
INTRODUCTION
Prostate cancer is the most frequently diagnosed male cancer and the second leading cause of cancer death in men in the United States [1]. However, only a fraction of cases of prostate cancer lead to death. Thus, reliably identifying individuals at higher risk of progression to metastatic disease is of great potential utility.
GWA (Genome-wide association) studies have been performed to identify more than 70 risk variants associated with overall risk of developing prostate cancer [2–13]. A few recent GWA studies showed that some of these risk variants may correlate with the progression of prostate cancer [14–19] and, thus, could be clinically useful given that the majority prostate cancer cases are indolent and not a threat to life. Individually, the identified risk variants are low-penetrance susceptibility loci and have little clinical utility. Almost all are located in non-protein-coding or intergenic regions with unknown mechanisms of influence on phenotype. GWA follow-up studies have identified possible associations between the risk variants and gene expression levels of nearby or local genes (cis-associations) [6, 10, 13]. Remote associations (trans-) between these SNP variants and significant expression changes are likely but unknown, as yet.
Prostate cancer risk alleles can ultimately manifest their phenotype in a variety of ways. A subset may do so by altering gene expression in one or more cell types, including prostate epithelium, prostate stroma, the immune system, or others. In addition, an overlapping subset of risk SNPs may also affect progression after cancer has arisen, mediated by expression in one or more cell types.
Independent of SNP data, progression risk within the tumors has been associated with differences in gene expression [20–23] and in DNA methylation [24–26]. However, the cell, genetic, and epigenetic heterogeneity within a tumor and between tumors is a barrier to developing biomarkers for reliable prognosis. The number of candidate biomarkers that agree across studies is very small [23]. Similarly, mapping the effect of SNPs on expression in tumor could also be noisy. Reactive stroma initiates during early prostate cancer development and coevolves with prostate cancer progression. Recent evidence suggests that prostate epithelium and stroma interact in a highly organ-specific, androgen-dependent, and temporally related manner [27, 28]. Meanwhile, few somatic genetic differences have been identified in the stroma of prostate cancer [29, 30] and we have shown that the stroma undergoes expression differences with diagnostic [31] and prognostic potential [32].
If SNP risk markers could be linked with diagnostic and prognostic expression data in the relatively genetically stable environment of the stroma then, together, these associations may increase the utility of low risk SNPs by identifying a subset of patients where an otherwise low penetrance risk SNP was relevant.
To make the first step towards this goal, we applied eQTL (Expression Quantitative Trait Loci) analysis to expression data from tumor-adjacent stroma, to define associations between gene activity and risk variants [33–36]. This is a powerful method that treats each gene as a quantitative trait and identifies loci whose genotype variation such as in SNPs is associated with gene expression difference. The identified loci are considered as eQTLs.
In the eQTL analysis, we modified a Bayesian clustering method [34] to analyze expressed prostate cancer-related genes and susceptible loci jointly in a single model. Applying the approach to stroma-enriched samples, we identified 47 eQTL associations. In particular, the variant rs10896449 is associated with 32 significant expression differences in the stroma. This is the first study to identify trans-eQTL associations occurring between multiple SNPs and multiple significant expression differences in cancer stroma. The possible relationships between the identified associations and clinical properties, including cancer outcome, is examined and trends are identified.
RESULTS
Identification of SNP-transcript associations
A literature search of prostate cancer-related genes was conducted through the electronic database PubMed [20–23, 31, 32, 37–39] including differential expressed genes of utility for diagnosis and prognosis and genes with local distance to susceptible loci as reported in GWA studies. A total of 4030 such genes were identified. A second literature search of GWA studies of prostate cancer was conducted through the electronic database PubMed [2–14, 18, 40–43] covering a total of ~40000 prostate cancer cases, identifying more than 70 susceptible SNPs that were reported to have significant association with prostate cancer. 35 SNPs were selected and summarized in Supplementary Tables S2 and S3 for eQTL analysis.
In eQTL analysis, we examined possible associations between the 35 SNPs and 4030 transcripts using 49 stroma-enriched samples. Four eQTL analyses were performed in total. Samples with stroma cell type percentage greater than 50%, 60%, 70%, and 80% were used for the four eQTL analyses. Table 1 shows the number of selected samples in the four eQTL analyses. In addition to the “80%” criterion described above, an eQTL was only accepted when the transcript-SNP association appeared in all four eQTL analyses.
A total of 47 associations including 8 SNPs and 46 transcripts were identified in all four eQTL analyses. Table 2 shows in detail the combinations of associations among 8 SNPs and 46 transcripts. For example, the SNP rs10896449 is associated with the most transcripts, 32 transcripts.
In order to test whether any of the 47 associations are false-positives, we carried out two extensive resampling studies in order to estimate false discovery rates for the associations. The first resampling study is a permutation test as follows:
Table 1: Number of samples used in four eQTL analyses on the basis on varying stroma cell type percentage.
Stroma percentage |
>50% |
>60% |
>70% |
>80% |
Number of samples |
49 |
41 |
33 |
25 |
Table 2: 47 associations between 8 SNPs and 46 transcripts in eQTL analysis.
Association ID |
Probe Set |
Gene Symbol |
Location |
SNP rs10896449 (G) 11q13 |
|||
1 |
209716_at |
CSF1 |
1p13 |
2 |
204175_at |
ZNF593 |
1p36 |
3 |
204197_s_at |
RUNX3 |
1p36 |
4 |
202546_at |
VAMP8 |
2p11-p12 |
5 |
205174_s_at |
QPCT |
2p22 |
6 |
218864_at |
TNS1 |
2q35-q36 |
7 |
226766_at |
ROBO2 |
3p12 |
8 |
219551_at |
EAF2 |
3q13 |
9 |
232099_at |
PCDHB16 |
5q31 |
10 |
205100_at |
GFPT2 |
5q34-q35 |
11 |
208583_x_at |
HIST1H2AJ |
6p22 |
12 |
223475_at |
CRISPLD1 |
8q21 |
13 |
204501_at |
NOV |
8q24 |
14 |
205041_s_at |
ORM1 /// ORM2 |
9q32 |
15 |
205127_at |
PTGS1 |
9q32-q33 |
16 |
213004_at |
ANGPTL2 |
9q34 |
17 |
203666_at |
CXCL12 |
10q11 |
18 |
204396_s_at |
GRK5 |
10q26 |
19 |
203835_at |
LRRC32 |
11q13-q14 |
20 |
211964_at |
COL4A2 |
13q34 |
21 |
201562_s_at |
SORD |
15q15 |
22 |
203151_at |
MAP1A |
15q15 |
23 |
214297_at |
CSPG4 |
15q24 |
24 |
224476_s_at |
MESP1 |
15q26 |
25 |
229730_at |
SMTNL2 |
17p13 |
26 |
218980_at |
FHOD3 |
18q12 |
27 |
37996_s_at |
DMPK |
19q13 |
28 |
222106_at |
PRND |
20p13 |
29 |
205439_at |
GSTT2 |
22q11 |
30 |
201787_at |
FBLN1 |
22q13 |
31 |
220663_at |
IL1RAPL1 |
Xp21-p22 |
32 |
204584_at |
L1CAM |
Xq28 |
SNP rs1859962 (G) 17q24 |
|||
33 |
238079_at |
TPM3 |
1q21 |
34 |
206307_s_at |
FOXD1 |
5q12-q13 |
35 |
203438_at |
STC2 |
5q35 |
36 |
205040_at |
ORM1 |
9q32 |
SNP rs401681 (C) 5p15 |
|||
37 |
206529_x_at |
SLC26A4 |
7q31 |
38 |
204846_at |
CP |
3q23-q25 |
39 |
203021_at |
SLPI |
20q12 |
SNP 9623117 (C) 22q13 |
|||
40 |
206307_s_at |
FOXD1 |
5q12-q13 |
41 |
220120_s_at |
EPB41L4A |
5q21 |
42 |
228256_s_at |
EPB41L4A |
5q21 |
43 |
214676_x_at |
MUC3A |
7q22 |
SNP rs12621278 (G) 2q31 |
|||
44 |
211734_s_at |
FCER1A |
1q23 |
SNP rs1465618 (A) 2p21 |
|||
45 |
205132_at |
ACTC1 |
15q14 |
SNP rs620861 (C) 8q24 |
|||
46 |
210452_x_at |
CYP4F2 |
19p13 |
SNP rs6983267 (G) 8q24 |
|||
47 |
223775_at |
HHIP |
4q28-q32 |
1. Permute SNP samples for the smallest data set (>80% stroma).
2. Move to the next smallest data set and permute the SNP samples which are not included in previous permutated data set.
3. Repeat step 2 until all four data sets are permuted.
After permutation, the modified BAYES method was used to perform eQTL analyses between the randomly permutated SNP data and original expression data. The associations that appear in all four eQTL analyses were considered as false discovered associations. A total of 1000 permutations were performed. No permutation was identified with associations greater than 47 associations. The average number of falsely discovered associations is 1.57 compared to the 47 observed, and a false discovery rate of 3.3% was estimated.
In the second resampling study, 35 SNPs were randomly selected from over 1 million SNPs represented on the Illumina SNP array and used to perform four eQTL analyses. Again a total of 1000 random selections were performed. Again no permutation was identified with associations greater than 47 associations. The average number of falsely discovered associations is 2.55 compared to 47 observed, and the false discovery rate was estimated as 5.4%. Thus, the two resampling studies are consistent and strongly indicate that only three of our identified 47 transcript-SNP associations could be considered to occur by chance. The genes of the significant expression differences linked to 8 SNPs are given in Table 2.
Association of SNP-transcript associations with clinical parameters
We followed a two-step procedure (see Supplementary methods for details) to boost the power of identifying possible associations with clinical properties. This was necessary because the power to identify highly significant correlations with clinical parameters is limited due to the small sample size which needs to be further divided into subgroups for such an analysis.
First, the 49 patients were subdivided into three risk groups (high, intermediate, and low risk group) defined by each transcript-SNP association. Supplementary Figure 1 shows the way that the three risk groups were defined. Then ordinal logistic regression analysis was performed to identify associations between risk groups and clinical properties. Supplementary Table S3 shows the results of ordinal logistic regression analysis. Association 40 (FOXD1 and rs9623117 C) is the most interesting association because the p-values in both univariate and multivariate models were less than 0.05. Furthermore, we examined the high and low risk groups only defined by association 40. A Kaplan-Meier survival analysis (Figure 1) has an estimated hazard ratio 2.23 and significant p-value of 0.034 (logrank test).
DISCUSSION
In previous studies, gene signatures were identified by gene expression profiling and genetic variants were identified by GWA studies. These results were used to develop biomarkers for prostate cancer diagnosis and prognosis. However, the low concordance among gene signatures of profiling studies and the marginal risk of the best loci found in GWA studies have reduced the utility of such markers, to date. We speculated that by combining expression data with known genotypes, the resulting associations would add value and provide new evidence of a mechanism for the association between SNP and risk of prostate cancer.
We have established significant associations between risk-associated SNPs and RNA expression levels in tumor-adjacent prostate stroma. We used tumor-adjacent stroma instead of tumor for the study because there are very few genetic alternations in stroma [29, 30, 44, 45]. Studying the stroma does not preclude observing eQTLs in the tumor however. On the contrary, variants affecting expression in the tumor can in turn affect expression in the stroma because there are many active interactions between tumor and tumor-adjacent stroma in prostate cancer [31, 46, 47]. Epithelial cells of prostate cancer infiltrate and propagate in a microenvironment consisting largely of myofibroblast cells as well as inflammatory cells and other supporting cells and structures. The mesenchymal component is not passive but responds to signals from the tumor component and, in turn, alters tumor properties, some of which are essential for tumor growth and progression [46, 48]. Similarly SNPs affecting the endocrine and immune system may also be detectable in the response of prostate stroma.
We modified the BAYES method to perform an eQTL study to identify potential associations between risk loci and genes differentially expressed in prostate cancer stroma. The comprehensive mapping between SNPs and transcripts is one of the advantages of the modified BAYES method, allowing the eQTL study to identify not only cis-linkages but also trans-linkages. We utilized an extensive amount of information that is available for prostate cancer from prior GWA studies, encompassing around 40,000 patients, in order to form a short list of 35 SNPs for further consideration. Similarly a short list of 4030 prostate cancer-related genes were selected based on previously reported prostate cancer diagnosis and prognosis studies. To reduce false discovery rate, we performed eQTL analysis on four data sets with increased stroma percentages. Our two permutation tests also showed that the false discovery rate is very low. We obtained both expression and SNP data for 49 patients, 27 non-relapse, 20 relapse, and 2 unknown (Supplemental Table S1 for list of parameters) and observed 47 associations between eight SNPs and 46 transcripts (45 genes).
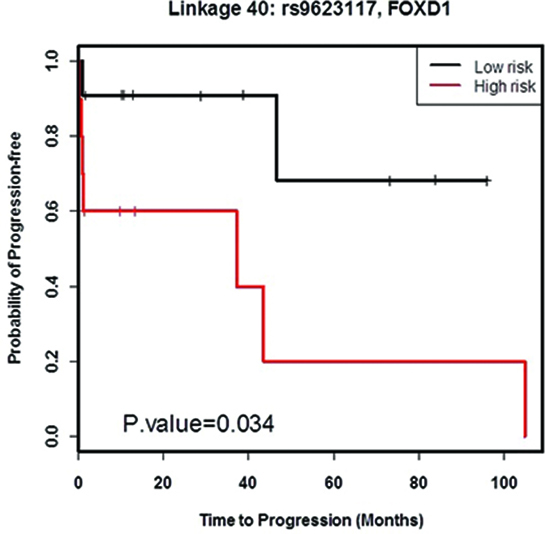
Figure 1: Survival analysis of high and low risk groups defined by FOXD1-rs9623117.
Next we examined if there was any evidence that the SNP-expression associations could better stratify patients for risk of progression of prostate cancer. Most SNPs associated with prostate cancer have been identified in case-control GWA studies. However, SNPs associated with disease progression are beginning to be identified. A recent study reported that two variants at 10q11 and one variant at 8q24 associated with susceptibility influence biochemical recurrence following radical prostatectomy [49]. Variants at 8q24 have been evaluated for risks of aggressiveness of prostate cancer based on age at onset, familial aggregation and tumor grades and stages. Some GWA studies have reported that the aggressive forms of the disease are influenced by the same variants associated with susceptibility to prostate cancer [15, 16, 18, 19]. A recent study also showed that variants at 8q24 act as enhancers of proto-oncogene c-MYC [50]. c-MYC is also located at 8q24, a known regulator of cell growth, and has a critical role in prostate cancer development and progression. It is likely that a subset of SNPs affecting the probability of getting prostate cancer will also affect the probability of progressing to advanced disease once prostate cancer has developed.
Using a two-step procedure described in supplemental method, we looked for prognostic correlation between clinical properties and SNP-expression associations. In one case, association 40 (FOXD1 and rs9623117 C), the penetrance of the association appears to extend to an association with survival. This association is consistent with the properties of FOXD1. FOXD1 is a member of Forkhead family of transcription factors involved in the Wnt pathway [51], which regulates epithelial-mesenchymal transition (EMT) in cancer. EMT is an important mechanism by which prostate cancer gains aggressive properties in cell migration, vascular invasion and early metastases. Moreover, Koga et al. characterized FOXD1 as a mediator and indicator of the cell reprogramming process, the prevention of FOXD1 expression resulted in a reduced number of iPSCs (induced pluripotent stem cells) [52]. Thus the properties of FOXD1 suggest one testable mechanism that explains the association of the FOXD1-rs9623117 C with survival observed here.
Multiple trans-associations between SNPs and gene expression in prostate cancer have not been observed before. Our results provide new ways for understanding the basis of risk for eight SNPs previously associated with occurrence of prostate cancer, namely involving altered expression among identified genetically unlinked genes. In future studies, it will be desirable to expand the number of samples in our study to more reliably test parameters associated with progression. It will also be desirable to obtain both expression and SNP data from archived normal stroma samples that have many years of follow-up data on the subsequent occurrence of prostate cancer. Such samples may reveal the role of SNPs in regulating expression in stroma not only after cancer has occurred, as we have studied here, but also during the onset of prostate cancer.
MATERIALS AND METHODS
Prostate cancer patient samples
Tissues for a total of 55 patients treated by radical prostatectomy (RP) for prostate cancer were analyzed. Informed consent was obtained in all cases following a protocol approved by the UCI Office Research Administration Institutional Review Board (IRB) as part of the NCI “SPECS” consortium at UCI for Strategic Partners for the Evaluation of Cancer Signatures for Prostate Cancer. All tissues were collected at surgery and escorted to pathology for expedited review, dissection and snap freezing in liquid nitrogen.
Demographic and clinical parameters such as Pre-operative Prostate Specific Antigen (Pre-PSA), surgical margin status, post-prostatectomy Gleason sum, age, T stage, are presented in Supplementary Table S1. Biochemical relapse was defined as post-operative PSA >0.2 ng/ml following a post-operative PSA nadir of undetectable for patients with surgical negative margins.
DNA and RNA preparation
The frozen tissue of each subject was manually microdissected while mounted in a cryostat into multiple sections for RNA/DNA preparation. Tissue for RNA preparation was monitored by frozen section preparation and examination of sections with hematoxylin and eosin stains to ensure the location of tumor enriched tissue. Frozen tissue was directly dissolved in TRIzol® Reagent. RNA was prepared from stroma adjacent to tumor and used for expression analysis as previously described [31, 53]. DNA was prepared from prostate tissue remote from tumor for hybridization to Illumina Human 1M-Duov3 B arrays exactly as recommended by Illumina Inc.
Expression, genotyping, and eQTL analysis
80 RNA samples from 55 patients, providing two or three biological replicates for some patients, were hybridized to Affymetrix GeneChip U133plus2 for gene expression analysis. The raw intensity data was background corrected, normalized, and summarized to be gene expression data through RMA (Robust Multi-array Average) algorithm [54]. The resulting Microarray data have been deposited in the publicly accessible Gene Expression Omnibus (GEO) database with accession number GSE17951. Purified DNA samples of 55 patients were applied to Illumina Human1M-Duov3_B SNP array for genotyping. In eQTL analysis, we selected 49 tissue samples containing more than 50% stroma. The tissue composition for each patient sample that has been used for array assay (tumor epithelial cells, stroma cells, epithelial cells of BPH) was determined by four pathologists [23, 55].
In order to identity associations between SNPs and transcript levels, we modified the BAYES [34] method to perform eQTL analysis. The BAYES method is a model-based iterative method that analyzes all expressed transcripts and SNPs jointly. The method combines multiple linear regression (MLR) with unsupervised clustering analysis in order to identify associations between transcripts and SNPs. The MLR method is the backbone of the model which describes the relationship between transcripts and SNPs with the assumption that expression level of each transcript is the sum of contributions of all possible relevant SNPs (Eqn. 1).
Where yij is the expression level of transcript j for the ith patient, βj is the intercept for transcript j, Zik is the genotype of SNP k for the ith patient, γjk is the effect of the SNP k on transcript j (i.e., a measure of SNP-expression association), εij is the residual error. In each iteration, the unsupervised clustering analysis is applied to cluster the transcripts into SNP-associated clusters and non-SNP-associated clusters for each SNP. A positive association is observed between a transcript and a SNP if the transcript is clustered into the SNP-associated group after n iterations with more than 80% of iterations supporting the association. The advantage of BAYES method is that a transcript may be simultaneously associated with multiple SNPs and vice versa. In order to reduce the computing time, we modified the BAYES method by applying the Stochastic Expectation-Maximization (SEM) algorithm instead of the Monte Carlo Markov Chain (MCMC) algorithm in the model. The simulation study showed that the two algorithms achieve the same result upon eQTL analysis.
Competing interests
D. Mercola and M. McClelland are board members of Proveri Inc. which has a license from the Regents of the University of California for the development of clinical assays for prostate cancer. The other authors declare they have no conflict of interest with this publication.
ACKNOWLEDGMENTS
This work was supported by NCI Early Detection Research Network (EDRN) Consortium grant U01 CA152738, the NCI Strategic Partner for the Evaluation of Cancer Signatures (SPECS) consortium UO1 CA114810, and by bridging funds from the UCI Chao Family Comprehensive Cancer Center Support Grant NCI CA P3062203. This work was also supported in part by Department of Defense Congressionally Directed Medical Research Programs grant W81XWH-08-1-0720 and by support from the California Women's Auxiliary to the Veterans of Foreign Wars.
REFERENCES
1. A.C.S. American Cancer Society: Cancer Facts & Figures 2014 [online]. 2014.
2. Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Cazier JB, Sainz J. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006; 38:652–658.
3. Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007; 39:631–637.
4. Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, Rafnar T, Gudbjartsson D, Agnarsson BA, Baker A. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007; 39:977–983.
5. Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007; 39:645–649.
6. Eeles RA, Kote-Jarai Z, Giles GG, Al Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008; 40:316–321.
7. Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, Gudbjartsson D, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Blondal T. Common sequence variants on 2p15 and Xp11. 22 confer susceptibility to prostate cancer. Nat Genet. 2008; 40:281–283.
8. Sun J, Zheng SL, Wiklund F, Isaacs SD, Purcell LD, Gao Z, Hsu F-C, Kim S-T, Liu W, Zhu Y. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008; 40:1153–1155.
9. Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008; 40:310–315.
10. Eeles RA, Kote-Jarai Z, Al Olama AA, Giles GG, Guy M, Severi G, Muir K, Hopper JL, Henderson BE, Haiman CA. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009; 41:1116–1121.
11. Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009; 41:1122–1126.
12. Sun J, Zheng SL, Wiklund F, Isaacs SD, Li G, Wiley KE, Kim S-T, Zhu Y, Zhang Z, Hsu F-C. Sequence variants at 22q13 are associated with prostate cancer risk. Cancer Res. 2009; 69:10–15.
13. Pomerantz MM, Shrestha Y, Flavin RJ, Regan MM, Penney KL, Mucci LA, Stampfer MJ, Hunter DJ, Chanock SJ, Schafer EJ. Analysis of the 10q11 cancer risk locus implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010; 6:e1001204.
14. Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, Wiklund F, Robbins C, Isaacs SD, Cheng Y, Li G. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007; 99:1836–1844.
15. Pal P, Xi H, Guha S, Sun G, Helfand BT, Meeks JJ, Suarez BK, Catalona WJ, Deka R. Common variants in 8q24 are associated with risk for prostate cancer and tumor aggressiveness in men of European ancestry. The Prostate. 2009; 69:1548–1556.
16. Cheng I, Plummer SJ, Neslund-Dudas C, Klein EA, Casey G, Rybicki BA, Witte JS. Prostate cancer susceptibility variants confer increased risk of disease progression. Cancer Epidemiol Biomarkers Prev. 2010; 19:2124–2132.
17. Xu J, Zheng SL, Isaacs SD, Wiley KE, Wiklund F, Sun J, Kader AK, Li G, Purcell LD, Kim S-T. Inherited genetic variant predisposes to aggressive but not indolent prostate cancer. Proc Natl Acad Sci USA. 2010; 107:2136–2140.
18. Pomerantz MM, Werner L, Xie W, Regan MM, Lee GSM, Sun T, Evan C, Petrozziello G, Nakabayashi M, Oh WK. Association of prostate cancer risk Loci with disease aggressiveness and prostate cancer-specific mortality. Cancer Prev Res. 2011; 4:719–728.
19. Al Olama AA, Kote-Jarai Z, Schumacher FR, Wiklund F, Berndt SI, Benlloch S, Giles GG, Severi G, Neal DE, Hamdy FC. A meta-analysis of genome-wide association studies to identify prostate cancer susceptibility loci associated with aggressive and non-aggressive disease. Hum Mol Genet. 2013; 22:408–415.
20. Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D'Amico AV, Richie JP. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002; 1:203–209.
21. Bismar TA, Demichelis F, Riva A, Kim R, Varambally S, He L, Kutok J, Aster JC, Tang J, Kuefer R. Defining aggressive prostate cancer using a 12-gene model. Neoplasia(New York, NY). 2006; 8:59.
22. Chen X, Xu S, Wang Y, McClelland M, Jia Z, Mercola D. Identification of biomarkers for prostate cancer prognosis using a novel two-step cluster analysis. Pattern Recognition in Bioinformatics. 2011; 7036:63–74.
23. Chen X, Xu S, McClelland M, Rahmatpanah F, Sawyers A, Jia Z, Mercola D. An Accurate Prostate Cancer Prognosticator Using a Seven-Gene Signature Plus Gleason Score and Taking Cell Type Heterogeneity into Account. PloS one. 2012; 7:e45178.
24. Jerónimo C, Henrique R, Hoque MO, Mambo E, Ribeiro FR, Varzim G, Oliveira J, Teixeira MR, Lopes C, Sidransky D. A quantitative promoter methylation profile of prostate cancer. Clin Cancer Res. 2004; 10:8472–8478.
25. Nelson WG, Yegnasubramanian S, Agoston AT, Bastian PJ, Lee BH, Nakayama M, De Marzo AM. Abnormal DNA methylation, epigenetics, and prostate cancer. Front Biosci. 2006; 12:4254–4266.
26. Henrique R, Ribeiro FR, Fonseca D, Hoque MO, Carvalho AL, Costa VL, Pinto M, Oliveira J, Teixeira MR, Sidransky D. High promoter methylation levels of APC predict poor prognosis in sextant biopsies from prostate cancer patients. Clin Cancer Res. 2007; 13:6122–6129.
27. Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol. 2001; 166:2472–2483.
28. Ayala G, Tuxhorn JA, Wheeler TM, Frolov A, Scardino PT, Ohori M, Wheeler M, Spitler J, Rowley DR. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin Cancer Res. 2003; 9:4792–4801.
29. McCarthy RP, Zhang S, Bostwick DG, Qian J, Eble JN, Wang M, Lin H, Cheng L. Molecular genetic evidence for different clonal origins of epithelial and stromal components of phyllodes tumor of the prostate. Am J Pathol. 2004; 165:1395–1400.
30. Ashida S, Orloff MS, Bebek G, Zhang L, Zheng P, Peehl DM, Eng C. Integrated analysis reveals critical genomic regions in prostate tumor microenvironment associated with clinicopathologic phenotypes. Clin Cancer Res. 2012; 18:1578–1587.
31. Jia Z, Wang Y, Sawyers A, Yao H, Rahmatpanah F, Xia XQ, Xu Q, Pio R, Turan T, Koziol JA. Diagnosis of prostate cancer using differentially expressed genes in stroma. Cancer Res. 2011; 71:2476–2487.
32. Jia Z, Rahmatpanah FB, Chen X, Lernhardt W, Wang Y, Xia XQ, Sawyers A, Sutton M, McClelland M, Mercola D. Expression Changes in the Stroma of Prostate Cancer Predict Subsequent Relapse. PloS one. 2012; 7:e41371.
33. Rockman MV, Kruglyak L. Genetics of global gene expression. Nature Reviews Genetics. 2006; 7:862–872.
34. Jia Z, Xu S. Mapping quantitative trait loci for expression abundance. Genetics. 2007; 176:611–623.
35. Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nature Reviews Genetics. 2009; 10:184–194.
36. Freedman ML, Monteiro ANA, Gayther SA, Coetzee GA, Risch A, Plass C, Casey G, De Biasi M, Carlson C, Duggan D. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet. 2011; 43:513–518.
37. Joesting MS, Perrin S, Elenbaas B, Fawell SE, Rubin JS, Franco OE, Hayward SW, Cunha GR, Marker PC. Identification of SFRP1 as a candidate mediator of stromal-to-epithelial signaling in prostate cancer. Cancer Res. 2005; 65:10423–10430.
38. Richardson AM, Woodson K, Wang Y, Rodriguez-Canales J, Erickson HS, Tangrea MA, Novakovic K, Gonzalez S, Velasco A, Kawasaki ES. Global expression analysis of prostate cancer-associated stroma and epithelia. Diagn Mol Pathol. 2007; 16:189–197.
39. Dakhova O, Ozen M, Creighton CJ, Li R, Ayala G, Rowley D, Ittmann M. Global gene expression analysis of reactive stroma in prostate cancer. Clin Cancer Res. 2009; 15:3979–3989.
40. FitzGerald LM, Kwon EM, Conomos MP, Kolb S, Holt SK, Levine D, Feng Z, Ostrander EA, Stanford JL. Genome-wide association study identifies a genetic variant associated with risk for more aggressive prostate cancer. Cancer Epidemiol Biomarkers Prev. 2011; 20:1196–1203.
41. Gallagher DJ, Vijai J, Cronin AM, Bhatia J, Vickers AJ, Gaudet MM, Fine S, Reuter V, Scher HI, Halldén C. Susceptibility loci associated with prostate cancer progression and mortality. Clin Cancer Res. 2010; 16:2819–2832.
42. Xu J, Zheng SL, Isaacs SD, Wiley KE, Wiklund F, Sun J, Kader AK, Li G, Purcell LD, Kim ST. Inherited genetic variant predisposes to aggressive but not indolent prostate cancer. Proc Natl Acad Sci USA. 2010; 107:2136–2140.
43. Zheng SL, Sun J, Cheng Y, Li G, Hsu F-C, Zhu Y, Chang B-L, Liu W, Kim JW, Turner AR. Association between two unlinked loci at 8q24 and prostate cancer risk among European Americans. J Natl Cancer Inst. 2007; 99:1525–1533.
44. Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005; 123:1001–1011.
45. Banerjee J, Mishra R, Li X, Jackson R, Sharma A, Bhowmick N. A reciprocal role of prostate cancer on stromal DNA damage. Oncogene. 2013; 33:4924–31.
46. Cunha GR, Hayward SW, Wang Y, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003; 107:1–10.
47. Chung LW, Huang W-C, Sung S-Y, Wu D, Odero-Marah V, Nomura T, Shigemura K, Miyagi T, Seo S, Shi C. Stromal-epithelial interaction in prostate cancer progression. Clin Genitourin Cancer. 2006; 5:162–170.
48. Cunha GR, Hayward SW, Wang Y. Role of stroma in carcinogenesis of the prostate. Differentiation. 2002; 70:473–485.
49. Huang S-P, Huang L-C, Ting W-C, Chen L-M, Chang T-Y, Lu T-L, Lan Y-H, Liu C-C, Yang W-H, Lee H-Z. Prognostic significance of prostate cancer susceptibility variants on prostate-specific antigen recurrence after radical prostatectomy. Cancer Epidemiol Biomarkers Prev. 2009; 18:3068–3074.
50. Ahmadiyeh N, Pomerantz MM, Grisanzio C, Herman P, Jia L, Almendro V, He HH, Brown M, Liu XS, Davis M. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci USA. 2010; 107:9742–9746.
51. Wada S, Hamada M, Kobayashi K, Satoh N. Novel genes involved in canonical Wnt/β-catenin signaling pathway in early Ciona intestinalis embryos. Dev Growth Differ. 2008; 50:215–227.
52. Koga M, Matsuda M, Kawamura T, Sogo T, Shigeno A, Nishida E, Ebisuya M. Foxd1 is a mediator and indicator of the cell reprogramming process. Nat Commun. 2014; 5:3197.
53. Wang Y, Xia X-Q, Jia Z, Sawyers A, Yao H, Wang-Rodriquez J, Mercola D, McClelland M. In silico estimates of tissue components in surgical samples based on expression profiling data. Cancer Res. 2010; 70:6448–6455.
54. Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003; 19:185–193.
55. Stuart RO, Wachsman W, Berry CC, Wang-Rodriguez J, Wasserman L, Klacansky I, Masys D, Arden K, Goodison S, McClell M. In silico dissection of cell-type-associated patterns of gene expression in prostate cancer. Proc Natl Acad Sci USA. 2004; 101:615–620.