary epithelial cells [26]. Our results indicate that, in addition to post-transcriptional regulation of gene expression, CBFβ plays important roles in transcriptional activity of DNA binding members of the RUNX family. Although unlikely, it should be noted that the pharmacological inhibitor used in our study may also impact non-canonical functions of CBFβ. Furthermore, the inhibitor will disrupt interaction of CBFβ with any RUNX family member expressed in breast epithelial cells. Because low levels of RUNX2 are detected in some breast epithelial cells, it will be important to further examine the individual contributions of CBFβ interaction with each of the RUNX proteins expressed in mammary epithelial cells. Future studies using precision genome editing will be valuable to address this question of mechanistic importance. Notably, our findings suggest that mitotic gene bookmarking by RUNX1 may contribute to regulation of the mammary epithelial phenotype by regulating target genes critical for cell proliferation, growth and phenotype. Because RUNX1 interacts with multiple co-activators and co-repressors, additional in-depth studies are required to determine contributions of RUNX1 co-regulatory proteins to mitotic gene bookmarking.
Another novel contribution of the current study is mitotic occupancy of lncRNAs by a transcription factor. RUNX1 was recently shown to regulate lncRNAs NEAT1 and MALAT1 [111, 113], which have critical roles in the onset and progression of breast cancer [114]. Our findings confirm these studies and further show that, in addition to occupying protein coding genes, RUNX1 bookmarks several lncRNAs for post-mitotic regulation. Mechanistically, it will be interesting to determine if RUNX1-bookmarked lncRNAs have G1-specific roles in maintaining the normal mammary epithelial phenotype and/or in the onset and progression of breast cancer.
In summary, this study shows that RUNX1 occupies RNA Pol I- and II-transcribed genes in interphase and during mitosis for potential regulation of normal mammary epithelial proliferation, growth, and phenotype. Disruption of RUNX1 DNA binding by interfering with the RUNX1-CBFβ interaction leads to the epithelial-to-mesenchymal transition, a key event in breast cancer onset. Our observation of RUNX1 retention on mitotic chromosomes at target genes involved in cell lineage determination and global protein synthesis implicates RUNX1 mitotic gene bookmarking as a potential epigenetic mechanism to physiologically sustain the mammary epithelial phenotype.
Materials and Methods
Cell culture techniques
Breast epithelial (MCF10A) cells were cultured in DMEM/F-12 50/50 mixture (Corning™, Corning, NY, USA). Culturing media was also supplemented with horse serum to 5% (GIBCO, Grand Island, NY, USA), human insulin to 10 μg/mL (Sigma Aldrich, St. Louis, MO, USA), human epidermal growth factor to 20 ng/mL (PeproTech, Rocky Hill, NJ, USA), cholera toxin to 100 ng/mL (Thomas Scientific, Swedesboro, NJ, USA), hydrocortisone to 500 ng/mL (Sigma Aldrich, St. Louis, MO, USA), Penicillin-Streptomycin to 100 U/mL (Thermo Fisher Scientific, Asheville, NC, USA), and L-Glutamine to 2 mM (Thermo Fisher Scientific, Asheville, NC, USA).
For mitotic arrest of parental MCF10A cells, culturing media was supplemented with 50 ng/mL of Nocodazole (Sigma Aldrich, St. Louis, MO, USA) and incubated with cells for 16 hrs. Supplementing culturing media with equivalent volumes of DMSO (Sigma Aldrich, St. Louis, MO, USA) served as a control. DMSO-treated and mitotically arrested populations of MCF10A cells were harvested following the 16 hr incubation. For G1 (released from mitotic arrest) populations of MCF10A cells, the nocodazole-supplemented media was replaced with normal media and cells were incubated for 3 hrs, after which the released cell population was harvested for subsequent analyses that include western blotting, qPCR, FACS, and ChIP-seq.
Western blot analyses
Protein lysates were prepared by incubating cells in RIPA buffer on ice for 30 min, followed by sonication using Q700 Sonicator (QSonica, Newtown, CT, USA). Proteins were resolved by SDS-PAGE and transferred to PVDF membrane using standard protocols. Following primary antibodies were used at a 1:1000 dilution (except Lamin B, which was used at 1:2000 dilution) in this study: UBF (sc-13125, Santa Cruz Biotechnology, Dallas, TX, USA); RUNX1 (4334S, Cell Signaling Technologies, Danvers, MA, USA); Cyclin B (4138S, Cell Signaling Technologies, Danvers, MA, USA); Beta-Actin (3700S, Cell Signaling Technologies, Danvers, MA, USA), and CDT1 (ab70829, AbCam, Cambridge, UK); Lamin B1 (ab16048, AbCam, Cambridge, UK). Horseradish peroxidase conjugated secondary antibodies used in this studies were: goat anti-mouse IgG at 1:5000 dilution (31460, Invitrogen, Carlsbad, CA, USA), goat anti-rabbit IgG HRP conjugated (31430, Thermo Fisher Scientific, Asheville, NC, USA) at 1:1000, 1:2000, or 1:5000 dilutions. Blots were developed using Clarity Western ECL Substrate (Bio-Rad, Hercules, CA, USA) and imaged using Molecular Imager® Chemi doc™ XRS+ Imaging System (Bio-Rad, Hercules, CA, USA) aided by Image Lab Software Version 5.1 (Bio-Rad, Hercules, CA, USA).
Confocal microscopy, image acquisition, processing, and analyses
MCF10A cells were plated on gelatin-coated coverslips in 6-well plates at 175,000 cells/mL and processed for immunofluorescence 24 hrs after plating using standard protocol. Briefly, cells were washed twice with sterile-filtered PBS on ice and cell were fixed in 1% MeOH-free Formaldehyde in PBS for 10 min. After permeabilization in 0.25% Triton X-100-PBS solution, cells were sequentially incubated with primary and Alexa fluorophore conjugated secondary antibodies for 1 hr each at 37°C in a humidified chamber with extensive washes after each incubation. Primary antibodies used were: RUNX1 at 1:10 dilution (4334S, Cell Signaling Technologies, Danvers, MA, USA), and Upstream Binding Transcription Factor (UBF) at 1:200 dilution (F-9 sc-13125, Santa Cruz Biotechnology, Dallas, TX, USA). Secondary antibodies used were goat anti-rabbit IgG conjugated with Alexa Fluor 488 (A-11070, Life Technologies, Carlsbad, CA, USA) and goat anti-mouse IgG conjugated with Alexa Fluor 594 (A-11005, Life Technologies, Carlsbad, CA, USA) diluted 1:800. Cells were counterstained with DAPI to visualize DNA and coverslips were mounted onto slides in ProLong Gold Antifade Mountant (Thermo Fisher Scientific, Asheville, NC, USA). Images were captured using a Zeiss Axio Imager. Z2 fluorescent microscope and Hamamatsu ORCA-R2 C10600 digital camera. Images were processed using ZEN 2012 software.
To examine mitotic localization of RUNX1 in unfixed cells, an expression plasmid containing RUNX1-EGFP was introduced using either nucleofection or Lipofectamine 3000 transfection reagent in actively proliferating MCF10A cells grown on gelatin-coated coverslips. After 16 hours of nucleofection, cells were washed once with 1× PBS and stained with Hoechst dye to visualized DNA. Coverslips were mounted using the ProLong Gold Antifade Mountant and subjected to confocal microscopy.
Cells were initially imaged with a Zeiss LSM 510 META confocal laser scanning microscope (Carl Zeiss Microscopy, LLC., Thornwood, NY, USA). The DAPI signal was excited with a 405 nm laser and collected with a 425–475 nm band pass filter, Alexa 488 was excited with a 488 nm laser and collected with a 500–550 nm band pass filter, and Alexa 568 with a 561 nm laser and collected with a 570–620 nm band pass filter. Images were captured with a Plan-Fluor 40× (1.3 NA) objective lens. The confocal pinhole was initially set to 1.2 Airy Unit diameter for the 561 nm excitation giving an optical section thickness of 0.41 μm. Images were acquired at 12-bit data depth, and all settings, including laser power, amplifier gain, and amplifier offset were established using a look up table to provide an optimal gray-scale intensities. All images were acquired using matching imaging parameters. The acquired images were subjected to colocalization analysis via Volocity version 6.3.0 (Perkin Elmer, Waltham, MA, USA). At least 15 interphase and 15 metaphase cells were identified in captured images and appropriate thresholds were manually determined to eliminate background fluorescence for calculating Pearsons and Manders correlation coefficients between RUNX1 and UBF.
To ensure the specificity of our observations, additional samples were imaged with a Nikon A1R-ER laser scanning confocal microscope (Nikon, Melville, NY, USA). Images were acquired with the galvano scanner at a frame size of 1024 × 1024 pixels with an Apo TIRF 60× objective lens (N.A. 1.49) zoom of 2 and 1.2 Airy Unit pinhole setting. Images were also viewed in NIS Elements version 5.02.01 and analyzed using the line profiling tool. Overlaying DAPI, RUNX1, and UBF fluorescent intensities from individual channels along the line profile revealed overlapping peak intensities between the RUNX1 and UBF channels, thus indicating colocalization.
RNA isolation, cDNA synthesis, and quantitative PCR
Total RNA was isolated from MCF10A cells using TRIzol™ Reagent (Invitrogen, Carlsbad, CA, USA) and Direct-Zol™ RNA MiniPrep isolation kit (Zymo Research, Irvine, CA, USA) following manufacturer instructions. cDNA was created using SuperScript IV® First-Strand Synthesis System for RT-PCR (Thermo Fisher, Asheville, NC, USA). Resulting samples were quantified on a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and diluted to 500 pg/μL. Equal amounts of DNA template were loaded for samples analyzed by qPCR. At least three independent biological replicates were analyzed for expression of RUNX1 bookmarked genes by qPCR. Student’s t-test was used to determine the significance of changes in transcript levels under different biological conditions.
Chromatin immunoprecipitation, library preparation, sequencing, and bioinformatics analyses
Asynchronous (Asynch), mitotically arrested (M), and released from mitosis (G1) MCF10A breast epithelial cells were subjected to chromatin immunoprecipitation using a modified Farnham protocol [120]. Sonication parameters for each population of cells was as follows: Peak Watt 140W, Duty Factor 10, Cycle/Burst 200 using a S220 focused ultra-sonicator (Covaris, Matthews, NC, USA). M and G1 populations of cells were sonicated for 28 min total, whereas asynchronous populations of cells were sonicated for 36 min. An aliquot of sonicated lysates was boiled in 100°C for 15 min with NaCl and elution buffer and DNA was purified using PureLink™ PCR Purification Kit (K310001, Thermo Fisher, Asheville, NC, USA). Purified DNA was resolved on a 1.5% agarose gel to confirm optimal sonication (bulk of fragments between 200–400 bp) prior to performing ChIP. In parallel, an aliquot was also quantified via Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and analyzed by using a High Sensitivity DNA Kit on a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA).
For chromatin immunoprecipitation (ChIP) reactions, 150 ug of sonicated chromatin was incubated with 10 ug of RUNX1 antibody (4336BF, Cell Signaling Technologies, Danvers, MA, USA), diluted 1:10 in IP dilution buffer, and incubated overnight (16–18 hrs) at 4°C with mild agitation. Following incubation, 150 uL of Protein A/G magnetic beads (Thermo Scientific: Pierce, Waltham, MA, USA) per ug of antibody used were added to each IP reaction and incubated for 2–4 hrs at 4°C with mild agitation. Beads were extensively washed with IP wash buffers and resuspended in Elution Buffer to extract immunoprecipitated chromatin, which was subsequently purified using PureLink™ PCR Purification Kit. At least 3 biological replicates were performed for each cell population and each antibody.
ChIP libraries were generated using Accel-NGS® 2S Plus DNA Library kit (Swift Biosciences, Ann Arbor, MI, USA) following manufacturers protocol. Input and RUNX1 ChIP samples were normalized to 1ng prior to library generation. Libraries were amplified in an optional PCR step for 12 total cycles. Finalized libraries were double size selected using AMPure XP beads (0.8× and 0.2× volume ratios to sample), resulting in the majority fragments sized between 250–400 bp. Next generation sequencing of pooled ChIP libraries was performed by the University of Vermont Cancer Center, Vermont Integrated Genomics Resource (VIGR).
Single end, 50 bp reads (SE50) were processed pre-alignment by removing adapter reads (Cutadapt v1.6) and trimming low quality base calls from both ends (FASTQ Quality Trimmer 1.0.0; min score ≥ 20, window of 10, and step size of 1). Because we were specifically investigating rDNA, a customized build of hg38 was constructed that included normally masked regions of rDNA (Gencode U13369). Since some (although not complete) rDNA sequence is present in the hg38 assembly, we masked all parts of hg38 that would normally be attributed to rDNA sequences (bedtools v2.25.0 maskfasta) based on alignment positions of 50 bp in silico reads generated across U13369. Finally, we appended the complete rDNA sequence as a distinct sequence (chrU13369.1) to the masked hg38 FASTA resulting in the hg38_rDNA assembly used for analysis.
Resulting reads were aligned to hg38_rDNA (STAR v2.4; splicing disabled with ‘–alignIntronMax 1’). Next, we called peaks and generated fold-enrichment (FE) bedGraph files (MACS2 v2.1.0.20140616; callpeak at p-value e-5; and bdgcmp with FE method) [121]. Irreproducible Discovery Rate (IDR) was conducted using unpooled replicates with all peaks in pooled samples passing an IDR cutoff of 0.5 [122]. To reduce artificial peaks, we calculated strand cross-correlation for all peaks at a shift of 95 bp (the mean observed fragment size of 180 bp minus the read size of 85 bp) and unshifted [123]. We eliminated peaks that exhibited low shifted correlation (shifted correlation < 0.7) and those that exhibited high unshifted correlation relative to shifted (shifted–unshifted correlation < 0.1). This increased retrieval of the RUNX1 motif and improved agreement with other RUNX1 datasets. Passing peaks were annotated separately to mRNA and lncRNA transcript start sites (TSSs) using Gencode v27 with a distance cutoff of 5000 bp. Regional distribution of peaks was determined using the same annotation reference limited to the “basic” tag for exons and promoters.
Inhibitor treatment and assessment of global protein synthesis
Core binding factor: Beta (CBFβ) inhibitors AI-4-88 and AI-14-91 were kindly provided by John H. Bushweller (University of Virginia, USA) and used to evaluate RUNX1 DNA-binding inhibition in MCF10A cells. Protein synthesis evaluation by immunofluorescence was conducted following manufacturer protocol (K715-100, BioVision, San Francisco, CA, USA). To examine effects of inhibiting the RUNX1-CBFβ interaction, MCF10A cells were treated with active or inactive compound for 48 hours. Culture medium containing the active or inactive compounds was replaced with fresh medium without the compounds and cells were harvested 4- and 7-days post medium change.
RNA-sequencing, differential expression analysis, and pathway analysis
RNA was isolated using Direct-zol RNA MiniPrep (Zymo Research, Irvine, CA, USA) and was quantified and assayed for RNA integrity by Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). Following the removal of ribosomal RNA, the RNA pool was reverse transcribed, amplified, purified, and bound to strand-specific adaptors following the manufacturer’s protocol (SMARTer Stranded Total RNA Sample Prep Kit, Takara Bio, Mountain View, CA, USA). cDNA libraries were assayed for quality control by Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). After cDNA quality validation, generated libraries were sequenced. 24 hour and 48-hour counts were grouped together into one “crisis” category and the day 4 recovery and day 7 recovery counts were grouped together into one “recovery” category. Treatment groups were compared with untreated MCF10A cells. After demultiplexing and quality filtering, reads were aligned to hg38 using Gencode (GRCh38. p13). As a reference, annotation with STAR (v2.5.2a) [124] aligned reads were then counted using HT-Seq [125]. Differential gene expression was analyzed using DESeq2 in R v.3.5.1 [126]. Parameters for significant differential expression were base mean expression greater than five, absolute log2 fold change greater than one, and a p-value less than 0.05. Pathway analysis was performed using Gene Set Enrichment Analysis v6.3 (Broad Institute, Inc., MIT, UC San Diego, CA, USA).
ACKNOWLEDGMENTS
The authors thank Dr. Prachi Ghule, Department of Biochemistry, University of Vermont, for assistance in confocal microscopy; Scott Tighe, Pheobe Kehoe, and Jessica Hoffman of the Vermont Integrative Genomics Resource for performing next generation sequencing of samples; Dr. Roxana del Rio-Guerra of the Flow Cytometry and Cell Sorting Facility for analysis of samples by FACS; and Dr. Alan Howe, Department of Pharmacology, University of Vermont, for providing Phalloidin reagent used in immunofluorescence microscopy experiments.
Data availability
GEO accession number for the sequencing data generated in this study is GSE121370.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
This work was supported by NIH grants P01 CA082834 (GSS & JLS), U01 CA196383 (JLS), U54 GM115516 (GSS), F32 CA220935 (to AJ Fritz), and the Charlotte Perelman Fund for Cancer Research (GSS). GS Stein is the Arthur J. Perelman Professor of Cancer Research. The confocal microscopy work described in this manuscript was supported by Award Number 1S10RR019246 from the National Center for Research Resources for purchase of the Zeiss 510 META confocal scanning laser microscope and NIH award number 1S10OD025030-01 for purchase of the Nikon A1R-ER point scanning confocal microscope from the National Center for Research Resources. FACS experiments performed at the Harry Hood Bassett Flow Cytometry and Cell Sorting Facility, University of Vermont College of Medicine were supported by NIH S10-ODO18175.
References
1. Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012; 486:405–409. https://doi.org/10.1038/nature11154. [PubMed].
2. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, et al, and TCGA Research Network. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell. 2015; 163:506–519. https://doi.org/10.1016/j.cell.2015.09.033. [PubMed].
3. Elsheikh SE, Green AR, Rakha EA, Powe DG, Ahmed RA, Collins HM, Soria D, Garibaldi JM, Paish CE, Ammar AA, Grainge MJ, Ball GR, Abdelghany MK, et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009; 69:3802–3809. https://doi.org/10.1158/0008-5472.CAN-08-3907. [PubMed].
4. Li Z, Herold T, He C, Valk PJ, Chen P, Jurinovic V, Mansmann U, Radmacher MD, Maharry KS, Sun M, Yang X, Huang H, Jiang X, et al. Identification of a 24-gene prognostic signature that improves the European LeukemiaNet risk classification of acute myeloid leukemia: an international collaborative study. J Clin Oncol. 2013; 31:1172–1181. https://doi.org/10.1200/JCO.2012.44.3184. [PubMed].
5. Widschwendter M, Jones PA. DNA methylation and breast carcinogenesis. Oncogene. 2002; 21:5462–5482. https://doi.org/10.1038/sj.onc.1205606. [PubMed].
6. Zhang M, Lee AV, Rosen JM. The Cellular Origin and Evolution of Breast Cancer. Cold Spring Harb Perspect Med. 2017; 7:a027128. https://doi.org/10.1101/cshperspect.a027128. [PubMed].
7. Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol. 2017; 232:3261–3272. https://doi.org/10.1002/jcp.25797. [PubMed].
8. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–1428. https://doi.org/10.1172/JCI39104. [PubMed].
9. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013; 342:1234850. https://doi.org/10.1126/science.1234850. [PubMed].
10. Prieto-Garcia E, Diaz-Garcia CV, Garcia-Ruiz I, Agullo-Ortuno MT. Epithelial-to-mesenchymal transition in tumor progression. Med Oncol. 2017; 34:122. https://doi.org/10.1007/s12032-017-0980-8. [PubMed].
11. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. 2017; 11:805–823. https://doi.org/10.1002/1878-0261.12095. [PubMed].
12. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–890. https://doi.org/10.1016/j.cell.2009.11.007. [PubMed].
13. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010; 15:117–134. https://doi.org/10.1007/s10911-010-9178-9. [PubMed].
14. Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009; 28:151–166. https://doi.org/10.1007/s10555-008-9179-y. [PubMed].
15. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010; 107:15449–15454. https://doi.org/10.1073/pnas.1004900107. [PubMed].
16. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008; 14:818–829. https://doi.org/10.1016/j.devcel.2008.05.009. [PubMed].
17. Ito Y, Bae SC, Chuang LS. The RUNX family: developmental regulators in cancer. Nat Rev Cancer. 2015; 15:81–95. https://doi.org/10.1038/nrc3877. [PubMed].
18. Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001; 20:723–733. https://doi.org/10.1093/emboj/20.4.723. [PubMed].
19. Qin X, Jiang Q, Matsuo Y, Kawane T, Komori H, Moriishi T, Taniuchi I, Ito K, Kawai Y, Rokutanda S, Izumi S, Komori T. Cbfb regulates bone development by stabilizing Runx family proteins. J Bone Miner Res. 2015; 30:706–714. https://doi.org/10.1002/jbmr.2379. [PubMed].
20. Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S, Ogata K. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001; 104:755–767. https://doi.org/10.1016/S0092-8674(01)00271-9. [PubMed].
21. Adya N, Castilla LH, Liu PP. Function of CBFbeta/Bro proteins. Semin Cell Dev Biol. 2000; 11:361–368. https://doi.org/10.1006/scdb.2000.0189. [PubMed].
22. Chin DW, Watanabe-Okochi N, Wang CQ, Tergaonkar V, Osato M. Mouse models for core binding factor leukemia. Leukemia. 2015; 29:1970–1980. https://doi.org/10.1038/leu.2015.181. [PubMed].
23. Komori T. Requisite roles of Runx2 and Cbfb in skeletal development. J Bone Miner Metab. 2003; 21:193–197. [PubMed].
24. Roudaia L, Cheney MD, Manuylova E, Chen W, Morrow M, Park S, Lee CT, Kaur P, Williams O, Bushweller JH, Speck NA. CBFbeta is critical for AML1-ETO and TEL-AML1 activity. Blood. 2009; 113:3070–3079. https://doi.org/10.1182/blood-2008-03-147207. [PubMed].
25. Wang CQ, Chin DW, Chooi JY, Chng WJ, Taniuchi I, Tergaonkar V, Osato M. Cbfb deficiency results in differentiation blocks and stem/progenitor cell expansion in hematopoiesis. Leukemia. 2015; 29:753–757. https://doi.org/10.1038/leu.2014.316. [PubMed].
26. Malik N, Yan H, Moshkovich N, Palangat M, Yang H, Sanchez V, Cai Z, Peat TJ, Jiang S, Liu C, Lee M, Mock BA, Yuspa SH, et al. The transcription factor CBFB suppresses breast cancer through orchestrating translation and transcription. Nat Commun. 2019; 10:2071. https://doi.org/10.1038/s41467-019-10102-6. [PubMed].
27. Bidet A, Laharanne E, Achard S, Migeon M, Moreau C, Lippert E. Analysis of RUNX1 rearrangements: insights into leukaemogenesis mechanisms. Br J Haematol. 2016; 175:738–740. https://doi.org/10.1111/bjh.13881. [PubMed].
28. Brettingham-Moore KH, Taberlay PC, Holloway AF. Interplay between Transcription Factors and the Epigenome: Insight from the Role of RUNX1 in Leukemia. Front Immunol. 2015; 6:499. https://doi.org/10.3389/fimmu.2015.00499. [PubMed].
29. De Braekeleer E, Douet-Guilbert N, Morel F, Le Bris MJ, Ferec C, De Braekeleer M. RUNX1 translocations and fusion genes in malignant hemopathies. Future Oncol. 2011; 7:77–91. https://doi.org/10.2217/fon.10.158. [PubMed].
30. Dowdy CR, Frederick D, Zaidi SK, Colby JL, Lian JB, van Wijnen AJ, Gerstein RM, Stein JL, Stein GS. A germline point mutation in Runx1 uncouples its role in definitive hematopoiesis from differentiation. Exp Hematol. 2013; 41:980–991.e1. https://doi.org/10.1016/j.exphem.2013.06.006. [PubMed].
31. Dowdy CR, Xie R, Frederick D, Hussain S, Zaidi SK, Vradii D, Javed A, Li X, Jones SN, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Definitive hematopoiesis requires Runx1 C-terminal-mediated subnuclear targeting and transactivation. Hum Mol Genet. 2010; 19:1048–1057. https://doi.org/10.1093/hmg/ddp568. [PubMed].
32. Downing JR, Higuchi M, Lenny N, Yeoh AE. Alterations of the AML1 transcription factor in human leukemia. Semin Cell Dev Biol. 2000; 11:347–360. https://doi.org/10.1006/scdb.2000.0183. [PubMed].
33. Durst KL, Hiebert SW. Role of RUNX family members in transcriptional repression and gene silencing. Oncogene. 2004; 23:4220–4224. https://doi.org/10.1038/sj.onc.1207122. [PubMed].
34. Friedman AD. Cell cycle and developmental control of hematopoiesis by Runx1. J Cell Physiol. 2009; 219:520–524. https://doi.org/10.1002/jcp.21738. [PubMed].
35. Hart SM, Foroni L. Core binding factor genes and human leukemia. Haematologica. 2002; 87:1307–23. [PubMed].
36. Lam K, Zhang DE. RUNX1 and RUNX1-ETO: roles in hematopoiesis and leukemogenesis. Front Biosci. 2012; 17:1120–1139. https://doi.org/10.2741/3977. [PubMed].
37. Lichtinger M, Ingram R, Hannah R, Müller D, Clarke D, Assi SA, Lie-A-Ling M, Noailles L, Vijayabaskar MS, Wu M, Tenen DG, Westhead DR, Kouskoff V, et al. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. EMBO J. 2012; 31:4318–4333. https://doi.org/10.1038/emboj.2012.275. [PubMed].
38. Lo Coco F, Pisegna S, Diverio D. The AML1 gene: a transcription factor involved in the pathogenesis of myeloid and lymphoid leukemias. Haematologica. 1997; 82:364–70. [PubMed].
39. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017; 129:2070–2082. https://doi.org/10.1182/blood-2016-10-687830. [PubMed].
40. Browne G, Dragon JA, Hong D, Messier TL, Gordon JA, Farina NH, Boyd JR, VanOudenhove JJ, Perez AW, Zaidi SK, Stein JL, Stein GS, Lian JB. MicroRNA-378-mediated suppression of Runx1 alleviates the aggressive phenotype of triple-negative MDA-MB-231 human breast cancer cells. Tumour Biol. 2016; 37:8825–8839. https://doi.org/10.1007/s13277-015-4710-6. [PubMed].
41. Browne G, Taipaleenmaki H, Bishop NM, Madasu SC, Shaw LM, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Runx1 is associated with breast cancer progression in MMTV-PyMT transgenic mice and its depletion in vitro inhibits migration and invasion. J Cell Physiol. 2015; 230:2522–2532. https://doi.org/10.1002/jcp.24989. [PubMed].
42. Chimge NO, Frenkel B. The RUNX family in breast cancer: relationships with estrogen signaling. Oncogene. 2013; 32:2121–2130. https://doi.org/10.1038/onc.2012.328. [PubMed].
43. Chimge NO, Little GH, Baniwal SK, Adisetiyo H, Xie Y, Zhang T, O’Laughlin A, Liu ZY, Ulrich P, Martin A, Mhawech-Fauceglia P, Ellis MJ, Tripathy D, et al. RUNX1 prevents oestrogen-mediated AXIN1 suppression and beta-catenin activation in ER-positive breast cancer. Nat Commun. 2016; 7:10751. https://doi.org/10.1038/ncomms10751. [PubMed].
44. Ferrari N, Mohammed ZM, Nixon C, Mason SM, Mallon E, McMillan DC, Morris JS, Cameron ER, Edwards J, Blyth K. Expression of RUNX1 correlates with poor patient prognosis in triple negative breast cancer. PLoS One. 2014; 9:e100759. https://doi.org/10.1371/journal.pone.0100759. [PubMed].
45. Janes KA. RUNX1 and its understudied role in breast cancer. Cell Cycle. 2011; 10:3461–3465. https://doi.org/10.4161/cc.10.20.18029. [PubMed].
46. Recouvreux MS, Grasso EN, Echeverria PC, Rocha-Viegas L, Castilla LH, Schere-Levy C, Tocci JM, Kordon EC, Rubinstein N. RUNX1 and FOXP3 interplay regulates expression of breast cancer related genes. Oncotarget. 2016; 7:6552–6565. https://doi.org/10.18632/oncotarget.6771. [PubMed].
47. Wang L, Brugge JS, Janes KA. Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proc Natl Acad Sci U S A. 2011; 108:E803–E812. https://doi.org/10.1073/pnas.1103423108. [PubMed].
48. Hong D, Messier TL, Tye CE, Dobson JR, Fritz AJ, Sikora KR, Browne G, Stein JL, Lian JB, Stein GS. Runx1 stabilizes the mammary epithelial cell phenotype and prevents epithelial to mesenchymal transition. Oncotarget. 2017; 8:17610–17627. https://doi.org/10.18632/oncotarget.15381. [PubMed].
49. Sokol ES, Sanduja S, Jin DX, Miller DH, Mathis RA, Gupta PB. Perturbation-expression analysis identifies RUNX1 as a regulator of human mammary stem cell differentiation. PLoS Comput Biol. 2015; 11:e1004161. https://doi.org/10.1371/journal.pcbi.1004161. [PubMed].
50. van Bragt MP, Hu X, Xie Y, Li Z. RUNX1, a transcription factor mutated in breast cancer, controls the fate of ER-positive mammary luminal cells. eLife. 2014; 3:e03881. https://doi.org/10.7554/eLife.03881. [PubMed].
51. Festuccia N, Gonzalez I, Owens N, Navarro P. Mitotic bookmarking in development and stem cells. Development. 2017; 144:3633–3645. https://doi.org/10.1242/dev.146522. [PubMed].
52. John S, Workman JL. Bookmarking genes for activation in condensed mitotic chromosomes. BioEssays. 1998; 20:275–279. https://doi.org/10.1002/(SICI)1521-1878(199804)20:4<275::AID-BIES1>3.0.CO;2-P. [PubMed].
53. Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics Chromatin. 2013; 6:6. https://doi.org/10.1186/1756-8935-6-6. [PubMed].
54. Lodhi N, Ji Y, Tulin A. Mitotic bookmarking: maintaining post-mitotic reprogramming of transcription reactivation. Curr Mol Biol Rep. 2016; 2:10–16. https://doi.org/10.1007/s40610-016-0029-3. [PubMed].
55. Sarge KD, Park-Sarge OK. Mitotic bookmarking of formerly active genes: keeping epigenetic memories from fading. Cell Cycle. 2009; 8:818–823. https://doi.org/10.4161/cc.8.6.7849. [PubMed].
56. Zaidi SK, Lian JB, van Wijnen A, Stein JL, Stein GS. Mitotic Gene Bookmarking: An Epigenetic Mechanism for Coordination of Lineage Commitment, Cell Identity and Cell Growth. Adv Exp Med Biol. 2017; 962:95–102. https://doi.org/10.1007/978-981-10-3233-2_7. [PubMed].
57. Zaidi SK, Young DW, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet. 2010; 11:583–589. https://doi.org/10.1038/nrg2827. [PubMed].
58. Zaret KS. Genome reactivation after the silence in mitosis: recapitulating mechanisms of development? Dev Cell. 2014; 29:132–134. https://doi.org/10.1016/j.devcel.2014.04.019. [PubMed].
59. Ali SA, Dobson JR, Lian JB, Stein JL, van Wijnen AJ, Zaidi SK, Stein GS. RUNX2-HDAC1 co-repressor complex regulates rRNA gene expression by modulating UBF acetylation. J Cell Sci. 2012; 125:2732–2739. https://doi.org/10.1242/jcs.100909. [PubMed].
60. Ali SA, Zaidi SK, Dacwag CS, Salma N, Young DW, Shakoori AR, Montecino MA, Lian JB, van Wijnen AJ, Imbalzano AN, Stein GS, Stein JL. Phenotypic transcription factors epigenetically mediate cell growth control. Proc Natl Acad Sci U S A. 2008; 105:6632–6637. https://doi.org/10.1073/pnas.0800970105. [PubMed].
61. Ali SA, Zaidi SK, Dobson JR, Shakoori AR, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Transcriptional corepressor TLE1 functions with Runx2 in epigenetic repression of ribosomal RNA genes. Proc Natl Acad Sci U S A. 2010; 107:4165–4169. https://doi.org/10.1073/pnas.1000620107. [PubMed].
62. Bakshi R, Zaidi SK, Pande S, Hassan MQ, Young DW, Montecino M, Lian JB, van Wijnen AJ, Stein JL, Stein GS. The leukemogenic t(8;21) fusion protein AML1-ETO controls rRNA genes and associates with nucleolar-organizing regions at mitotic chromosomes. J Cell Sci. 2008; 121:3981–3990. https://doi.org/10.1242/jcs.033431. [PubMed].
63. Pande S, Ali SA, Dowdy C, Zaidi SK, Ito K, Ito Y, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Subnuclear targeting of the Runx3 tumor suppressor and its epigenetic association with mitotic chromosomes. J Cell Physiol. 2009; 218:473–479. https://doi.org/10.1002/jcp.21630. [PubMed].
64. Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007; 445:442–446. https://doi.org/10.1038/nature05473. [PubMed].
65. Young DW, Hassan MQ, Yang XQ, Galindo M, Javed A, Zaidi SK, Furcinitti P, Lapointe D, Montecino M, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Mitotic retention of gene expression patterns by the cell fate-determining transcription factor Runx2. Proc Natl Acad Sci U S A. 2007; 104:3189–3194. https://doi.org/10.1073/pnas.0611419104. [PubMed].
66. Zaidi SK, Young DW, Pockwinse SM, Javed A, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Mitotic partitioning and selective reorganization of tissue-specific transcription factors in progeny cells. Proc Natl Acad Sci U S A. 2003; 100:14852–14857. https://doi.org/10.1073/pnas.2533076100. [PubMed].
67. Pallier C, Scaffidi P, Chopineau-Proust S, Agresti A, Nordmann P, Bianchi ME, Marechal V. Association of chromatin proteins high mobility group box (HMGB) 1 and HMGB2 with mitotic chromosomes. Mol Biol Cell. 2003; 14:3414–3426. https://doi.org/10.1091/mbc.e02-09-0581. [PubMed].
68. Arampatzi P, Gialitakis M, Makatounakis T, Papamatheakis J. Gene-specific factors determine mitotic expression and bookmarking via alternate regulatory elements. Nucleic Acids Res. 2013; 41:2202–2215. https://doi.org/10.1093/nar/gks1365. [PubMed].
69. Blobel GA, Kadauke S, Wang E, Lau AW, Zuber J, Chou MM, Vakoc CR. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol Cell. 2009; 36:970–983. https://doi.org/10.1016/j.molcel.2009.12.001. [PubMed].
70. Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 2013; 27:251–260. https://doi.org/10.1101/gad.206458.112. [PubMed].
71. Deluz C, Friman ET, Strebinger D, Benke A, Raccaud M, Callegari A, Leleu M, Manley S, Suter DM. A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes Dev. 2016; 30:2538–2550. https://doi.org/10.1101/gad.289256.116. [PubMed].
72. Festuccia N, Dubois A, Vandormael-Pournin S, Gallego Tejeda E, Mouren A, Bessonnard S, Mueller F, Proux C, Cohen-Tannoudji M, Navarro P. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat Cell Biol. 2016; 18:1139–1148. https://doi.org/10.1038/ncb3418. [PubMed].
73. Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012; 150:725–737. https://doi.org/10.1016/j.cell.2012.06.038. [PubMed].
74. Lake RJ, Tsai PF, Choi I, Won KJ, Fan HY. RBPJ, the major transcriptional effector of Notch signaling, remains associated with chromatin throughout mitosis, suggesting a role in mitotic bookmarking. PLoS Genet. 2014; 10:e1004204. https://doi.org/10.1371/journal.pgen.1004204. [PubMed].
75. Lerner J, Bagattin A, Verdeguer F, Makinistoglu MP, Garbay S, Felix T, Heidet L, Pontoglio M. Human mutations affect the epigenetic/bookmarking function of HNF1B. Nucleic Acids Res. 2016; 44:8097–8111. https://doi.org/10.1093/nar/gkw467. [PubMed].
76. Liu Y, Pelham-Webb B, Di Giammartino DC, Li J, Kim D, Kita K, Saiz N, Garg V, Doane A, Giannakakou P, Hadjantonakis AK, Elemento O, Apostolou E. Widespread Mitotic Bookmarking by Histone Marks and Transcription Factors in Pluripotent Stem Cells. Cell Rep. 2017; 19:1283–1293. https://doi.org/10.1016/j.celrep.2017.04.067. [PubMed].
77. Lodhi N, Kossenkov AV, Tulin AV. Bookmarking promoters in mitotic chromatin: poly(ADP-ribose)polymerase-1 as an epigenetic mark. Nucleic Acids Res. 2014; 42:7028–7038. https://doi.org/10.1093/nar/gku415. [PubMed].
78. Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. A dynamic mode of mitotic bookmarking by transcription factors. eLife. 2016; 5:e22280. https://doi.org/10.7554/eLife.22280. [PubMed].
79. Xing H, Wilkerson DC, Mayhew CN, Lubert EJ, Skaggs HS, Goodson ML, Hong Y, Park-Sarge OK, Sarge KD. Mechanism of hsp70i gene bookmarking. Science. 2005; 307:421–423. https://doi.org/10.1126/science.1106478. [PubMed].
80. Carlton AL, Illendula A, Gao Y, Llaneza DC, Boulton A, Shah A, Rajewski RA, Landen CN, Wotton D, Bushweller JH. Small molecule inhibition of the CBFbeta/RUNX interaction decreases ovarian cancer growth and migration through alterations in genes related to epithelial-to-mesenchymal transition. Gynecol Oncol. 2018; 149:350–360. https://doi.org/10.1016/j.ygyno.2018.03.005. [PubMed].
81. Illendula A, Gilmour J, Grembecka J, Tirumala VS, Boulton A, Kuntimaddi A, Schmidt C, Wang L, Pulikkan JA, Zong H, Parlak M, Kuscu C, Pickin A, et al. Small Molecule Inhibitor of CBFbeta-RUNX Binding for RUNX Transcription Factor Driven Cancers. EBioMedicine. 2016; 8:117–131. https://doi.org/10.1016/j.ebiom.2016.04.032. [PubMed].
82. Choi JY, Pratap J, Javed A, Zaidi SK, Xing L, Balint E, Dalamangas S, Boyce B, van Wijnen AJ, Lian JB, Stein JL, Jones SN, Stein GS. Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc Natl Acad Sci U S A. 2001; 98:8650–8655. https://doi.org/10.1073/pnas.151236498. [PubMed].
83. Javed A, Afzal F, Bae JS, Gutierrez S, Zaidi K, Pratap J, van Wijnen AJ, Stein JL, Stein GS, Lian JB. Specific residues of RUNX2 are obligatory for formation of BMP2-induced RUNX2-SMAD complex to promote osteoblast differentiation. Cells Tissues Organs. 2009; 189:133–137. https://doi.org/10.1159/000151719. [PubMed].
84. Lian JB, Stein JL, Stein GS, van Wijnen AJ, Montecino M, Javed A, Gutierrez S, Shen J, Zaidi SK, Drissi H. Runx2/Cbfa1 functions: diverse regulation of gene transcription by chromatin remodeling and co-regulatory protein interactions. Connect Tissue Res. 2003; 44:141–148. https://doi.org/10.1080/03008200390152232. [PubMed].
85. Stein GS, Lian JB, Stein JL, van Wijnen AJ, Choi JY, Pratap J, Zaidi SK. Temporal and spatial parameters of skeletal gene expression: targeting RUNX factors and their coregulatory proteins to subnuclear domains. Connect Tissue Res. 2003; 44:149–153. https://doi.org/10.1080/03008900390152241. [PubMed].
86. Vradii D, Zaidi SK, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Point mutation in AML1 disrupts subnuclear targeting, prevents myeloid differentiation, and effects a transformation-like phenotype. Proc Natl Acad Sci U S A. 2005; 102:7174–7179. https://doi.org/10.1073/pnas.0502130102. [PubMed].
87. Zaidi SK, Dowdy CR, van Wijnen AJ, Lian JB, Raza A, Stein JL, Croce CM, Stein GS. Altered Runx1 subnuclear targeting enhances myeloid cell proliferation and blocks differentiation by activating a miR-24/MKP-7/MAPK network. Cancer Res. 2009; 69:8249–8255. https://doi.org/10.1158/0008-5472.CAN-09-1567. [PubMed].
88. Zaidi SK, Javed A, Choi JY, van Wijnen AJ, Stein JL, Lian JB, Stein GS. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J Cell Sci. 2001; 114:3093–3102. [PubMed].
89. Zaidi SK, Javed A, Pratap J, Schroeder TM, Westendorf JJ, Lian JB, van Wijnen AJ, Stein GS, Stein JL. Alterations in intranuclear localization of Runx2 affect biological activity. J Cell Physiol. 2006; 209:935–942. https://doi.org/10.1002/jcp.20791. [PubMed].
90. Zaidi SK, Medina RF, Pockwinse SM, Bakshi R, Kota KP, Ali SA, Young DW, Nickerson JA, Javed A, Montecino M, van Wijnen AJ, Lian JB, Stein JL, Stein GS. Subnuclear localization and intranuclear trafficking of transcription factors. Methods Mol Biol. 2010; 647:77–93. https://doi.org/10.1007/978-1-60761-738-9_4. [PubMed].
91. Zaidi SK, Pande S, Pratap J, Gaur T, Grigoriu S, Ali SA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc Natl Acad Sci U S A. 2007; 104:19861–19866. https://doi.org/10.1073/pnas.0709650104. [PubMed].
92. Zaidi SK, Young DW, Javed A, Pratap J, Montecino M, van Wijnen A, Lian JB, Stein JL, Stein GS. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer. 2007; 7:454–463. https://doi.org/10.1038/nrc2149. [PubMed].
93. Zeng C, McNeil S, Pockwinse S, Nickerson J, Shopland L, Lawrence JB, Penman S, Hiebert S, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Intranuclear targeting of AML/CBFalpha regulatory factors to nuclear matrix-associated transcriptional domains. Proc Natl Acad Sci U S A. 1998; 95:1585–1589. https://doi.org/10.1073/pnas.95.4.1585. [PubMed].
94. Zeng C, van Wijnen AJ, Stein JL, Meyers S, Sun W, Shopland L, Lawrence JB, Penman S, Lian JB, Stein GS, Hiebert SW. Identification of a nuclear matrix targeting signal in the leukemia and bone-related AML/CBF-alpha transcription factors. Proc Natl Acad Sci U S A. 1997; 94:6746–6751. https://doi.org/10.1073/pnas.94.13.6746. [PubMed].
95. Chen CCL, Goyal P, Karimi MM, Abildgaard MH, Kimura H, Lorincz MC. H3S10ph broadly marks early-replicating domains in interphase ESCs and shows reciprocal antagonism with H3K9me2. Genome Res. 2018; 28:37–51. https://doi.org/10.1101/gr.224717.117. [PubMed].
96. Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, Inagaki M. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999; 274:25543–25549. https://doi.org/10.1074/jbc.274.36.25543. [PubMed].
97. Shrivastava T, Mino K, Babayeva ND, Baranovskaya OI, Rizzino A, Tahirov TH. Structural basis of Ets1 activation by Runx1. Leukemia. 2014; 28:2040–2048. https://doi.org/10.1038/leu.2014.111. [PubMed].
98. Gébrane-Younès J, Fomproix N, Hernandez-Verdun D. When rDNA transcription is arrested during mitosis, UBF is still associated with non-condensed rDNA. J Cell Sci. 1997; 110:2429–40. [PubMed].
99. Lopez-Camacho C, van Wijnen AJ, Lian JB, Stein JL, Stein GS. CBFbeta and the leukemogenic fusion protein CBFbeta-SMMHC associate with mitotic chromosomes to epigenetically regulate ribosomal genes. J Cell Biochem. 2014; 115:2155–2164. https://doi.org/10.1002/jcb.24892. [PubMed].
100. Kim W, Barron DA, San Martin R, Chan KS, Tran LL, Yang F, Ressler SJ, Rowley DR. RUNX1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc Natl Acad Sci U S A. 2014; 111:16389–16394. https://doi.org/10.1073/pnas.1407097111. [PubMed].
101. Wu D, Ozaki T, Yoshihara Y, Kubo N, Nakagawara A. Runt-related transcription factor 1 (RUNX1) stimulates tumor suppressor p53 protein in response to DNA damage through complex formation and acetylation. J Biol Chem. 2013; 288:1353–1364. https://doi.org/10.1074/jbc.M112.402594. [PubMed].
102. Ozaki T, Nakagawara A, Nagase H. RUNX Family Participates in the Regulation of p53-Dependent DNA Damage Response. Int J Genomics. 2013; 2013:271347. https://doi.org/10.1155/2013/271347. [PubMed].
103. Satoh Y, Matsumura I, Tanaka H, Harada H, Harada Y, Matsui K, Shibata M, Mizuki M, Kanakura Y. C-terminal mutation of RUNX1 attenuates the DNA-damage repair response in hematopoietic stem cells. Leukemia. 2012; 26:303–311. https://doi.org/10.1038/leu.2011.202. [PubMed].
104. Xu L, Gu ZH, Li Y, Zhang JL, Chang CK, Pan CM, Shi JY, Shen Y, Chen B, Wang YY, Jiang L, Lu J, Xu X, et al. Genomic landscape of CD34+ hematopoietic cells in myelodysplastic syndrome and gene mutation profiles as prognostic markers. Proc Natl Acad Sci U S A. 2014; 111:8589–8594. https://doi.org/10.1073/pnas.1407688111. [PubMed].
105. Cargnello M, Tcherkezian J, Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis. 2015; 30:169–176. https://doi.org/10.1093/mutage/geu045. [PubMed].
106. Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014; 40:862–871. https://doi.org/10.1016/j.ctrv.2014.03.004. [PubMed].
107. Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007; 104:14718–14723. https://doi.org/10.1073/pnas.0706933104. [PubMed].
108. Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A. 2006; 103:2196–2201. https://doi.org/10.1073/pnas.0510974103. [PubMed].
109. Mueller SO, Clark JA, Myers PH, Korach KS. Mammary gland development in adult mice requires epithelial and stromal estrogen receptor alpha. Endocrinology. 2002; 143:2357–2365. https://doi.org/10.1210/endo.143.6.8836. [PubMed].
110. Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010; 30:3943–3955. https://doi.org/10.1128/MCB.00118-10. [PubMed].
111. Barutcu AR, Hong D, Lajoie BR, McCord RP, van Wijnen AJ, Lian JB, Stein JL, Dekker J, Imbalzano AN, Stein GS. RUNX1 contributes to higher-order chromatin organization and gene regulation in breast cancer cells. Biochim Biophys Acta. 2016; 1859:1389–1397. https://doi.org/10.1016/j.bbagrm.2016.08.003. [PubMed].
112. Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013; 23:12–22. https://doi.org/10.1101/gr.139469.112. [PubMed].
113. Gutschner T, Hammerle M, Diederichs S. MALAT1–a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl). 2013; 91:791–801. https://doi.org/10.1007/s00109-013-1028-y. [PubMed].
114. Yu X, Li Z, Zheng H, Chan MT, Wu WK. NEAT1: A novel cancer-related long non-coding RNA. Cell Prolif. 2017; 50:e12329. https://doi.org/10.1111/cpr.12329. [PubMed].
115. Li X, Cao Y, Li M, Jin F. Upregulation of HES1 Promotes Cell Proliferation and Invasion in Breast Cancer as a Prognosis Marker and Therapy Target via the AKT Pathway and EMT Process. J Cancer. 2018; 9:757–766. https://doi.org/10.7150/jca.22319. [PubMed].
116. Strom A, Arai N, Leers J, Gustafsson JA. The Hairy and Enhancer of Split homologue-1 (HES-1) mediates the proliferative effect of 17beta-estradiol on breast cancer cell lines. Oncogene. 2000; 19:5951–5953. https://doi.org/10.1038/sj.onc.1203990. [PubMed].
117. Weyemi U, Redon CE, Sethi TK, Burrell AS, Jailwala P, Kasoji M, Abrams N, Merchant A, Bonner WM. Twist1 and Slug mediate H2AX-regulated epithelial-mesenchymal transition in breast cells. Cell Cycle. 2016; 15:2398–2404. https://doi.org/10.1080/15384101.2016.1198864. [PubMed].
118. Kageyama R, Ohtsuka T, Kobayashi T. The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007; 134:1243–1251. https://doi.org/10.1242/dev.000786. [PubMed].
119. Rani A, Greenlaw R, Smith RA, Galustian C. HES1 in immunity and cancer. Cytokine Growth Factor Rev. 2016; 30:113–117. https://doi.org/10.1016/j.cytogfr.2016.03.010. [PubMed].
120. O’Geen H, Frietze S, Farnham PJ. Using ChIP-seq technology to identify targets of zinc finger transcription factors. Methods Mol Biol. 2010; 649:437–455. https://doi.org/10.1007/978-1-60761-753-2_27. [PubMed].
121. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. https://doi.org/10.1186/gb-2008-9-9-r137. [PubMed].
122. Li QH, Brown JB, Huang HY, Bickel PJ. Measuring Reproducibility of High-Throughput Experiments. Ann Appl Stat. 2011; 5:1752–1779. https://doi.org/10.1214/11-AOAS466.
123. Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012; 22:1813–1831. https://doi.org/10.1101/gr.136184.111. [PubMed].
124. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. https://doi.org/10.1093/bioinformatics/bts635. [PubMed].
125. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. https://doi.org/10.1093/bioinformatics/btu638. [PubMed].
126. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. https://doi.org/10.1186/gb-2010-11-10-r106. [PubMed].