

Introduction
1,25-Dihydroxyvitamin D3 (1,25D3), the high affinity ligand for the nuclear VDR, regulates multiple cancer processes (cell cycle, apoptosis, migration, invasion) in vivo and in vitro, however the specific gene targets and mechanisms that mediate these effects are unclear. We previously established invasive mammary tumor cell lines from wild-type (WT) and VDR knockout (KO) mice and demonstrated that the VDR is necessary for 1,25D3 mediated anti-cancer signaling in vitro and in vivo [1–3]. Genomic profiling in this model system [4, 5] identified 35 transcripts that were altered by the 1,25D3-VDR complex, only four of which were down-regulated. One of the VDR down-regulated genes was Has2 (hyaluronan synthase-2), an enzyme that synthesizes the polysaccharide hyaluronic acid (HA). 1,25D3 treatment (100 nM, 24 h) reduced Has2 expression 50–70% in VDR positive cells but was without effect in VDR negative cells. Has2 encodes one of three integral membrane proteins (Has1, Has2, and Has3) that synthesize and extrude HA into the pericellular space [6]. Mature HA is a heterogeneous polysaccharide (MW ranges from 5,000 to 20,000,000 Da in vivo) that is ubiquitously present in normal tissues. However, considerable evidence indicates that virtually all human epithelial tumors exhibit elevated amounts of HA and that tumor HA content negatively correlates with disease progression and survival [7–12]. HA is a linear chain of glucuronic acid and N-acetyl-D-glucosamine units which are derived from the hexosamine biosynthesis pathway. High activity of the hexosamine pathway in breast cancer cells leads to HA accumulation and pro-tumorigenic signaling, making this pathway a relevant therapeutic target [13]. HAS2 and HA promote epithelial-mesenchymal transition (EMT), survival, invasion and metastasis in vitro and in vivo [14–21]. Many of these effects result from HA-mediated activation of CD44, a pro-survival receptor enriched on the surface of cancer stem cells [17, 22–26]. Collectively, these data suggest that survival and outgrowth of CD44+ cancer stem cells are dependent on continued HA synthesis through HAS2 activity. This concept predicts that disruption of HA-CD44 signaling would inhibit disease progression in patients whose tumors overexpress HAS2. In the studies reported here we assessed whether 1,25D3 regulates HAS2 in cellular models of human breast cancer, and whether suppression of HAS2 by 1,25D3 is sufficient to inhibit HA synthesis in the context of aggressive disease.
Results
Has2 mRNA is down-regulated by 1,25D3 in murine mammary carcinoma cells
In previous studies we demonstrated that 1,25D3 down-regulated mRNA expression of the HA synthesizing enzyme Has2 in a VDR-dependent manner after 24 hours [4]. Here we have extended these findings to assess whether regulation of Has2 mRNA by 1,25D3 alters HA production and/or phenotype of breast cancer cells. We first examined the kinetics of Has2 mRNA down-regulation by 1,25D3 in KO240, WT145, and KOhVDR cells. RT-qPCR was conducted in samples harvested 6, 12, 24, and 48 hours after treatment with 100 nM 1,25D3 or vehicle (Figure 1A). In KO240 cells lacking VDR, Has2 mRNA was variable with up and down trends over the time course and no consistent effect of 1,25D3. In contrast, 1,25D3 reduced Has2 expression at all time points tested in WT145 cells (which express murine Vdr) and in KOhVDR cells (KO240 cells engineered to ectopically express human VDR). Both VDR positive cell lines displayed down-regulation of Has2 within 6 hours of 1,25D3 treatment, with the peak decrease (approximately 25% of control values) at 24 hours and suppression sustained through 48 hours.
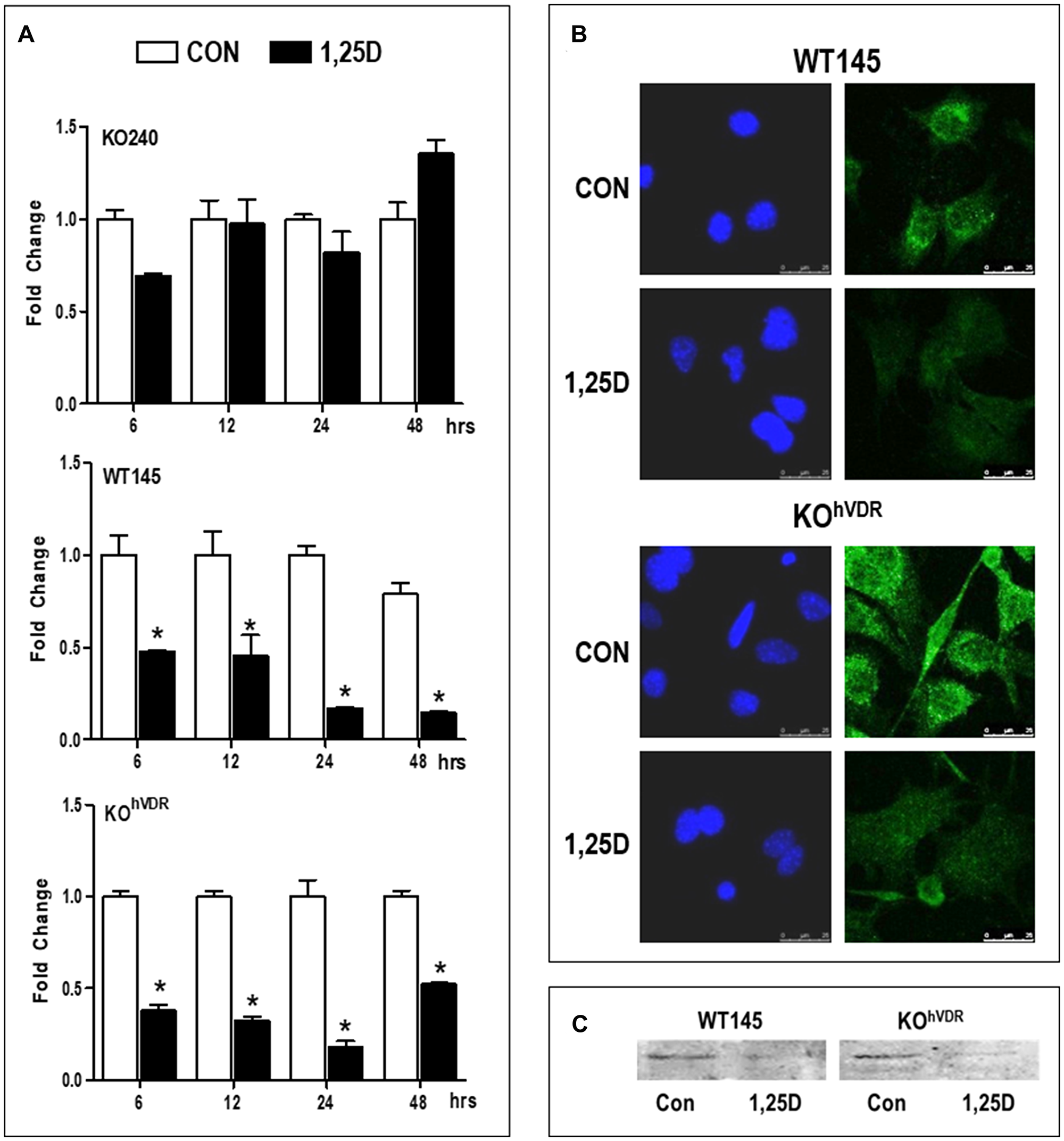
Figure 1: VDR is required for 1,25D3 mediated down-regulation of Has2 mRNA and protein. (A) RNA was isolated from KO240, WT145 and KOhVDR cells treated with 100 nM 1,25D3 for 6, 12, 24, or 48 hours. Has2 mRNA in control and 1,25D3 treated samples was assessed by the ΔΔCt method and values were normalized against 18S and expressed as fold change (1,25D3 vs control) for each cell line. Bars represent mean ± standard deviation, *p < 0.05 control vs 1,25D3 treated at each time point as evaluated by Student’s t test. (B) Immunofluorescence for HAS2 (green) in WT145 and KOhVDR cells treated with 100 nM 1,25D3 or vehicle for 48 hours. Nuclei were stained with DAPI (blue). Images were acquired on a Leica DMI6000 microscope with a TCS SP5 confocal laser scanner using Leica Application Suite software. (C) Lysates from WT145 and KOhVDR cells treated with 100 nM 1,25D3 for 48 hours were blotted with antibodies against HAS2.
We assessed HAS2 protein expression by immunofluorescent staining of WT145 and KOhVDR cells that were treated with 1,25D3 or vehicle for 48 hours. As shown in Figure 1B, confocal imaging localized punctate staining of HAS2 on cell surfaces, and treatment with 1,25D3 reduced staining intensity in both WT145 and KOhVDR cells. Western blotting confirmed down-regulation of HAS2 protein in VDR positive cells treated with 1,25D3 for 48 h (Figure 1C). Collectively, these data demonstrate that the down-regulation of Has2 mRNA by 1,25D3 requires VDR and is of sufficient magnitude to reduce HAS2 protein expression.
1,25D3 reduces cell-associated and secreted HA
To examine if the reduction of Has2 in response to 1,25D3 treatment translated to a reduction in HA production, we assessed both cell-associated and secreted HA. In the majority of cell types, newly synthesized HA is extruded at the plasma membrane and associates with cell surface proteins, forming an extensive pericellular coat. This pericellular matrix can be imaged by particle exclusion assays which employ red blood cells that are repelled by HA [27]. As shown in Figure 2A, distinct exclusion areas surround control KOhVDR cells, allowing visualization of the HA-coated cell surfaces. In contrast, the surfaces of 1,25D3 treated KOhVDR cells are barely visible, indicating the absence of an HA-rich pericellular coat. To quantitate HA synthesis, conditioned media samples were collected from KO240, WT145, and KOhVDR cells at time 0 and 12, 24, and 48 hours after treatment with 100 nM 1,25D3. Total HA was evaluated with a solid-phase sandwich ELISA employing biotinylated HA binding protein (bHABP) which detects both low and high MW HA chains. As shown in Figure 2B, all three cell lines secreted HA which accumulated in media over time. However, only VDR positive cells (WT145 and KOhVDR) exhibited a decrease in HA accumulation in response to 1,25D3 treatment. The amount of secreted HA normalized for final cell numbers at 72 hours was significantly reduced in VDR positive cells but not in VDRKO cells (Figure 2C). These data are consistent with the observed VDR dependent effects of 1,25D3 on Has2 mRNA expression (Figure 1) and indicate that Has2 down-regulation by 1,25D3 is of sufficient magnitude to disrupt HA synthesis and secretion.
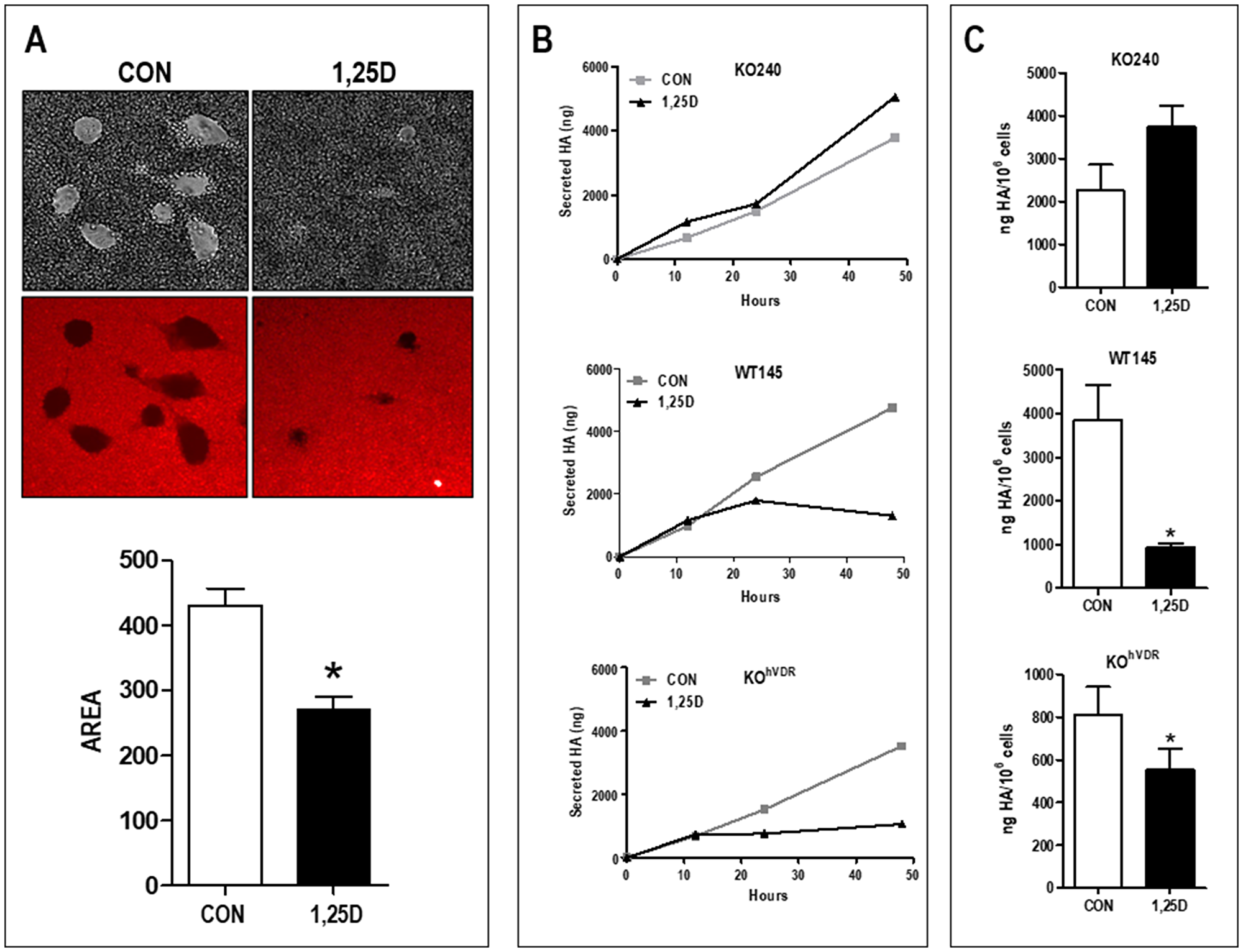
Figure 2: 1,25D3 reduces both cell surface and secreted HA. (A) Cell associated HA was imaged by particle exclusion assay in which sheep erythrocytes are overlaid onto cell monolayers. Shown are KOhVDR cells treated for 48 hours with vehicle or 100 nM 1,25D3 prior to assay. Areas in which erythrocytes (captured as autofluorescence in the FITC channel) are excluded represent the HA-rich pericellular matrix. Graph depicts quantitation of the exclusion area in 19–21 fields for each treatment (mean ± standard error, *p < 0.0001; Student’s t test). (B) Secreted HA was quantitated by ELISA of conditioned media of KO240, WT145 and KOhVDR cells sampled at 0, 12, 24 or 48 hours after treatment with 100 nM 1,25D3 or vehicle. (C) Total secreted HA after 72 hours normalized to final cell number (mean ± standard error, *p < 0.05; Student’s paired t test).
HA rescues cells from 1,25D3 mediated growth inhibition
To assess the contribution of HA to cell growth in this model system, we first examined the effect of the HA synthesis inhibitor 4-methylumbelliferone (4MU) on cell density. 4MU, which binds UDP-glucuronic acid and prevents its incorporation into HA [28], significantly reduced the amount of HA secreted from WT145 cells (Supplementary Figure 1). Since 4MU does not block HA synthesis via targeting Has2 expression, we anticipated that combining 4MU with 1,25D3 would additively inhibit cell growth. In WT145 cells, both 1,25D3 (100 nM) and 4MU (500 μM) inhibited culture density, and density was lowest in cultures treated simultaneously with 1,25D3 and 4MU (Figure 3). We next examined whether exogenous addition of HA could rescue 1,25D3 treated cells from growth inhibition. Because HA can exert unique effects depending on molecular size, we tested both High Molecular Weight HA (HMW, 1.54 × 106 kDa) and Low Molecular Weight HA (LMW, 29 kDa) in these assays. Cell density was assessed in KO240, WT145, and KOhVDR cells treated for 96 hours with 100 nM 1,25D3, 500 μg/mL HA (LMW or HMW), the combination of both, or vehicle. As shown in Figure 4A, growth of all three cell lines was unaffected by LMW HA but was increased by HMW HA. As expected, the effects of HA on KO240 cells lacking VDR were not altered in the presence of 1,25D3. However, growth of both WT145 and KOhVDR cells was inhibited by 1,25D3 and co-treatment with HMW HA (but not LMW HA) abrogated this growth inhibition. The morphology of WT145 cells after 96 h treatment with 1,25D3 in the presence or absence of HMW HA is shown in Figure 4B. We observed a clear increase in cell density with no change in overall morphology in cultures treated with 1,25D3 and HMW HA relative to those treated with 1,25D3 alone. These data suggest that HMW HA can rescue 1,25D3 mediated growth inhibition, supporting the concept that suppression of Has2 and HA synthesis contributes to VDR dependent growth regulation.
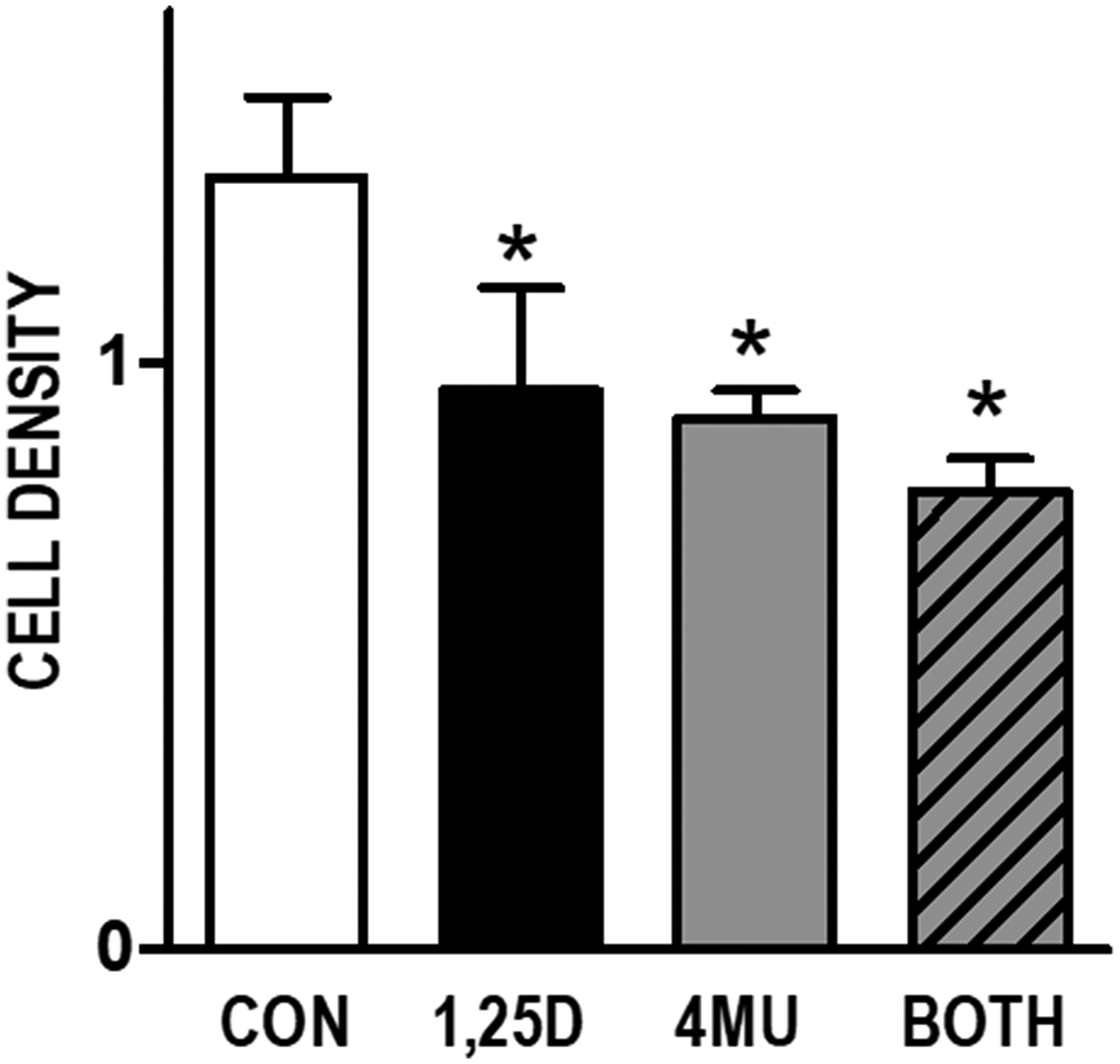
Figure 3: Effects of HA synthesis inhibitor 4MU and 1,25D3 on growth of WT145 cultures. WT145 cells were treated with 500 μM 4-methylumberellifone (4MU) in the presence or absence of 100 nM 1,25D3 for 96 hours. Adherent cell growth was measured by crystal violet absorbance. Bars represent mean ± standard error of at least 2 independent samples analyzed in quadruplicate. p < 0.05, control vs treatments as assessed by one-way ANOVA and Bonferroni’s Multiple Comparison test.
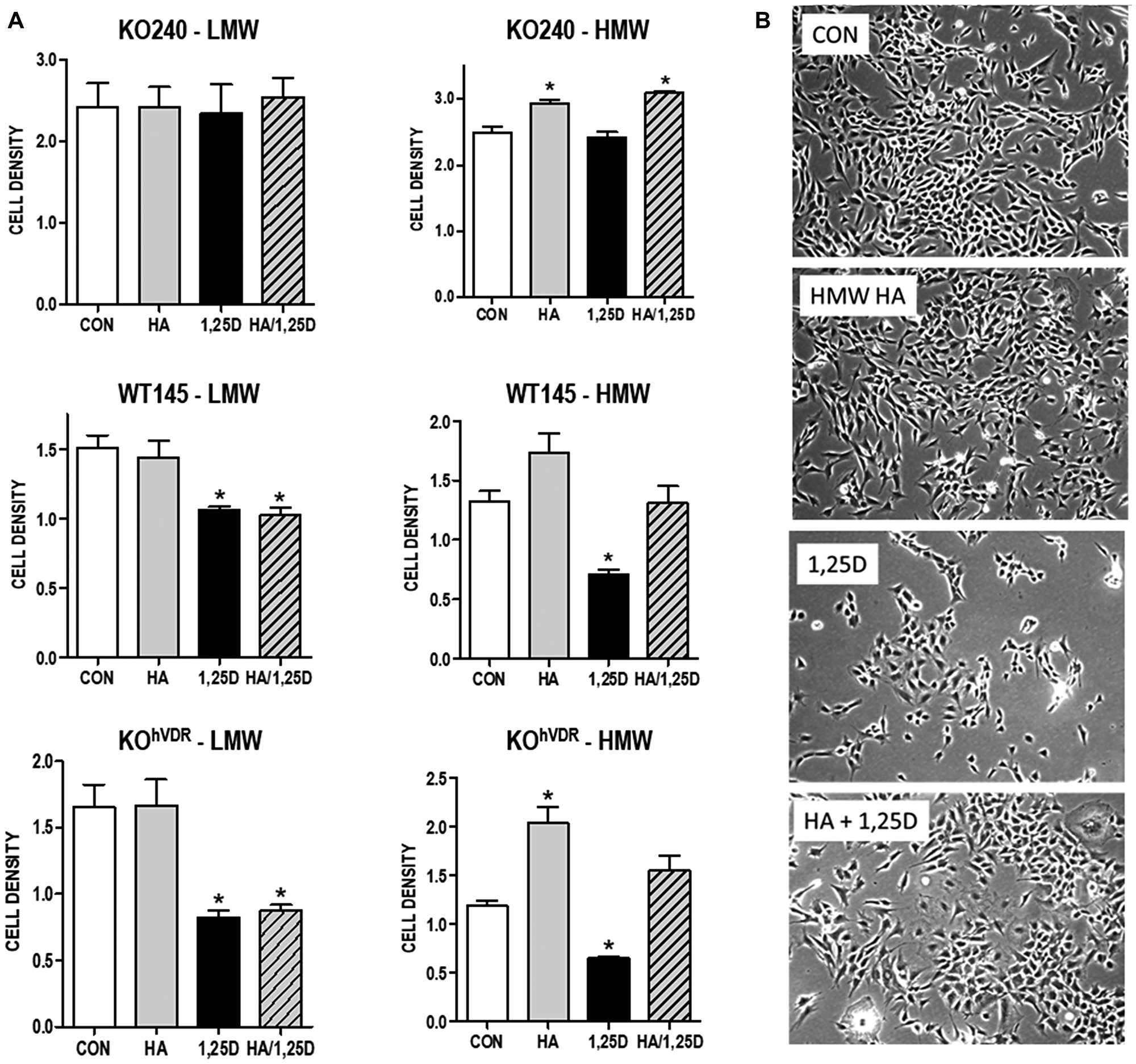
Figure 4: Effect of exogenous HA ± 1,25D3 on growth and morphology of mammary tumor cells. (A) KO240, WT145 and KOhVDR cells were treated with 500 mg/mL high molecular weight (HMW) or low molecular weight (LMW) HA in the presence or absence of 100 nM 1,25D3 for 96 hours. Adherent cell growth was measured by crystal violet absorbance. Bars represent mean ± standard error of 2 independent experiments analyzed in quadruplicate. *p < 0.05, control vs individual treatments assessed by one-way ANOVA. (B) Phase contrast images of WT145 cells 96 hours after treatment with vehicle, 500 mg/mL HMW HA, 100 nM 1,25D3 or the combination of 1,25D3 and HA.
Effect of 1,25D3 on additional genes involved in HA synthesis and degradation
Having determined that 1,25D3 down-regulates expression of Has2 mRNA in a VDR dependent manner, genes coding for the remaining two HA synthesizing enzymes (Has1 and Has3) and three enzymes involved in HA degradation (Hyal1, Hyal2, and Hyal3) were selected for further analysis. RT-qPCR was performed on WT145 and KOhVDR cells treated with 100 nM 1,25D3 or ethanol vehicle for 24 hours. Has1 and Has3 expression were not altered by 1,25D3 treatment in WT145 or KOhVDR cells (Supplementary Figure 2). Similarly, 1,25D3 did not significantly alter Hyal1 or Hyal3 expression in WT145 or KOhVDR cells. 1,25D3 treated KOhVDR cells displayed a non-significant increase in Hyal2 expression compared to vehicle treated cells. Thus, of the genes involved in HA synthesis or degradation that were analyzed, only Has2 is consistently regulated by 1,25D3/VDR signaling. Similar results were observed in tumor associated fibroblasts isolated from WT mice (Supplementary Figure 3), with the exception that 1,25D3 up-regulated Hyal3 in fibroblasts. No effects of 1,25D3 were observed in tumor associated fibroblasts isolated from VDRKO mice (not shown). These data suggest that VDR dependent suppression of Has2 by 1,25D3 may extend to the tumor microenvironment.
HAS2 and the HA pathway is up-regulated in tissues from VDRKO mice
In previous studies we demonstrated that ablation of Vdr alters mammary gland development and tumorigenesis [29, 30]. Using archived paraffin embedded sections from those previous studies, we assessed HAS2 and HA abundance in tissues from adult female WT and VDRKO mice. Very few HAS2 positive cells were identified in mammary tissue of WT mice, whereas HAS2 positive cells were frequent in the stroma of glands from VDRKO mice (Figure 5A). Similarly, Alcian blue staining for glycosaminoglycans was minimal in WT tissue but prominent in stroma surrounding mammary ducts of VDRKO mice (Figure 5B). Specific localization of HA with bHABP also indicated stronger staining around and within ducts of glandular tissue from VDRKO mice relative to WT mice (Figure 5C). Similar results were observed in skin of VDRKO mice (Supplementary Figure 5) which also displays enhanced susceptibility to carcinogenesis relative to WT mice [31]. In xenografts composed of Vdr positive WT145 cells, Alcian blue staining presented as small clusters between tumor cells, whereas in xenografts of KO240 cells staining was more extensive, with long filamentous strands visible in close association with tumor cells (Figure 5D). Collectively, these data support the concept that VDR suppresses HAS2 and HA production in both normal mammary gland and in tumor cells in vivo.
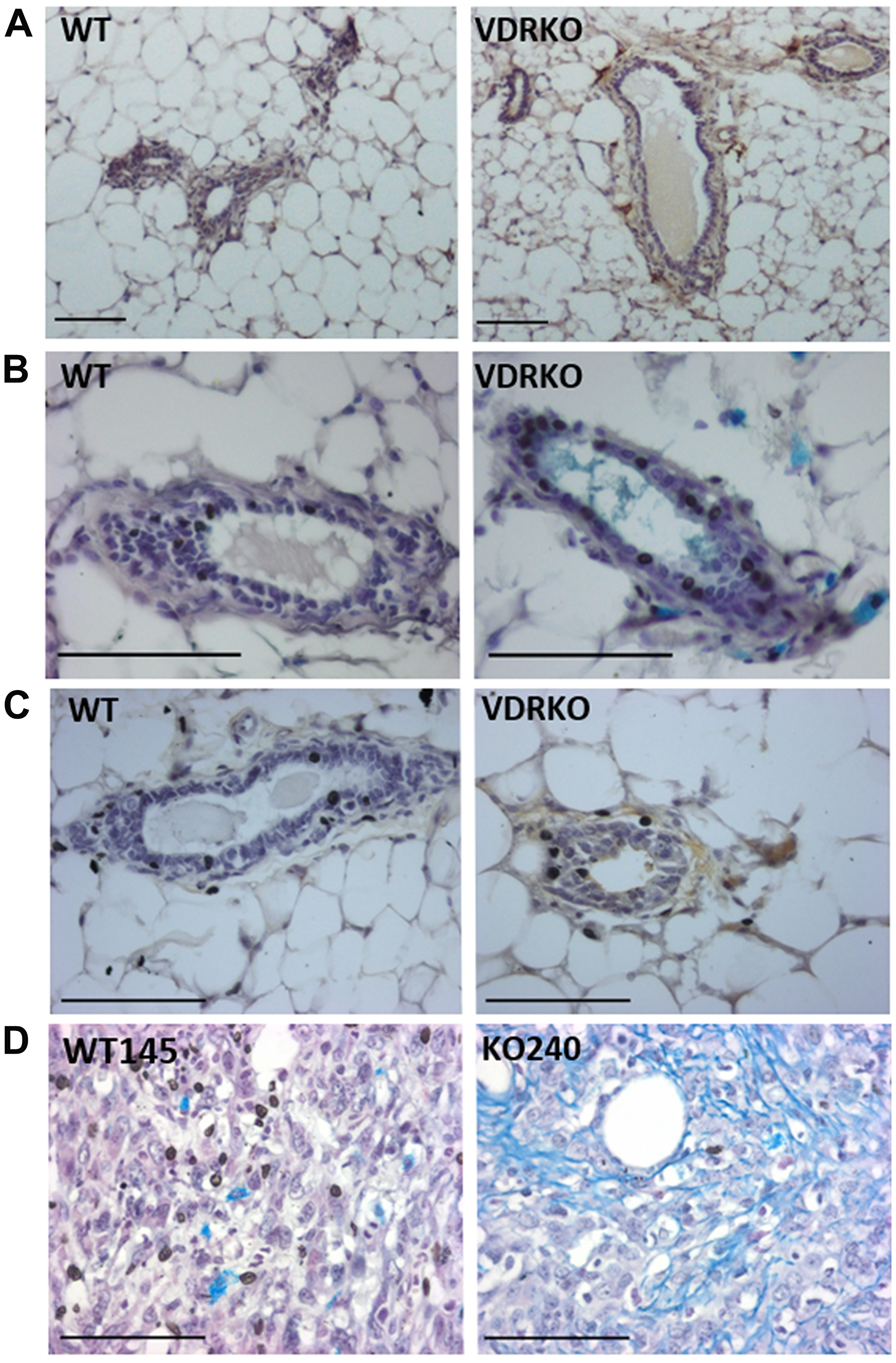
Figure 5: HAS2, glycosaminoglycans and HA localization in wild-type and VDRKO mice. (A) Mammary glands from female wild-type (WT) and VDRKO mice maintained on high calcium rescue diet were formalin fixed, paraffin embedded and processed for immunohistochemistry with HAS2 antibody. Brown staining indicates HAS2 positivity. (B) Glycosaminoglycans including HA were detected by alcian blue staining of glandular tissue from WT and VDRKO mice. (C) HA was localized by bHABP and detected with DAB chromogen in archived glandular tissue [30]. (D). Alcian blue staining of xenografts composed of WT145 and KO240 cell lines [1].
HA pathway is elevated during human mammary epithelial cell transformation
Although many studies have demonstrated elevated HA in aggressive tumors, little is known about changes in the HA pathway during tumor development. We used a well characterized model of EMT [32] to assess whether the HA pathway is altered during this stage of carcinogenesis. VDR and HA related gene expression was compared in immortalized human mammary epithelial cells (HMLE) expressing GFP (control cells) to HMLE cells expressing factors known to induce EMT (TGFβ, SNAIL or TWIST). As shown in Figure 6, mRNA expression of VDR is dramatically reduced, while HAS2 is strongly increased, in cells that have undergone EMT regardless of the trigger (TGFβ, SNAIL, or TWIST). In addition, mRNA for HAS2-AS1, the natural antisense transcript of HAS2 which induces transcription of the HAS2 gene [33], was increased in parallel with HAS2 mRNA during EMT. Concomitant with the changes in HAS2 and HAS2-AS1 expression, secretion of HA into conditioned media was significantly elevated in TGFβ, SNAIL and TWIST expressing cells. 1,25D3 suppressed HA secretion from TGFβ, SNAIL, and TWIST expressing cells suggesting residual 1,25D3 signaling despite VDR down-regulation. In support of an important role for HA in sustaining EMT, TGFβ, SNAIL, and TWIST expressing cells were more sensitive to 4MU mediated growth inhibition than parental (GFP) cells (Figure 7). In addition, 1,25D3 cooperated with 4MU to inhibit growth of cells that had undergone EMT but not that of control cells.
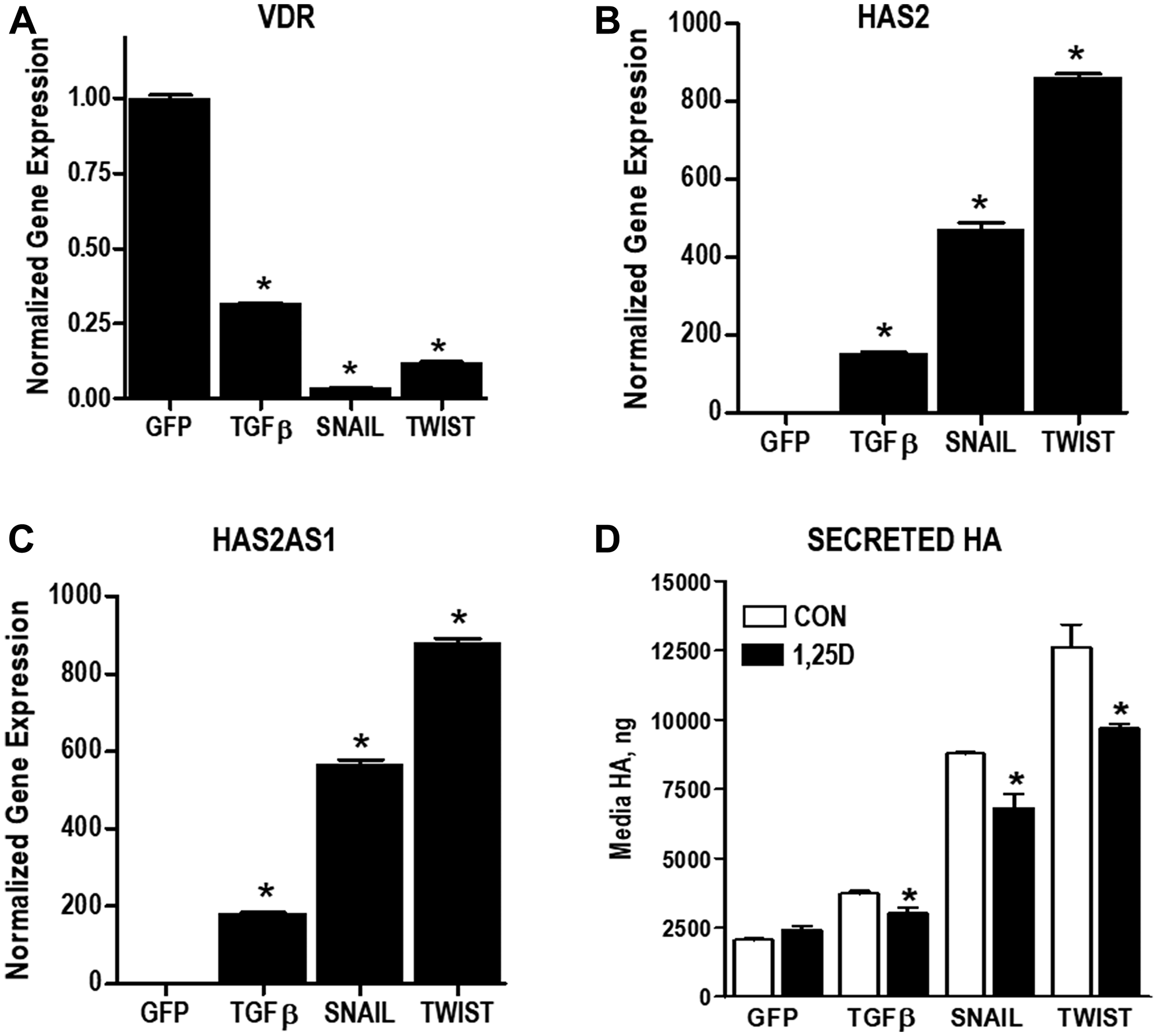
Figure 6: Changes in VDR and HA pathway during EMT. (A–C) RNA was isolated from immortalized human mammary epithelial (HMLE) cells stably expressing GFP (control), TGFβ, SNAIL or TWIST. Expression of VDR (A), HAS2 (B) and HAS2AS1(C) were evaluated by RT-qPCR, normalized to 18S expression and expressed relative to GFP control cell line. Bars represent mean ± standard deviation of at 3 independent samples analyzed in triplicate. *p < 0.05 vs expression in HMLE cells as determined by one-way ANOVA and Dunnett’s post hoc test. (D) Secreted HA was evaluated in conditioned media of HMLE cells stably expressing GFP, TGFβ, SNAIL or TWIST by ELISA. *p < 0.05, 1,25D3 treated vs control for each cell line as evaluated by Student’s t test.
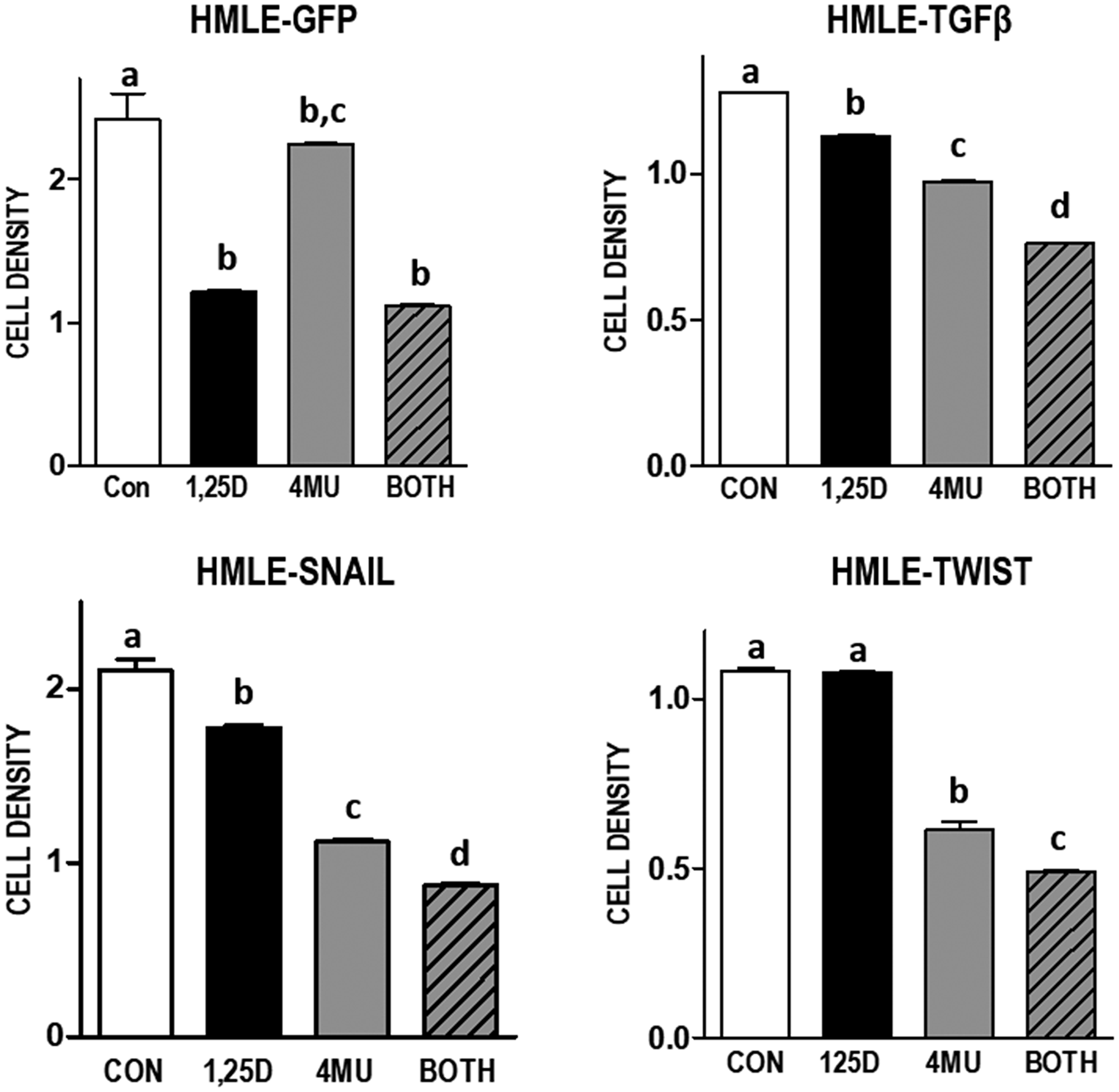
Figure 7: Effect of 1,25D3 and HA synthesis inhibitor 4MU on culture density in mammary epithelial cell models of EMT. Indicated cell lines were treated with 100 μM 4-methylumberellifone (4MU) in the presence or absence of 100 nM 1,25D3 for 96 hours. Adherent cell growth was measured by crystal violet absorbance and expressed relative to untreated samples for each cell line. Data are mean ± standard deviation, n = 6. Bars annotated with different letters are significant at p < 0.05 by one-way ANOVA and Bonferroni post -test.
In a second model of HMLE transformation we assessed the impact of H-RASV12, an oncogenic form of RAS, on the HA pathway in the presence and absence of 1,25D3. In non-transformed HMLE cells, 1,25D3 significantly suppressed HAS2 and HAS1 mRNA expression but was without effect on HAS3 (Figure 8). Upon H-RASV12 transformation, HAS2 was significantly increased while both HAS1 and HAS3 were strongly reduced. Consistent with previous reports that RAS transformation abrogates VDR signaling [34–36], 1,25D3 failed to suppress HAS2 mRNA in HMLE-RAS cells and did not alter the expression of HAS1 or HAS3. Of the hyaluronidase genes, HYAL1 was increased while HYAL2 was decreased in HMLE-RAS cells relative to HMLE parental cells, but no consistent effects of 1,25D3 on these genes were noted (data not shown).
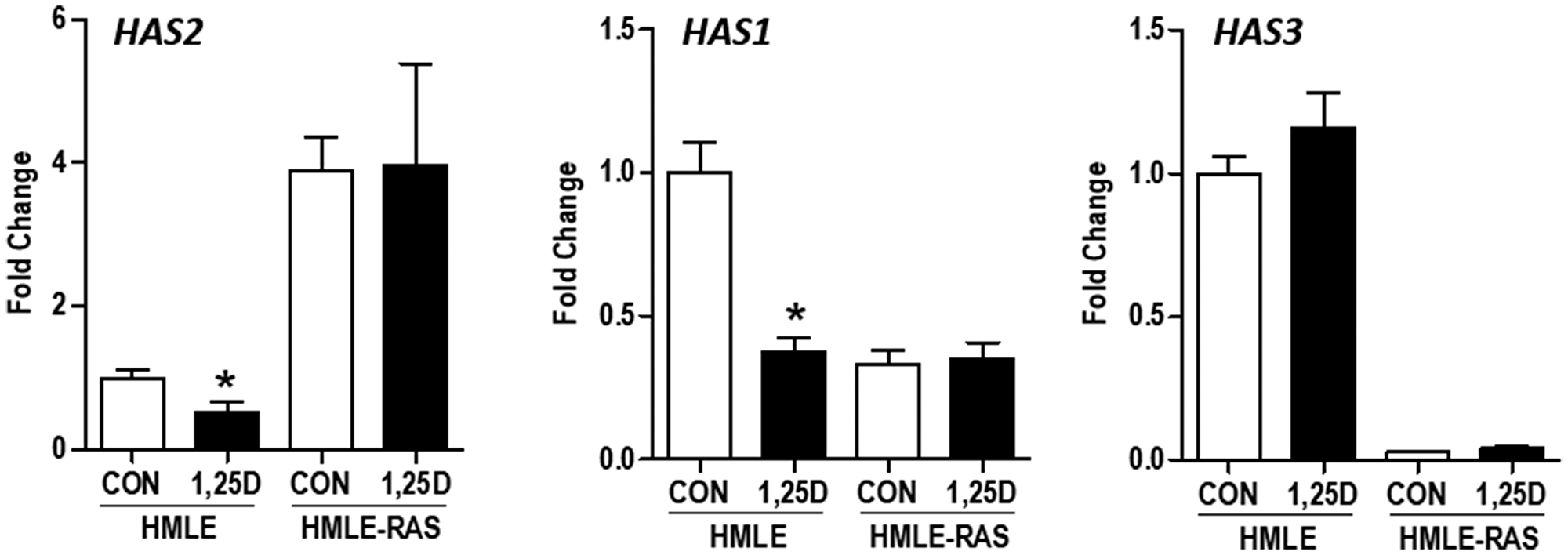
Figure 8: Effect of H-RASV12 transformation on HAS gene expression in immortalized human mammary epithelial cells. RNA was isolated from control human mammary epithelial (HMLE) cells and those expressing oncogenic H-RASV12 (HMLE-RAS) 24 hours after treatment with 100 nM 1,25D3 or vehicle. HAS2, HAS1, and HAS3 expression was evaluated by RT-qPCR, normalized to 18S mRNA and expressed relative to untreated HMLE cells. Bars represent mean ± standard deviation of 3 independent samples analyzed in quadruplicate. *p < 0.05, 1,25D3 treated vs control in each cell line as evaluated by Student’s t test.
1,25D3 reduces HAS2 and HA in human breast cancer cells
Given the effects of 1,25D3 on the HA pathway in murine mammary tumor cells and in models of mammary cell transformation, we questioned whether similar responses would be observed in human breast cancer cells. The human triple negative breast cancer (TNBC) cell line Hs578T highly expresses HAS2 and produces high molecular weight HA which drives cell survival and invasion [21]. We thus assessed VDR function and the ability of 1,25D3 to regulate the HA pathway in this cell line. VDR protein expression was detected in the nucleus of Hs578T cells, whereas HAS2 and HA (visualized with bHABP) co-localized at cell surface protrusions (Figure 9A). Consistent with the results from murine cells, western blots (Figure 9B) demonstrated that 1,25D3 (100 nM, 48 h) enhanced VDR expression while reducing HAS2 expression in in Hs578T cells. 1,25D did not alter HAS3, HYAL1 or HYAL2 expression but significantly increased HYAL3, in Hs578T cells (Supplementary Figure 4). HAS1 was not detected in control or treated Hs578T cells (data not shown). VDR transcriptional activity was confirmed by RT-qPCR measurement of CYP24A1 (a well characterized Vdr target gene), which was up-regulated > 800-fold after 1,25D3 treatment (Figure 9C). The change in HAS2 protein in 1,25D3 treated Hs578T cells corresponded to reduction in HAS2 mRNA and translated to a decrease in secreted HA (Figure 9C). Hs578T cells also highly express the breast cancer stem cell marker CD44, which when activated by HA promotes survival [37]. Notably, 1,25D3 treatment reduced CD44 expression (Figure 9C), induced morphological changes indicative of differentiation (Figure 9D) and reduced both culture growth (Figure 9E) and migration (Supplementary Figure 6). Treatment with HA inhibitor 4MU or the glutamine analog 6-diazo-5-oxo-l-norleucine (DON), which inhibits the hexosamine pathway and generation of UDP-hexoses, strongly reduced density of Hs578T cultures, confirming that Hs578T cell growth/survival is dependent on HA (Figure 9F). Flow cytometric determination of cell cycle, cell number and annexin positivity (Figure 9G–9I) indicated that 1,25D3 cooperated with 4MU to inhibit entry into mitosis, reduce cell viability and enhance apoptosis. Collectively, these data suggest that, like murine mammary tumor cells and HMLE model systems, 1,25D3 suppresses HAS2 and HA synthesis in association with growth inhibition of human TNBC cells.
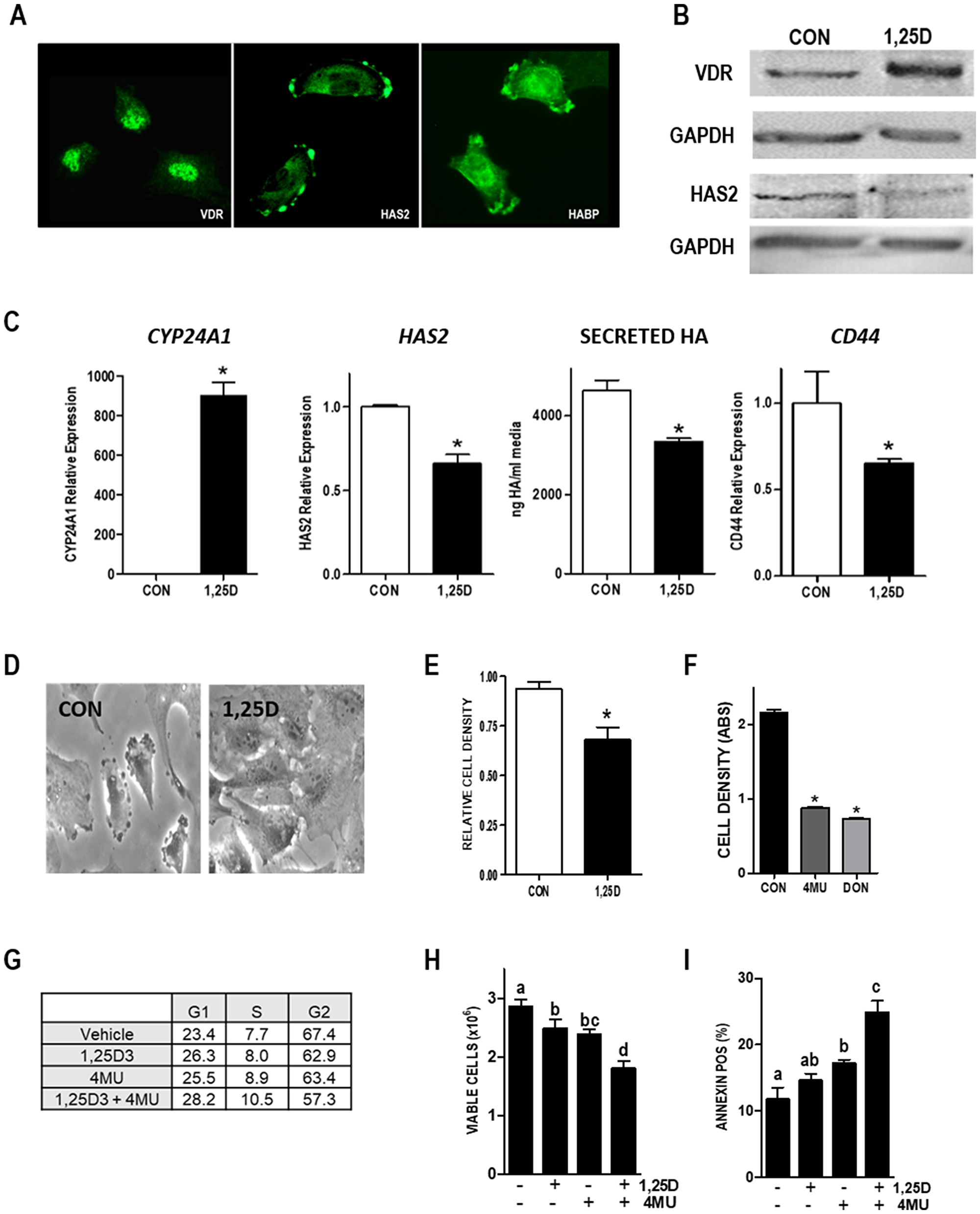
Figure 9: Vitamin D and HA pathways in Hs578T human TNBC cells. (A) Images of VDR, HAS2, and HA localization in Hs578T cells. (B) Lysates from Hs578T cells treated with vehicle or 100 nM 1,25D3 for 48 hours were blotted with antibodies against VDR, HAS2, or loading control GAPDH. (C) RNA was isolated from Hs578T cells treated with 100 nM 1,25D3 for 24 hours. CYP24A1, HAS2 and CD44 were quantitated by RT-qPCR, normalized to 18S and expressed relative to vehicle treated cells. Secreted HA was evaluated by ELISA of conditioned media removed 48 hours after 1,25D3 or vehicle treatment. Bars represent mean ± standard error, with quadruplicates for RT-qPCR and triplicates for ELISA. *p < 0.05, control vs 1,25D3 treated in each cell line as evaluated by Student’s t test. (D) Phase contrast images of Hs578T cells treated with 100 nM 1,25D3 or vehicle for 96 hours. (E) Crystal violet assay for culture density was conducted in HS578T cells treated with 100 nM 1,25D3 for 144 hours. (F) Crystal violet assay for culture density was conducted in HS578T cells treated with HA synthesis inhibitors 4MU or DON for 96 hours. Data from growth assays are mean ± standard error of quadruplicates. *p < 0.05, control vs treated as evaluated by Student’s t test. (G) Percentages of cells in G1, S and G2 phases of the cycle after 96 hours treatment with 100 nM 1,25D ± 100 μM 4MU. Data are mean of duplicates with 5000 cells analyzed per run. (H) Viable cell count (mean ± standard deviation, n = 4) after 96 hours treatment with 100 nM 1,25D ± 100 μM 4MU. Bars annotated with different letters are significant at p < 0.05 by one-way ANOVA and Bonferroni post -test. (I) Percentage of Annexin positive cells indicative of apoptosis (mean ± standard deviation, n = 4) after 96 hours treatment with 100 nM 1,25D ± 100 μM 4MU. Bars annotated with different letters are significant at p < 0.05 by one-way ANOVA and Bonferroni post -test.
Clinical relevance of HAS2 to breast cancer
The clinical relevance of HAS2 in human breast cancer was assessed in the METABRIC dataset accessed via cBIO Portal [38, 39]. We annotated genomic alterations including mutations based on exome sequencing, copy number changes based on the Genomic Identification of Significant Targets in Cancer (GISTIC) algorithm and mRNA abundance based on RNA-Seq (Z score threshold ± 2). We found the overall frequency of genomic alterations in HAS2 was 27% in 1904 cases of Breast Invasive Carcinoma with complete data (Figure 10A). Remarkably, all but two of the 523 tumor-associated changes in HAS2 were amplifications or up-regulations suggesting that enhanced HA synthesis is frequent in breast cancer. Alterations in HAS1 (< 2%) and HAS3 (6%) were rare, suggesting that HAS2 is the most relevant HA synthase in breast tumors. Of note, genomic alterations in VDR were also rare (5% of cases) and the majority of these were amplifications or mRNA up-regulations, suggesting retention of vitamin D signaling in the majority of breast cancers. Kaplan–Meier analysis indicated that the presence of HAS2 alterations in tumors correlated with significant reduction in median overall survival from 164 months to 143 months (Figure 10B). This finding is consistent with the observation that HAS2 alterations were more common in the aggressive subtypes HER2+, Luminal B and Basal-Like (Figure 10C). Given that HAS2 activity depends on the availability of UDP-hexoses, it is not surprising that GFPT2, the rate limiting enzyme of the hexosamine pathway which is inhibited by DON (Figure 10D), was strongly associated with HAS2 expression in this dataset (Figure 10D). Surprisingly, the association of HAS2 alterations with survival was more pronounced in pre-menopausal women and in those whose tumors are estrogen receptor positive (Supplementary Figure 7) suggesting that HA production may identify a more aggressive subset of hormone dependent disease. Collectively, our studies indicate that HAS2 and GFPT2 represent attractive drug targets in human breast cancer.
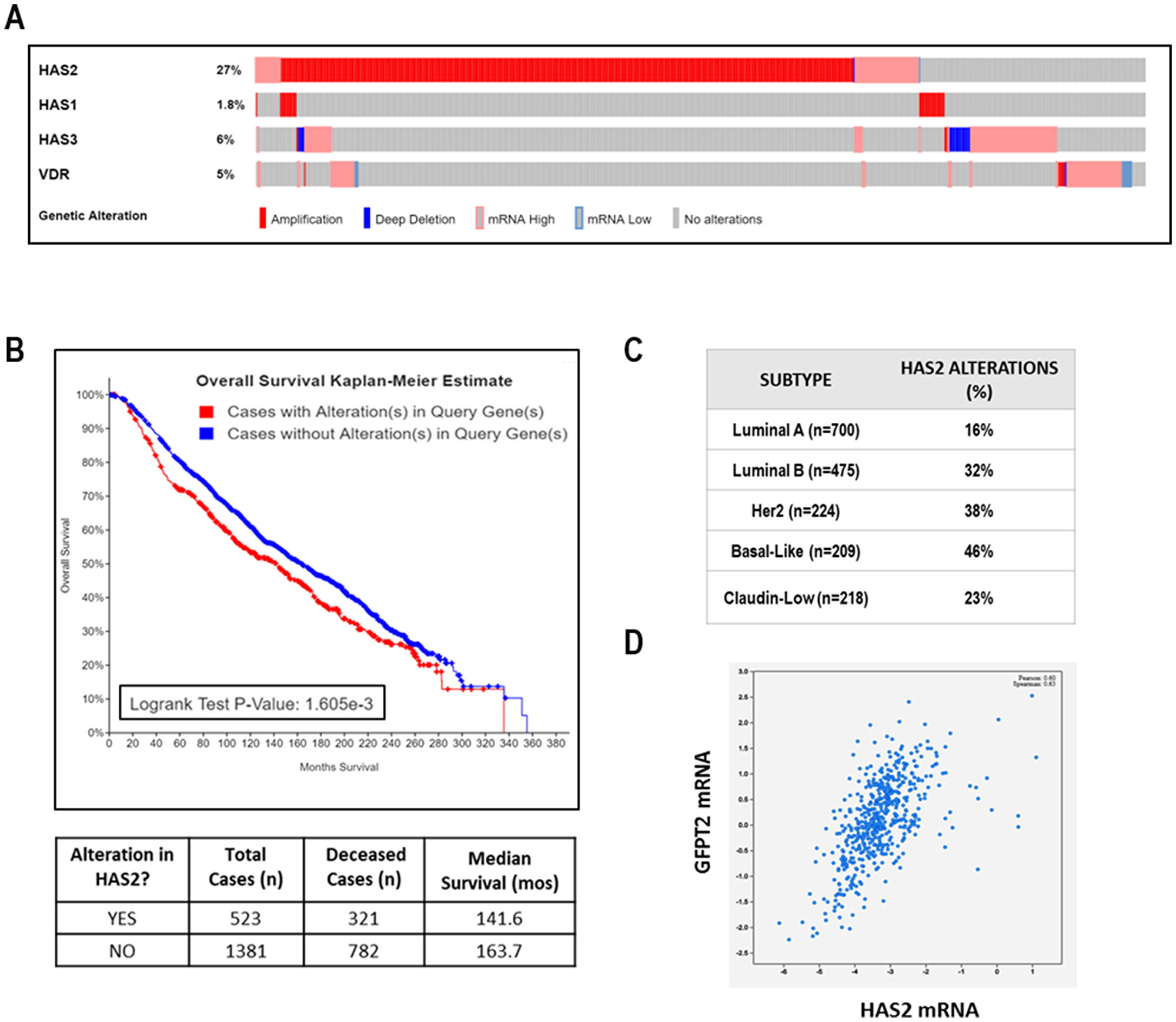
Figure 10: Clinical relevance of HAS2 in TCGA METABRIC dataset of human breast cancer. (A) Oncoprint showing genomic alterations in VDR, HAS1, HAS2, and HAS3 in 1904 human breast tumors. Each column of light grey bars in this scheme represents one case of human breast cancer; colored bars indicate specific types of genomic alterations as indicated in the legend. (B) Kaplan Meier analysis of overall survival in METABRIC dataset stratified by HAS2 alterations. (C) HAS2 alterations according to breast cancer subtype. (D) Correlation between HAS2 and GFPT2 expression. All data were calculated from publicly accessible METABRIC database at cBio Cancer Genomics Portal.
DISCUSSION
This work defines a novel role for 1,25D3 and the VDR in control of HA synthesis in epithelial tissues that likely contributes to its anti-cancer actions. We evaluated the relationship between 1,25D3, VDR, and HAS2 function in several model systems. In murine mammary tumor cells, we observed rapid and sustained down-regulation of Has2 by 1,25D3. Notably, the decreased expression of Has2 in VDR positive cells was sufficient to significantly reduce both cell-associated and secreted HA. More importantly, exogenous addition of high molecular weight HA (similar in size to that produced by HAS2) rescued VDR positive cells from 1,25D3 mediated growth inhibition. Low molecular weight HA, similar in size to fragments generated by hyaluronidase enzymes, was unable to rescue cells from 1,25D3 mediated growth inhibition. These data provide evidence that down-regulation of Has2 leading to reduced synthesis of high molecular weight HA may be an important contributor to 1,25D3 mediated growth inhibition. Our data is consistent with reports that 1,25D and other endogenous vitamin D steroids inhibited TGF-β1-induced HAS2 expression and HA synthesis in association with anti-fibrotic actions, reduced HA accumulation in UV-treated mouse skin and decreased serum HA in a rat fibrosis model [40–42]. Furthermore, vitamin D supplementation of hepatitis patients (15,000 IU/week for 48 weeks) significantly reduced serum HA at all three time points studied (12, 24, and 48 weeks) [43]. Collectively, these data suggest that vitamin D comprehensively inhibits the HA pathway both in vitro and in vivo, supporting the translational relevance of our work.
We have also extended these findings in model systems of EMT and oncogenesis as well as in an established human TNBC cell line. We established that HAS2 expression and HA production are elevated in normal human mammary epithelial cells induced to undergo EMT (through stable expression of TGFβ, SNAIL or TWIST) and in those expressing oncogenic H-RASV12, indicating that deregulation of HA production may be an early and frequent event in breast tumorigenesis. 1,25D3 reduced HAS2 and HA secretion and acted additively with an HA synthesis inhibitor to slow growth of cells expressing TGFβ, SNAIL and TWIST. Similar results were observed in Hs578T cells, an aggressive TNBC cell line characterized by high HAS2 expression and production of high molecular weight HA [21]. VDR expression and transcriptional activity was confirmed in Hs578T cells, as 1,25D3 treatment induced the canonical VDR target gene CYP24A1 over 800-fold. In Hs578T cells, 1,25D3 also reduced expression of HAS2 and CD44 and decreased HA secretion in association with phenotypic evidence of differentiation, inhibition of cell cycle progression and induction of apoptosis.
Using cells derived from VDRKO mice, we established that Vdr was necessary and sufficient for regulation of Has2 by 1,25D3. No changes in Has2 expression, HA secretion, or cell density were observed in response to 1,25D3 treatment of KO240 cells which were derived from VDRKO mice [1]. However, when KO240 cells were engineered to express human VDR (KOhVDR cells), all of these responses were triggered by 1,25D3. These data suggest that both murine and human VDR are capable of repressing Has2 when bound by 1,25D3. It is worth noting that 1,25D3 did not suppress HAS2 in mammary epithelial cells expressing H-RASV12, likely because this oncogene has consistently been shown to corrupt VDR signaling [34–36]. Collectively these data establish that 1,25D3 regulation of Has2 is mediated through VDR rather than through alternative receptors such as the retinoic acid receptor-related orphan receptors (RORs) which can be activated by other dihydroxyvitamin D3 metabolites including 20,23(OH)2D3 [44].
Although the regulation of HAS2 by 1,25D3 is dependent on VDR expression, no consensus VDREs have been identified in its promoter region by in silico analysis [45]. A potential alternative mechanism for this down-regulation is through the actions of microRNAs. Interestingly, miR-498, which has been shown to contain a consensus VDRE in its 5′ regulatory region, can target both HAS2 and CD44 [46]. Additional studies will be necessary to determine whether miR-498 or other microRNAs contribute to down-regulation of HAS2 by 1,25D3.
Previous studies have shown that VDRKO mice are more susceptible to DMBA-induced breast cancer than WT mice [30]. HA interacts with several receptors including CD44 and RHAMM, leading to activation of signaling cascades that promote cell migration and proliferation [47]. Here we have demonstrated that Vdr ablation leads to increased HAS2 protein and accumulation of HA in normal tissues and in tumors. Since forced expression of HAS2 in MMTV-Neu mice enhances tumorigenesis [19], it is plausible that the HA accumulation observed in VDRKO mice contributes to their increased sensitivity to carcinogen induced tumors, however further studies would be needed to directly address this possibility.
To assess the translational relevance of our studies we interrogated the METABRIC TCGA dataset of > 1900 cases of human breast cancer. Surprisingly, few human breast tumors (5%) displayed genomic alterations in VDR, and most of the alterations that were detected were amplifications. Although VDR may frequently be retained in breast tumors, many women with breast cancer are vitamin D deficient [48, 49]. Therefore, attention to vitamin D status and/or vitamin D supplementation may be necessary to ensure appropriate VDR transcriptional activity in tumors. In support of a beneficial effect of vitamin D in cancer, the most recent meta-analysis of vitamin D trials including VITAL [50] indicated a significant reduction in cancer mortality with vitamin D supplementation. However, additional research is needed to determine the patient subsets most likely to benefit from attention to vitamin D status. Our data suggest that the anti-tumor actions of vitamin D may be mediated in part through suppression of HA signaling. In the METABRIC dataset, HAS2 was overexpressed in 27% of all breast tumors, with highest frequency in those subtypes with poor prognosis (Luminal B, HER2+ and Basal-like). Kaplan–Meier curves of overall survival indicate significantly reduced median survival of patients whose tumors highly expressed HAS2. In this dataset, we also found a strong positive correlation between HAS2 and GFPT2, suggesting that up-regulation of the hexosamine metabolic pathway may be necessary to generate UDP-hexoses for incorporation into HA by HAS2. If so, this metabolic vulnerability could be targeted with drugs that block GFPT2 activity. Since HA content is increased during breast cancer progression and elevations in HAS2 and HA correlate with poor prognosis, further studies to evaluate the association of vitamin D signaling and the HA pathway in aggressive human breast cancers are of significant interest. Our data suggest that combining vitamin D supplementation with therapies that target HA signaling (i.e., CD44) or the hexosamine pathway (GFPT2) may be therapeutically beneficial.
Materials and Methods
Cellular model systems
Murine mammary tumor cells with differential VDR expression (WT145 and KO240) were generated in our lab from wild-type and VDRKO mice [1]. The KOhVDR cells were KO240 cells engineered to stably express human VDR (hVDR), the clone used here was KOhVDR-4 as described in LaPorta and Welsh [4]. All murine cell lines were maintained in DMEM/F12 media (50:50) supplemented with 5% charcoal-stripped FBS [1]. Fibroblasts isolated from DMBA-induced tumors of WT and VDRKO mice by collagenase digestion were adapted to culture and maintained in DMEM/F12 media (50:50) supplemented with 5% charcoal-stripped FBS. The effects of EMT were examined in engineered immortalized human mammary epithelial (HMLE) cells stably expressing GFP (control), SNAIL, TWIST or TGFβ (obtained from Dr. Sendurai Mani, MD Anderson Cancer Center, TX, USA) which were maintained in DMEM/F12/Media 171 (1:1) with mammary epithelial growth supplements (MEGS), insulin (5 ug/mL), EGF (5 ng/mL) and hydrocortisone (0.25 mg/mL) [32]. The effect of oncogenic transformation was evaluated in HMLE cells stably expressing H-RASV12 (obtained from Dr. Robert Weinberg, MIT, MA, USA) which were maintained in Media 171 with MEGS [51]. The established human TNBC cell line Hs578T was obtained from ATCC and cultured in high-glucose DMEM with HEPES, 10% FBS and human insulin (10 ug/mL). All cell lines were maintained in a 37°C and 5% CO2 incubator with antibiotics and passaged every 3–4 days.
Culture density, cell cycle, viability, and apoptosis assays
Culture density was assessed in cells plated in 24-well plates and treated the following day with indicated treatments or vehicle (ethanol for 1,25D3; methanol for 4MU; H2O or PBS for DON). After 96 hours, cells were fixed with 1% glutaraldehyde in PBS, stained with 0.1% crystal violet and air-dried overnight. The following day, stain was resuspended in 0.2% Triton X-100 and absorbance was read at 590 nm as an indicator of adherent cell density. Treatments included 1,25D3, 4MU, DON or vehicles as indicated in figure legends (all from Sigma). Low (29 kDa) and High (1.54 × 106 kDa) molecular weight HA (R&D Systems, Minneapolis, MN) were suspended in H2O and added to media at a final concentration of 500 μg/mL. Cell cycle, viability and annexin positivity was assessed on a Millipore MUSE cell analyzer using Muse® Count and Viability kit, Cell Cycle kit and Annexin V & Dead Cell Kit according to manufacturer’s directions.
Reverse-Transcriptase quantitative PCR (RT-qPCR)
Cells were typically treated with 100 nM 1,25D3 or vehicle for 24 hours, but some experiments involved extended time courses as indicated in the figure legends. mRNA was isolated with the RNeasy Mini Kit (Qiagen) according to manufacturer’s instructions and used for cDNA synthesis. RT-qPCR was performed for HAS1, HAS2, HAS3, HYAL1, HYAL2, HYAL3, VDR, CYP24A1, and CD44 gene expression using primers from Origene and SYBR green master mix (Life Technologies Applied Biosystems). Fold change was calculated as ΔΔCt after normalizing to 18S and expressed relative to the ΔΔCt of vehicle treated cells (for 1,25D3 treatments) or parental cell lines (for EMT and H-RASV12 model systems).
Analysis of HA
Secreted HA was quantitated by solid-phase sandwich ELISA of conditioned media removed after treatment (1,25D3, 4MU or respective vehicles). Cells were trypsinized and counted after the last time point for normalization. The low molecular weight (15–40 kDa), medium molecular weight (75–350 kDa), and high molecular weight (> 950 kDa) forms of HA are equally detected in this assay. Briefly, Nunc Maxisorp 96-well Immunoplates (ThermoFisher Scientific) were coated with HABP (non-biotinylated; Millipore) overnight at 4°C. Next day, the plate was washed with PBS containing 0.1% Tween 20 (PBST) and blocked with PBST containing 2% BSA for 1 hour at 37°C. After another series of washes, samples and HA standards (10 to 320 ng/mL; R&D Systems, Minneapolis, MN, USA) were added and incubated for 1 hour at 37°C. After another series of washes, bHABP was added and incubated for 1 hour at 37°C. After washing to remove unbound bHABP, and incubation with streptavidin-HRP (R&D Systems) for 1 hour at 37°C, TMB peroxidase substrate (SureBlue TMB; SeaCare Life Sciences, Milford, MA, USA) was added. Reactions were stopped with 1 M HCl and absorbance was measured at 450 nm with a background correction at 590 nm on a Victor3 V microtiter plate reader (PerkinElmer, Waltham, MA, USA). The amount of HA extrapolated from the standard curves was corrected for media blanks and dilution factors and expressed as absolute values in total media (for time courses) or per 106 cells (for end point measurements). For particle exclusion assays to detect cell surface HA [27], cells were treated with 100 nM 1,25D3 for 48 hours prior to removal of media, PBS washes and addition of glutaraldehyde stabilized sheep erythrocytes (Fitzgerald Industries International, Acton, MA, USA). Images were obtained after erythrocytes settled on the plate and quantitation of the excluded areas was on an INCell 2200 high content cell analyzer.
Western blotting
Whole cell lysates from cells treated for 48 hours with 100 nM 1,25D3 or EtOH were separated on SDS-PAGE gels, transferred to PVDF, and probed with mouse monoclonal VDR antibody (D6, Santa Cruz) or goat polyclonal HAS2 (Y-14) antibody (Santa Cruz). Blots were developed with ECL Plus substrate (Pierce) for 10 minutes and processed on a Storm 860 Molecular Imaging System using the 450 nm (blue) light source.
Immunofluorescence
WT145 and KOhVDR cells were seeded into 4-well chamber slides at a density of 20,000 cells/well and treated the next day with 100 nM 1,25D3 or vehicle. 48 hours after treatment, slides were fixed and permeabilized with ice cold methanol for 10 minutes, rinsed with PBS, and blocked with Dako Protein Block for 1 hour at room temperature. Slides were stained with rabbit polyclonal HAS2 antibody (H-60, SantaCruz Biotechnology) overnight at 4°C. After washing with PBS, slides were incubated with AlexaFluor 488 secondary antibody (Life Technologies), washed and coverslipped with ProLong Gold with DAPI (Invitrogen). Hs578T cells were seeded in chamber slides, fixed in ice cold methanol and immunostained for HAS2 (4E7, Abcam), VDR (D6, Santa Cruz) or HA (bHABP). After primary antibody incubations, slides were incubated with appropriate secondary antibodies (Life Technologies) and coverslipped with ProLong Gold with DAPI (Invitrogen). Imaging was performed on a Leica DMI6000 microscope with a TCS SP5 confocal laser scanner using Leica Application Suite AF version 2.6.0.7266 software or on a Zeiss Axioskop 2 microscope equipped with Axiovision software.
Tissue analyses
In a previous study [52], inguinal mammary glands and dorsal skin were harvested from female WT and VDRKO mice maintained on a high calcium “rescue” diet and treated with DMBA. Xenografts of WT145 and KO240 cells grown in nude mice supplemented with estrogen were available from another previous study [1]. These published studies were conducted at the University of Notre Dame under IACUC-approved protocols. Archived samples, which had been formalin fixed, were sectioned at 5μ and interrogated for abundance of mucins, HAS2, and HA. Alcian blue was used as a general stain for acidic mucins which include HA. Slides were deparaffinized, rehydrated and stained with alcian blue (pH 2.5) for 5 minutes. For detection of HAS2 or HA, slides were deparaffinized and rehydrated as above prior to quenching of endogeneous peroxidase activity with 3% hydrogen peroxide. Non-specific binding was blocked with the Avidin/Biotin blocking kit (Vector Labs) followed by Dako protein block. For HA, slides were incubated with bHABP (Calbiochem) overnight at 4°C, rinsed with PBS, developed with the VECTASTAIN Elite ABC kit (Vector Labs) and detected with DAB chromogen as per manufacturer’s directions. For HAS2, slides were sequentially incubated with rabbit polyclonal HAS2 (H-60, Santa Cruz Biotechnology) and biotinylated secondary antibody prior to development with the VECTASTAIN Elite ABC kit as for HABP. For all stains, slides were counterstained with hematoxylin, rinsed, dehydrated and mounted with Permount. Images were acquired on a Zeiss Axioskop 2 microscope equipped with Axiovision software.
Statistical analyses
GraphPad Prism software (La Jolla, CA, USA) was used for statistical analysis. Significant outliers as identified with the Grubbs test were removed. Significance was determined by Student’s t test (for 2 group comparisons) or one-way ANOVA (for 3 or more group comparisons) Post-hoc testing was with either Dunnetts or Bonferroni analysis as indicated in figure legends. Differences between means were considered significant when p < 0.05.
Abbreviations
1,25D3: 1,25-dihydroxyvitamin D3; 4MU: 4-methylumbelliferone; bHABP: biotinylated HA binding protein; DAB: 3,3′-Diaminobenzidine; DON: 6-diazo-5-oxo-l-norleucine; ELISA: enzyme linked immunosorbent assay; EMT: epithelial mesenchymal transition; GISTIC: Genomic Identification of Significant Targets in Cancer algorithm; HA: hyaluronic acid; HABP: HA binding protein; HAS 1, 2, 3: Hyaluronan synthases; HMLE: immortalized human mammary epithelial cell line; HMW: high molecular weight; H-RASV12: oncogenic form of H-RAS; HYAL: hyaluronidase; KO: knockout; KO240: cell line derived from DMBA mammary tumor induced in VDRKO mice; KOhVDR: KO240 cell line engineered for stable expression of human VDR; LMW: low molecular weight; MW: molecular weight; RT-PCR: reverse transcriptase polymerase chain reaction; TNBC: triple negative breast cancer; VDR: vitamin D receptor; WT: wild type; WT145: cell line derived from DMBA mammary tumor induced in WT mice.
ACKNOWLEDGMENTS
The authors are grateful to Drs Weinberg and Mani for provision of HMLE model cell lines.
CONFLICTS OF INTEREST
Authors have no conflicts of interest to declare.
FUNDING
NIH RO1CA194500 to J Welsh.
References
1. Zinser GM, McEleney K, Welsh J. Characterization of mammary tumor cell lines from wild type and vitamin D3 receptor knockout mice. Mol Cell Endocrinol. 2003; 200:67–80. https://doi.org/10.1016/S0303-7207(02)00416-1. [PubMed].
2. Valrance ME, Brunet AH, Acosta A, Welsh J. Dissociation of growth arrest and CYP24 induction by VDR ligands in mammary tumor cells. J Cell Biochem. 2007; 101:1505–1519. https://doi.org/10.1002/jcb.21263. [PubMed].
3. Valrance ME, Brunet AH, Welsh J. Vitamin D receptor-dependent inhibition of mammary tumor growth by EB1089 and ultraviolet radiation in vivo. Endocrinology. 2007; 148:4887–4894. https://doi.org/10.1210/en.2007-0267. [PubMed].
4. LaPorta E, Welsh J. Modeling vitamin D actions in triple negative/basal-like breast cancer. J Steroid Biochem Mol Biol. 2014; 144:65–73. https://doi.org/10.1016/j.jsbmb.2013.10.022. [PubMed].
5. Keith ME, LaPorta E, Welsh J. Stable expression of human VDR in murine VDR-null cells recapitulates vitamin D mediated anti-cancer signaling. Mol Carcinog. 2014; 53:286–99. https://doi.org/10.1002/mc.21975. [PubMed].
6. Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, Shinomura T, Hamaguchi M, Yoshida Y, Ohnuki Y, Miyauchi S, Spicer AP, McDonald JA, Kimata K. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem. 1999; 274:25085–92. https://doi.org/10.1074/jbc.274.35.25085. [PubMed].
7. Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res. 2011; 317:383–391. https://doi.org/10.1016/j.yexcr.2010.11.017. [PubMed].
8. Saito T, Tamura D, Nakamura T, Makita Y, Ariyama H, Komiyama K, Yoshihara T, Asano R. 4-methylumbelliferone leads to growth arrest and apoptosis in canine mammary tumor cells. Oncol Rep. 2013; 29:335–342. https://doi.org/10.3892/or.2012.2100. [PubMed].
9. Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008; 18:288–295. https://doi.org/10.1016/j.semcancer.2008.03.005. [PubMed].
10. Boregowda RK, Appaiah HN, Siddaiah M, Kumarswamy SB, Sunila S, Thimmaiah KN, Mortha K, Toole B, Banerjee S. Expression of hyaluronan in human tumor progression. J Carcinog. 2006; 5:2. https://doi.org/10.1186/1477-3163-5-2. [PubMed].
11. Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, Hirvikoski P, Eskelinen M, Kosma VM. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol. 2000; 156:529–536. https://doi.org/10.1016/S0002-9440(10)64757-8. [PubMed].
12. Auvinen PK, Parkkinen JJ, Johansson RT, Agren UM, Tammi RH, Eskelinen MJ, Kosma VM. Expression of hyaluronan in benign and malignant breast lesions. Int J Cancer. 1997; 74:477–81. https://doi.org/10.1002/(SICI)1097-0215(19971021)74:5<477::AID-IJC1>3.0.CO;2-0. [PubMed].
13. Chokchaitaweesuk C, Kobayashi T, Izumikawa T, Itano N. Enhanced hexosamine metabolism drives metabolic and signaling networks involving hyaluronan production and O-GlcNAcylation to exacerbate breast cancer. Cell Death Dis. 2019; 10:803. https://doi.org/10.1038/s41419-019-2034-y. [PubMed].
14. Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003; 278:45801–45810. https://doi.org/10.1074/jbc.M308168200. [PubMed].
15. Bernert B, Porsch H, Heldin P. Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J Biol Chem. 2011; 286:42349–42359. https://doi.org/10.1074/jbc.M111.278598. [PubMed].
16. Porsch H, Bernert B, Mehic M, Theocharis AD, Heldin CH, Heldin P. Efficient TGFbeta-induced epithelial-mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene. 2013; 32:4355–65. https://doi.org/10.1038/onc.2012.475. [PubMed].
17. Udabage L, Brownlee GR, Nilsson SK, Brown TJ. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp Cell Res. 2005; 310:205–217. https://doi.org/10.1016/j.yexcr.2005.07.026. [PubMed].
18. Udabage L, Brownlee GR, Waltham M, Blick T, Walker EC, Heldin P, Nilsson SK, Thompson EW, Brown TJ. Antisense-mediated suppression of hyaluronan synthase 2 inhibits the tumorigenesis and progression of breast cancer. Cancer Res. 2005; 65:6139–6150. https://doi.org/10.1158/0008-5472.CAN-04-1622. [PubMed].
19. Koyama H, Hibi T, Isogai Z, Yoneda M, Fujimori M, Amano J, Kawakubo M, Kannagi R, Kimata K, Taniguchi S, Itano N. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: possible involvement of versican/PG-M. Am J Pathol. 2007; 170:1086–1099. https://doi.org/10.2353/ajpath.2007.060793. [PubMed].
20. Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, Xing F, Liu W, Pandey PR, Fukuda K, Modur V, Ghosh A, Wilber A, Watabe K. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res. 2012; 72:537–547. https://doi.org/10.1158/0008-5472.CAN-11-1678. [PubMed].
21. Li Y, Li L, Brown TJ, Heldin P. Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. Int J Cancer. 2007; 120:2557–2567. https://doi.org/10.1002/ijc.22550. [PubMed].
22. Misra S, Heldin P, Hascall VC, Karamanos NK, Skandalis SS, Markwald RR, Ghatak S. Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 2011; 278:1429–1443. https://doi.org/10.1111/j.1742-4658.2011.08071.x. [PubMed].
23. Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest. 2011; 121:1064–1074. https://doi.org/10.1172/JCI44540. [PubMed].
24. Afify A, Purnell P, Nguyen L. Role of CD44s and CD44v6 on human breast cancer cell adhesion, migration, and invasion. Exp Mol Pathol. 2009; 86:95–100. https://doi.org/10.1016/j.yexmp.2008.12.003. [PubMed].
25. Heldin P, Karousou E, Bernert B, Porsch H, Nishitsuka K, Skandalis SS. Importance of hyaluronan-CD44 interactions in inflammation and tumorigenesis. Connect Tissue Res. 2008; 49:215–218. https://doi.org/10.1080/03008200802143323. [PubMed].
26. Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011; 11:254–267. https://doi.org/10.1038/nrc3023. [PubMed].
27. Simpson MA. Hyaluronan Pericellular Matrix: Particle Exclusion Assay. Methods Mol Biol. 2019; 1952:103–110. https://doi.org/10.1007/978-1-4939-9133-4_9. [PubMed].
28. Nagy N, Kuipers HF, Frymoyer AR, Ishak HD, Bollyky JB, Wight TN, Bollyky PL. 4-methylumbelliferone treatment and hyaluronan inhibition as a therapeutic strategy in inflammation, autoimmunity, and cancer. Front Immunol. 2015; 6:123. https://doi.org/10.3389/fimmu.2015.00123. [PubMed].
29. Zinser GM, Welsh J. Effect of Vitamin D3 receptor ablation on murine mammary gland development and tumorigenesis. J Steroid Biochem Mol Biol. 2004; 89–90:433–6. https://doi.org/10.1016/j.jsbmb.2004.03.012. [PubMed].
30. Zinser GM, Suckow M, Welsh J. Vitamin D receptor (VDR) ablation alters carcinogen-induced tumorigenesis in mammary gland, epidermis and lymphoid tissues. J Steroid Biochem Mol Biol. 2005; 97:153–64. https://doi.org/10.1016/j.jsbmb.2005.06.024. [PubMed].
31. Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002; 23:2103–2109. https://doi.org/10.1093/carcin/23.12.2103. [PubMed].
32. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704–15. https://doi.org/10.1016/j.cell.2008.03.027. [PubMed].
33. Vigetti D, Deleonibus S, Moretto P, Bowen T, Fischer JW, Grandoch M, Oberhuber A, Love DC, Hanover JA, Cinquetti R, Karousou E, Viola M, D’Angelo ML, et al. Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J Biol Chem. 2014; 289:28816–28826. https://doi.org/10.1074/jbc.M114.597401. [PubMed].
34. Solomon C, Kremer R, White JH, Rhim JS. Vitamin D resistance in RAS-transformed keratinocytes: mechanism and reversal strategies. Radiat Res. 2001; 155:156–162. https://doi.org/10.1667/0033-7587(2001)155[0156:vdrirt]2.0.co;2. [PubMed].
35. Zhang Z, Kovalenko P, Cui M, Desmet M, Clinton SK, Fleet JC. Constitutive activation of the mitogen-activated protein kinase pathway impairs vitamin D signaling in human prostate epithelial cells. J Cell Physiol. 2010; 224:433–442. https://doi.org/10.1002/jcp.22139. [PubMed].
36. DeSmet ML, Fleet JC. Constitutively active RAS signaling reduces 1,25 dihydroxyvitamin D-mediated gene transcription in intestinal epithelial cells by reducing vitamin D receptor expression. J Steroid Biochem Mol Biol. 2017; 173:194–201. https://doi.org/10.1016/j.jsbmb.2017.01.008. [PubMed].
37. Herrera-Gayol A, Jothy S. Effects of hyaluronan on the invasive properties of human breast cancer cells in vitro. Int J Exp Pathol. 2001; 82:193–200. https://doi.org/10.1111/j.1365-2613.2001.iep196.x. [PubMed].
38. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
39. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7:11479. https://doi.org/10.1038/ncomms11479. [PubMed].
40. Slominski A, Janjetovic Z, Tuckey RC, Nguyen MN, Bhattacharya KG, Wang J, Li W, Jiao Y, Gu W, Brown M, Postlethwaite AE. 20S-hydroxyvitamin D3, noncalcemic product of CYP11A1 action on vitamin D3, exhibits potent antifibrogenic activity in vivo. J Clin Endocrinol Metab. 2013; 98:E298–E303. https://doi.org/10.1210/jc.2012-3074. [PubMed].
41. Koshiishi I, Mitani H, Sumita T, Imanari T. 1,25-Dihydroxyvitamin D(3) prevents the conversion of adipose tissue into fibrous tissue in skin exposed to chronic UV irradiation. Toxicol Appl Pharmacol. 2001; 173:99–104. https://doi.org/10.1006/taap.2001.9178. [PubMed].
42. Huang GR, Wei SJ, Huang YQ, Xing W, Wang LY, Liang LL. Mechanism of combined use of vitamin D and puerarin in anti-hepatic fibrosis by regulating the Wnt/beta-catenin signalling pathway. World J Gastroenterol. 2018; 24:4178–4185. https://doi.org/10.3748/wjg.v24.i36.4178. [PubMed].
43. Sabry D, Al-Ghussein MA, Hamdy G, Abul-Fotouh A, Motawi T, El Kazaz AY, Eldemery A, Shaker M. Effect of vitamin D therapy on interleukin-6, visfatin, and hyaluronic acid levels in chronic hepatitis C Egyptian patients. Ther Clin Risk Manag. 2015; 11:279–288. https://doi.org/10.2147/TCRM.S66763. [PubMed].
44. Slominski AT, Kim TK, Hobrath JV, Oak ASW, Tang EKY, Tieu EW, Li W, Tuckey RC, Jetten AM. Endogenously produced nonclassical vitamin D hydroxy-metabolites act as “biased” agonists on VDR and inverse agonists on RORalpha and RORgamma. J Steroid Biochem Mol Biol. 2017; 173:42–56. https://doi.org/10.1016/j.jsbmb.2016.09.024. [PubMed].
45. Wang TT, Tavera-Mendoza LE, Laperriere D, Libby E, MacLeod NB, Nagai Y, Bourdeau V, Konstorum A, Lallemant B, Zhang R, Mader S, White JH. Large-scale in silico and microarray-based identification of direct 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol. 2005; 19:2685–2695. https://doi.org/10.1210/me.2005-0106. [PubMed].
46. Kasiappan R, Shen Z, Tse AK, Jinwal U, Tang J, Lungchukiet P, Sun Y, Kruk P, Nicosia SV, Zhang X, Bai W. 1,25-Dihydroxyvitamin D3 suppresses telomerase expression and human cancer growth through microRNA-498. J Biol Chem. 2012; 287:41297–41309. https://doi.org/10.1074/jbc.M112.407189. [PubMed].
47. Caon I, Bartolini B, Parnigoni A, Carava E, Moretto P, Viola M, Karousou E, Vigetti D, Passi A. Revisiting the hallmarks of cancer: The role of hyaluronan. Semin Cancer Biol. 2020; 62:9–19. https://doi.org/10.1016/j.semcancer.2019.07.007. [PubMed].
48. Yao S, Ambrosone CB. Associations between vitamin D deficiency and risk of aggressive breast cancer in African-American women. J Steroid Biochem Mol Biol. 2013; 136:337–341. https://doi.org/10.1016/j.jsbmb.2012.09.010. [PubMed].
49. Peppone LJ, Rickles AS, Janelsins MC, Insalaco MR, Skinner KA. The association between breast cancer prognostic indicators and serum 25-OH vitamin D levels. Ann Surg Oncol. 2012; 19:2590–2599. https://doi.org/10.1245/s10434-012-2297-3. [PubMed].
50. Manson JE, Bassuk SS, Buring JE, and VITAL Research Group. Principal results of the VITamin D and OmegA-3 TriaL (VITAL) and updated meta-analyses of relevant vitamin D trials. J Steroid Biochem Mol Biol. 2020; 198:105522. https://doi.org/10.1016/j.jsbmb.2019.105522. [PubMed].
51. Kemmis CM, Welsh J. Mammary epithelial cell transformation is associated with deregulation of the vitamin D pathway. J Cell Biochem. 2008; 105:980–988. https://doi.org/10.1002/jcb.21896. [PubMed].
52. Zinser G, Packman K, Welsh J. Vitamin D(3) receptor ablation alters mammary gland morphogenesis. Development. 2002; 129:3067–3076. [PubMed].