
Introduction
Head and neck squamous cell carcinoma (HNSCC) is an aggressive epithelial cancer that derives from mucosa linings of the oral cavity, oropharynx, hypopharynx or larynx. It is also a major public health problem with it being the sixth most incident cancer worldwide, responsible of more than 700 000 cases every year and around 350 000 deaths [1]. Different methods are employed by this aggressive disease to avoid immune recognition, including direct T-cell suppression with soluble or surface inhibitory factors like Programmed-death ligand 1 (PDL1), and the recruitment of immuno-suppressive cell populations [2].
Allison first hypothesized that overcoming the anergic state of T-lymphocytes is possible via the blockade of co-inhibitory signals [3] and research has initially been focused mainly on the PDL1-PDL2/PD1 axis. Immune checkpoint inhibitors (ICI) have since completely revolutionized the treatment of Recurrent/Metastatic (R/M) HNSCC [4–6].
Primary resistance to ICI is frequent across all tumor types, including HNSCC, and concerns almost 60% of patients overall [7]. In a worrying manner, it was suggested that some patients even experience an acceleration of tumor growth kinetics (TGK) on immunotherapy [hyperprogressive disease (HPD)] [8]. Medical charts of 34 patients treated with PD1/PDL1 inhibitors from four different institutions were retrospectively reviewed in 2017 and HPD was found to be frequent (29%) and associated with a worse outcome [8]. Other studies found HPD in different tumor types with varying rates [9, 10] but no consistent predictive genomic or clinical characteristic was associated. All were of a retrospective nature, without a control arm. In consequence, a causal relationship of HPD to ICI has not been proven and a natural evolution of the disease cannot be excluded in the observed cases. Many preclinical studies have hypothesized mechanisms, but no clear biological explanation has seen the day.
Furthermore, after failure of ICI, salvage chemotherapy (SCT) is usually the treatment of choice, but not a lot of data is available on the outcomes in this context. When facing an initial RECIST (Response Evaluation Criteria In Solid Tumors) progressive disease (PD) on ICI, physicians are often confronted with the choice between starting subsequent SCT or continuing ICI in hope for a late response, especially if patients do not experience a worsening general condition. The utility of TGK in this situation is unknown.
Our aim in this study was to know if HPD patients had slower tumor growth before treatment with ICI compared to patients with a naturally exponential growing disease, in which case a strong argument for a causal relationship between ICI and HPD can be made. For this, we used TGK to determine the rate of hyperprogression in clinical trial patients and then compared tumor growth before the onset of immunotherapy with rapidly deteriorating screen-failure (SF) patients. The impact of TGK on outcomes with SCT after initial PD with ICI was also evaluated.
Results
In total, there were 192 patients in 9 clinical trials testing anti-PD1/PDL1 agents alone or in combination with anti-CTLA4 or anti-KIR antibodies. Among those, 158 patients were treated with ICI (the other 34 patients received exclusive chemotherapy) and 120/158 patients were eligible for HPD analysis. The remaining 38 patients were not included in the final analysis because of the absence of available pre-baseline imaging (2 patients) and the absence of post treatment imaging (36 patients) (Figure 1). Median follow-up time since the start of immunotherapy was 34.3 months (95% CI, 32.2 to 35.5).
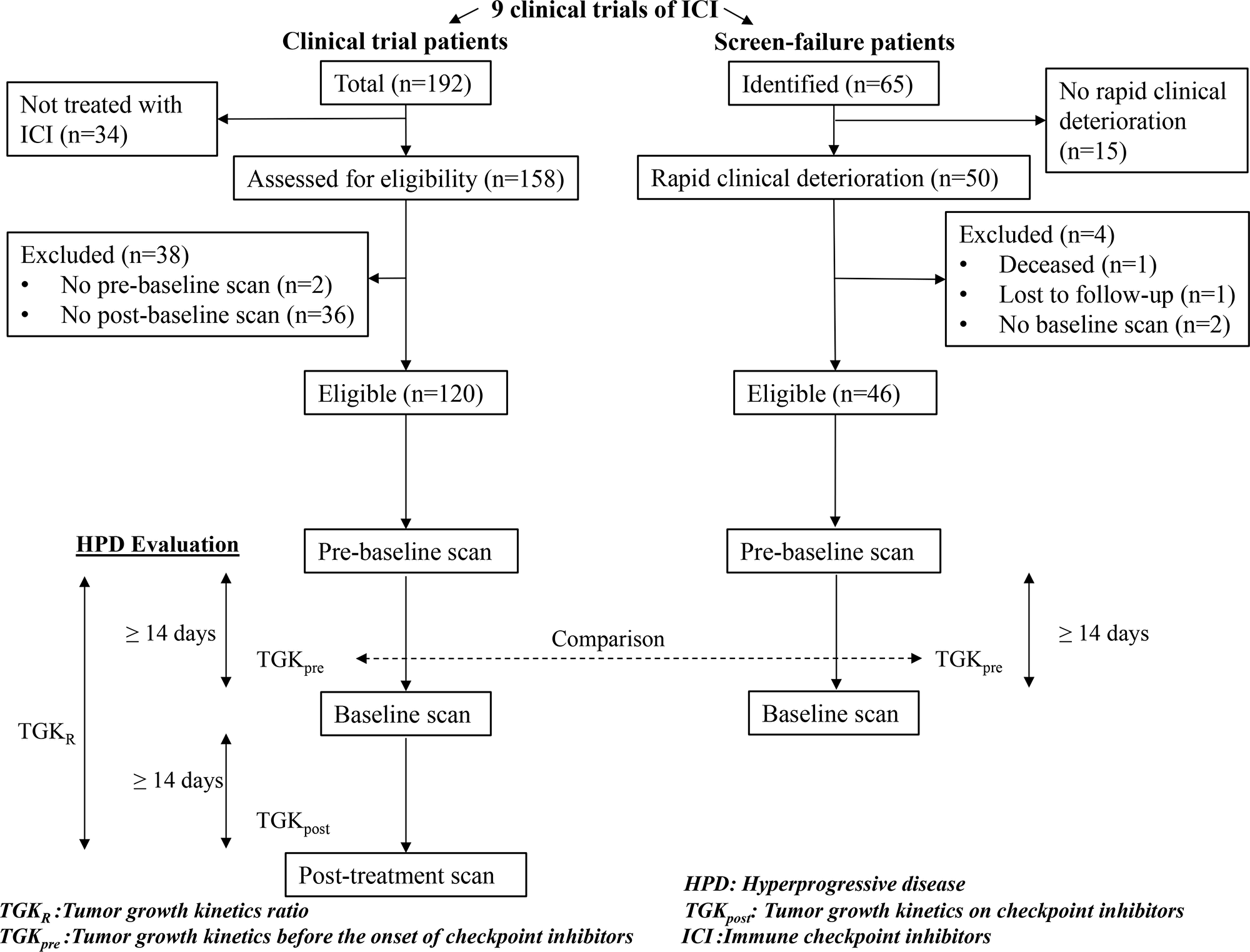
Figure 1: Study flowchart. Hyperprogressive disease (HPD) was evaluated in the clinical trial patients using Tumor Growth Kinetics ratio (TGKR). Tumor Growth Kinetics before the onset of immunotherapy (TGKpre) was compared with screen-failure patients.
Hyperprogressive disease and comparison with screen failure
Hyperprogressive disease rate
22/120 (18%) patients had HPD. Median TGKR was 3.2 (95% CI, 2.5 to 4.5). Median TGKpre was 4.8 (95% CI, 1.7 to 7.4) and median TGKpost was 17.1 (95% CI, 7.7 to 25).
HPD was associated with high NLR (p < 0.04) (Table 1). No correlation was found with the use of antibiotics, PDL1 or HPV status, elderly age, performance status, disease site, smoking or gender (Table 1). The median PFS was 1.9 months (95% CI, 1.8 to 2.3) in the HPD group vs 3.9 months (95% CI, 3.6 to 5.4). PFS was significantly lower for the HPD group (HR, 2.8; 95% CI, 1.4 to 5.6; p < 0.0001) (Figure 2). The median OS was 3.8 months (95% CI, 2.8 to 7.8) in the HPD group vs 14.6 months (95% CI, 10.1 to 18.7). OS was significantly lower for the HPD group (HR, 2.2; 95% CI, 1.1 to 4.3; p = 0.0018) (Figure 3).
Table 1: Baseline clinical and biological characteristics
| Clinical or biological characteristic | HPD (n = 22) N (%) | Non-HPD (n = 98) N (%) | P |
|---|---|---|---|
| Neutrophil-to-lymphocyte ratio | 0.04 | ||
| High | 13 (59) | 48 (48) | |
| Low/Intermediate | 7 (32) | 49 (49) | |
| Unknown | 2 (9) | 1 (1) | |
| Antibiotic use | 0.07 | ||
| Yes | 5 (23) | 15 (15) | |
| No | 16 (73) | 83 (85) | |
| Unknown | 1 (4) | 0 (0) | |
| PDL1 status | 0.9 | ||
| Negative | 4 (18) | 16 (16) | |
| Positive | 10 (46) | 42 (43) | |
| Unknown | 8 (36) | 40 (41) | |
| Checkpoint inhibitor | 0.62 | ||
| PD1 based | 14 (64) | 68 (69) | |
| PDL1 based | 8 (36) | 30 (31) | |
| Checkpoint inhibition regimen | 1.0 | ||
| Monotherapy | 6 (27) | 26 (27) | |
| Combination | 16 (73) | 72 (73) | |
| Immunotherapy line | 0.16 | ||
| 1st line | 7 (22) | 48 (49) | |
| ≥ 2nd line | 15 (68) | 50 (51) | |
| Age | 0.35 | ||
| ≥ 65 years | 8 (36) | 47 (48) | |
| < 65 years | 14 (64) | 51 (52) | |
| Previous radiation therapy | 0.51 | ||
| No | 4 (18) | 13 (13) | |
| Yes | 18 (82) | 85 (87) | |
| Human papillomavirus | 0.82 | ||
| Negative | 12 (55) | 46 (47) | |
| Positive | 2 (9) | 10 (10) | |
| Unknown | 8 (36) | 42 (43) | |
| Performance status | 0.45 | ||
| 0 | 4 (18) | 31 (32) | |
| 1 | 17 (77) | 64 (65) | |
| ≥ 2 | 1 (5) | 3 (3) | |
| Gender | 1.0 | ||
| Male | 18 (82) | 79 (81) | |
| Female | 4 (18) | 19 (19) | |
| Smoking status | 0.75 | ||
| Non-smoker | 4 (18) | 15 (15) | |
| Previous/current smoker | 18 (82) | 83 (85) |
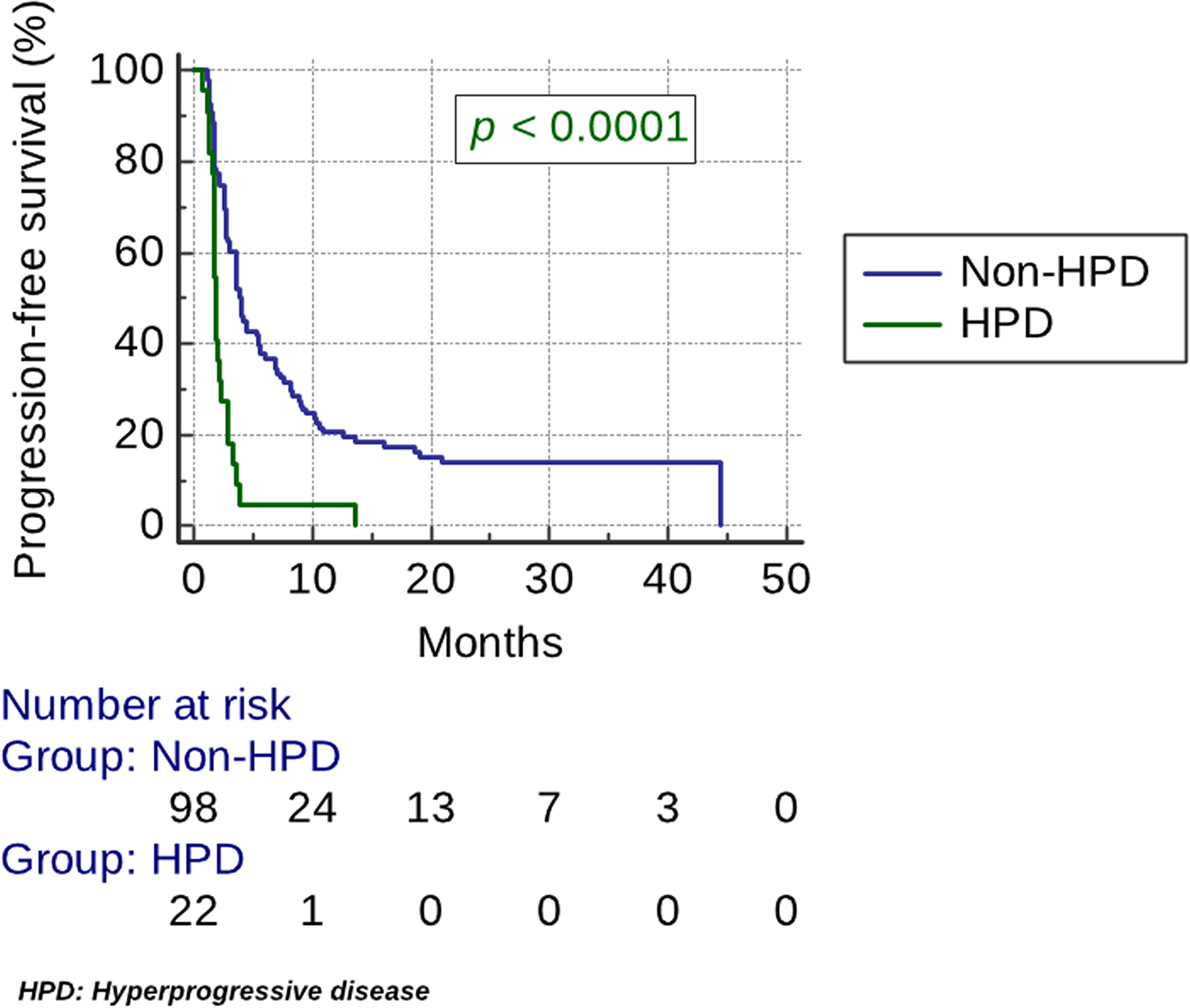
Figure 2: Kaplan–Meier estimates of progression-free survival (PFS). The median PFS was 1.9 months (95% CI, 1.8 to 2.3) in the HPD group vs 3.9 months (95% CI, 3.6 to 5.4). PFS was significantly lower for the HPD group (HR, 2.8; 95% CI, 1.4 to 5.6; p < 0.0001).
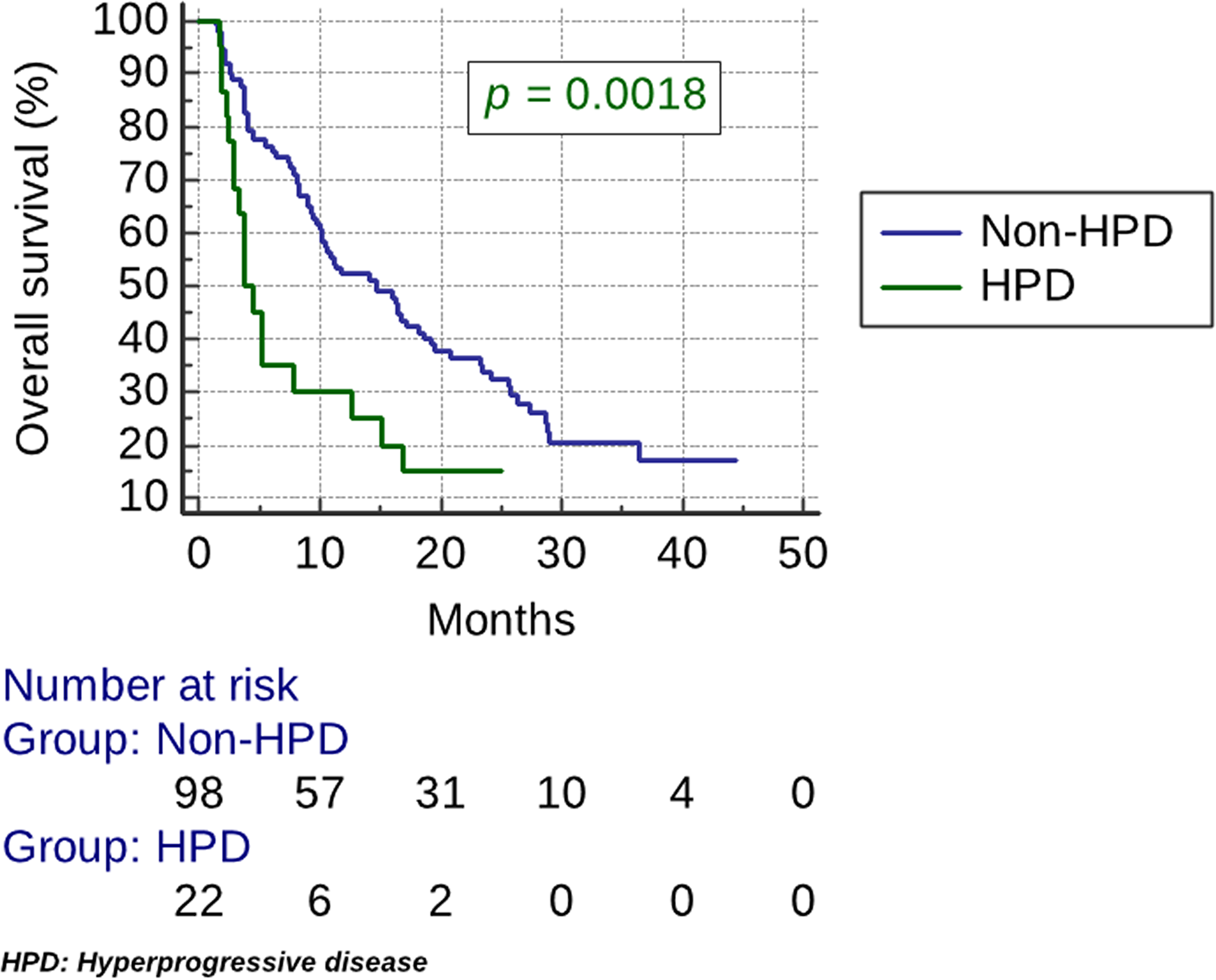
Figure 3: Kaplan–Meier estimates of overall survival (OS). The median OS was 3.8 months (95% CI, 2.8 to 7.8) in the HPD group vs 14.6 months (95% CI, 10.1 to 18.7). OS was significantly lower for the HPD group (HR, 2.2; 95% CI, 1.1 to 4.3; p = 0.0018).
Hyperprogressive disease rate with total tumor burden
When calculating TGKR with TTB, HPD was found in 21/120 (17.5%) patients. Median TGKR was 3.2 (95% CI, 2.4 to 4.7). HPD was concordant between RECIST 1.1 and total tumor burden evaluation for 16/22 (73%) patients.
SF tumor growth kinetics comparison
In total, 65 patients were screen-failed in the 9 clinical trials. Of these, 50 SF cases were attributed to rapid clinical deterioration and were included in the final analysis (Figure 1). The following reasons were the cause of SF in the included patients: death, symptomatic cerebral metastases, elevated liver enzymes attributed to metastatic disease, corticosteroid use for disease control and worsening general condition. 46/50 patients were eligible for TGKpre assessment as 1 patient was deceased, 1 patient was lost to follow up and 2 patients didn’t have an available CT-scan.
Median TGKpre was 2.7 (95% CI, 2 to 3.3). No significant difference in TGKpre with HPD patients was found using a Mann–Whitney test (p = 0.17) (Figure 4).
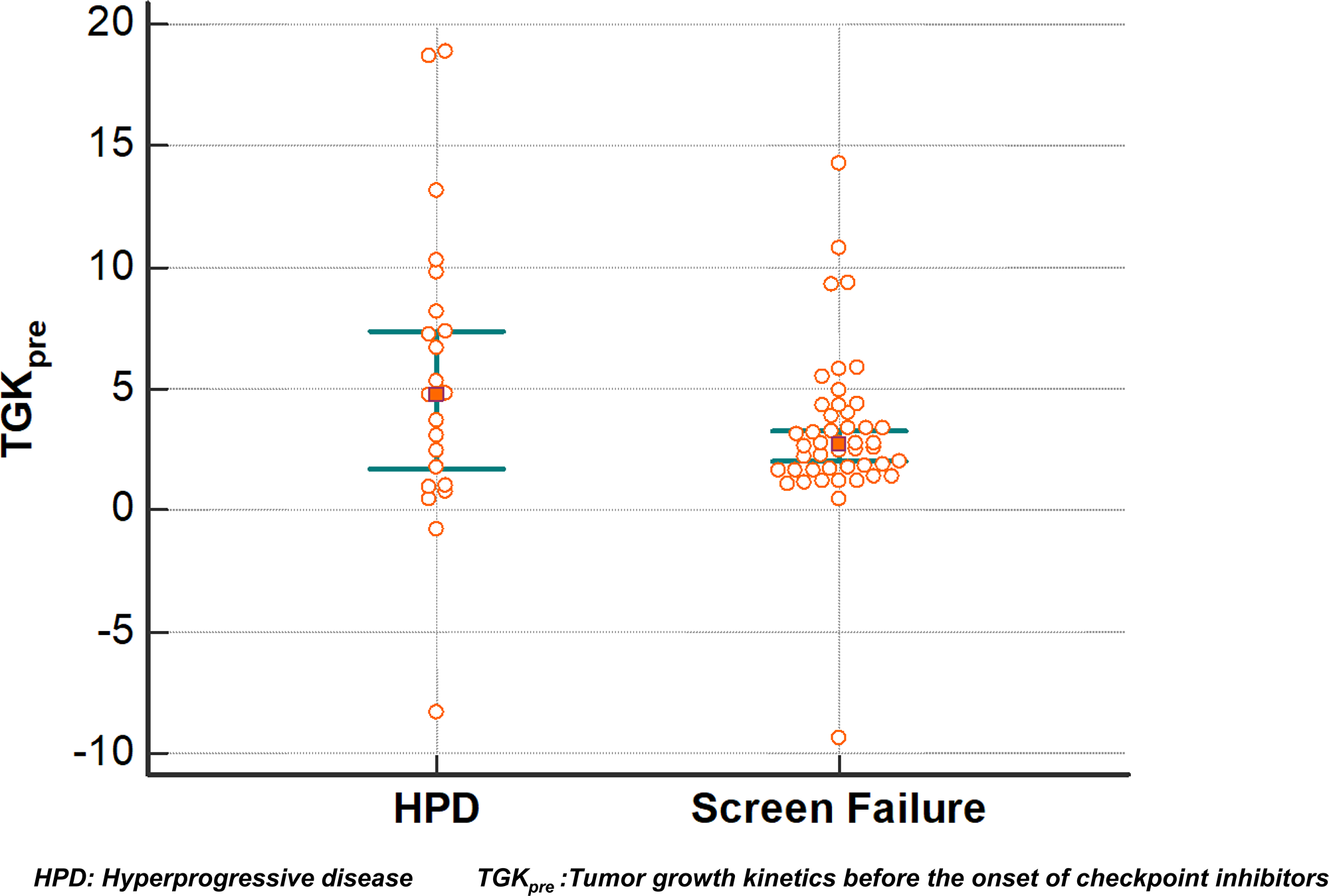
Figure 4: Tumor growth kinetics before the onset of immunotherapy (TGKpre). Each dot represents a distinct TGKpre value. Overlapping confidence intervals of this dot plot show that distribution is similar.
Tumor growth kinetics and salvage chemotherapy
Outcomes on salvage chemotherapy
Out of 158 patients treated with ICI, 67 patients were eligible. ICI were given as monotherapy in 31% of patients or as combination in 69%. Salvage chemotherapy included platinum-based regimen (55%), taxane-based regimen (21%), capecitabine (3%), cetuximab (8%), vinorelbine (1%) and methotrexate (12%). Cetuximab was administered in combination with platinum or taxanes in 14% of patients. The median number of prior treatment lines was 2 (range 1–5). The ORR (Objective response rate) was 28%. 6 patients (9%) presented CR (4 with platinum-based chemotherapy, 1 with Docetaxel and 1 with cetuximab) and 13 patients (19%) had PR. The DCR was 61%. The median PFS was 3.5 months (95% CI, 2.5 to 4.9) and the median OS was 9 months (95% CI, 7.2 to 13.8).
TGKR after initial progression on checkpoint inhibitors
Out of 39 patients who presented initial RECIST 1.1 PD with ICI and were subsequently treated with salvage chemotherapy, 32 patients were eligible for TGKR assessment. 7 patients were ineligible because of the absence of pre-baseline scan. Seven (7) out of 14 (50%) patients with disease deceleration (TGKR < 1) on ICI had PR or CR and 5 (36%) had SD. DCR was 86%. 3 out of 18 (17%) patients with disease acceleration (TGKR > 1) on ICI had PR and 4 (22%) had SD. DCR was 39% (Figure 5). Median time from last ICI administration to first imaging on salvage chemotherapy was 2.3 months (95% CI, 2 to 2.7).
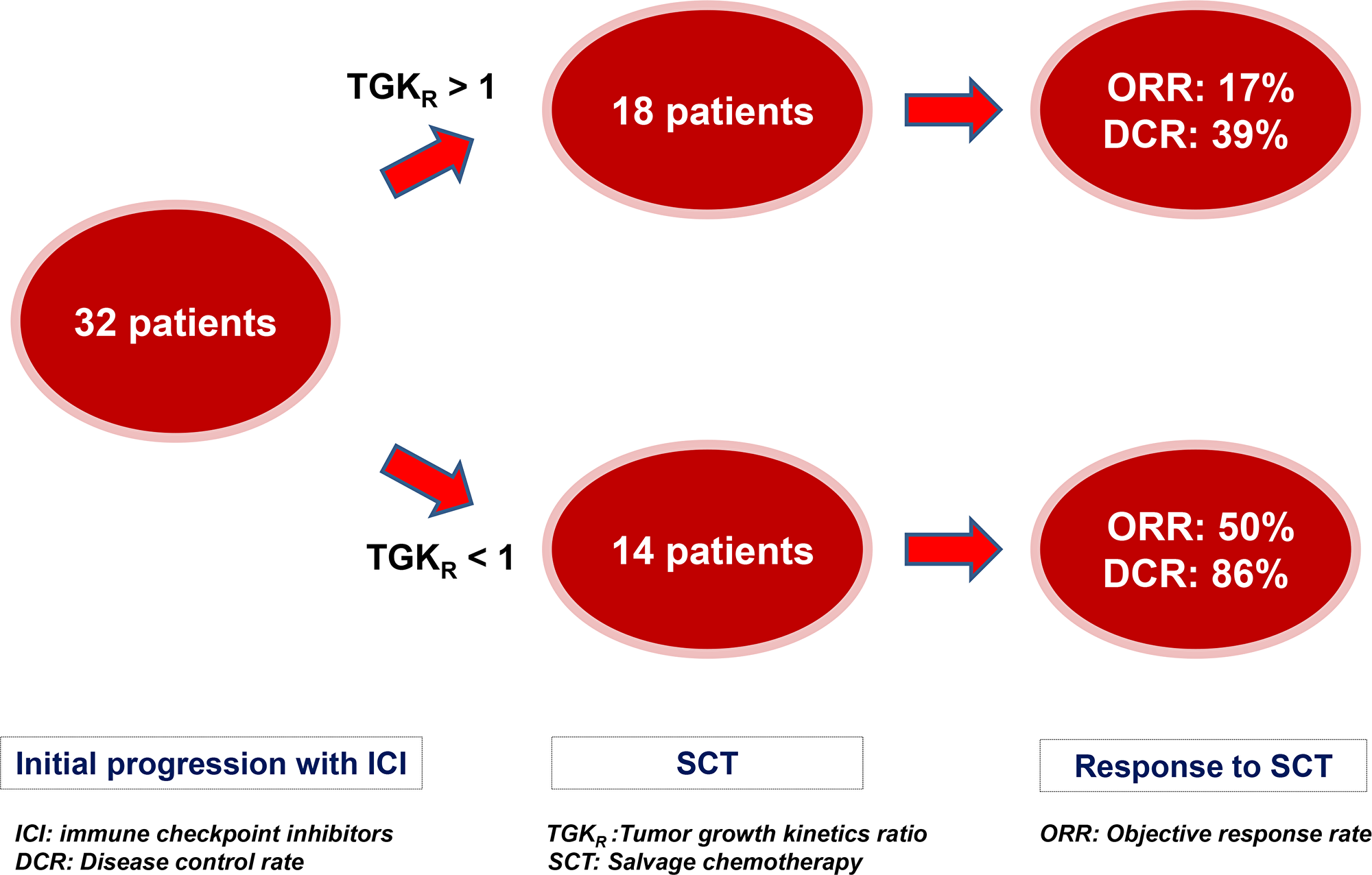
Figure 5: Impact of Tumor Growth Kinetics ratio (TGKR) on outcomes with salvage chemotherapy (SCT) after initial RECIST 1.1 progression with immune checkpoint inhibitors (ICI). Patients were more likely to respond to SCT in case of tumor growth deceleration (0 < TGKR < 1) and had higher disease control rate (DCR) than those with tumor growth acceleration (TGKR > 1).
DISCUSSION
Hyperprogression: a real phenomenon?
TGK did not differ between rapidly deteriorating SF and ICI-treated HPD patients, strongly suggesting that there is no relevant causal relationship between HPD and ICI, at least in some cases. In other words, patients that are considered to have ICI-induced hyperprogression have a tumor growth before ICI treatment that is similar to patients that have a rapidly evolving disease, not treated with ICI. Exponentially growing tumors are expected to continue to grow exponentially when untreated.
In our study, we used TGK to evaluate hyperprogression as this was the methodology used in the first published work on HPD in HNSCC by Saâda-Bouzid et al [8]. If another definition was used [e.g., 50% Tumor growth ratio (TGR) in 4 weeks], results might have been different, highlighting the need of an international consensus to define hyperprogression. More recently, HPD evaluation in non-small-cell lung cancer (NSCLC) patients was highly concordant using TGK and TGR which suggests the interchangeability of these two methods [11].
Multiple mechanisms have been raised: oncogenic signaling activation [11], upregulation of alternative immune checkpoints [12], major immune reactions caused by PD1/PDL1 inhibitors, previous irradiation [8], tumor proliferation via a direct (DNA damage with free radicals) or indirect (angiogenesis and tissue remodeling promotion) effect [13–15], expansion of PD1-expressing T-regulatory cells and modulation of tumour-promoting cells [16]. Interaction with tumor-associated macrophages via the Fc domain of the anti-PD1 antibody with reprogramming into immunosuppressive M2-like macrophages has also been incriminated [17]. No consistent predictors have yet been validated, further supporting our results.
On the other hand, 36 patients in our cohort were excluded because of the absence of post-baseline imaging that might be due to in some cases to hyperprogression-related death. This may lead to underestimating the phenomenon, although some of these deaths might also be due to disease natural behavior.
The absence of randomization and the difference in screening windows could of course be a source of bias, but we believe that our comparison with rapidly deteriorating SF patients is pertinent as those are the patients that might best represent a naturally exponential growing disease.
Furthermore, the heterogeneity of the immunotherapy regimens in this study could also be a confounding factor, but a previous work evaluating HPD in early-phase immunotherapy trials found hyperprogression with combination ICI as well as monotherapy, with no significant difference [18], mirroring our results.
We chose to compare growth kinetics before starting immunotherapy (TGKpre) between the two groups (and not the ratio of TGKpost to TGKpre), as we thought that this was the most appropriate time interval to show similarity in the speed of disease progression. SF patients were often lost to follow-up, received ICI in another setting or had variable timing in CT-scan evaluation after screen-failure, so comparison of the ratio of TGKpost to TGKpre would have been less appropriate.
In addition, we have found that HPD evaluation with total tumor burden and with RECIST 1.1 was concordant for about seventy percent of patients only, further emphasizing the need to find a consensus in HPD diagnostic criteria.
This is the largest cohort evaluating HPD in HNSCC to date. Our findings underscore the difficulties in interpreting the evolution of the disease under ICI and present an important message to clinicians that are treating patients with R/M HNSCC.
We found that hyperprogression was frequent (18%) and correlated only with a high NLR. Previous retrospective studies (albeit with less included patients) have already shown a correlation of high NLR with poor outcomes [19, 20], and so our results confirm this literature data, even if the relative small number of HPD patients might have weakened the statistical power of the comparison.
Impact of tumor growth kinetics
Some chemotherapeutic agents have been shown to exert immune-reactive events such as upregulation of MHC (major histocompatibility complex) class molecules and increased antigen presentation [21, 22]. Chemotherapy can augment tumor immunity by decreasing the number of Immunosuppressive cells like myeloid-derived suppressor cells (MDSC) or T-regulator cells in the microenvironment which can lead to an accumulation of helper T-cells on site [23, 24] and by promoting anti-tumor CD4+ T-cell phenotype [25]. In fact, Improved responses to chemotherapy has been reported after vaccination immunotherapy in various tumor types [26].
Our results confirm that salvage chemotherapy seems to be more effective with better outcomes (ORR, PFS, and OS), suggesting that chemotherapy is enhanced by immunotherapy. Indeed, ORR was 28%; which is higher than the historical controls before the era of immunotherapy; varying between 6 and 24% [27–29].
By showing that patients who presented initial PD on ICI associated to a tumor deceleration (TGKR < 1) had higher ORR that those with tumor acceleration (TGKR > 1), we think that some of the responses in our study were due/enhanced by circulating anti-PD1/PDL1 antibodies causing delayed onset of response. The median time from the last immunotherapy administration and the first evaluation on salvage chemotherapy was 2.3 months, which is compatible with the half-life of these agents [30, 31]. We by consequence also believe that TGKR might be a useful tool to help select potential late responders to ICI and thus avoid toxic and possibly inefficient chemotherapy.
Of course, this hypothesis should be evaluated in other data sets and if confirmed, it could potentially be tested prospectively by randomizing to continued immunotherapy versus switch to salvage chemotherapy in case of tumor growth decelaration.
In conclusion, HPD was found in 18% of cases and correlated with high NLR. Growth kinetics before ICI-treated HPD patients were similar to SF patients, suggesting that the published rates of HPD might be due to natural disease behavior, at least in some cases. After initial RECIST PD with ICI, tumor growth deceleration was associated with better outcomes compared to tumor growth acceleration, indicating that TGKR might be useful to detect late responders and avoid SCT, meriting prospective investigations.
Materials and Methods
Patient selection and data collection
All patients with a histologically confirmed R/M HNSCC treated in Léon Bérard cancer center in a clinical trial testing PD1/PDL1 antibodies alone or in combination with an anti-Cytotoxic T-Lymphocyte Antigen 4 (CTLA4) or an anti-Killer Immunoglobulin-like Receptor (KIR) antibody between March 2014 and November 2018; were included (Figure 1). All imaging was retrospectively reviewed by medical oncologists and independent radiologists. SCT was defined as the first line of chemotherapy administered after failure of ICI. The following data was collected and recorded: age, gender, primary tumor location, tobacco use, human papilloma virus (HPV) status (p16 immunostaining and/or DNA in situ hybridization), previous multimodal therapy at the initial stage, neutrophil-to-lymphocyte ratio (NLR) (0 to 72 hours before start of treatment; with a cut-off ≥5 defining high NLR), antibiotics intake in the 3 months prior to immunotherapy, the number and dates of previous and following lines of systemic therapy. Also collected was the best overall response on and after failure of immunotherapy using RECIST 1.1, progression-free survival (PFS) and overall survival (OS).
PD-L1 expression in archival formalin-fixed, paraffin-embedded (FFPE) samples was assessed. Samples were provided by the local Biological Resources Center (BB-0033-00050, CRB Centre Léon Bérard, Lyon France). 4-μm thick tissue sections of FFPE tissue were prepared according to conventional procedures. For each sample, hematoxylin and eosin (HES) staining was performed to determine the number of tumor cells. Immunohistochemistry (IHC) was performed on an automated immunostainer (Ventana Benchmark ultra, Roche, Meylan, France) using Ultra View DAB Kit according to the manufacturer’s instructions. Sections were incubated with a rabbit monoclonal human anti-PDL1 Ab (diluted at 1:50, Quartett, Berlin, Germany) clone QR1. The Ventana amplification kit was used and an anti-rabbit-HRP was applied on sections. Staining was visualized with DAB solution with 3,3′-diaminobenzidine as a chromogenic substrate. Finally, the sections were counterstained with Gill’s hematoxylin. All samples were examined by a qualified anatomopathologist for combined positive score (CPS), defined as the number of PD-L1-positive cells (tumour cells, macrophages and lymphocytes) divided by the number of tumour cells × 100 (a minimum of 100 viable tumour cells must have been present for the specimen to be considered evaluable). CPS ≥ 1 was the cut-off for PDL1 positivity.
The data collection cutoff point was December 4, 2019.
Hyperprogressive disease definition
In order to be eligible for TGKR assessment, patients had to have a pre-baseline scan, a baseline scan and post-treatment scan. Minimal delay between 2 CT-scans was 14 days and patients had to have started ICI therapy in the 6 weeks following baseline scan (Figure 1). TGK before (TGKpre) and after (TGKpost) anti-PD1/PDL1 therapy were evaluated. TGKpre was defined as the difference of the sum of the largest diameters of the target lesions per unit of time between pre-baseline and baseline imaging [(S0-Spre)/(T0-Tpre)]. TGKpost was defined in the same manner between on immunotherapy and baseline imaging [(SPOST-S0)/(TPOST-T0)]. HPD was defined as TGKR (ratio of TGKpost to TGKpre) ≥ 2. TGKR > 1 indicated tumor growth acceleration, while 0 < TGKR < 1 indicated tumor deceleration. TGKR < 0 indicated tumor shrinkage.
TGKR was calculated with RECIST 1.1 for all patients. Since TGK only evaluates the variation of target lesions and does not include new lesions in the assessment of tumor growth, TGKR was also calculated using total tumor burden (TTB).
Screen-failure inclusion
All patients that were screened for the same clinical trials and were subsequently ineligible were analyzed. Patients that were considered screen-failed because of rapid clinical deterioration attributed to disease progression were included in the ‘screen failure’ (SF) group. Patients were not included if they were subsequently treated with ICI in another clinical trial or setting. TGKpre was calculated in this group in the same manner in order to compare tumor growth with HPD patients before the onset of immunotherapy (Figure 1).
Statistical analysis
Descriptive statistics were used to summarize the baseline characteristics of the patients. Chi-squared or Fisher’s exact test was used for statistical comparisons of categorical data. Mann–Whitney test was used to compare distribution. Partial response (PR) and complete response (CR) defined objective response (OR). Disease control rate (DCR) was defined as the sum of CR, PR, and stable disease (SD). PFS time was defined as the period from the date of initial treatment administration to the date of clinical disease progression, mortality from any cause or the last follow-up. OS time was defined as the period from the date of initial treatment administration to the date of mortality from any cause or the last follow-up. The Kaplan–Meier method was used to assess PFS and OS. Comparison was done using the log-rank test. Data of patients who were lost to follow-up were censored at the time of last contact. Statistical analysis was done using MedCalc 18.11.6 statistical software (MedCalc Software, Mariakerke, Belgium).
Abbreviations
HNSCC: Head and neck squamous cell carcinoma; PDL1: Programmed-death ligand 1; ICI: Immune checkpoint inhibitors; R/M: Recurrent/metastatic; TGK: Tumor growth kinetics; HPD: Hyperprogressive disease; SCT: Salvage chemotherapy; RECIST: Response Evaluation Criteria In Solid Tumors; PD: Progressive disease; SF: Screen-failure; TGR: Tumor growth ratio; NSCLC: Non-small-cell lung cancer; MHC: Major histocompatibility complex; MDSC: Myeloid derived suppressor cells; CTLA4: Cytotoxic T-Lymphocyte Antigen 4; KIR: Killer Immunoglobulin-like Receptor; HPV: Human papilloma virus; NLR: Neutrophil-to-lymphocyte ratio; PFS: Progression-free survival; OS: Overall survival; FFPE: Formalin-fixed, paraffin-embedded; HES: Hematoxylin and eosin; IHC: Immunohistochemistry; CPS: Combined positive score; TGKpre: TGK before anti-PD1/PDL1 therapy; TGKpost: TGK after anti-PD1/PDL1 therapy; TTB: Total tumor burden; PR: Partial response; CR: Complete response; OR: Objective response; DCR: Disease control rate; SD: Stable disease.
ACKNOWLEDGMENTS
The authors thank the clinical research associates of the Léon Bérard cancer center and the local Biological Resources Center (BB-0033-00050, CRB Centre Léon Bérard, Lyon, France) for their help in this study.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68:394–424. https://doi.org/10.3322/caac.21492. [PubMed].
2. Horton JD, Knochelmann HM, Day TA, Paulos CM, Neskey DM. Immune Evasion by Head and Neck Cancer: Foundations for Combination Therapy. Trends Cancer. 2019; 5:208–32. https://doi.org/10.1016/j.trecan.2019.02.007. [PubMed].
3. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015; 348:56–61. https://doi.org/10.1126/science.aaa8172. [PubMed].
4. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington KJ, Kasper S, Vokes EE, Even C, Worden F, Saba NF, Docampo LC, et al. Nivolumab vs investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. 2018; 81:45–51. https://doi.org/10.1016/j.oraloncology.2018.04.008. [PubMed].
5. Cohen EE, Soulières D, Le Tourneau C, Dinis J, Licitra L, Ahn MJ, Soria A, Machiels JP, Mach N, Mehra R, Burtness B, Zhang P, Cheng J, et al, and KEYNOTE-040 investigators. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019; 393:156–67. https://doi.org/10.1016/S0140-6736(18)31999-8. [PubMed].
6. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr, Psyrri A, Basté N, Neupane P, Bratland Å, Fuereder T, Hughes BG, Mesía R, et al, and KEYNOTE-048 Investigators. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet. 2019; 394:1915–28. https://doi.org/10.1016/S0140-6736(19)32591-7. [PubMed].
7. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54. https://doi.org/10.1056/NEJMoa1200690. [PubMed].
8. Saâda-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, Even C, Fayette J, Guigay J, Loirat D, Peyrade F, Alt M, Gal J, Le Tourneau C. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2017; 28:1605–11. https://doi.org/10.1093/annonc/mdx178. [PubMed].
9. Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res. 2017; 23:4242–50. https://doi.org/10.1158/1078-0432.CCR-16-3133. [PubMed].
10. Matos IS, Martin-Liberal J, Hierro C, de Olza MO, Viaplana C, Costa M, Felip-Falg’s E, Mur-Bonet G, Vieito M, Brana I, Azaro A, Perez-Gago C, Rodríguez-Freixinós V, et al. Incidence and clinical implications of a new definition of hyperprogression (HPD) with immune checkpoint inhibitors (ICIs) in patients treated in phase 1 (Ph1) trials. J Clin Oncol. 2018; 36:3032–3032. https://doi.org/10.1200/JCO.2018.36.15_suppl.3032.
11. Kim CG, Kim KH, Pyo KH, Xin CF, Hong MH, Ahn BC, Kim Y, Choi SJ, Yoon HI, Lee JG, Lee CY, Park SY, Park SH, et al. Hyperprogressive disease during PD-1/PD-L1 blockade in patients with non-small-cell lung cancer. Ann Oncol. 2019; 30:1104–13. https://doi.org/10.1093/annonc/mdz123. [PubMed].
12. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016; 7:10501. https://doi.org/10.1038/ncomms10501. [PubMed].
13. Guo X, Zhai L, Xue R, Shi J, Zeng Q, Gao C. Mast cell tryptase contributes to pancreatic cancer growth through promoting angiogenesis via activation of angiopoietin-1. Int J Mol Sci. 2016; 17:E834. https://doi.org/10.3390/ijms17060834. [PubMed].
14. Manson G, Norwood J, Marabelle A, Kohrt H, Houot R. Biomarkers associated with checkpoint inhibitors. Ann Oncol. 2016; 27:1199–206. https://doi.org/10.1093/annonc/mdw181. [PubMed].
15. DeNardo DG, Andreu P, Coussens LM. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev. 2010; 29:309–16. https://doi.org/10.1007/s10555-010-9223-6. [PubMed].
16. Champiat S, Ferrara R, Massard C, Besse B, Marabelle A, Soria JC, Ferté C. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat Rev Clin Oncol. 2018; 15:748–62. https://doi.org/10.1038/s41571-018-0111-2. [PubMed].
17. Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, Ferro S, Ganzinelli M, Gasparini P, Huber V, Milione M, Porcu L, Proto C, et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin Cancer Res. 2019; 25:989–99. https://doi.org/10.1158/1078-0432.CCR-18-1390. [PubMed].
18. Kanjanapan Y, Day D, Wang L, Al-Sawaihey H, Abbas E, Namini A, Siu LL, Hansen A, Razak AA, Spreafico A, Leighl N, Joshua AM, Butler MO, et al. Hyperprogressive disease in early-phase immunotherapy trials: clinical predictors and association with immune-related toxicities. Cancer. 2019; 125:1341–49. https://doi.org/10.1002/cncr.31999. [PubMed].
19. Foster CC, Kochanny S, Khattri A, Acharya R, Dekker A, Tan YH, Klema E, Brisson RJ, Saloura V, Pearson AT, Vokes EE, Leidner RS, Mehanna HM, Seiwert TY. Association of a baseline neutrophil-to-lymphocyte ratio (NLR) with progression-free and overall survival in head and neck cancer patients receiving anti-PD-1 therapy. J Clin Oncol. 2018; 36:6038. https://doi.org/10.1200/JCO.2018.36.15_suppl.6038.
20. Yasumatsu R, Wakasaki T, Hashimoto K, Nakashima K, Manako T, Taura M, Matsuo M, Nakagawa T. Monitoring the neutrophil-to-lymphocyte ratio may be useful for predicting the anticancer effect of nivolumab in recurrent or metastatic head and neck cancer. Head Neck. 2019; 41:2610–18. https://doi.org/10.1002/hed.25737. [PubMed].
21. Ohtsukasa S, Okabe S, Yamashita H, Iwai T, Sugihara K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J Cancer Res Clin Oncol. 2003; 129:719–26. https://doi.org/10.1007/s00432-003-0492-0. [PubMed].
22. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS One. 2012; 7:e32542. https://doi.org/10.1371/journal.pone.0032542. [PubMed].
23. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005; 11:6713–21. https://doi.org/10.1158/1078-0432.CCR-05-0883. [PubMed].
24. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004; 34:336–44. https://doi.org/10.1002/eji.200324181. [PubMed].
25. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res. 2015; 3:436–43. https://doi.org/10.1158/2326-6066.CIR-15-0064. [PubMed].
26. Antonia SJ, Mirza N, Fricke I, Chiappori A, Thompson P, Williams N, Bepler G, Simon G, Janssen W, Lee JH, Menander K, Chada S, Gabrilovich DI. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006; 12:878–87. https://doi.org/10.1158/1078-0432.CCR-05-2013. [PubMed].
27. Argiris A, Ghebremichael M, Gilbert J, Lee JW, Sachidanandam K, Kolesar JM, Burtness B, Forastiere AA. Phase III randomized, placebo-controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: an eastern cooperative oncology group trial. J Clin Oncol. 2013; 31:1405–14. https://doi.org/10.1200/JCO.2012.45.4272. [PubMed].
28. Soulières D, Faivre S, Mesía R, Remenár É, Li SH, Karpenko A, Dechaphunkul A, Ochsenreither S, Kiss LA, Lin JC, Nagarkar R, Tamás L, Kim SB, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017; 18:323–35. https://doi.org/10.1016/S1470-2045(17)30064-5. [PubMed].
29. Martinez-Trufero J, Isla D, Adansa JC, Irigoyen A, Hitt R, Gil-Arnaiz I, Lambea J, Lecumberri MJ, Cruz JJ. Phase II study of capecitabine as palliative treatment for patients with recurrent and metastatic squamous head and neck cancer after previous platinum-based treatment. Br J Cancer. 2010; 102:1687–91. https://doi.org/10.1038/sj.bjc.6605697. [PubMed].
30. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010; 28:3167–75. https://doi.org/10.1200/JCO.2009.26.7609. [PubMed].
31. Syed YY. Durvalumab: First Global Approval. Drugs. 2017; 77:1369–76. https://doi.org/10.1007/s40265-017-0782-5. [PubMed].