
Introduction
Triple negative breast cancer (TNBC) is the most aggressive subtype of breast cancer and is associated with a poor prognosis [1]. TNBC is defined by the absence of oestrogen and progesterone receptors and the absence of HER2 overexpression. There is a lack of targeted therapy for TNBC due to wide heterogeneity for this subtype, which contributes to increased drug resistance, virulence and difficult to treat nature [2]. A key event promoting tumour cell proliferation and migration is metabolic reprogramming, where in response to oncogenic signals increased nutrient supplies drive cancer progression [3]. Several pathways have been associated with tumour progression including the phosphoinositide-3 kinase (PI3K)/serine/threonine-specific protein kinase (Akt) and the Ras/extracellular-signal-related kinase (ERK) pathway (also known as the Ras-Raf-MEK-ERK pathway). These pathways are controlled through the import of nutrients into cells by growth factors, such as insulin-like growth factor 1 (IGF-1), epidermal growth factor (EGF) and insulin, which mediate a cascade of events that regulate numerous cellular processes including cell growth and proliferation [4]. Upregulation of key signalling proteins of these pathways such as PI3K and ERK have been linked to several cancers including lung, thyroid and pancreatic cancer [5].
More recently the branched chain aminotransferase proteins (BCAT), which catalyse the transamination of the branched chain amino acids (BCAA), leucine, valine and isoleucine to their respective α-keto acids and glutamate (reviewed in [6]) have been identified as regulators of cell proliferation and migration [7, 8]. This cytosolic protein, BCATc, regulated by c-Myc, was found to be upregulated in a variety of cancers including gliomas [7, 9] ovarian, colorectal and breast [8, 10, 11]. The BCAA, particularly leucine, are potent nutrient signals and together with growth factors such as the IGFs and the hormone insulin regulate PI3K/Akt signalling [12]. Nutrient status is also sensed by the general control non-derepressible 2 kinase (GCN2) co-ordinates cell growth, proliferation and cell survival [13]. Therefore, by upregulating membrane transporters and nutrient uptake, cancer cells support the increased demand for macromolecules needed for cell growth [14].
Emerging evidence has indicated that the BCAT proteins, traditionally assigned metabolic roles, may have additional ‘moonlighting’ cellular functions, which are regulated through their peroxide sensitive-redox active CXXC motif [15]. We have recently established that the BCAT proteins can form thiol-dependent interactions with key signalling molecules such as those in the PI3K/Akt and the Ras/ERK pathways (unpublished observations) and show that phosphorylation of BCAT by PKC kinases is redox-dependent [16]. We showed that BCATc regulated autophagy through an interdependence of redox-regulated phosphorylation, supporting a ‘moonlighting’ role for this metabolic protein.
In this study, we show that BCATc regulates cross-talk between the PI3K/Akt and the Ras/ERK pathway. Using the TNBC cell model MDA-MB-231, we demonstrate that knockdown of BCATc results in a significant decrease of IGF-1 and insulin-mediated cell proliferation and migration. Subsequent analysis demonstrated that cell proliferation was associated with activation of the PI3K/Akt pathway that resulted in increased activation of FOXO3a, a transcription factor known to be involved in cell proliferation. Importantly, BCATc at the same time suppressed phosphorylation of ERK, indicating that regulation of proliferation through BCATc is primarly through the PI3K/Akt pathway rather than through ERK signalling, thus highlighting the plasticity of tumours to advance and adapt to changing environments.
Results and discussion
BCATc regulates proliferation, migration and invasion through the IGF-1 and insulin signalling pathway
Using siRNA we show that BCATc is intrinsically involved in mediating cell proliferation, migration and invasion of MDA-MB-231 cells (Figure 1A–1D). Similar studies have also reported a decrease in proliferation, metastasis and invasion of tumour cells in breast, colon and hepatocellular cancer in response to BCAT1 knockdown [8, 11, 17]. The oncogene c-Myc not only upregulates BCAT1 but transporters associated with glutamine and the neutral amino acid transporter 5 (SN5) [18]. Accumulation of glutamine and upregulation of glutaminase, which converts glutamine to glutamate, enhances glutathione synthesis, TCA cycle activation together with lipid and protein synthesis promoting cell growth and proliferation [19]. Moreover, leucine (a key substrate for BCAT), when restricted, has been shown to reduce cell proliferation, in several cancer cell lines including malignant melanoma (A375), lung cancer (A549), ovarian cancer (A2780) and breast cancer (MCF-7 and MDA-MB-231) [20] supporting a role for BCAA metabolism in regulating cell proliferation. Leucine together with glutamine is required for the activation of mTOR, as it relies on glutamine export for the intracellular transport of leucine through the bidirectional SLC7A5/SLC3A2 transporter [21]. In acute lymphoblastic leukaemia (ALL) deletion of SLC7A5 (a high affinity transporter for glutamine) impaired T-cell tumour progression suggesting that several aspects of BCAA metabolism are important in regulating cell proliferation [22].
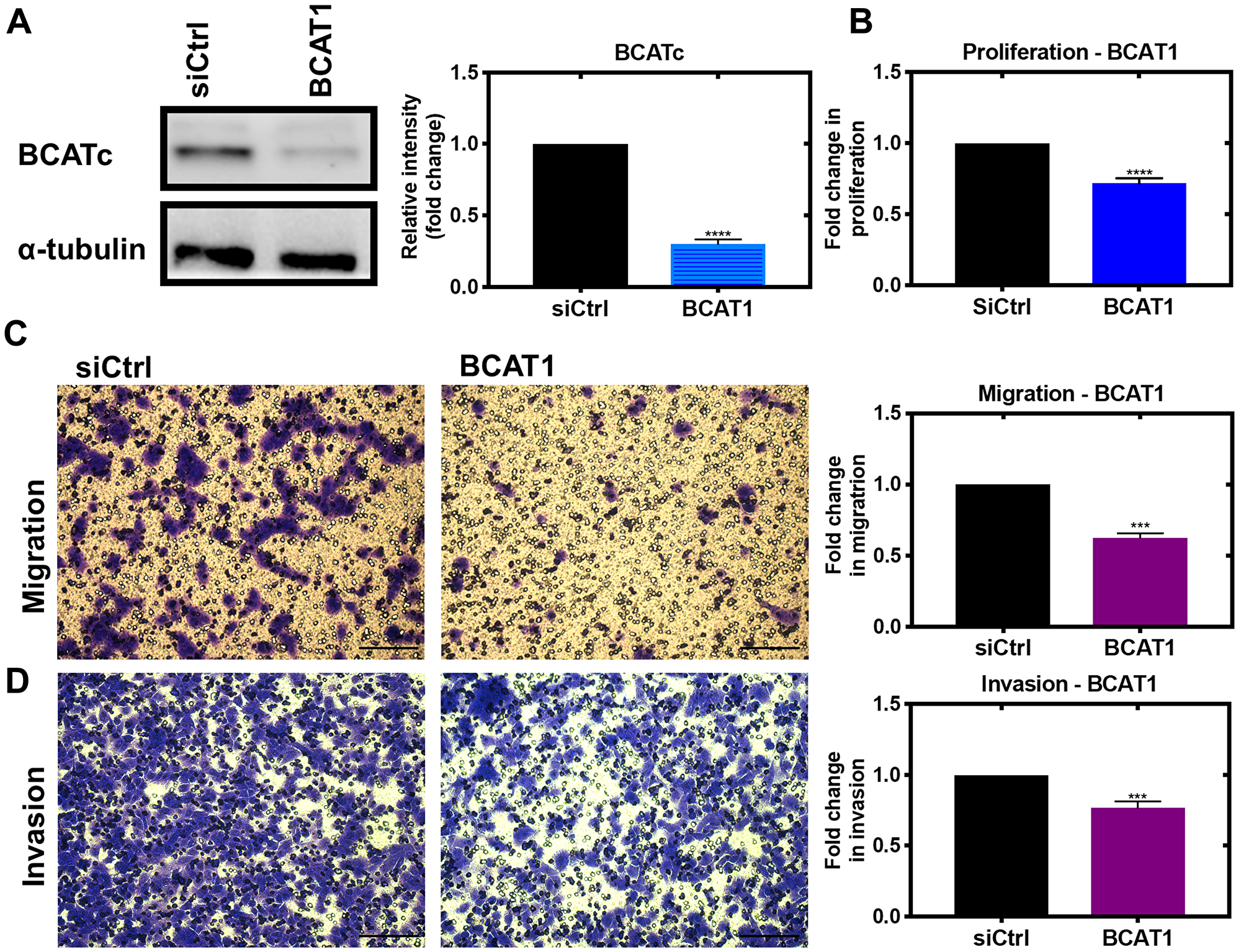
Figure 1: Knockdown of BCAT1 significantly reduces proliferation, migration and invasion of MDA-MB-231 cells. Cells were treated with 20 nM BCAT1 siRNA for 72 hours and the effect on proliferation assessed using the tritiated thymidine incorporation assay, migration was assessed using cells seeded onto 8 μm Transwell inserts (Greiner Bio-One) coated with collagen and after 24 hours, migrated were fixed and stained with 0.2% Crystal Violet, solubilised and absorbance measured and to assess invasion Matrigel added to the inserts as described above (A) Respective densitometric analysis of fold changes of protein expression relative to ɑ-tubulin are presented to the right of immunoblots. (B) Fold change in disintegrations per minute (DPM) and representative images of (C) migrated cells and (D) invaded cells with fold changes in absorbance at 590 nm ± SEM presented (n = 3) ***p < 0.001 and ****p < 0.0001 (scale bars = 100 μm).
We next showed that the IGF-1 and insulin-mediated increase in proliferation and migration of MDA-MB-231 cells was significantly attenuated by BCAT1 knockdown indicating that BCATc controls proliferation and migration through the IGF-1 and insulin pathway (Figure 2A–2C). This was also observed in the SKOV-3, ovarian cell line (Supplementary Figure 1A–1D). The IGF-1/insulin pathway facilitates an orchestrated activation of numerous cell signalling events initiated through phosphorylation of insulin receptor substrates (IRS1/2) [23]. Numerous studies support a role for this pathway in tumorigenesis (reviewed in [24]) with overexpression of key proteins such as the IGF-1 receptor tyrosine kinase reported in breast cancer [25]. Leucine signalling is intrinsically linked with insulin with a suggestion that plasma BCAA levels play a role in insulin-mediated regulation through the Akt/mTOR pathway (as reviewed by [26]).
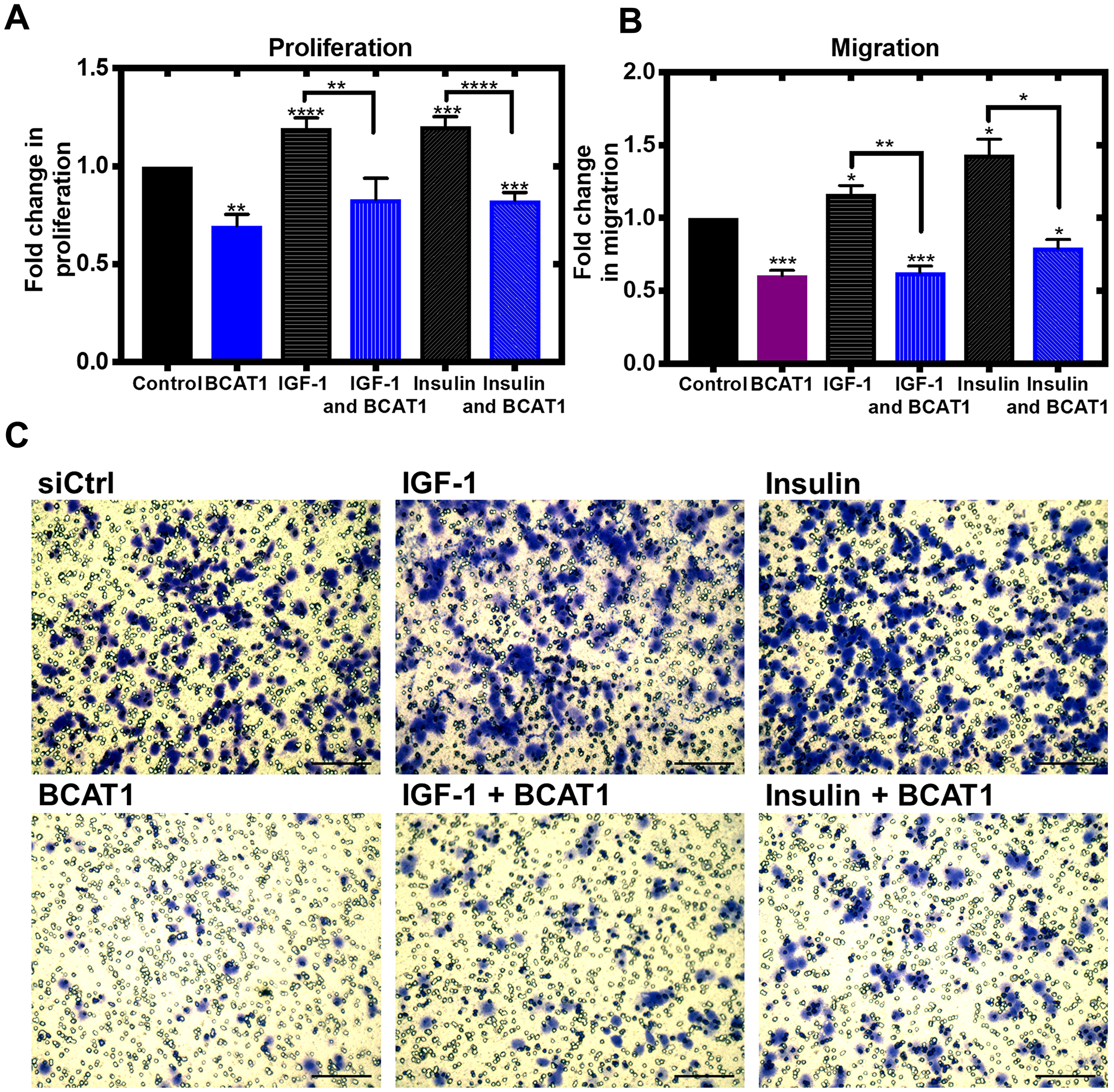
Figure 2: Knockdown of BCAT1 significantly reduces insulin and IGF-1-mediated migration of MDA-MB-231 cells. Cells were treated with 20 nM BCAT1 siRNA, 100 nM insulin and 100 ng/mL IGF-1 accordingly cell proliferation measured using the thymidine incorporation (TTI) assay and migration was assessed using cells seeded onto 8 μm Transwell inserts (Greiner Bio-One) coated with collagen and after 24 hours, migrated were fixed and stained with 0.2% Crystal Violet, solubilised and absorbance measured (A) Fold changes of mean values of disintegrations per minute (DPM) ± SEM, relative to control (B) Data presented as mean fold changes of absorbance at 590 nm. *p < 0.05, **p < 0.01 and ***p < 0.001 (C) Representative images of migrated cells (scale bars = 100 μm).
BCATc significantly reduces IGF-1 mediated activation of ERK
Knockdown of BCATc led to a significant decrease in the levels of the IGF-1Rβ (Figure 3A and 3B). Elevated levels of BCAA have been reported to inhibit the activation of IGF-1R [27], indicating that BCATc upregulation together with BCAA, allows activation of the IGF-1R signalling cascade. Activation of the IGF-1/insulin pathway triggers two predominant signalling pathways, the PI3K/Akt and Ras/ERK pathway. ERK proteins are central transducers of extracellular signals from hormones, growth factors, cytokines and environmental stress; they regulate many cellular processes throughout the cell and phosphorylate transcription factors, cytoskeletal proteins and other protein kinases and enzymes [28–30].
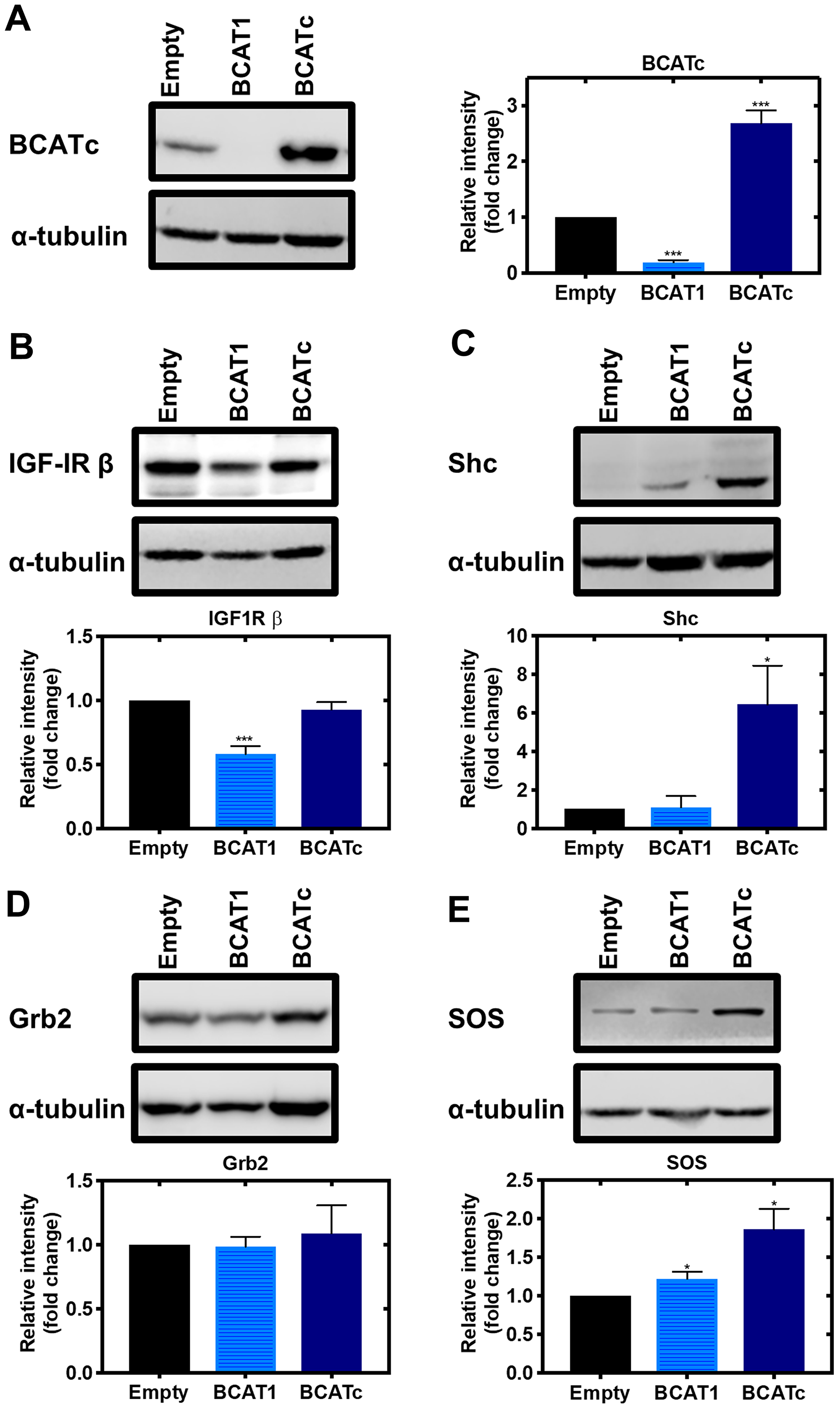
Figure 3: Knockdown and overexpression of BCAT1 regulates IGF-1/insulin signalling cascade in MDA-MB-231 cells. Cells were treated with stable shRNA transfection for BCAT1 knockdown (BCAT1) and overexpression (BCATc) for 8 days. (A) Western blot analysis confirmed BCAT1 knockdown and overexpression. Western blot analysis was also used to assess changes in the protein expression of IGF-1 downstream targets (B) IGF-1Rβ (C) Shc (D) Grb2 (E) SOS. Respective densitometric analysis of fold changes of protein expression relative to ɑ-tubulin are presented. Data representative of mean ± SEM (n = 3) *p < 0.05, ***p < 0.001 and ****p < 0.0001.
Recruitment and phosphorylation of insulin receptor substrate 1/2 (IRS1/2) via IGF-1 acting on its receptor activates the Src homology 2 domain-containing (Shc) protein, which was shown to be significantly increased in response to BCATc overexpression (Figure 3C). The Shc substrate binds to the adaptor protein growth factor receptor-bound 2 (Grb2), levels of which were unaffected by BCATc and the associated guanine nucleotide exchange protein, son of sevenless (SOS), that we showed was increased when BCATc was overexpressed (Figure 3D and 3E, respectively). Interestingly, Grb2 has been assigned a regulatory role in the activation of the Ras/ERK pathway, through monomer/dimer interchange, that controls binding to SOS [31]. SOS allows the exchange of nucleotide guanosine diphosphate (GDP) bound to Ras with nucleotide guanosine triphosphate (GTP) in the cytosol. GTP-bound Ras allows membrane recruitment and activation of the serine/threonine protein kinase Raf to the plasma membrane [32]. Normally, this would enhance and activate mitogen-activated protein kinase (MAPK), which we expected to see in response to overexpression of BCAT. However, we show that knockdown of BCAT1 increased phosphorylation of MAPK, even in the presence of IGF-1 or insulin and the opposite when BCATc was overexpressed (Figure 4A and 4B). Using confocal microscopy, we show that knockdown of BCAT1 increased MAPK translocation to the nucleus indicating that BCATc may play an important role as a chaperone or scaffold protein for MAPK (Figure 5), a role previously reported for BCATc [33]. This response was significantly increased in response to insulin, which will affect downstream nuclear and cytoplasmic targets.
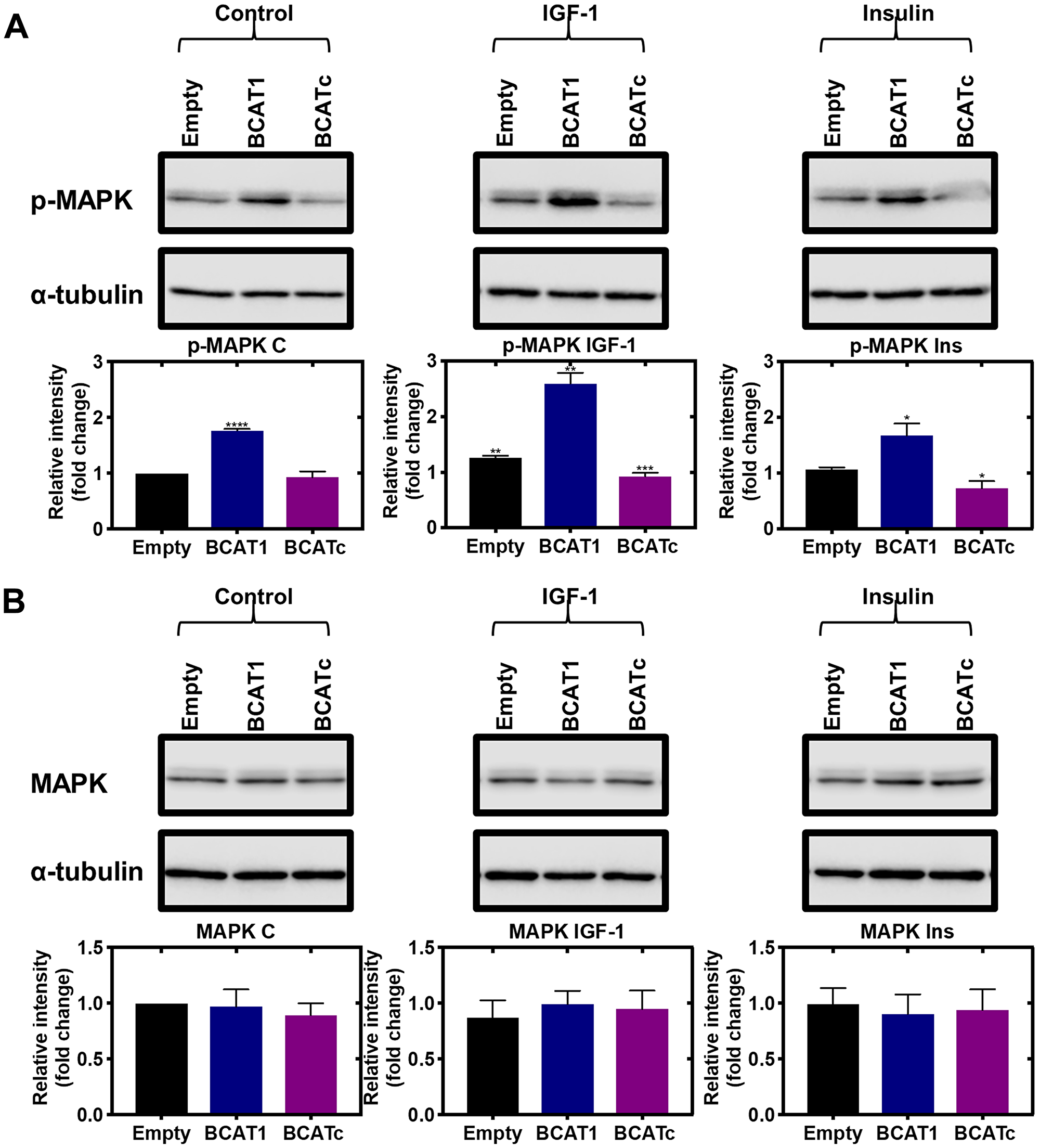
Figure 4: Knockdown and overexpression of BCAT1 negatively regulates phosphorylation of MAPK in MDA-MB-231 cells. Cells were treated with stable shRNA transfection for BCAT1 knockdown (BCAT1) and overexpression (BCATc) or unmodified pULTRA plasmid with no insertions as a control (Empty) for 8 days and treated with 100 nM insulin and 100 ng/mL IGF-1. (A) Western blot analysis was used to assess changes in the protein expression of phospho-MAPK and (B) MAPK. Respective densitometric analysis of fold changes of protein expression relative to ɑ-tubulin are presented below the immunoblots. Data representative of mean ± SEM (n = 3) *p < 0.05, **p < 0.01 and ***p < 0.001.
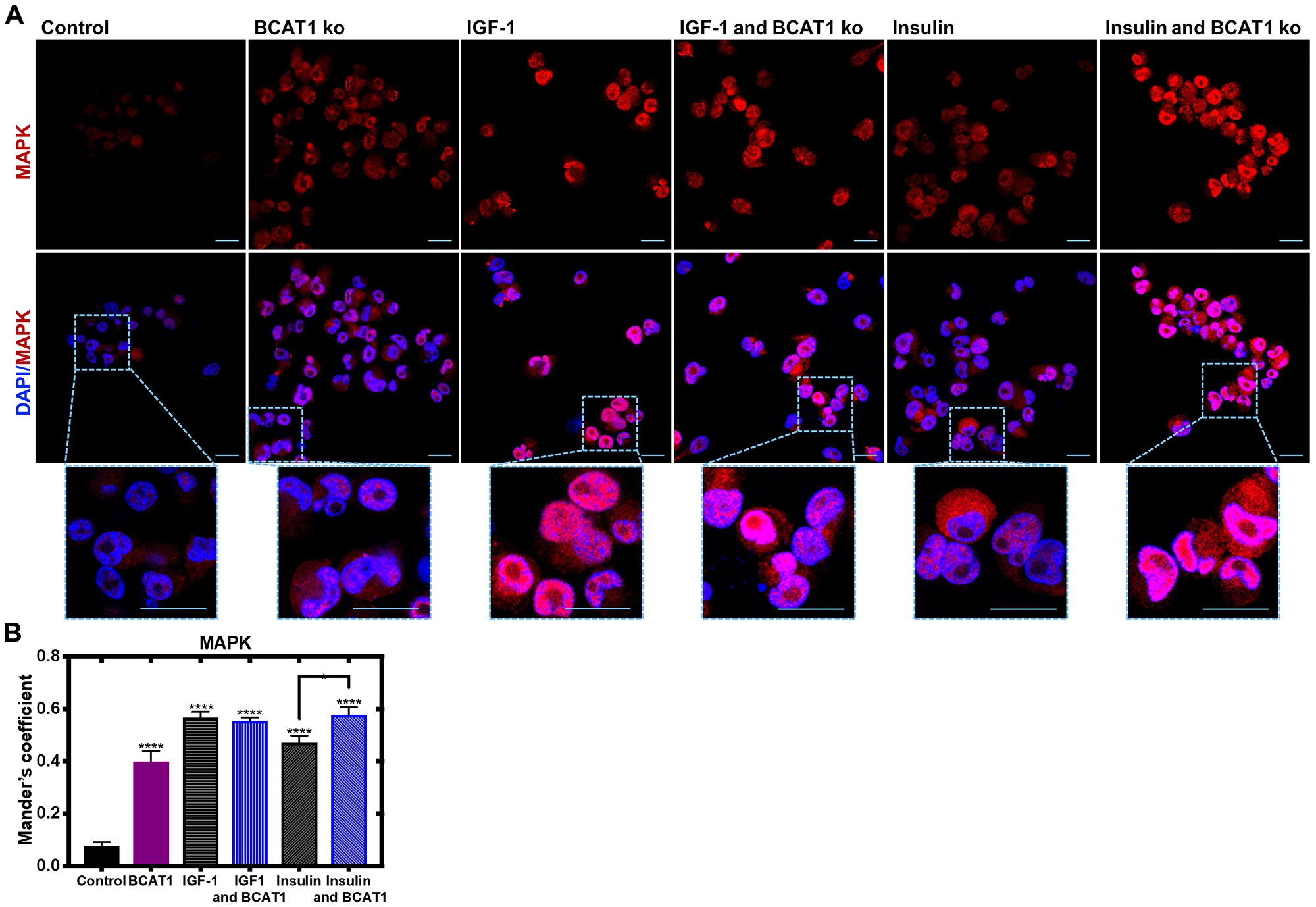
Figure 5: Insulin-mediated nuclear translocation of MAPK is significantly increased with BCAT1 knockdown in MDA-MB-231 cells. MDA-MB-231 cells were cultured on sterile glass coverslips (treated with poly-L-lysine) and treated with 20 nM BCAT1 siRNA, 100 nM insulin and 100 ng/mL IGF-1. Cells were fixed with 4% paraformaldehyde and permeabilised with Triton ×. (A) Immunofluorescence Z stacked images of MDA-MB-231 cells probed with anti-MAPK (1:100) overnight at 4C, and then anti-rabbit Alexa Fluor® 568 (1:250) for 1.5 hours. The coverslips were then mounted in VECTASHIELD HardSet antifade mounting medium with DAPI and imaged using a Leica SP8 confocal microscope. (B) Respective velocity analysis of Mander’s coefficient as compared to control. Data representative of mean ± SEM (n = 3) *p < 0.05 and ***p < 0.001. Blue - DAPI; Red - MAPK.
We have previously shown that BCATc is phosphorylated through redox-regulated PKC activation [16]. The BCAT proteins also have various phosphorylation motifs for MAPK defined using the Motif Scan program (http://scansite.mit.edu) (Figure 6A), highlighting 3 structurally accessible consensus sequences for MAPK, T33, S188 and T278 (Figure 6B). We show that BCATc is a substrate for MAPK phosphorylation where the redox sensor is important for phosphorylation. Interestingly, we show that when both proteins are oxidised then MAPK-mediated phosphorylation of BCATc is enhanced (Figure 6C). Therefore, we propose that the association between MAPK and BCATc is controlled through redox regulated phosphorylation governed by insulin and redox signalling, such that under conditions that promote tumour growth, BCATc levels not only increase but a change in function from transaminase to chaperone occurs. We evidence that BCATc shows both positive and negative regulation of key proteins involved in the Ras/ERK pathway with the ultimate decrease in IGF-mediated activation of ERK, suggesting that BCATc-induced regulation of cell proliferation and metastasis is unlikely to occur via this pathway.
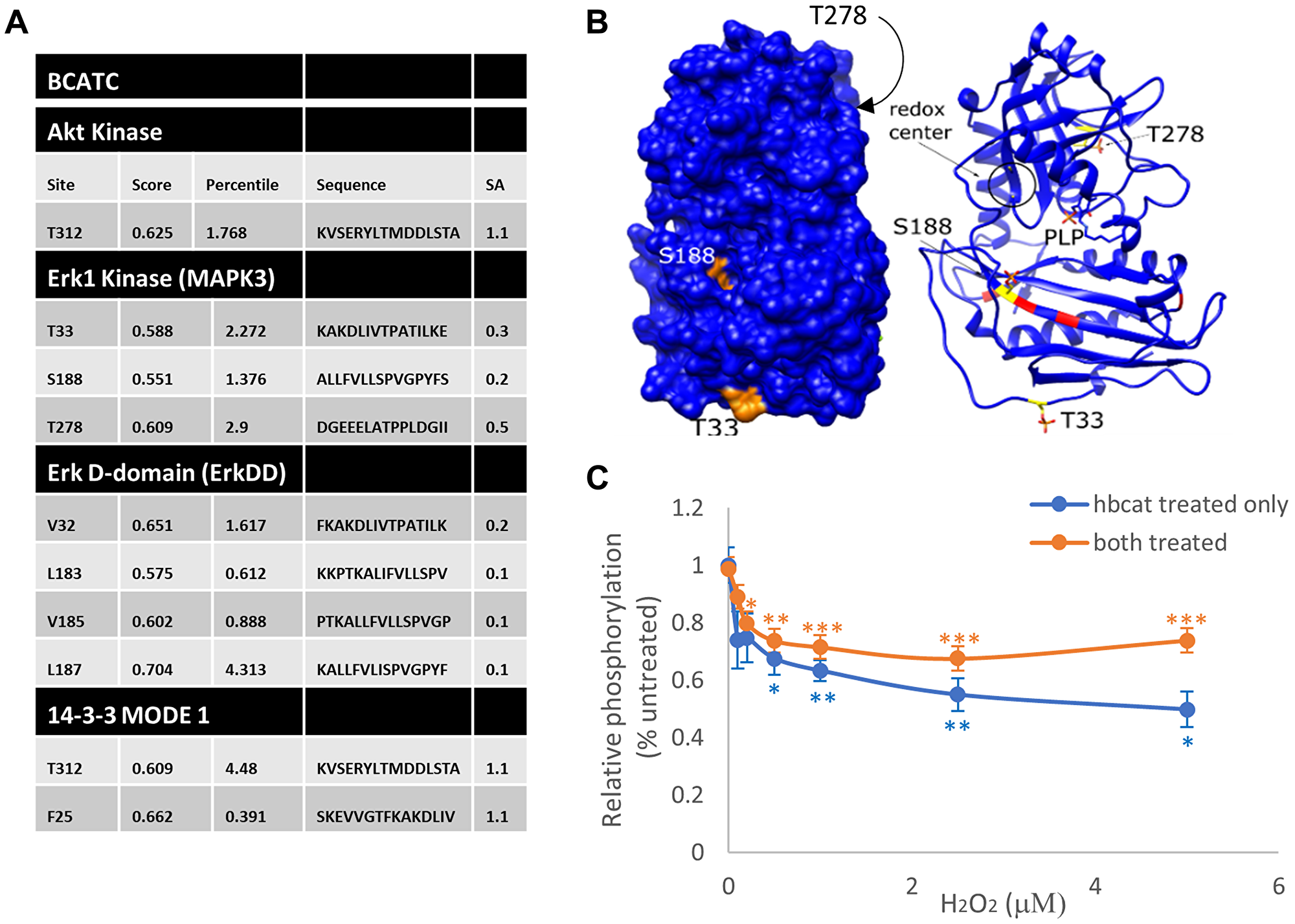
Figure 6: Putative phosphorylation sites for BCAT and its interaction with ERK (A) The BCAT protein sequence (accession #) was analysed using the Motif Scan program, Scansite 4 (http://scansite.mit.edu). Putative Akt and ERK1/2 phosphorylation sites on BCAT, as well as regions conforming to the ERK1/2 D-recognition site (DRS) substrate docking site (ERK-DD) and 14-3-3 phosphoamino-acid binding motif (14-3-3) are shown. (B) Phospho-Thr residues (TPO) and Phospho-Ser (SEP) were mapped onto the BCATc structure (PDB ID: BCAT) using UCSF Chimera. Putative phosphosites (S188, T33 and T278) are shown in the surface display (left) and ribbon display (right). The redox center of BCAT, which is composed of C335 and C338, is shown in the circle. Likewise, the predicted DRS binding site on BCATc is labelled red. (C) H2O2 titration curves of phosphorylation of BCATc by ERK2. ERK2 and BCATc were either both pre-treated with the indicated concentrations of H2O2 for 10 min at room temperature (orange) or only BCAT was pre-treated (blue). Excess H2O2 was then scavenged by catalase and the enzymes were combined in reaction buffer and incubated at 30°C for 30 min. The extent of phosphorylation was measured using the ADP-Glo assay. All activities were background corrected and normalized to that of the untreated controls. Error bars represent standard error (n = 6 for each dataset; *p < 0.05, **p < 0.01 and ***p < 0.001 relative to the untreated control).
BCATc regulates cross-talk between the Ras/ERK and PI3K/Akt pathways
Since increased activation of MAPK in response to BCAT1 knockdown does not contribute to cell proliferation or migration, we next considered if these outcomes were through the PI3K/Akt signalling cascade. In response to IGF-1 and insulin signalling, overexpression of BCATc increased phosphorylation of Akt (Figure 7), whereas a significant increase in mammalian target of rapamycin (mTOR) was not reported (Supplementary Figure 2). Under normal cellular conditions, the metabolite α-ketoglutarate, the product of leucine transamination, activates mTOR. However, BCAT is regulated through phosphorylation and changes in the redox environment, which can prompt BCATc to associate with other signalling pathways, dependent on the stimuli, such as nutrient load, insulin/IGF-1 or changes in the redox environment [16]. Therefore, in response to IGF-1 or insulin the role of BCATc may switch from supporting mTOR activation to associate with proteins from the PI3K/Akt pathway. The dynamics and vectorality of these mechanisms are not entirely clear but more than likely will involve redox-regulated phosphorylation, such as that described for BCATc.
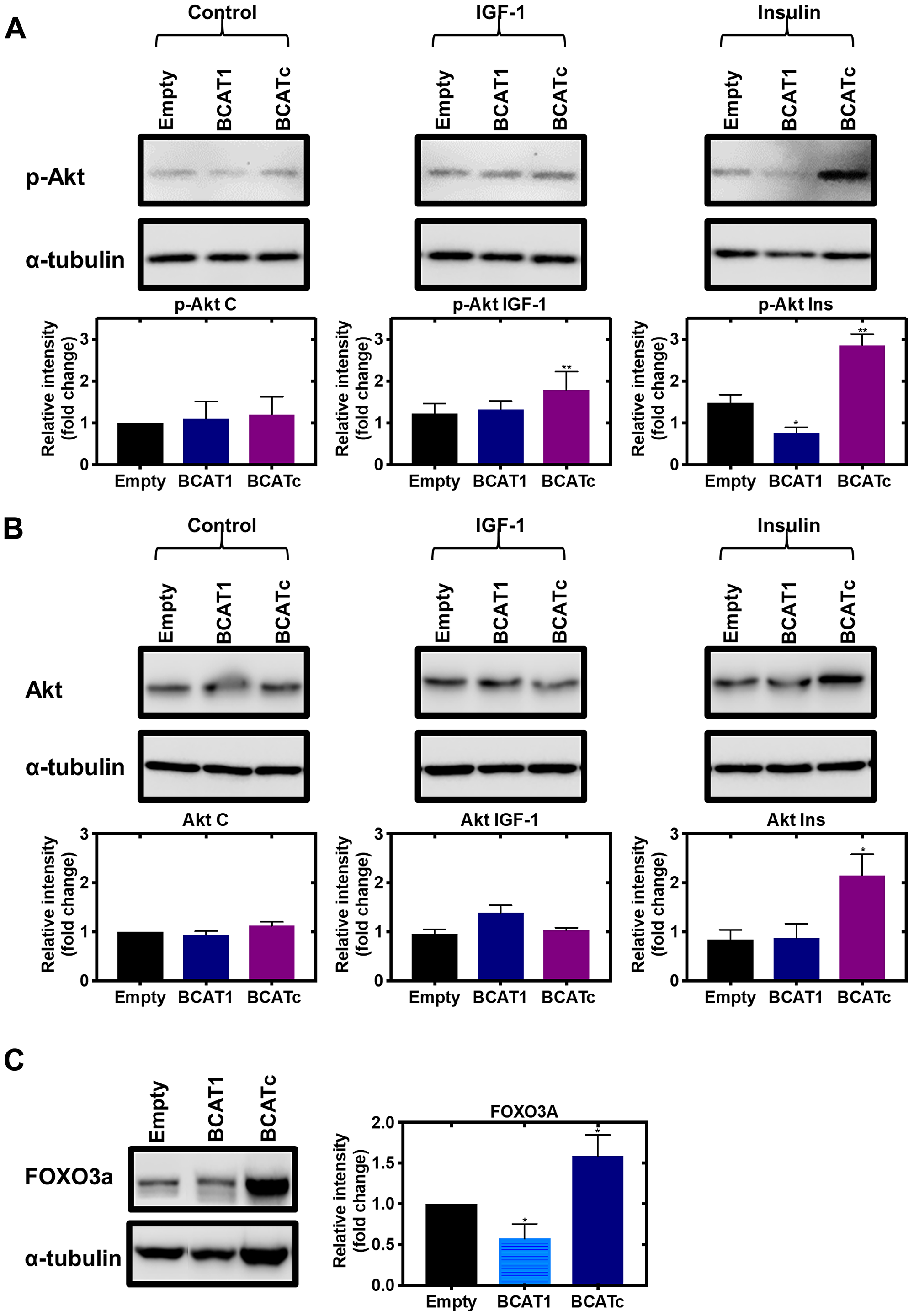
Figure 7: Knockdown and overexpression of BCAT1 regulates the PI3K/Akt axis in MDA-MB-231 cells. Cells were treated with stable shRNA transfection for BCAT1 knockdown (BCAT1) and overexpression (BCATc) or unmodified pULTRA plasmid with no insertions as a control (Empty) for 8 days and treated with 100 nM insulin and 100 ng/mL IGF-1. (A) Western blot analysis was used to assess changes in the protein expression of phospho-Akt (B) Akt and (C) FOXO3a. Respective densitometric analysis of fold changes of protein expression relative to ɑ-tubulin are presented below the immunoblots. Data representative of mean ± SEM (n = 3) *p < 0.05 and **p < 0.01.
As discussed above, the levels of Shc and SOS were increased in response to increased BCATc expression (Figure 3C and 3D). Whilst it is accepted that the activation of IGF-1Rβ, Shc, Grb2 and SOS leads to MAPK activation [34, 35], SOS can also activate Ras [36]. It is known that Ras, which is activated with increased SOS, can also activate PI3K, Rac and Rho proteins associated with the regulation of the cytoskeleton and invasiveness of tumour cells [37]. Shc has been demonstrated to increase Grb2 interaction with PI3K on the p85 regulatory subunit leading to activation of the catalytic subunit of p110 [38, 39]. Moreover, Grb2 in a complex with the scaffolding protein Grb2-associated binder 1 (GAB) directly binds to p85 and enhances PI3K activation and subsequent Akt signalling [34, 40]. Thus, in this instance the increased level of these proteins may be associated with increased activation of the PI3K/Akt axis rather than increased activation of ERK.
Cross-talk of the RAS/ERK and PI3K/Akt pathways in the IGF-1/insulin signalling cascade allows tumour cells to utilise different effector molecules to promote tumour survival and progression (reviewed by [41]). One mechanism details that ERK phosphorylation of GAB1 inhibits GAB-1-mediated membrane recruitment of PI3K which in turn leads to the suppression of Akt signalling [42]. On the other hand, at high doses of IGF-1, Akt phosphorylates RAF at Ser259 and thus suppresses the activity of the RAS/ERK signalling pathway [43, 44], which may be related to the role of BCATc. Downstream of the PI3K/Akt pathway, we found that the transcription factor forkhead box O3 (FOXO3a) was upregulated in response to BCATc overexpression and knockdown of BCAT1 led to reduced levels (Figure 7C). This effect was repeated in SKOV-3 cells (Supplementary Figure 3A). This member of the FOXO subfamily of forkhead transcription factors, which mediate a variety of cellular processes including cell cycle progression and apoptosis [45]. The role of FOXO3a in breast cancer has been suggested to be ERα+-dependent whereby FOXO3a upregulation in ERα+ MCF-7 cells reduced cell migration, however, in TNBC MDA-MB-231 cells FOXO3a was found to promote tumour cell migration [46, 47]. FOXO3a can act on cyclin-dependent kinase inhibitor 1B (p27Kip1) directly by disrupting cyclin D/CDK4 and cyclin E/CDK2 complexes to promote cell proliferation [48]. Therefore, positive regulation of this transcription factor by BCATc in TNBC may contribute to BCATc-mediated proliferation, migration and invasion. IGF-1R signalling and subsequent Akt phosphorylation of FOXO3a leads to its translocation from nucleus to cytoplasm, where it associates with 14.3.3 protein [49]. The 14.3.3 proteins act as scaffolds to integrate signalling proteins with targets involved in biological processes, including cell cycle regulation [50]. Interestingly, BCATc also has predicted binding motifs for Akt and 14-3-3 phosphorylation (Figure 6A), which like ERK may be the site of regulation for the interaction of these proteins. Overexpression of 14-3-3z has been associated with breast cancer recurrence indicating a role in therapy resistance, mediated via the downregulation of the pro-apoptotic proteins Bcl-2-associated death promoter (BAD) and Bcl-2-like protein (BIM) [51, 52]. Knockdown of BCAT1 was found to increase the percentage of cells in the early apoptotic stage and reduce the percentage of live cells (Figure 8A and 8B), indicating that BCATc expression allows TNBC cells to evade apoptosis. The underlying mechanism by which hBCATc regulates cell apoptosis has not yet been elucidated. Hence, regulation of these Akt downstream effector proteins indicates an intrinsic role for BCATc for tumour progression mediated by cell proliferation, migration, cell cycle control and evasion of apoptosis through the regulation of FOXO3a.
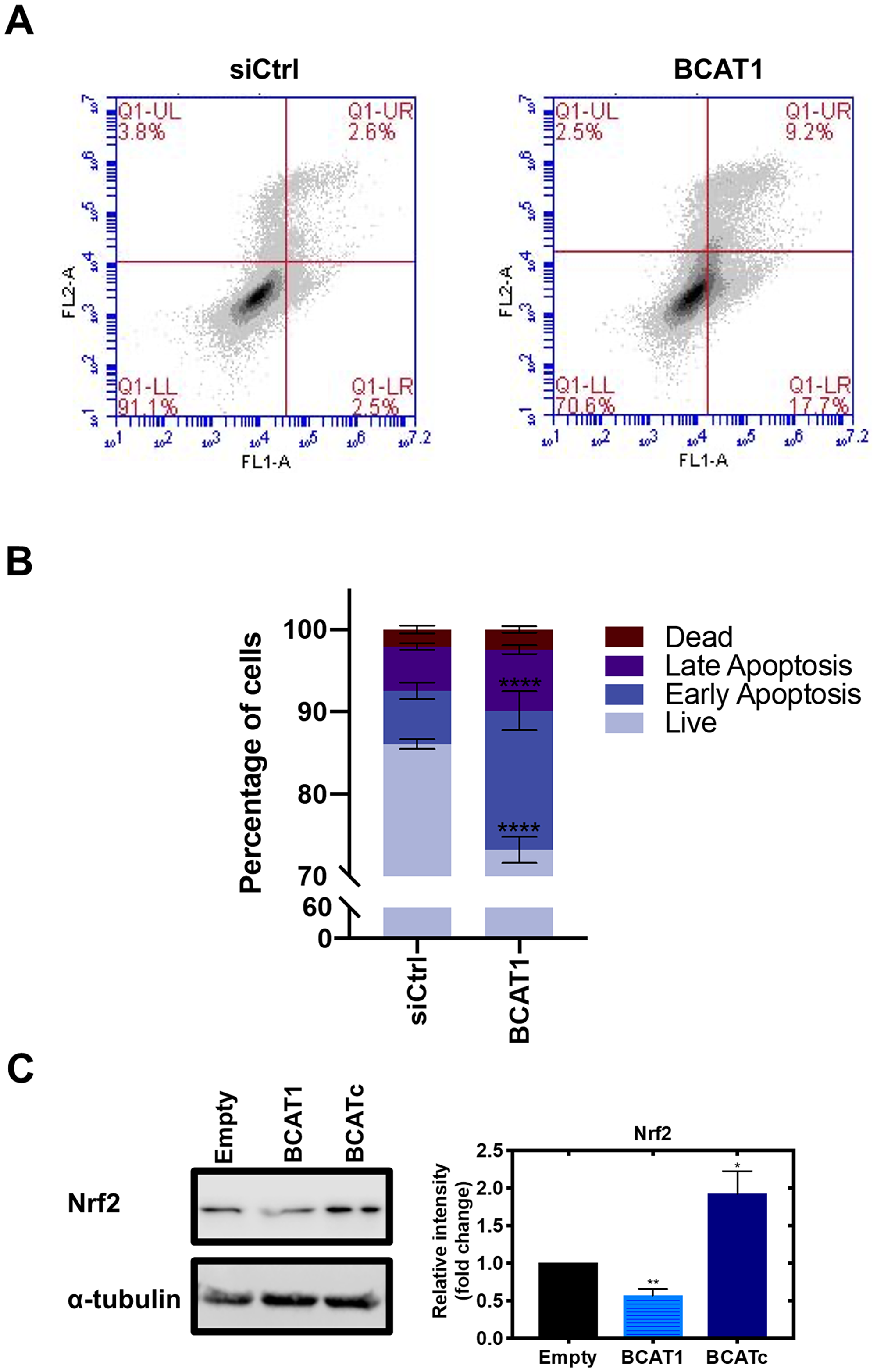
Figure 8: BCAT1 promotes tumour cell survival by evasion of apoptosis. Cells were treated with 20 nM BCAT1 siRNA for 72 hours or transfection control, and cell apoptosis was determined by FITC-Annexin V and propidium iodide staining using flow cytometry. (A) Representative flow cytometric analysis (B) Data representative of mean ± SEM (n = 3) *p < 0.05 and **p < 0.01 (C) Western blot analysis was used to assess changes in the protein expression of Nrf2. Respective densitometric analysis of fold changes of protein expression relative to ɑ-tubulin are presented. Data representative of mean ± SEM (n = 3) *p < 0.05 and **p < 0.01.
BCATc regulates cellular redox
We were interested to further elucidate the role of BCATc for tumour cell survival as increased glutamate levels contribute to glutathione (GSH) biosynthesis by facilitating the uptake of cystine via the xc-cysteine transporter, which is coupled to the efflux of glutamate [53]. GSH plays an important role in redox homeostasis and tumour cell survival by protecting cells from damage caused by reactive oxygen species (ROS) generated during oxidative stress [19]. Redox status is predominantly regulated by the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) and its repressor protein kelch-like ECH-associated protein 1 (Keap1) in response to redox cellular status [54]. PI3K/Akt activation is essential for the nuclear translocation of Nrf2, whereby Akt inhibitors, but not MAPK inhibitors, were found to reduce Nrf2 transcriptional activation of antioxidant genes [55]. Akt can also increase the stability of Nrf2 by activating p21 which disrupts the interaction between Keap1 and Nrf2, reducing Nrf2 phosphorylation, thereby preventing its nuclear export and ubiquitination [56]. Under low concentrations of ROS, Nrf2 is bound to the E3 ubiquitin ligase Keap1 in the cytosol and degraded by the proteasome [57]. In response to oxidative stress, this interaction is inhibited, by the oxidation of cysteine residues on Keap1, thus allowing Nrf2 translocation to the nucleus to induce the expression of antioxidant genes such as the oxidoreductases, thioredoxin (Trx) and glutaredoxin (Grx) [58]. Here, levels of Nrf2 were increased with BCATc overexpression and decreased with knockdown of BCAT1 (Figure 8C), suggesting that levels of BCATc positively regulate Nrf2 expression. Knockdown of BCAT1 also showed a decreased expression of Nrf2 in SKOV-3 cells, which resulted in an increase in ROS (Supplementary Figure 3B). Nrf2 modulates the expression of oxidoreductases, thioredoxin (Trx) and glutaredoxin (Grx) which are important in regulating cellular redox state [58]. Trx is a pro-oxidant with anti-apoptotic effects, including inactivation of caspase-3 [59], a primary player in the apoptotic signalling pathway, via a redox reactive nitrosothiol transfer [60]. Similarly, Grx1 inhibits apoptosis through Akt activation [61]. Therefore, whilst the role of BCATc mediates tumour survival by activating IGF-1/insulin PI3K/AKT signalling pathway, its expression may also influence Nrf2 regulation of the cellular redox state.
Summary
IGF-1 is secreted by the liver in response to growth hormone, and its circulating levels remain constant via its unique interaction with its IGF binding proteins (IGFBPs). Unlike insulin, IGFs are also made in most cells of the body, where they play a key role in growth, survival and metabolism and are upregulated during cancer development. Insulin and IGF-1 are intricately linked systemically, for example during an insulin-resistant state the usual normalising processes are inhibited, leading to increased levels of circulating insulin and glucose. It also leads to a stimulation of hepatic IGF-1 synthesis, and downregulation of IGFBP-1 and IGFBP-2, resulting in an increased bioavailability of IGF-1. The high circulating insulin and IGFs, in an insulin-resistant state, is thought to be one of the mechanisms underlying the link between disturbed metabolism and cancer progression.
Deregulation of nutrient signalling that affects the PI3K/Akt and Ras/ERK pathways potentiates proliferation and metastasis of malignant cells including breast cancer [5]. Our findings suggest that BCATc behaves as a molecular chaperone or molecular scaffold that influences cell survival through the regulation and crosstalk of several key pathways. We have shown BCATc to increase activation of the IGF-1R signalling cascade with subsequent activation of the PI3K/Akt axis whilst subsequently down-regulating the RAS/ERK pathway. Moreover, BCATc promoted tumour cell survival through evasion of apoptosis mediated potentially through regulation of the redox status of the cells. In summary, BCATc provides TNBC cells with metabolic plasticity to alter dependence on the RAS/ERK and PI3K/Akt signalling cascades, in response to IGF-1/insulin, to promote tumour survival and progression (Figure 9).
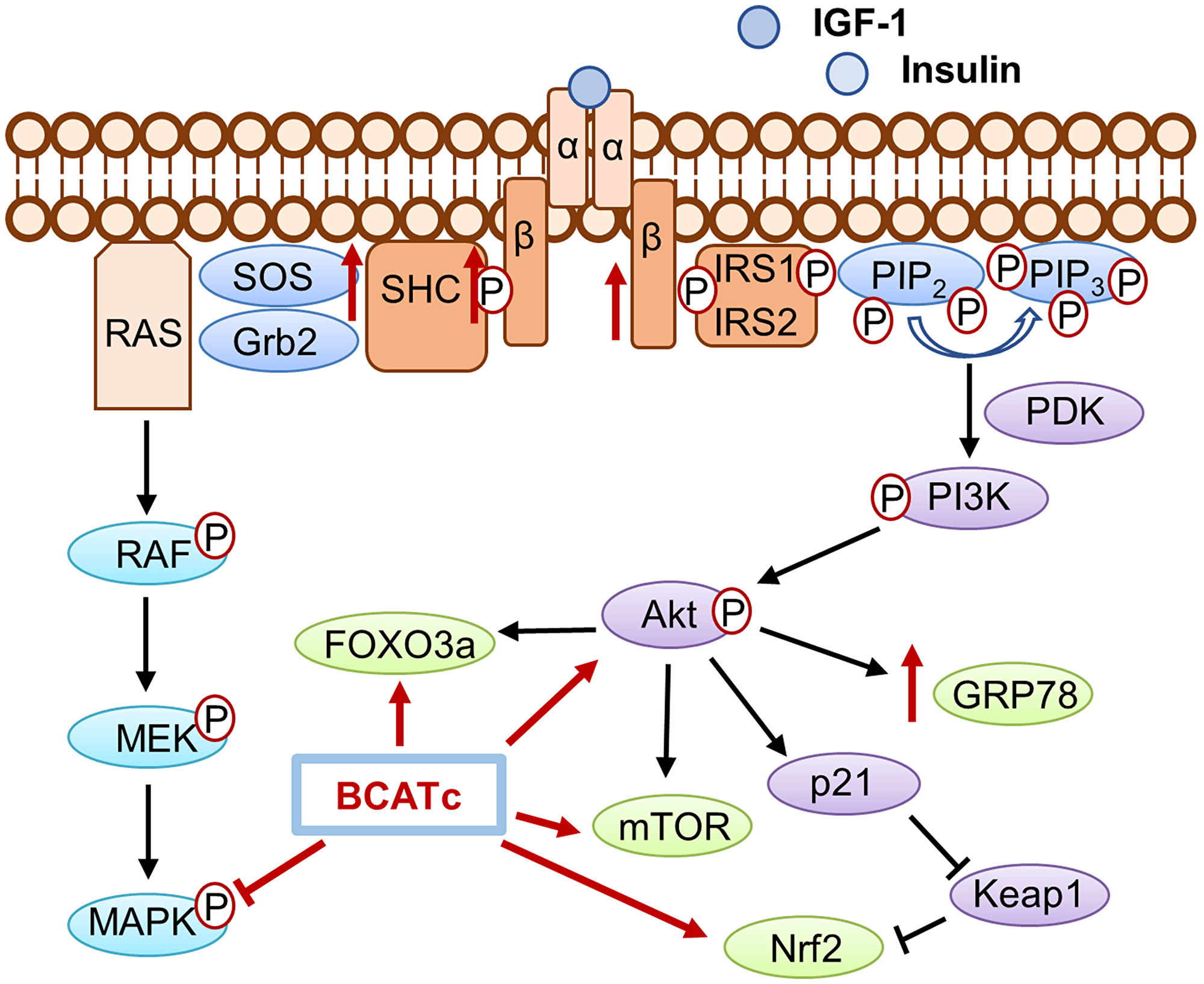
Figure 9: The role of BCATc on the insulin/IGF-1 signalling cascades. Activation of IGF-1R and insulin receptor, mediated by ligand binding of IGF-1 or insulin leads to the subsequent activation of the PI3K/AKT pathways and the Ras/MAPK pathways. Increased expression of BCATc increased levels of IGF-1Rβ, Shc and SOS however MAPK activation was increased with knockdown of BCAT1 suggesting BCATc inhibits MAPK activation. Increased levels of hBCATc increased phosphorylation of Akt and the expression levels of the downstream effector proteins; FOXO3a, and Nrf2 thereby promoting cell proliferation, migration, and evasion of apoptosis in TNBC.
Materials and Methods
Contact of reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Myra Conway (
Experimental model and subject details
Cell lines and cell culture
MDA-MB-231 (triple negative breast cancer) were purchased from ATCC and cultured in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% foetal bovine serum (FBS) in a humidified incubator with 5% CO2 at 37°C.
Method details
siRNA transfection
The siRNA sequences for BCAT1 (Sense 5′CAUUAUCUACUGCUUCACAUU and antisense 5′ UGUGAAGCAGUAGAUAAUGUU) were previously designed and validated by Dr. Tom Forshaw (primers were synthesised by Eurofins Genomics, Germany). Cells were transfected with 20 nM BCAT1 siRNA using Lipofectamine RNAimax or transfection solution prepared in Opti-MEM for 72 hours (Invitrogen, Paisley, UK).
Lentiviral shRNA transfection
The pULTRA-BCAT1 plasmids were prepared. In brief, an H1 promoter-BCAT1 shRNA sequence cassette was assembled in the pSUPER plasmid (VEC-PBS-0002, Oligoengine, Seattle, WA, a kind gift by Dr. Tim Craig) by ligation into the BglII and XhoI restriction sites. This was then amplified by PCR and the new plasmid assembled by ligation into the SnaBI restriction site of the pULTRA lentiviral vector (Addgene 24127, Cambridge, MA). For overexpression of BCAT1, the human gene was PCR amplified from our pET28a (Novagen, Madison, WI) bacterial overexpression vectors (Davoodi et al., 1998) and the plasmid assembled by ligation into the BamHI and EcoRI restriction sites of pULTRA. Unmodified pULTRA plasmid with no insertions was included as a control. The pULTRA vector design included enhanced green fluorescent protein (GFP) expression, which was used to confirm uptake of the plasmid into transfected cells.
Lentiviral particles were produced by co-transfection of Sigma Mission lentiviral packaging mix to manufacturers guidelines and the respective pULTRA plasmid into HEK293T cells using the PEI method (PMC4246624) and supplementation of viral media with 1 mM sodium butyrate to enhance viral yield (PMC3830501/). The lentiviral particles were harvested 48 hours and 72 hours post-transfection of the HEK293T cells. For viral transfection of the MDA-MB-231 cells, harvested lentiviral particles were incubated with the cells for at least 8 days. To improve transfection efficiency 8 μg/mL polybrene was added to the shRNA transfected cells. Transfection efficiency was assessed by fluorescent microscopy as indicated by GFP activation.
Western blot analysis
Cultured cells were lysed with radioimmune precipitation assay (RIPA) buffer supplemented with protease inhibitor and the protein concentration for each sample determined by Bradford assay. Aliquots of cell lysates (20 μg of protein) were resuspended in NuPAGE® LDS (4% β-mercaptoethanol) and denatured at 95°C for 10 minutes. Proteins samples were separated on a 4–12% polyacrylamide gel and transferred onto activated (100% methanol) PVDF membrane. Membrane was blocked with 5% milk in TBST (50 mM Tris-HCl, 150 mM NaCl, 0.1% Tween-20) for 1 hour, washed with TBST, and incubated with appropriate primary antibodies overnight at 4°C. Next, membranes were washed with TBST and incubated with an appropriate secondary antibody at room temperature for 1 hour. Luminata Forte ECL solution was used to visualise the bands using the Li-Cor Odyssey system and densitometric analysis preformed using IMAGE STUDIO Lite software.
3H thymidine incorporation proliferation assay
Radioactively labelled (tritium) thymidine was used to measure the proliferation of MDA-MB-231 cells by the incorporation of [3H] thymidine into the DNA of dividing cells. Cells were seeded at a seeding density of 2 × 104 cells in 24 well plates. Following treatment, cells were labelled with 0.1 μCi [3H] thymidine per well, incubated with 5% trichloroacetic acid at 4°C for 10 minutes, followed by a 1 hour incubation with 1 M sodium hydroxide. The resulting suspension was added to a vial containing 2 mL ultima gold liquid scintillation cocktail (Perkin Elmer Beaconsfield, Bucks, UK) and incorporated counts were measured using a Beckman Scintillation Counter LS6500. Data were recorded as disintegrations per minute (DPM).
Migration and invasion assay
Thincert™ cell culture inserts (8 μm) (Greiner Bio-One) were coated with 10 μg/ml Collagen-I to measure migration and Matrigel® to assess invasion 24 hours prior to cell seeding at room temperature. Cells were seeded in the upper chambers of the inserts at a density of 1 × 105 MDA-MB-231 cells/mL in serum-free DMEM and 10% FBS DMEM placed in the lower chambers, in a humidified incubator at 37°C with 5% CO2. Following 24 hours, cells on the upper surface of the membrane were removed using an ethanol coated cotton swab and cells on the lower chamber fixed in 4% paraformaldehyde and stained with 0.2% Crystal Violet (Sigma). For each insert, representative images in 3 evenly distributed ×40 fields of view were captured using a light microscope (Zeiss AX10). To quantify migration and invasion of cells, 0.1% SDS in PBS was added to the lower chambers for 1 hour at room temperature and absorbance intensity measured on a plate reader at an excitation of 590 nm.
ERK2 activation and purification
Activated doubly-phosphorylated MAPK1/ERK2 (pERK2) was generated and purified essentially as described by [62]. Briefly, 5× ATP solution (50 mM HEPES-KOH pH 7.38, 100 mM MgCl2, 20 mM DTT, 20 mM ATP) and dilution buffer (10 mM HEPES-KOH pH 7.38, 0.1% BME, 0.01% Triton X-100) were combined in a 1:4 ratio for a final volume of 25 mL. Inactive ERK2 and constitutively active MEK1 were then added to the solution before the reaction mix was divided into 0.5 mL aliquots. Reactions were then incubated at 30°C for 60 min. The solution was then dialyzed against ERK2 dialysis buffer (20 mM Tris-HCl, pH 8, 1 mM DTT) at 4°C overnight. Dialyzed ERK2 was purified by 1 mL HiTrap Q HP anion exchange column (GE Healthcare) that was preequilibrated with equilibration buffer (20 mM Tris-HCl, pH 8.0, 1 mM DTT, 0.1 M KCl, 10% glycerol). The reaction mix was then loaded onto the column and the column was washed with buffer A (20 mM Tris-HCl (pH 8.0), 1 mM DTT, 10% glycerol) with a step gradient of buffer A supplemented with 0 to 0.3 M KCl. Immobilized pERK2 eluted at about 0.2 M KCl. To assess the purity of the products, each fraction was resolved on a 5/10% SDS-PAGE gel and stained with Simply Blue Coomasie reagent. To determine which elution fractions contained the most protein, a Bradford total protein assay was completed. Briefly, elution fractions were assayed on a 96-well plate and incubated with Coomassie brilliant blue G-250 dye for 10 min at room temperature. Absorbance (595 nm) was measured using a TECAN Infinite F500 Pro microplate reader and negative control readings were subtracted from all readings to obtain background-corrected values. Likewise, the activity of each pERK2 fraction was determined by in vitro kinase assays, as described below, using the modular ERK2 peptide substrate, Sub-D. Fractions containing active pERK2 were pooled and then concentrated using an Amicon Ultra-15 (30K MWCO) before being dialyzed into ERK2 storage buffer (20 mM Tris-HCl, pH 8.0, 1 mM DTT, 1 mM EDTA, 0.2 M KCl, 10% glycerol) overnight at 4°C.
H2O2-dependent oxidation of pERK2 and BCAT
pERK2 and the BCAT isoforms was diluted to concentrations of 465 nM and 2 μM, respectively. The proteins were then treated with various concentrations of H2O2 (0–5 μM) and incubated for 10 minutes at room temperature. Untreated controls were incubated with diH2O in place of H2O2. The remaining H2O2 was then scavenged by one-minute exposure to catalase (2.5 units). Next, 2.5 μL of pERK2 (treated and untreated) was incubated with 2.5 μL 4× kinase reaction buffer (8 mM MOPS, pH 7.2, 4 mM Beta-glyc-phosphate, 20 μM ATP, 8 mM MgCl2, 1.6 mM EGTA, 0.64 mM EDTA, 64 ng/μL BSA) and 5 μL of BCAT substrate (treated and untreated) at 30°C for 30 minutes. Negative control reactions contained 2.5 μL of diH2O in place of active ERK2. Following the 30°C incubation, the ERK2 selective inhibitor, SCH772984, was added to the reaction to quench the reaction. The effects of H2O2 on pERK2’s ability to phosphorylate the BCAT isoforms was then detected using the ADP Glo® chemiluminescent kinase assay (Promega, Madison, WI) according to the manufacturer’s protocol. Briefly, the trans-phosphorylation reaction (10 μL) was transferred into a 96-well plate (Greiner, solid white, low-binding assay plates) and 10 μL of ADP Glo reagent was added to each well. The reaction was then incubated at room temperature for 40 min to deplete all remaining ATP [63]. The ADP produced by the enzyme/substrate interaction was then converted to ATP by adding 20 μL/well of Kinase Detection Reagent to yield a total assay volume of 40 μL/well. After the addition of Kinase Detection Reagent, the total reaction was incubated for 40 min and luminescence was detected with the TECAN Infinite F500-PRO microplate reader. Untreated controls received TBS vehicle at the same volume.
DCFDA – ROS assay
To evaluate the effect of BCAT1 on the generation of ROS, BCAT1-siRNA (20 nM) transfected SKOV-3 cells were seeded overnight. Here, cells were treated with 100 μL of 2× 30 μM DCFDA solution and incubated in the dark at 37°C for 45 minutes. The fluorescent intensity was then measured on a fluorescence plate reader at excitation of 485 nm and emission of 535 nm.
Apoptosis assay
To determine the effect of BCAT1 siRNA knockdown on cell apoptosis, MDA-MB-231 cells were seeded at a density of 1 × 106 cells per T-25 flask and treated with 20 nM BCAT1 siRNA for 72 hours. Cells were detached and washed twice with ice cold PBS and resuspended in 1× binding buffer. Annexin V-FITC (Biolegend, San Diego) and PI (Fisher-Scientific) were added into the binding buffer and incubated for 10 min at room temperature in the dark. Analysis was performed on a BD Accuri™ C6 Flow Cytometer (BD Biosciences) to identify the subpopulations of the apoptotic cells.
Statistical analysis
All data were expressed as means ± standard error of the mean (SEM). Significance levels for comparisons between groups were determined with unpaired two-tailed Student’s t-test using GraphPad Prism 8 (GraphPad Software). P values of < 0.05 were considered statistically significant. For Western blots, protein levels were normalised to α-tubulin and relative band density reported as a fold change to control.
Abbreviations
BCAT: branched-chain aminotransferase; BCATm: mitochondrial branched-chain aminotransferase; BCATc: cytosolic branched-chain aminotransferase; BCAA: branched-chain amino acids; BCKD complex: branched-chain α-keto dehydrogenase complex; HER2: human epidermal growth factor receptor-2; PR: progesterone receptor; TNBC: triple negative breast cancer; IGF-1: insulin-like growth factor; EGF: epidermal growth factor; PI3K: phosphatidyl 3-kinase; MAPK: mitogen-activated protein kinase; mTOR: mammalian target of rapamycin; mTOR: mammalian target of rapamycin complex 1; GCN2: general control nonderepressible 2 kinase; FOXO3a: forkhead box O3; EGFR: epithelial growth factor receptor; IRS1/2: insulin receptor substrates; GLUT1: glucose transporter 1; SHC: Src homology 2 domain-containing; Grb2: growth factor receptor-bound 2; SOS: son of sevenless; Nrf2: nuclear factor (erythroid-derived 2)-like 2; GDP: nucleotide guanosine diphosphate; GTP: nucleotide guanosine triphosphate; siRNA: small interfering RNA; shRNA: short hairpin RNA; TTI: tritiated thymidine incorporation; ROS: reactive oxygen species; Grx: glutaredoxin; Trx: thioredoxin; H2O2: hydrogen peroxide; PVDF: polyvinylidene difluoride; TCA: tricarboxylic acid.
ACKNOWLEDGMENTS AND FUNDING
This work was supported from an Alliance/UWE, Bristol PhD bursary and NIH/NIGMS grant numbers 1SC1GM130545 and 1SC2GM113784 (to R.H.N).
CONFLICTS OF INTEREST
None.
References
1. Foulkes WD, Smith IE. Triple-negative breast cancer. N Engl J Med. 2010; 363:1938–48. https://doi.org/10.1056/NEJMra1001389. [PubMed].
2. Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486:395–9. https://doi.org/10.1038/nature10933. [PubMed].
3. Yoshida GJ. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015; 34:111. https://doi.org/10.1186/s13046-015-0221-y. [PubMed].
4. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016; 2:e1600200. https://doi.org/10.1126/sciadv.1600200. [PubMed].
5. Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY). 2011; 3:192–222. https://doi.org/10.18632/aging.100296. [PubMed].
6. Conway ME, Hutson SM. BCAA Metabolism and NH3 Homeostasis. Adv Neurobiol. 2016; 13:99–132. https://doi.org/10.1007/978-3-319-45096-4_5. [PubMed].
7. Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AHC, Karra D, Piro RM, Felsberg J, Addington A, Lemke D, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013; 19:901–8. https://doi.org/10.1038/nm.3217. [PubMed].
8. Thewes V, Simon R, Hlevnjak M, Schlotter M, Schroeter P, Schmidt K, Wu Y, Anzeneder T, Wang W, Windisch P, Kirchgäßner M, Melling N, Kneisel N, et al. The branched-chain amino acid transaminase 1 sustains growth of antiestrogen-resistant and ERα-negative breast cancer. Oncogene. 2017; 36:4124–34. https://doi.org/10.1038/onc.2017.32. [PubMed].
9. Conway ME, Hull J, El Hindy M, Taylor SC, El Amraoui F, Paton-Thomas C, White P, Williams M, Ellis HP, Bertoni A, Radlwimmer B, Hutson SM, Kurian KM. Decreased expression of the mitochondrial BCAT protein correlates with improved patient survival in IDH-WT gliomas. Brain Pathol. 2016; 26:789–91. https://doi.org/10.1111/bpa.12385. [PubMed].
10. Wang ZQ, Gobeil S, Gregoire J, Guillemette C, Bachvarova M, Bachvarov D, Vanderhyden B, Plante M, Faddaoui A, Sebastianelli A, Macdonald E, Renaud MC. BCAT1 expression associates with ovarian cancer progression: possible implications in altered disease metabolism. Oncotarget. 2015; 6:31522–43. https://doi.org/10.18632/oncotarget.5159. [PubMed].
11. Yoshikawa R, Yanagi H, Shen CS, Fujiwara Y, Noda M, Yagyu T, Gega M, Oshima T, Yamamura T, Okamura H, Nakano Y, Morinaga T, Hashimoto-Tamaoki T. ECA39 is a novel distant metastasis-related biomarker in colorectal cancer. World J Gastroenterol. 2006; 12:5884–9. https://doi.org/10.3748/wjg.v12.i36.5884. [PubMed].
12. Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-Kinase/Akt/mTOR Signaling Pathway. Surgical Oncology Clinics of North America. 2013; 22:641–64. https://doi.org/10.1016/j.soc.2013.06.008. [PubMed].
13. Anthony TG, McDaniel BJ, Byerley RL, McGrath BC, Cavener DR, McNurlan MA, Wek RC. Preservation of liver protein synthesis during dietary leucine deprivation occurs at the expense of skeletal muscle mass in mice deleted for eIF2 kinase GCN2. J Biol Chem. 2004; 279:36553–61. https://doi.org/10.1074/jbc.M404559200. [PubMed].
14. Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, McSkimming D, Kannan N, Tojo A, Edison AS, Ito T. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature. 2017; 545:500–4. https://doi.org/10.1038/nature22314. [PubMed].
15. Conway ME, Lee C. The redox switch that regulates molecular chaperones. Biomol Concepts. 2015; 6:269–84. https://doi.org/10.1515/bmc-2015-0015. [PubMed].
16. Harris M, El Hindy M, Usmari-Moraes M, Hudd F, Shafei M, Dong M, Hezwani M, Clark P, House M, Forshaw T, Kehoe P, Conway ME. BCAT-induced autophagy regulates Aβ load through an interdependence of redox state and PKC phosphorylation-implications in Alzheimer’s disease. Free Radic Biol Med. 2020 Jan 23. [Epub ahead of print]. https://doi.org/10.1016/j.freeradbiomed.2020.01.019. [PubMed].
17. Xu M, Liu Q, Jia Y, Tu K, Yao Y, Liu Q, Guo C. BCAT1 promotes tumor cell migration and invasion in hepatocellular carcinoma. Oncol Lett. 2016; 12:2648–56. https://doi.org/10.3892/ol.2016.4969. [PubMed].
18. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017; 36:1302–15. https://doi.org/10.15252/embj.201696151. [PubMed].
19. Ming Yue A, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017; 21:3819–32. https://doi.org/10.1016/j.celrep.2017.12.002. [PubMed].
20. Xiao F, Wang C, Yin H, Yu J, Chen S, Fang J, Guo F. Leucine deprivation inhibits proliferation and induces apoptosis of human breast cancer cells via fatty acid synthase. Oncotarget. 2016; 7:63679–89. https://doi.org/10.18632/oncotarget.11626. [PubMed].
21. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009; 136:521–34. https://doi.org/10.1016/j.cell.2008.11.044. [PubMed].
22. Grzes KM, Swamy M, Hukelmann JL, Emslie E, Sinclair LV, Cantrell DA. Control of amino acid transport coordinates metabolic reprogramming in T-cell malignancy. Leukemia. 2017; 31:2771–9. https://doi.org/10.1038/leu.2017.160. [PubMed].
23. Boucher J, Kleinridders A, Ronald Kahn C. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014; 6:a009191. https://doi.org/10.1101/cshperspect.a009191. [PubMed].
24. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017; 168:960–76. https://doi.org/10.1016/j.cell.2017.02.004. [PubMed].
25. Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, Siddle K, Goldfine ID, Belfiore A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: Evidence for a second mechanism of IGF-I signaling. Clin Cancer Res. 1999; 5:1935–44. [PubMed].
26. Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014; 10:723–36. https://doi.org/10.1038/nrendo.2014.171. [PubMed].
27. Wubetu GY, Utsunomiya T, Ishikawa D, Ikemoto T, Yamada S, Morine Y, Iwahashi S, Saito Y, Arakawa Y, Imura S, Arimochi H, Shimada M. Branched chain amino acid suppressed insulin-initiated proliferation of human cancer cells through induction of autophagy. Anticancer Res. 2014; 34:4789–96. [PubMed].
28. Zakrzewska M, Haugsten EM, Nadratowska-Wesolowska B, Oppelt A, Hausott B, Jin Y, Otlewski J, Wesche J, Wiedlocha A. ERK-mediated phosphorylation of fibroblast growth factor receptor 1 on Ser 777 inhibits signaling. Sci Signal. 2013; 6:ra11. https://doi.org/10.1126/scisignal.2003087. [PubMed].
29. Han S, Ren Y, He W, Liu H, Zhi Z, Zhu X, Yang T, Rong Y, Ma B, Purwin TJ, Ouyang Z, Li C, Wang X, et al. ERK-mediated phosphorylation regulates SOX10 sumoylation and targets expression in mutant BRAF melanoma. Nat Commun. 2018; 9:28. https://doi.org/10.1038/s41467-017-02354-x. [PubMed].
30. Soond SM. ERK-mediated phosphorylation of Thr735 in TNF -converting enzyme and its potential role in TACE protein trafficking. Journal of Cell Science. 2005; 118:2371–80.
31. Pan XL, Ren RJ, Wang G, Tang HD, Chen SD. The Gab2 in signal transduction and its potential role in the pathogenesis of Alzheimer’s disease. Neurosci Bull. 2010; 26:241–6. https://doi.org/10.1007/s12264-010-1109-7. [PubMed].
32. Girnita L, Worrall C, Takahashi SI, Seregard S, Girnita A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci. 2014; 71:2403–27. https://doi.org/10.1007/s00018-013-1514-y. [PubMed].
33. El Hindy M, Hezwani M, Corry D, Hull J, El Amraoui F, Harris M, Lee C, Forshaw T, Wilson A, Mansbridge A, Hassler M, Patel VB, Kehoe PG, et al. The branched-chain aminotransferase proteins: Novel redox chaperones for protein disulfide isomerase-implications in Alzheimer’s disease. Antioxid Redox Signal. 2014; 20:2497–513. https://doi.org/10.1089/ars.2012.4869. [PubMed].
34. Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. Scaffolding Protein Gab1 Sustains Epidermal Growth Factor-Induced Mitogenic and Survival Signaling by Multiple Positive Feedback Loops. J Biol Chem. 2006; 281:19925–38. https://doi.org/10.1074/jbc.M600482200. [PubMed].
35. Pronk GJ, McGlade J, Pelicci G, Pawson T, Bos JL. Insulin-induced phosphorylation of the 46- and 52-kDa Shc proteins. J Biol Chem. 1993; 268:5748–53. [PubMed].
36. Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998; 394:337–43. https://doi.org/10.1038/28548. [PubMed].
37. Castellano E, Downward J. Ras interaction with PI3K: More than just another effector pathway. Genes Cancer. 2011; 2:261–74. https://doi.org/10.1177/1947601911408079. [PubMed].
38. Radhakrishnan Y, Maile LA, Ling Y, Graves LM, Clemmons DR. Insulin-like growth factor-I stimulates Shc-dependent phosphatidylinositol 3-kinase activation via Grb2-associated p85 in vascular smooth muscle cells. J Biol Chem. 2008; 283:16320–31. https://doi.org/10.1074/jbc.M801687200. [PubMed].
39. Jiménez C, Hernández C, Pimentel B, Carrera AC. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J Biol Chem. 2002; 277:41556–62. https://doi.org/10.1074/jbc.M205893200. [PubMed].
40. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001; 98:6074–9. https://doi.org/10.1073/pnas.111114298. [PubMed].
41. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem Sci. 2011; 36:320–8. https://doi.org/10.1016/j.tibs.2011.03.006. [PubMed].
42. Lehr S, Kotzka J, Avci H, Sickmann A, Meyer HE, Herkner A, Muller-Wieland D. Identification of major ERK-related phosphorylation sites in Gab1. Biochemistry. 2004; 43:12133–40. https://doi.org/10.1021/bi049753e. [PubMed].
43. Arkun Y. Dynamic Modeling and Analysis of the Cross-Talk between Insulin/AKT and MAPK/ERK Signaling Pathways. PLoS One. 2016; 11:e0149684. https://doi.org/10.1371/journal.pone.0149684. [PubMed].
44. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002; 277:31099–106. https://doi.org/10.1074/jbc.M111974200. [PubMed].
45. Farhan M, Wang H, Gaur U, Little PJ, Xu J, Zheng W. FOXO signaling pathways as therapeutic targets in cancer. Int J Biol Sci. 2017; 13:815–27. https://doi.org/10.7150/ijbs.20052. [PubMed].
46. Sisci D, Maris P, Cesario MG, Anselmo W, Coroniti R, Trombino GE, Romeo F, Ferraro A, Lanzino M, Aquila S, Maggiolini M, Mauro L, Morelli C, et al. The estrogen receptor α is the key regulator of the bifunctional role of FoxO3a transcription factor in breast cancer motility and invasiveness. Cell Cycle. 2013; 12:3405–20. https://doi.org/10.4161/cc.26421. [PubMed].
47. Storz P, Doppler H, Copland JA, Simpson KJ, Toker A. FOXO3a Promotes Tumor Cell Invasion through the Induction of Matrix Metalloproteinases. Mol Cell Biol. 2009; 29:4906–17. https://doi.org/10.1128/MCB.00077-09. [PubMed].
48. Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005; 24:7410–25. https://doi.org/10.1038/sj.onc.1209086. [PubMed].
49. Hao X, Ding W, Wang Y, Yu W, Wu W, Ponnusamy M, Ao X, Li P, Liu Y, Wang J. Critical role of FOXO3a in carcinogenesis. Mol Cancer. 2018; 17:104. https://doi.org/10.1186/s12943-018-0856-3. [PubMed].
50. Fu H, Subramanian RR, Masters SC. 14-3-3 Proteins: Structure, Function, and Regulation. Annu Rev Pharmacol Toxicol. 2002; 40:617–47. https://doi.org/10.1146/annurev.pharmtox.40.1.617. [PubMed].
51. Thistle JE, Hellberg Y, Mortensen K, Hamilton-Dutoit S, Kjærsgaard A, Cronin-Fenton D, Sørensen HT, Lash TL. The effect of 14-3-3ζ expression on tamoxifen resistance and breast cancer recurrence: a Danish population-based study. Breast Cancer Res Treat. 2017; 165:633–43. https://doi.org/10.1007/s10549-017-4289-2. [PubMed].
52. Niemantsverdriet M, Wagner K, Visser M, Backendorf C. Cellular functions of 14-3-3ζ in apoptosis and cell adhesion emphasize its oncogenic character. Oncogene. 2008; 27:1315–9. https://doi.org/10.1038/sj.onc.1210742. [PubMed].
53. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padró M, Daemen A, Hu M, Chan DA, Ethier SP, Van’tVeer LJ, Polyak K, McCormick F, Gray JW. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell. 2013; 24:450–65. https://doi.org/10.1016/j.ccr.2013.08.020. [PubMed].
54. Denicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475:106–9. https://doi.org/10.1038/nature10189. [PubMed].
55. Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett. 2003; 546:181–4. https://doi.org/10.1016/s0014-5793(03)00517-9. [PubMed].
56. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009; 34:663–73. https://doi.org/10.1016/j.molcel.2009.04.029. [PubMed].
57. Taguchi K, Yamamoto M. The KEAP1-NRF2 System in Cancer. Front Oncol. 2017; 7:85. https://doi.org/10.3389/fonc.2017.00085. [PubMed].
58. Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu Rev Pharmacol Toxicol. 2013; 53:401–26. https://doi.org/10.1146/annurev-pharmtox-011112-140320. [PubMed].
59. Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, Tora AD, Mehta K. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene. 2002; 21:8843–51. https://doi.org/10.1038/sj.onc.1206044. [PubMed].
60. Li H, Wan A, Xu G, Ye D. Small changes huge impact: The role of thioredoxin 1 in the regulation of apoptosis by S-nitrosylation. Acta Biochim Biophys Sin (Shanghai). 2013; 45:153–61. https://doi.org/10.1093/abbs/gms103. [PubMed].
61. Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, Kondo T. Glutaredoxin Exerts an Antiapoptotic Effect by Regulating the Redox State of Akt. J Biol Chem. 2003; 278:50226–33. https://doi.org/10.1074/jbc.M310171200. [PubMed].
62. Kaoud TS, Devkota AK, Harris R, Rana MS, Abramczyk O, Warthaka M, Lee S, Girvin ME, Riggs AF, Dalby KN. Activated ERK2 is a monomer in vitro with or without divalent cations and when complexed to the cytoplasmic scaffold PEA-15. Biochemistry. 2011; 50:4568–78. https://doi.org/10.1021/bi200202y. [PubMed].
63. Davis MI, Auld DS, Inglese J. Bioluminescence methods for assaying kinases in quantitative high-throughput screening (qHTS) format applied to yes1 tyrosine kinase, glucokinase, and PI5P4Kα lipid kinase. Methods Mol Biol. 2016; 1360:47–58. https://doi.org/10.1007/978-1-4939-3073-9_4. [PubMed].