
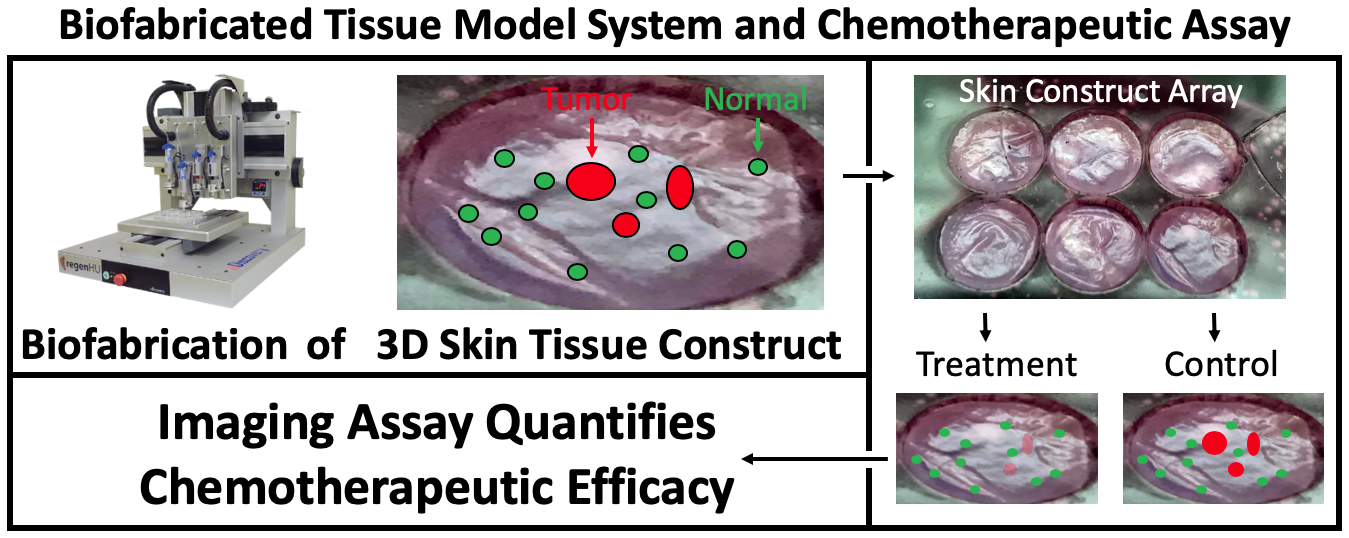
Introduction
Global incidence of cSCC is 2.2 million people [1, 2] and accounts for most of the ~10,000 annual non-melanoma skin cancer deaths in the United States [3, 4]. cSCC treatments include excision, radiation therapy, photodynamic therapy, and/or topical treatment, including 5-fluorouracil (5FU) [5–7]. Drug discovery for small molecule therapies to treat locally advanced/inoperable or metastatic cSCC and other cancers can be accelerated using patient-specific, physiologically relevant models amenable to high-throughput screening. Models should mimic the tumor microenvironment, given its influence on tumor progression and metastasis [8], and should reproduce in vivo tumor cell physiochemical signaling and mechanical cues from the surrounding tissue extracellular matrix [9]. Animal models may not be readily translatable to human cancer treatment [10], and three dimensional (3D) tissue culture models offer a viable alternative for pre-clinical screening of small molecule therapeutics. 3D models using human derived cell lines offer increased complexity and physiological fidelity compared with two dimensional (2D) monocultures [11] and have been developed for several cancer models, including melanoma, pancreatic cancer, and cervical cancer [12–15]. Recently a human glioblastoma-on-a-chip model was described as more accurately capturing the tumor microenvironment and predicting patient-specific therapeutic response, as compared to 2D monoculture [16]. 2D and spheroid-only systems are unusable for the prediction of changes at the epithelial mesenchymal transition unlike the 3D system which can reflect important interactions between SCC, therapeutic molecules, and a collagen matrix. 2D cultures and spheroid models will not demonstrate collagen invasion and/or degradation and the diffusion limited size of an SCC spheroid with central necrosis is not a factor in monolayer cultures. 3D systems more accurately model diffusion limits of nutrients causing potential micro-metastases that have not yet acquired a vascular supply. In addition, the use of human specific 3D models has the potential for scalability, standardization and adaptability to high-throughput assay techniques.
Engineered 3D skin tissue models produced using biofabrication techniques have been described [17, 18]. These tissues (Figure 1) are made with hydrogel scaffolding for dermal fibroblasts co-cultured with a surface layer of keratinocytes, and result in a 3D bi-layer model with structural similarities to human skin [19]. 3DBPS achieves precise architectures and better cellular placement than hydrogel models and can provide high-throughput, reproducible specimens [20]. It includes a 3D-printed fibroblast embedded collagen-based dermis and an epidermal layer of normal keratinocytes. The protocols for the 3-D printed fibroblast are also versatile and allow users, to readily introduce features of diseases. In the disease model presented here, A431 cSCC spheroids was introduced into the tissue, and histopathology and cDNA microarray analysis were used to confirm the biological fidelity of the cancer model.
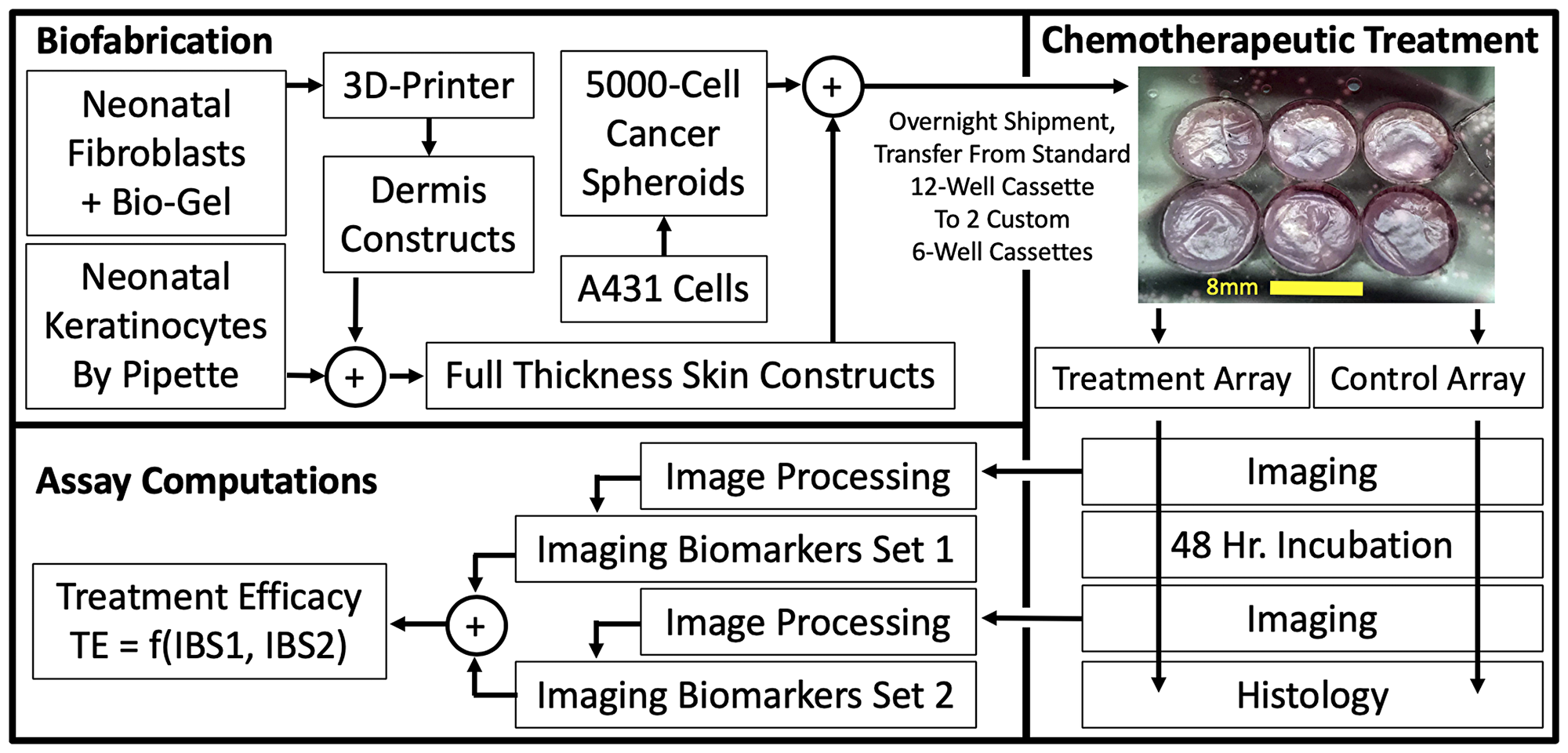
Figure 1: 3D, Biofabricated tissue model system and chemotherapeutic assay using multi-channel confocal microscopy imaging biomarkers. From aliquots of cryopreserved cells, full thickness skin samples were biofabricated that mimic normal human skin morphology in vivo. After 4 days of media submersion, cancer spheroids were introduced that displaced and invaded the normal skin constructs. Constructs were transferred to our custom transwell apparatus to form construct arrays. Array cassettes were compatible with both incubation and imaging so they were imaged, treated by addition of 1μM 5FU to their nutrient media and re-imaged after incubation.
Imaging biomarkers are quantifiable features of microscopy images that correlate with underlaying physiological processes of interest. The imaging/image processing assay computed imaging biomarkers (number of fluorescent cells and total amount of fluorescence) to define differential values before and after treatment that specified chemotherapeutic treatment efficacy, demonstrating different treatment effects on A431 cSCC cells versus normal (neonatal) keratinocytes.
Our objective was to quantify the therapeutic efficacy of a standard of care treatment for an cSCC skin tissue model that recapitulates the microenvironment in which this cancer grows. The 3D skin tissue cSCC assay was both scalable using current HT plate readers (i.e. fluorescence signal summation) and analytic on a cellular segmentation level, which may be more reliable due to optical turbidity in tissues [21]. Reflectance confocal microscopy (RCM) has been used for volumetric, in vivo imaging. In this protocol, multimodal confocal microscopy assayed our model system by tracking fluorescent cell populations, within reflective constructs, to quantify viability of normal and diseased tissue before and after chemotherapeutic treatment. Here we present a high-throughput-compatible, 3D-biofabricated cSCC skin tissue model system for efficacy testing of chemotherapeutics.
Results
A complete protocol for in vitro therapeutic screening was developed using 3DBPS cSCC construct with dermal and epidermal layers biofabricated in 12-well transwell plates using a modification of a previously described technique [18]. Tissues were fabricated with A431 cSCC cells transduced with tdTomato red fluorescent protein (tdT-RFP) and 1% of normal primary keratinocyte transduced ZsGreen green fluorescent protein (Zs-GFP) were spiked into the epidermis as an internal control for non-desired toxic effects of the treatments. RCM confirmed morphological similarities between human and biofabricated skin (Figure 2).
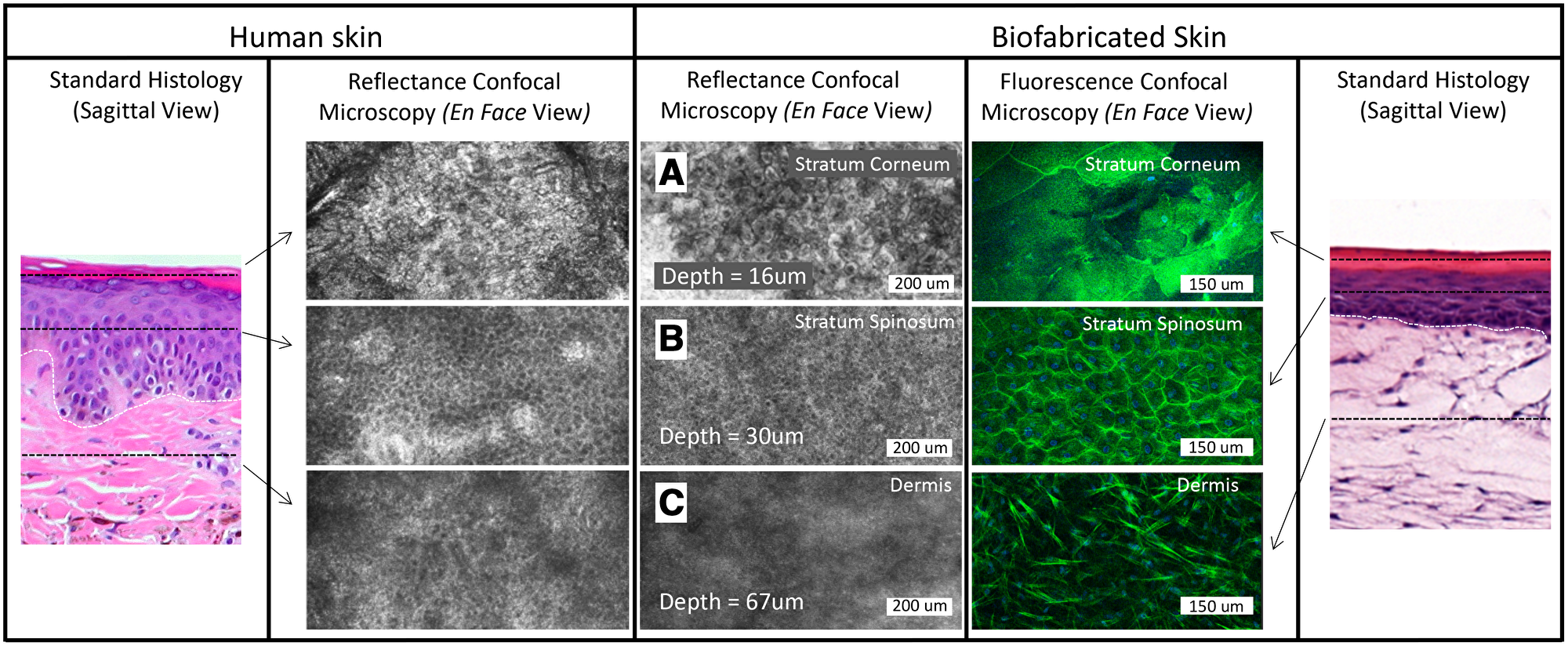
Figure 2: Stratum corneum (A), stratum spinosum (B), and dermis (C) of human skin and biofabricated skin at different depth layers in en face, confocal reflectance and fluorescence microscopy. The dermal-epidermal junction is shown as a dotted white line in the standard histology of skin and biofabricated skin images.
En face RCM of a typical skin model biofabricated during this study showed a stratified model with well-formed stratum corneum, stratum spinosum, and dermis (Figure 2A–2C). The superficial stratified layer is 5–10 μm thick, with reflective cells under RCM. This is followed by a ~50 μm-thick layer of repeating honeycomb patterned granular dark nuclei cells resembling the stratum spinosum of human skin [18, 24]. RCM of the dermis is similar to that of human papillary dermis, but lacks the papillary network [24]. Fluorescence confocal microscopy (1.08 μm lateral resolution, 1.52–5.33 μm slice separation) confirmed integration of the A431 cells into the tissues, which produced a disruption of epidermal stratification in an otherwise well-differentiated epidermal layer (Figure 3A, 3B), by visualizing tdT-RFP while Zs-GFP labeled 1% of the keratinocytes were spiked into the epidermal layer (Figure 3C).
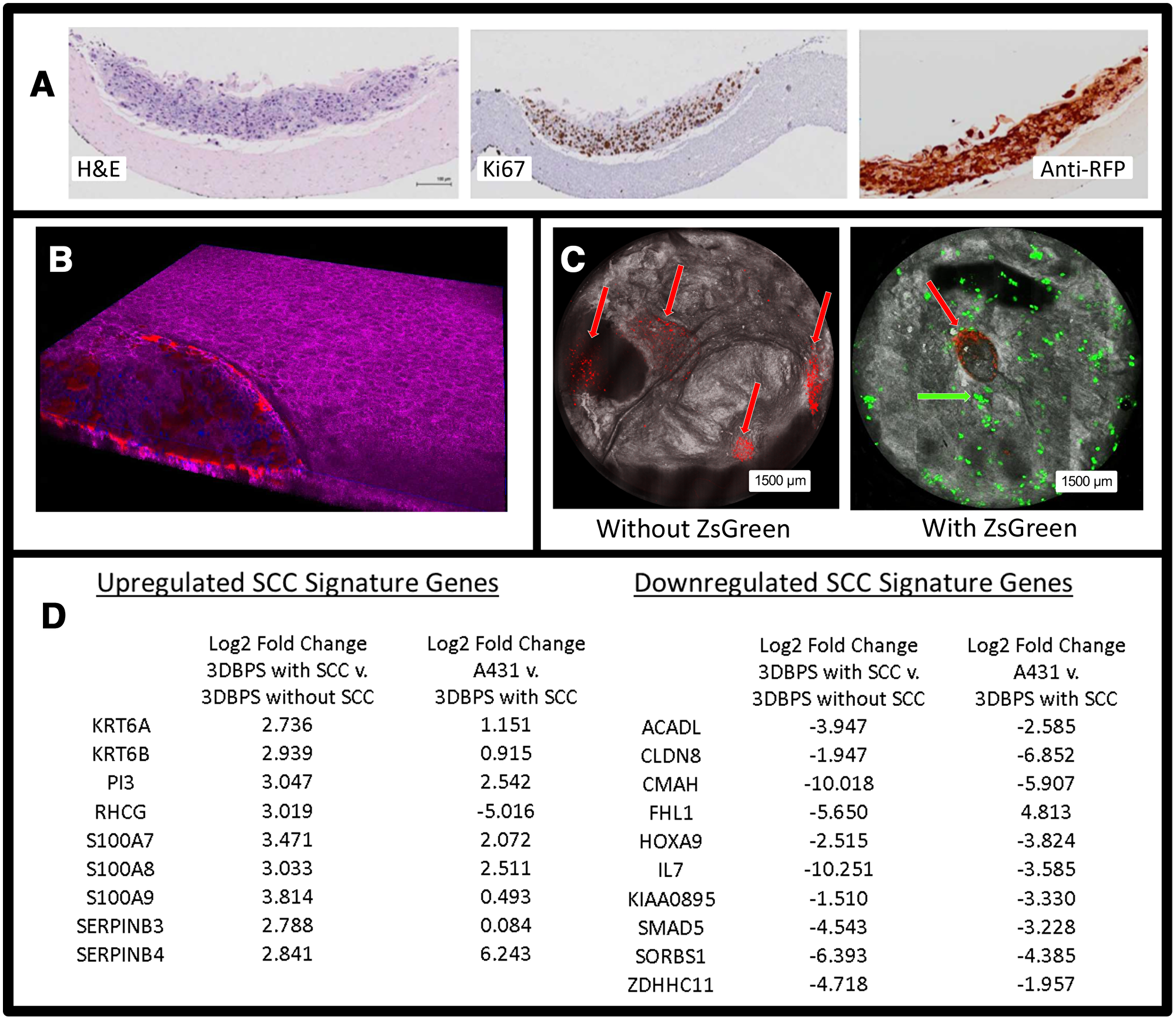
Figure 3: (A) H&E, Ki67, and Anti-RFP stained histology of 3DBPS model. (B) 3D confocal image of skin tissue with SCC tumor (purple: phalloidin staining, red: tdTomato red fluorescent protein (RFP) labeled SCC, blue: Hoescht staining). (C) Bimodal en face confocal microscopy of tdT-RFP-labeled cSCC (red arrows) in 3DBPS constructs with and without Zs-GFP-labeled neonatal keratinocytes (green arrow). All cSCC A431 cells are labeled with RFP while only 1% of keratinocytes are labeled with Zs-GFP resulting in sparsity of the green signal. (D) SCC signature genes that are up- and down-regulated in A431 cells grown in vitro and untreated full thickness 3DBPS without SCC spheroids compared to 3DBPS with SCC spheroids. Log2 fold change is normalized to Keratin 10.
Both Zs-GFP and tdT-RFP images were segmented, enabling automatic, volumetric cell counting. We formulated the first imaging biomarker as the number of cells in the 3D tissue. Our second imaging biomarker was the total Zs-GFP fluorescence signal within each construct and total tdT-RFP florescence signal within each tumor spheroid. RNA-Seq analysis on 3DBPS-SCC (A431) vs. 3DBPS alone and 3DBPS-SCC (A431) vs. A431 assessed model integrity at the level of gene expression, showing increased expression in 3DBPS-SCC which corresponds to our previous finding of increased expression at the leading edge of invasive human cSCC in vivo [25]. These included S100A7, S100A8, S100A9, KRT6A, SERPINB3, SERPINB4, and PI3. Gene expression was normalized to keratin 10 for increased specificity to cSCC vs. increased numbers of activated keratinocytes. Downregulated genes included IL-7 which was also downregulated at the leading edge of invasive cSCC in vivo (Figure 3D). We observed that tumor cells proliferated into the surrounding epidermis prior to 5FU treatment and formed a crater in the epidermis. To quantify therapeutic effect, we calculated a ratio of tumor fluorescence signal at the end of the treatment period between wells with mock treatment and those in which the drug was added. In 5FU-treated tumors, tdT-RFP fluorescence was reduced after treatment, especially in some tumor cores (Figure 4A). Ki67 staining indicated greater cSCC proliferation in controls compared to treated samples, which had fewer Ki67+ nuclei. Anti-RFP immunohistochemistry showed thicker untreated tumors with ~10 layers of cells compared to ~5 layers for treated tumors (Figure 4B).
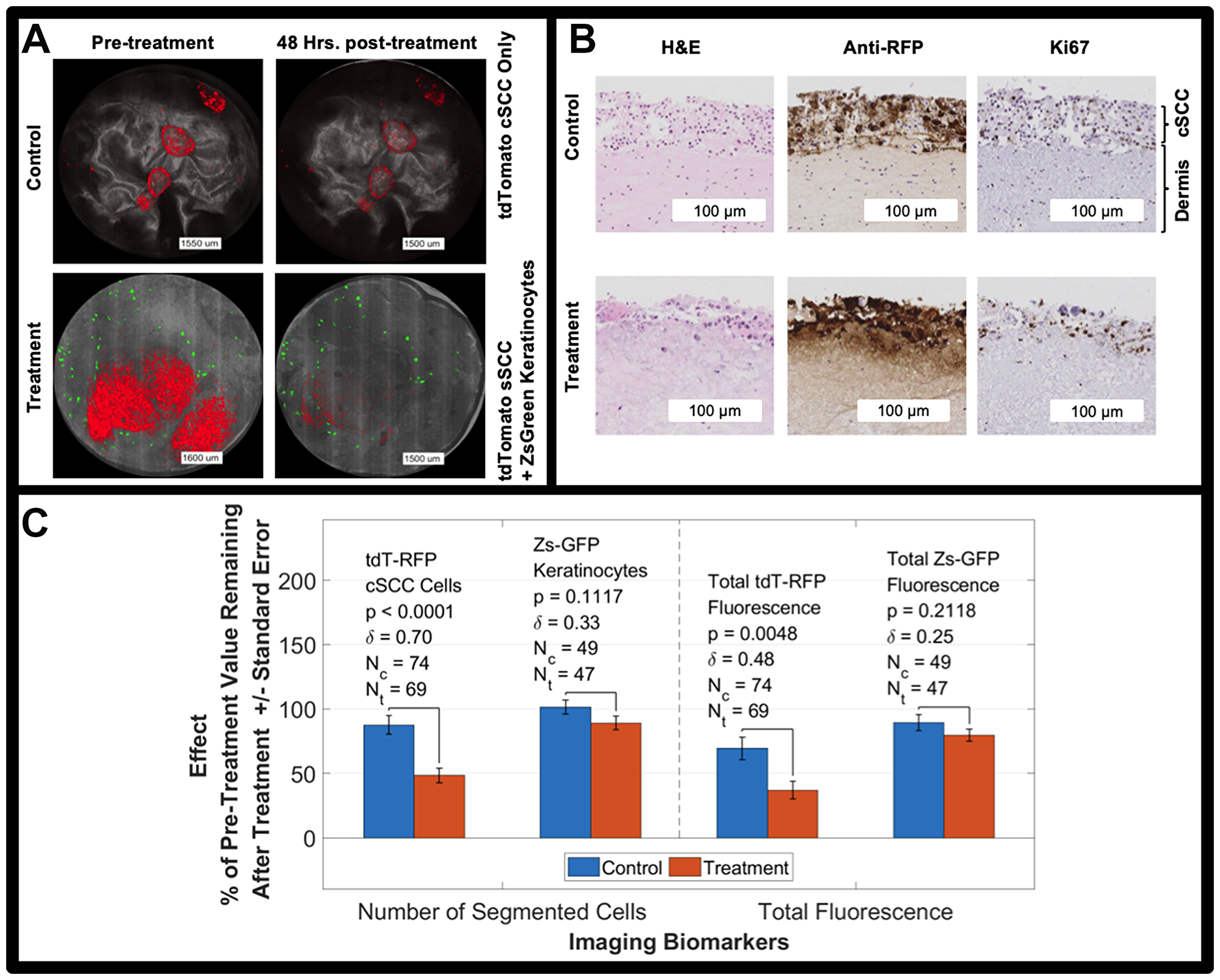
Figure 4: (A) Bimodal imaging examples of control and treated tumors (red) before and after the treatment period. cSCC tumors are red, reflectance is grayscale, and keratinocytes are green. All cSCC A431 cells are labeled with RFP while only 1% of keratinocytes are labeled with Zs-GFP resulting in sparsity of the green signal. (B) H&E, Ki67, and Anti-RFP antibody staining of untreated (control) tumor model and tumor model treated with 5FU for 48 hours. (C) 5FU treatment effect on tdT-RFP-labeled cSCC spheroids and Zs-GFP-labeled keratinocytes including p-value and effect size, δ. Two imaging biomarkers are shown, number of segmented objects and total fluorescence of the size-thresholded segmented objects. NOTE: the fluorescence in panel A is intentionally saturated for display.
We counted cells by fluorescence segmentation as our primary imaging biomarker before and after the 5FU treatment period [1] for samples fed by 5FU-treated media versus control samples fed by pure media and [2] for tdT-RFP cSCC cells and Zs-GFP using different fluorescence imaging channels. Table 1 shows our results. A two-way ANOVA for unbalanced design with cell type (A431 cSCC vs. keratinocytes) and treatment (untreated vs. treated) as factors resulted in a cell type-treatment interaction p-value of 0.039 indicating that 5FU differentially changed outcome for cSCC versus keratinocytes. A dose-response study showed increased reduction in the number of segmented cells for a 2 μM 5FU treatment compared to untreated controls versus a 1 μM treatment compared to untreated controls: 71.8 ± 47.2%, n = 15; 52.0 ± 41.3%, n = 18; and 34.7±36.1%, n = 20, respectively (Supplementary Figure 1).
Table 1: Imaging biomarker values: number of cells and total fluorescence as a post treatment percentage of their pre-treatment values
| Control Group | Treatment Group | |||||
|---|---|---|---|---|---|---|
| Mean±SD | N | Mean ± SD2 | N 3 | P-value | Hedges g | |
| %tdT-RFP cells | 87 ± 62 | 74 | 48 ± 46 | 69 | 4.29E-05 | 0.703 |
| %Zs-GFP cells | 101 ± 38 | 49 | 89 ± 36 | 47 | 0.1117 | 0.3251 |
| %Total tDT-RFP | 69 ± 75 | 74 | 37 ± 57 | 69 | 0.0048 | 0.4773 |
| %Total Zs-GFP | 89 ± 42 | 49 | 79 ± 31 | 47 | 0.2118 | 0.2546 |
DISCUSSION
The present studies are remarkable for the following: (1) we developed and validated a morphologically and genomically accurate 3D skin cSCC tissue model that enables study of the effect of chemotherapeutics on cancer cells growing in the context of their native tissue microenvironment; (2) we developed a nondestructive, 3D fluorescence confocal imaging assay to test both efficacy and general toxicity of anti-cancer chemotherapeutics; (3) we derived imaging biomarkers to quantify therapeutic effect in our system; and (4) we pharmacologically validated our assay by demonstrating different effects of 5-flurouracil on A431 SCC cells versus normal keratinocytes in the 3D model system.
Thereare currently several approaches to test the cytotoxic effects ofchemotherapeutics on cancer cells in vitro, from 2D monolayer systems to patient-derived organoids. Our novel3DBPS cSCC model provides several advantages over current methods: (1) it provides a relevant tissue context to the growth of cSCC cancer cells; (2) it enables non-destructive cellular monitoring during drug therapy on cSCC growth within skin tissue; (3) it recapitulates native cSCC biology, exhibiting similar histology and gene expression to invasive cSCC in vivo; and (4) it can be adapted as a HTS platform for evaluation of small molecule therapy intreatment of locally advanced, inoperable or metastatic cSCC. The 3DBPS model has been validated on morphological, molecular, and functional bases through investigation of pathology and confocal imaging, gene expression, and response to treatment respectively.
The model performed exceptionally well on the basis of morphology as determined by light and confocal microscopy. H&E evaluation of 3DBPS demonstrated a collagen-rich dermis with proliferative fibroblasts and a confluent layer of basal keratinocytes. Although SCC was seeded as spheroids within the 3DBPS, it exhibited vertical and radial growth. H&E, Ki67, and anti-RFP staining of treated and untreated 3DBPS cSCC skin models illustrated pharmacological effects after 48-hour 1 μM 5FU treatment, including decreased cSCC cells, and more compact architecture of tumor cells in the treatment group. Bimodal confocal visualization showed a proliferating perimeter of cSCC cells, often surrounding a non-fluorescent tumor core visualized by RCM. This suggested central necrosis. Prior to treatment, cSCC spheroids proliferated into and disrupted the normal epidermis. cSCC spheroids typically caused craters in the epidermis and extended into the dermis.
En face confocal imaging showed that 3DBPS resembled human skin, with three layers: stratum corneum, stratum spinosum, and dermis. The stratum spinosum of the 3DBPS showed the dark nuclei honeycomb pattern of keratinocytes which transitioned into larger, flatter, bright nuclei cells in the stratum corneum, indicating differentiation of a stratified epithelium consistent with human skin and high-level structural fidelity of the model.
We evaluated the model at the level of gene expression using RNA-Seq analysis, demonstrating increased expression of genes upregulated in cSCC in vivo, including keratin self-signaling genes. We identified eight genes that showed increased expression in spheroids that overlapped with genes we isolated at the leading edge of cSCC including S100A7, S100A8, S100A9, KRT6A, SERPINB3, SERPINB4, PI3, and RHCG. Of note, SERPINB3 (SCC antigen) was highly expressed in 3DBPS cSCC spheroids while showing low expression for 3DBPS without cSCC. SERPINB3 expression by 3DBPS with cSCC approached levels observed in A431 cells, supporting fidelity of our model at the gene expression level. SERPINB3 is highly expressed in other hyper-proliferative skin disorders, particularly psoriasis [26]. S100A7, which was expressed highly by 3DBPS with cSCC, is known to promote migration, invasion, and metastasis of cervical cancer and is highly expressed in human cSCC [25, 26]. S100A8/A9 form a calcium binding heterodimer that plays a role in leukocyte recruitment and myeloid cell function. KRT6A is highly expressed in aggressive SCC of the lung and leading edge of cSCC invadopodia [25, 27]. SERPINB3 (cSCC antigen) and B4 are highly expressed in cSCC and benign hyperproliferation (i. e., psoriasis). Rh family glycoproteins are highly expressed in SCC of the head and neck (SCCHN), are poor prognostic indicators for lung cancer [28]. Expression of these genes by A431 spheroids in the 3DBPS cSCC model demonstrates consistency with gene expression observed at the leading edge of invasive cSCC. Downregulation of IL-7 is consistent with blunting of immune response, particularly T cell immunity at leading edge of invasive carcinoma.
Our assay and model system were evaluated using imaging biomarkers. We evaluated functional performance of the model via total tumor fluorescence and number of cells, where the total fluorescence was quantified as the sum of the fluorescent intensity over the entire 3D confocal fluorescence image of the construct. We saw the same trend for both imaging biomarkers: quantified values decreased with treatment when computed using the red fluorescence from the tumor cells and did not change the Zs-GFP keratinocytes in a statistically significant way.
2-way ANOVA showed that that 5FU differentially changed the outcome for cSCC versus keratinocytes indicating selective killing of cSCC cells. The response to 5FU was 50% decrease in tumor cells post-treatment compared to <10% for untreated controls. Zs-GFP normal keratinocytes increased in untreated control samples and decreased in treated samples, although this trend wasn’t significant (p = 0.0553). Increasing dose of 5FU for a smaller set of samples (Ncontrol = 15, N1× dose = 18, N2 × dose = 20) showed an expected increase in treatment effect. This novel 3DBPS cSCC model can be used for non-destructive efficacy assessment of candidate therapies, more closely recapitulates invasive cSCC biology than current methods, and can be adapted for HTS of small molecule therapy for cSCC.
The model described provides a higher degree of clinical relevance because it enables the testing of chemotherapeutics against tumor cell growth in a tissue specific context, thus capturing any potential interactions between the tumor and its microenvironment. We envision that this model could be adopted in a “bedside” manner and applied to cells from cSCC patient tumor biopsies.
Materials and Methods
Cell culture
Neonatal human dermal fibroblasts (HDFN, Zen Bio DFN-F) were cultured at 37° C, 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Gibco 11965), 10% fetal bovine serum (Hyclone), and 1% penicillin-streptomycin (Gibco). Neonatal keratinocytes (NHEKN, ScienCell 2100) were cultured at 37° C, 5% CO2 in Keratinocyte Media (Lonza 192060). SCC A431cells (ATCC® CRL-1555) were cultured at 37° C, 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Gibco 11965), 10% fetal bovine serum (Hyclone), and 1% penicillin-streptomycin (Gibco). NHEKN and A431 were transduced with Zs-GFP and tdT-RFP lentiviral vectors (Vectalys 0010VCT, 0008VCT), respectively, following the vendor provided protocol (MOI = 10, 4 μg/mL polybrene, 4-hour incubation at 37° C).
Biofabricated cSCC skin models
Skin models were biofabricated in 12-well transwell plates as described previously [18] with the only modification being that the basement membrane layer (Laminin/Entactin solution) and the NHEKN cell suspension were applied by hand pipetting. Zs-GFP-expressing NHEKN were mixed homogenously with non-fluorescent NHEKN at 1% prior to application of the cell suspension. After 1.5-hour room temperature incubation, tissues were submerged and incubated at 37° C. On day three after tissues were printed, tdT-RFP-expressing A431 cSCC cells were pipetted at 5000 cells/well in 50 μL into a 384-well low-attachment plate (Nexcelom ULA-3840-020) to promote spheroid formation. On day four, A431 spheroids were pipetted onto the top surface of tissue (five spheroids per tissue). Tissues were subsequently incubated for three more days submerged and seven days with the air-liquid interface as described [18].
Histology
3DBPS samples were bisected and fixed in 10% neutral formalin for 72 hours, transferred to 70% ethanol and processed to paraffin. Scaffolds were embedded with the bisected surface down, collected at 4μm on Plus slides (Fisher Scientific, Cat # 22-042-924) and stored at room temperature prior to use. One section was stained with standard H&E. Chromogenic immunohistochemistry was performed on a Ventana Medical Systems Discovery XT instrument with online deparaffinization using Ventana’s reagents and detection kits unless otherwise noted (Ventana-Roche Diagnostics, Indianapolis IN, USA). tdTomato-expressing cells were detected with polyclonal rabbit anti-RFP (Rockland Cat# 600-401-379 Lot# 35055 RRID: AB_2335885) raised against a fusion protein corresponding to the full-length amino acid sequence from mushroom coral Discosoma. Proliferating cells were detected with rabbit anti-mouse Ki67, clone SP6 (Spring Biosciences Cat# M3062 Lot# 160726LVS RRID: AB_11219741).
Sections were incubated for 1 hour at 60° C followed by online deparaffinization. tdTomato was antigen-retrieved using protease-3 (Ventana Medical Systems) for 12 minutes. Ki67 was heat retrieved for 36 minutes in Ventana Cell Conditioner 1 (Tris-Borate-EDTA pH 8.5). Endogenous peroxidase activity was blocked with hydrogen peroxide. tdTomato was diluted 1:1600 in Tris-BSA (25 mM Tris, 15 mM NaCL, 1% BSA, pH 7.2) and incubated for 12 hours at room temperature. Ki67 was diluted 1:400 in Ventana Antibody Dilution Buffer (Catalog# 251-018) and incubated for 1 hour at 37° C. tdTomato was detected with biotinylated, goat anti-rabbit (Vector Laboratories Cat#Ba-1000 Lot#ZA0324 RRID: AB_2313606) diluted 1:200 in Tris-BSA and incubated for 30 minutes at 37° C. This was followed by the application of streptavidin-horseradish-peroxidase conjugate. Ki67 was detected with goat anti-rabbit horseradish peroxidase conjugated multimer and incubated for eight minutes. Both antibodies were visualized with 3,3′-diaminobenzidene and enhanced with copper sulfate. Slides were washed in distilled water followed by counterstaining with hematoxylin, dehydrated, and mounted with permanent media. Appropriate positive and negative controls were run with study samples. These procedures were performed by the NYU Center for Biospecimen Research and Development (CBRD) core facility.
cDNA gene profiling
RNA was prepared from samples using RNeasy Mini Kit (Qiagen). RNA quality was assessed on the Agilent Bioanalyzer 2100 platform (Agilent, Santa Clara, CA). Libraries were prepared with the TruSeq RNA Sample Preparation Kit (Illumina). Sequencing (RNA-Seq) was done with the Illumina Next-Gen Sequencing HiSeq (Illumina, San Diego, CA, USA) with 30–45 million 50-bp, paired-end reads at the NYU Genome Technology Core. RNA-Seq raw reads were normalized using STAR software. Differential expression data were obtained using the DEseq algorithm. Analyses were completed with the Basepair (New York, NY) software.
Sample preparation
Samples were transferred (12 samples in a 12-well cassette, Product 3460, Corning Life Sciences, Corning NY, USA) via overnight courier with a cool pack in a polystyrene foam container from the National Center for Advancing Translational Sciences site to The Rockefeller University for processing, treatment, and imaging. Samples were separated into control and treatment groups and visually examined to guide an 8-mm punch biopsy to the area appearing to contain the most uniform epidermis and highest number of tumors, which typically occupied the central half of the 12-mm diameter. A fresh biopsy punch was used every three samples. The 8mm punches shrunk slightly so we LASER-cut circular wells (7.60-mm diameter = 7.50 mm cutting pattern +0.10 mm dilation due to cutting line width) in a circular slab of 1.02-mm thick acrylic (Product ACRYCLR0.040PM12 × 36, ePlastics, San Diego, CA, USA) to insert into a 75-mm transwell (Product 3419, Corning Life Sciences, Corning, NY, USA) to form wells (Figure 1). Supplementary Figure 2 demonstrates the optical coupling to submerge samples just beneath cover glass in the LASER-cut well structure for a 3-mm construct version that we test prepped to investigate HT compatibility in the 384-well format. Both the 8-mm and 3-mm custom well structures allowed sample preparation that is compatible with printing, incubation and imaging. Ki67 histological analysis showed that untreated cSCC cells from control samples were proliferative after two imaging sessions.
Confocal imaging
Reflectance and fluorescence confocal 3D images were collected under 488nm and 532 nm LASER excitation for a total of 4 imaging modes using a 10× air-immersion lens (RS-G4, Caliber ID Inc., Rochester NY, USA retrofitted with 488- and 532-nm LASERs from Toptica Photonics Inc., Farmington NY, USA). Lateral pixel resolution was 1.1 μm, and stack spacing ranged from 3.1 to 5.3 μm. Confocal images (~500GB) were acquired for 2 × 3 arrays of 6 specimens (e. g., Figure 1) (24 mm × 16 mm × 0.5 mm). Imaging and analysis were performed on seventeen sequential sample batches, four of which were excluded based on the criteria that in these batches, an incorrect optical filter setting caused image bleed-through between the fluorescence channels of the microscope, misregistering keratinocytes as A431 cSCC cells.
Image analysis
SCC cells (red fluorescence) and keratinocytes (green fluorescence) were overlaid in semi-opaque masks on grayscale RCM. Opacity of masks was linearly proportional to signal, and signal was multiplied by a constant for the purposes of visualization. Because tdT-RFP and Zs-GFP images were each acquired simultaneously with a corresponding RCM stack, the two fluorescent channels were spatially registered for rotation and translation using their respective z-sum RCM images and the function imregtform in MATLAB (MATLAB 2018b, The Mathworks, Natick, MA, USA). 3D fluorescent image stacks were segmented using custom software written in MATLAB (Supplementary Figure 3). Fluorescence signal for each 3D voxel was normalized for LASER power and detector (photomultiplier) gain to a standard image. LASER and detector settings were consistent between pre- and post-images for each sample but varied between sample batches.
Segmentation used a Gaussian smoothing filter with a kernel size of 8, binarization used Otsu’s method [22] and removal of connected components containing 4 or fewer voxels with 26-connectivity (i. e., adjacent corners, edges, and faces connect voxels). Image smoothing and small object elimination reduced background noise in images. Imaging biomarkers were calculated for entire samples in the case of Zs-GFP-labeled keratinocytes and for individual tumors for tdTomato-labeled SCC. The differences in pre- and post-imaging biomarkers between treated samples and untreated controls were compared using an unpaired two-sample t-test. Hedges’ g was calculated as a measure of effect size, δ, using the open source Measure of Effect Size Toolbox [23]. Treatment effect for keratinocytes and cSCC tumors was calculated as the post treatment percent of pre-treatment values for the imaging biomarkers (Table 1). Two-way ANOVA for unbalanced design with cell type (A431 cSCC vs. keratinocytes) and treatment (untreated vs. treated) as factors used the anovan function in MATLAB.
5-Fluorouracil treatment
The control and treatment groups were each seated in separate transwells, wherein 8 mL of media fed the constructs over the 48-hour treatment course. The control group contained pure media, while the treatment group contained media with 1 μM 5FU (Product F6627, Sigma Aldrich, St Louis, MO, USA). For three batches of samples, an additional treatment group was seated in a transwell assembly containing a double (2 μM 5FU) dose.
Abbreviations
cSCC: Cutaneous Squamous Cell Carcinoma; 3DBPS: 3 Dimensional Bio-Printed Skin; H&E: Hematoxylin and Eosin; 5FU: 5-Fluorouracil; Zs-GFP: Zs Green Fluorescent Protein; tdT-RFP: tdTomato Red fluorescent Protein; RCM: Reflectance Confocal Microscopy; HTS: High Throughput Screening.
ACKNOWLEDGMENTS AND FUNDING
We wish to acknowledge the support of a Cooperative Agreement 1U18TR002312 from the National Center for Advancing Translational Sciences and The Robertson Therapeutics Development Fund.
We wish to also acknowledge The Rockefeller University Laboratory of Investigative Dermatology and Center for Clinical and Translational Sciences is supported by National Institutes of Health. Grant Number: UL1TR001866.
The New York University Center for Biospecimen Research and Development Laboratory is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant NIH /NCI P30CA016087 and the National Institutes of Health S10 Grants NIH/ORIP, S10OD01058, and S10OD018338.
National Center for Advancing Translational Sciences was supported by the NIH Intramural Research Program and the Cures Acceleration Network program.
CONFLICTS OF INTEREST
The authors state no conflicts of interest.
References
1. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1545–602. https://doi.org/10.1016/S0140-6736(16)31678-6. [PubMed].
2. Stratigos A, Garbe C, Lebbe C, Malvehy J, del Marmol V, Pehamberger H, Peris K, Becker JC, Zalaudek I, Saiag P, Middleton MR, Bastholt L, Testori A, Grob JJ, and European Dermatology Forum (EDF), and European Association of Dermato-Oncology (EADO), and European Organization for Research and Treatment of Cancer (EORTC). Diagnosis and treatment of invasive squamous cell carcinoma of the skin: european consensus-based interdisciplinary guideline. Eur J Cancer. 2015; 51:1989–2007. https://doi.org/10.1016/j.ejca.2015.06.110. [PubMed].
3. Que SK, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018; 78:237–47. https://doi.org/10.1016/j.jaad.2017.08.059. [PubMed].
4. Weinberg AS, Ogle CA, Shim EK. Metastatic cutaneous squamous cell carcinoma: an update. Dermatol Surg. 2007; 33:885–99. https://doi.org/10.1097/00042728-200708000-00001. [PubMed].
5. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006; 354:567–78. https://doi.org/10.1056/NEJMoa053422. [PubMed].
6. Kennedy JC, Pottier RH, Pross DC. Photodynamic therapy with endogenous protoporphyrin IX: basic principles and present clinical experience. J Photochem Photobiol B. 1990; 6:143–48. https://doi.org/10.1016/1011-1344(90)85083-9. [PubMed].
7. Sadek H, Azli N, Wendling JL, Cvitkovic E, Rahal M, Mamelle G, Guillaume JC, Armand JP, Avril MF. Treatment of advanced squamous cell carcinoma of the skin with cisplatin, 5-fluorouracil, and bleomycin. Cancer. 1990; 66:1692–96. https://doi.org/10.1002/1097-0142(19901015)66:8<1692::AID-CNCR2820660807>3.0.CO;2-Y. [PubMed].
8. Clark AG, Vignjevic DM. Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol. 2015; 36:13–22. https://doi.org/10.1016/j.ceb.2015.06.004. [PubMed].
9. Spill F, Reynolds DS, Kamm RD, Zaman MH. Impact of the physical microenvironment on tumor progression and metastasis. Curr Opin Biotechnol. 2016; 40:41–48. https://doi.org/10.1016/j.copbio.2016.02.007. [PubMed].
10. Mak IW, Evaniew N, Ghert M. Lost in translation: animal models and clinical trials in cancer treatment. Am J Transl Res. 2014; 6:114–18. [PubMed].
11. Stock K, Estrada MF, Vidic S, Gjerde K, Rudisch A, Santo VE, Barbier M, Blom S, Arundkar SC, Selvam I, Osswald A, Stein Y, Gruenewald S, et al. Capturing tumor complexity in vitro: comparative analysis of 2D and 3D tumor models for drug discovery. Sci Rep. 2016; 6:28951. https://doi.org/10.1038/srep28951. [PubMed].
12. Müller I, Kulms D. A 3D Organotypic Melanoma Spheroid Skin Model. J Vis Exp. 2018; e57500. https://doi.org/10.3791/57500. [PubMed].
13. Bourland J, Fradette J, Auger FA. Tissue-engineered 3D melanoma model with blood and lymphatic capillaries for drug development. Sci Rep. 2018; 8:13191. https://doi.org/10.1038/s41598-018-31502-6. [PubMed].
14. Zhao Y, Yao R, Ouyang L, Ding H, Zhang T, Zhang K, Cheng S, Sun W. Three-dimensional printing of Hela cells for cervical tumor model in vitro. Biofabrication. 2014; 6:035001. https://doi.org/10.1088/1758-5082/6/3/035001. [PubMed].
15. Puls TJ, Tan X, Husain M, Whittington CF, Fishel ML, Voytik-Harbin SL. Development of a Novel 3D Tumor-tissue Invasion Model for High-throughput, High-content Phenotypic Drug Screening. Sci Rep. 2018; 8:13039. https://doi.org/10.1038/s41598-018-31138-6. [PubMed].
16. Yi HG, Jeong YH, Kim Y, Choi YJ, Moon HE, Park SH, Kang KS, Bae M, Jang J, Youn H, Paek SH, Cho DW. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat Biomed Eng. 2019; 3:509–19. https://doi.org/10.1038/s41551-019-0363-x. [PubMed].
17. Klicks J, von Molitor E, Ertongur-Fauth T, Rudolf R, Hafner M. In vitro skin three-dimensional models and their applications. J Cell Biotechnol. 2017; 3:21–39. https://doi.org/10.3233/JCB-179004.
18. Derr K, Zou J, Luo K, Song MJ, Sittampalam GS, Zhou C, Michael S, Ferrer M, Derr P. Fully Three-Dimensional Bioprinted Skin Equivalent Constructs with Validated Morphology and Barrier Function. Tissue Eng Part C Methods. 2019; 25:334–43. https://doi.org/10.1089/ten.TEC.2018.0318. [PubMed].
19. Bell E, Ehrlich HP, Buttle DJ, Nakatsuji T. Living tissue formed in vitro and accepted as skin-equivalent tissue of full thickness. Science. 1981; 211:1052–54. https://doi.org/10.1126/science.7008197. [PubMed].
20. Lee V, Singh G, Trasatti JP, Bjornsson C, Xu X, Tran TN, Yoo SS, Dai G, Karande P. Design and fabrication of human skin by three-dimensional bioprinting. Tissue Eng Part C Methods. 2014; 20:473–84. https://doi.org/10.1089/ten.tec.2013.0335. [PubMed].
21. Noller CM, Boulina M, McNamara G, Szeto A, McCabe PM, Mendez AJ. A Practical Approach to Quantitative Processing and Analysis of Small Biological Structures by Fluorescent Imaging. J Biomol Tech. 2016; 27:90–97. https://doi.org/10.7171/jbt.16-2703-001. [PubMed].
22. Otsu N. A Threshold Selection Method from Gray-Level Histograms. IEEE Trans Syst Man Cybern. 1979; 9:62–66. https://doi.org/10.1109/TSMC.1979.4310076.
23. Hentschke H, Stüttgen MC. Computation of measures of effect size for neuroscience data sets. Eur J Neurosci. 2011; 34:1887–94. https://doi.org/10.1111/j.1460-9568.2011.07902.x. [PubMed].
24. Gareau D. Automated identification of epidermal keratinocytes in reflectance confocal microscopy. J Biomed Opt. 2011; 16:030502. https://doi.org/10.1117/1.3552639. [PubMed].
25. Mitsui H, Suárez-Fariñas M, Gulati N, Shah KR, Cannizzaro MV, Coats I, Felsen D, Krueger JG, Carucci JA. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J Invest Dermatol. 2014; 134:1418–27. https://doi.org/10.1038/jid.2013.494. [PubMed].
26. Haider AS, Peters SB, Kaporis H, Cardinale I, Fei J, Ott J, Blumenberg M, Bowcock AM, Krueger JG, Carucci JA. Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia. J Invest Dermatol. 2006; 126:869–81. https://doi.org/10.1038/sj.jid.5700157. [PubMed].
27. Chang HH, Dreyfuss JM, Ramoni MF. A transcriptional network signature characterizes lung cancer subtypes. Cancer. 2011; 117:353–60. https://doi.org/10.1002/cncr.25592. [PubMed].
28. Han KH, Mekala K, Babida V, Kim HY, Handlogten ME, Verlander JW, Weiner ID. Expression of the gas-transporting proteins, Rh B glycoprotein and Rh C glycoprotein, in the murine lung. Am J Physiol Lung Cell Mol Physiol. 2009; 297:L153–63. https://doi.org/10.1152/ajplung.90524.2008. [PubMed].