
Introduction
Oral squamous cell carcinoma (OSCC) is the sixth most frequent cancer that affects the human population worldwide [1]. Despite significant developments in the current treatment modalities for OSCC, the disease-free survival rate has remained poor for the last several decades. Cisplatin, though is a first-line chemotherapeutic drug used for the treatment of OSCC patients [2, 3], acquisition of cisplatin-resistance to date remains a major impediment in its efficacious clinical application. There is, therefore, an urgent need to better understand the underlying mechanisms of cisplatin resistance as well as identifying reliable targets/mediators which can facilitate the restoration of sensitivity towards cisplatin.
Exosomes (the membrane secretory vesicle) acts as cargo for various molecular content (miRNA, LncRNA, msRNA, DNA, protein, enzyme) that depend on their origin and status; exosomes thus mediate cell-cell communication and convey biological signals to regulate cellular processes [4]. Emerging studies have shown that cancer cells release exosomes at a higher rate compared to the normal cells, and contribute significantly in instituting a milieu that supports neoplastic proliferation, angiogenesis, invasion, and metastasis [5]. In the past decade, the role of tumor cell derived-exosomes in inhibiting immune-surveillance and hence bestowing oncogenic transformation has gathered increasing attention [6]. They have also been implicated in the development of drug-resistance [7], and therefore now being considered as promising candidates both for cancer diagnostics and therapeutics [8].
An increasing amount of evidence has indicated that exosomes-derived miRNA plays an essential role in mediating cellular crosstalk within tumor-microenvironment [4], therefore are capable of moderating tumor progression and chemoresistance in many cancer types [7, 9]. Pioneering studies have shown the role of pancreatic cancer-derived exosomes in transferring miRNAs (miR212-3p, miR 203, mir 21) to dendritic cells leading to enhanced invasiveness and drug resistance [10–13]. Exosome-mediated miR155 delivery was found to cause EMT and associated chemoresistance in breast cancer [14]. On the other hand, exosome-mediated delivery of miR-151a and miR-128-3p has been shown to sensitize glioblastoma and colorectal cancer cells to temozolomide and oxaliplatin respectively [15–18].
miRNAs have also been shown to moderate chemoresistance via regulating autophagy [19], an approach adopted by tumor cells to survive the metabolic stress imposed by tumor-microenvironment [20, 21]. miR-30a has been shown to play a role in mediating cisplatin-chemoresistance in melanoma and pancreatic adenocarcinoma via IGFR1 and SNAI1 [17, 18]. However, in some other studies, the reverse role of miR30a on drug-resistance has been documented. Zou et al. showed a significant reduction in miR-30a expression in HeLa, MCF7 and HepG2 cancer cells following cisplatin treatment, whilst the forced expression of miR-30a sensitizes them to cisplatin via blocking beclin 1-mediated autophagy [22]. Similarly, miR30a induction in cisplatin-resistant lung cancer cells led to inhibition of Beclin 1-mediated autophagy and a concomitant increase in apoptosis [23]. The author thus suggested that enhancing miR-30a expression in breast, liver, and lung tumor cells offers a promising approach to combat chemoresistance. Besides, miR-30a has proven its function in regulating epithelial-mesenchymal transition, impeding proliferation and metastasis in multiple cancers and autophagy in CML [24]. Notably, reduced expression of miR-30a has recently been reported in OSCC, and it has also been associated with decreased cell proliferation, migration, and invasion [25, 26]. Cisplatin chemoresistance is also a big hurdle in OSCC treatment. Hence, it is worth determining if exogenously increasing miR-30a in OSCC has any role in combating cisplatin chemoresistance, regulating autophagy, a process known to influence chemoresistance and if exosomal-mediated miR30a delivery can be exploited as an approach to enhance the therapeutic efficacy.
In the present study, we show significantly decreased expression of miR-30a in oral cancer patients with disease recurrence post cisplatin treatment and OSCC cultured cells having cisplatin resistance. Herein, we present the first evidence that exosomal-mediated miR-30a delivery in the cisplatin-resistant OSCC cells led to decreased autophagic response via Beclin1 while it augments apoptosis by inhibiting Bcl2, hence mediating reversal of cisplatin sensitivity. Our results thus establish the significance of exosome-mediated miR-30a delivery in combating chemoresistance in oral cancer cells and open up new avenues for designing of exosomes-based clinical management of oral cancer.
Results
Beclin1 acts as a target for miR-30a in OSCC
Using three web-based tools, we predicted the gene targets of miR-30a in OSCC. TargetScan and DIANA-micro-T-CDS predicted 431 and 1782 gene-targets respectively (data not shown). Out of these, the top 100 gene-targets selected from targetscan (based on total context ++ score percentile) and DIANA-micro-T-CDS (based on miTG score) were considered for further screening. Next, following their known implication in cancer chemoresistance, 21 gene-targets were further shortlisted (Supplementary Table 1). Notably, Beclin1, an autophagy marker, showed the highest score with both TargetScan and DIANA-micro-T-CDS predictions (Supplementary Table 1 and Figure 1A and 1B). We further validated the selected gene-target using RNAhybrid tool wherein minimum free energy of binding of Beclin1 with miR-30a was found to be –23 kcal/mol (Figure 1C). Finally, based on the consensus obtained from all the three web-based tools, Beclin1 was selected as a potential target for miR-30a in cisplatin-resistant OSCC.
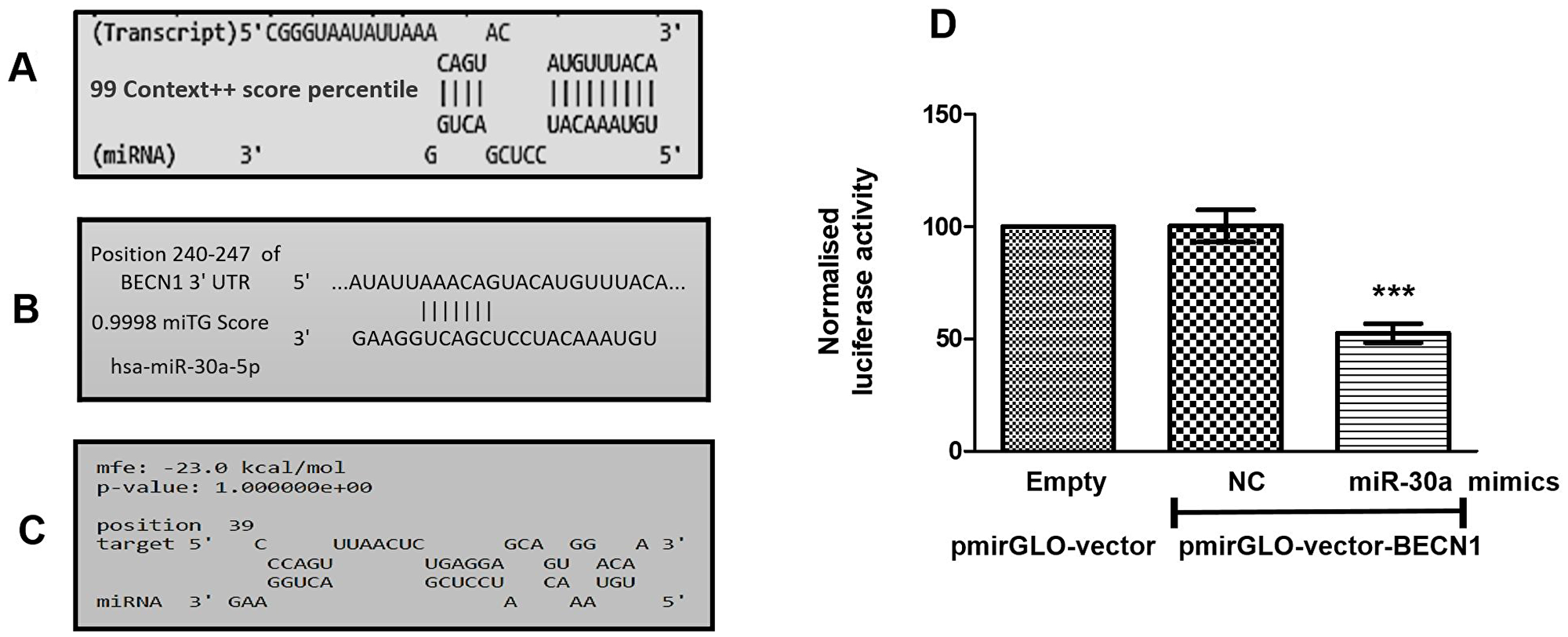
Figure 1: (A) Binding position prediction of miR-30a with Beclin1 using TargetScan web-based tool. (B) Binding position prediction of miR-30a with Beclin1 using the DIANA microT-CDS tool. (C) Binding energy prediction of miR-30a with Beclin1 by RNAhybrid. (D) BECN1 luciferase activity in cisRes cells co-transfected with either empty vector or pmirGLO-Becn1 vector having miR-30a target sequence and either NTC or miR30a mimics. Data are expressed as the mean +/– SD. ***P < 0.001, significant difference vs. control group (n = 3). Two independent experiments gave similar results.
We next validated in-silico predictions in cultured cisplatin-resistant OSCC cells using a luciferase reporter assay. Of note, mimics-mediated forced-expression of miR-30a in cisRes cells significantly decreased the miR-30a -3′UTR-Beclin1 luciferase activity compared to the cisRes cells transfected with NTC or empty vector control, thus demonstrating miR-30a-mediated regulatory control of Beclin1 (Figure 1D).
Exosomal miR-30a is downregulated, while Beclin1 is up-regulated in cisRes OSCC cells and patients
We next determined if miR-30a has any role in acquired cisplatin-resistance in OSCC. Interestingly, miR-30a expression was found to be significantly reduced in exosomes isolated from the serum of OSCC patients vs healthy volunteers (Figure 2A). Exosomes from tobacco smokers showed aberrant miR-30a expression compared to healthy volunteers. Of special relevance, is the highest and significant reduction in miR-30a expression observed in the exosomes from OSCC patients with tumor recurrence post cisplatin treatment vs healthy controls. We next corroborated our finding in clinical samples depicting an association of reduced miR-30a levels with disease recurrence, in vitro using cisRes OSCC cells. Notably, as observed in OSCC patients, both cellular and exosomal miR-30a expression was significantly lower in cisRes OSCC cells compared to cissens cells (Figure 2B and 2C).
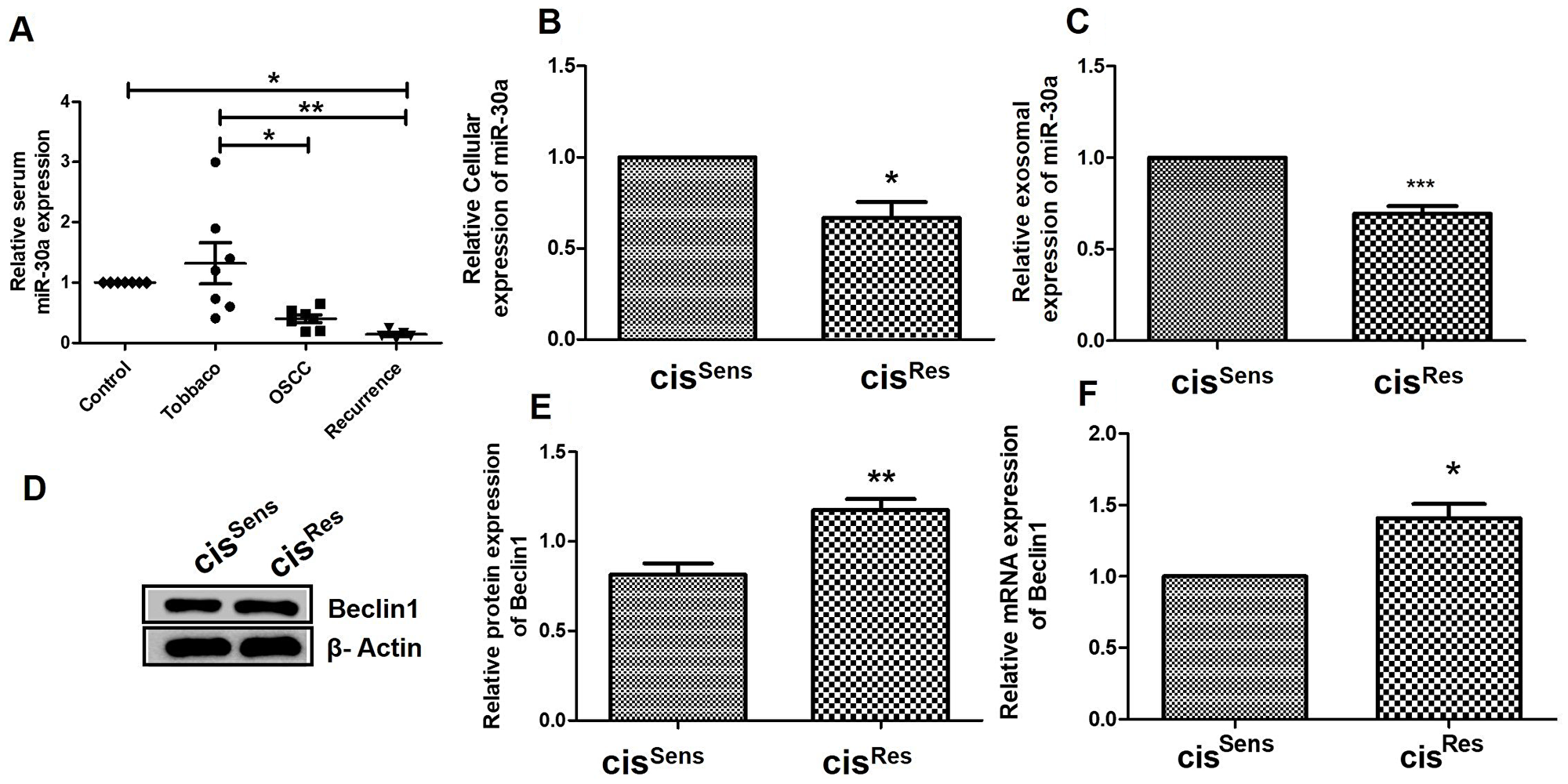
Figure 2: (A) miR-30a expression in clinical samples was quantified by qRT-PCR and normalized to U6 as a housekeeping gene. miR-30a expression profiling in cisRes and cisSens oral cancer cells at the (B) total cellular and (C) exosomal level. (D) Western blot analysis for Beclin1 expression in cisRes and cisSens oral cancer cells. (E) Densitometry of Beclin 1 Western blot normalized to actin as the loading control. (F) Beclin 1 mRNA expression as analyzed by qRT-PCR. 18S was used as a housekeeping gene.
We further evaluated the importance of miR-30a in cisRes OSCC by measuring the expression of its target gene, beclin1 (Figure 1). In concordance with the reduced miR-30a expression, cisRes cells showed enhancements in Beclin1 (an autophagy marker) both at the protein (Figure 2D, 2E) and mRNA levels (Figure 2F), when compared to the cisSens cells.
miR-30a restoration inhibited Beclin1 expression and conferred cisplatin sensitivity in cisRes OSCC cells
We identified Beclin1 as a miR-30a target in OSCC (Figure 1). However, its association with cisplatin-resistance in OSCC thus far remains undetermined. Hence, we next investigated if the decreased expression of miR-30a also modulates Beclin1 in cisRes OSCC cells. For this, using a mimics-transfection approach, we first restored the miR-30a expression in cisRes OSCC cells (Figure 3A). Notably, miR-30a restoration in cisRes cells (referred hereafter as CisRes, miR-30a-mimic) significantly decreased the Beclin1 expression (Figure 3B, 3C). cisSens cells were employed in the study as a control.
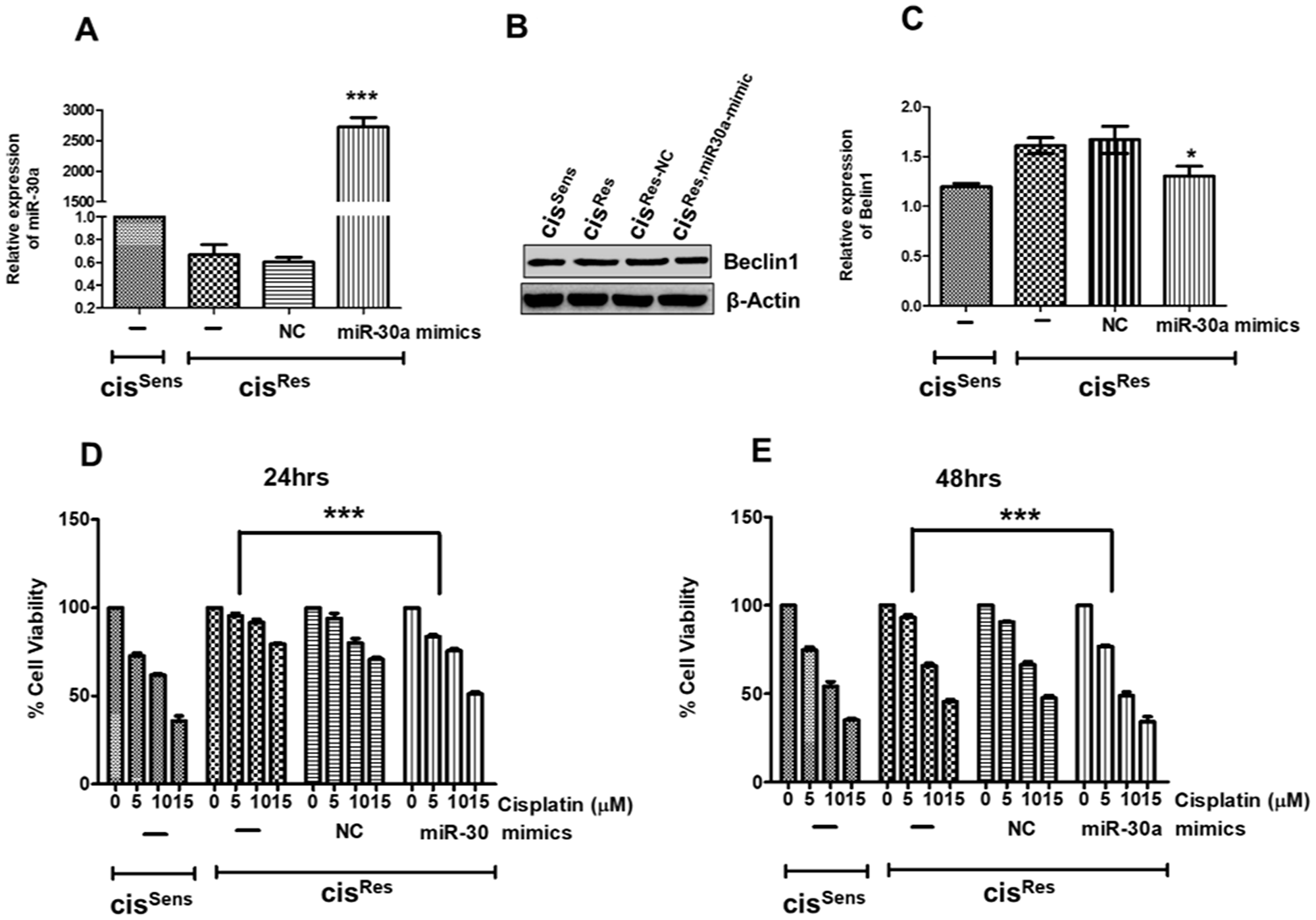
Figure 3: cisRes oral cancer cells were transfected with either miR-30a mimics or NC (negative control) and after 24 h, the following effects were analyzed. cisSens cells were employed for the comparison. (A) miR-30a expression as measured by qRT-PCR. (B) Beclin1 protein expression as determined by the Western blot analysis. (C) Densitometry analysis of Beclin1 Western blot was carried out by normalization to β-actin employed as the loading control. Following miR30a restoration, cells were treated with different concentrations of cisplatin. Cell viability was measured by MTT assay after (D) 24 h and (E) 48 h. Error bars represent the mean ± SD of three independent experiments. *p < 0.05, ***p < 0.001.
We next evaluated if the observed miR-30a effect on autophagy marker influences OSCC cell chemosensitivity. Remarkably, mimics-mediated miR-30a restoration in cisRes OSCC cells resulted in regaining of their cisplatin sensitivity when compared to the cells transfected with NC (Figure 3D, 3E). Notably, cisplatin resistance was significantly reduced at all tested doses (0–15 μM) and time-points (24–48 h). The IC50 value of cisRes-30a-mimics cells was found to decrease by two-fold compared to cisRes cells. This regains in cisplatin-sensitivity is comparable with the parental cisSens OSCC cells, especially at 48 h time-point. Our results thus argue for miR-30a implication in conferring a cisplatin-sensitive phenotype to the chemo-resistant OSCC cells.
miR-30a overexpression induces apoptosis in cisRes OSCC cells
BCl2 has been documented as a miR-30a target in various cancers, including non-small cell lung carcinoma, renal carcinoma, cutaneous squamous cell carcinoma [27, 28]. However, their association in OSCC remains elusive. Herein, we show that mimics-mediated miR-30a restoration in cisRes OSCC cells led to downregulation of anti-apoptotic protein Bcl2, while it enhanced the caspase3 expression compared to NC transfected cells (Figure 4A–4C); increment in caspase3 expression followed cisplatin dosage. miR-30a role in conferring an apoptotic phenotype was further confirmed by Annexin-V-PI staining. As expected, majority of the cisRes OSCC cells were viable (81–90%) even after treatment with two increasing concentrations of cisplatin (3 and 10 μM) (Figure 4D, 4E). Comparable results were obtained in NC-transfected cisRes cells (cisRes-NC). Of note, when cisRes-30a-mim OSCC cells were treated with increasing concentrations of cisplatin, 76–51% of Annexin V–PI– cells were observed. An increase in Annexin V+PI+ population was observed in cells receiving cisplatin treatment (3 or 10 μM, 17.19 and 43.52% respectively), indicative of late apoptotic or dead cells. Our results thus indicate that miR-30a restoration activates apoptosis in cisRes OSCC cells.
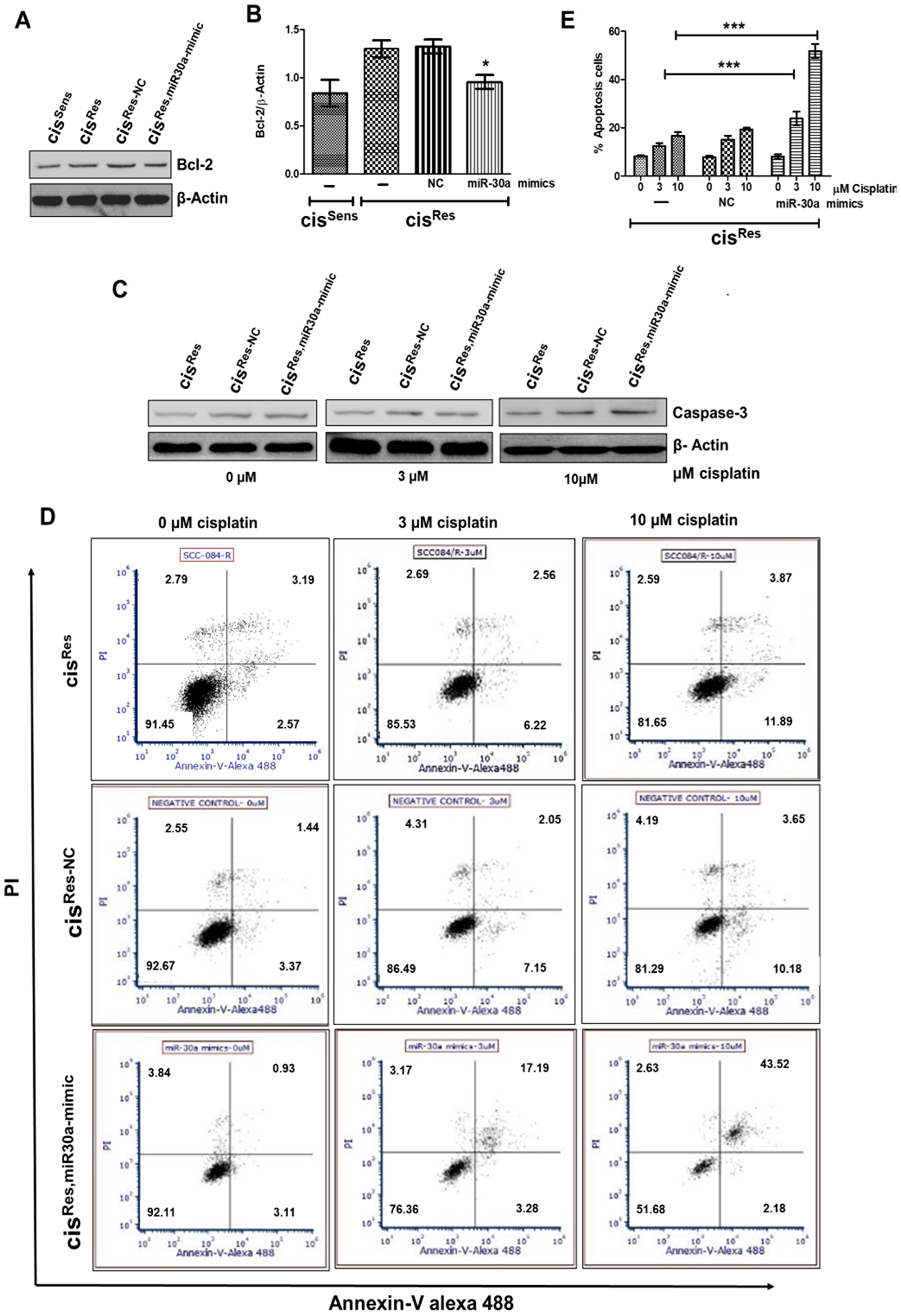
Figure 4: cisRes oral cancer cells were transfected with either miR-30a mimics or NC (negative control) and after 24 h, the following effects were analyzed. cisSens cells were employed for the comparison. (A) Bcl2 protein expression by Western blot. (B) Densitometry analysis of Bcl2 Western blot normalized to β-actin as the loading control. (C) Mimics or NC transfected cells were treated with or without cisplatin and caspase-3 expression was analyzed by Western blot analysis. (D) Cells were treated as described in (C) and after 72 h of cisplatin treatment, cells were collected and stained with FITC-conjugated annexin-V and PI and analyzed by flow cytometry. (E) Graphical representation of the percentage of apoptotic cells as analyzed by FACS analysis. Error bars represent the mean ± SD of three independent experiments. *p < 0.05, ***p < 0.001.
miR-30a restoration impedes migration of cisRes OSCC cells
Numerous studies have indicated that the development of chemoresistance influences cancer metastasis [29]. Having observed the effect of miR-30a on cisplatin-sensitization, we next intended to determine if miR-30a restoration modulates the migratory potential of cisRes OSCC cells. As analyzed by wound assay, cisplatin treatment in a dose-dependent manner enhanced the migration of cisRes OSCC cells, as evident from the faster wound-closure, when compared to cisSens cells (Figure 5). The wound closure percentage is comparable in cisRes-NC and cisRes OSCC cells. Notably, little or no closure was observed in the cisRes-30a-mim OSCC cells at all the cisplatin concentration tested, thereby suggesting that forced expression of miR-30a impaired the motile characteristic of cisRes cells.
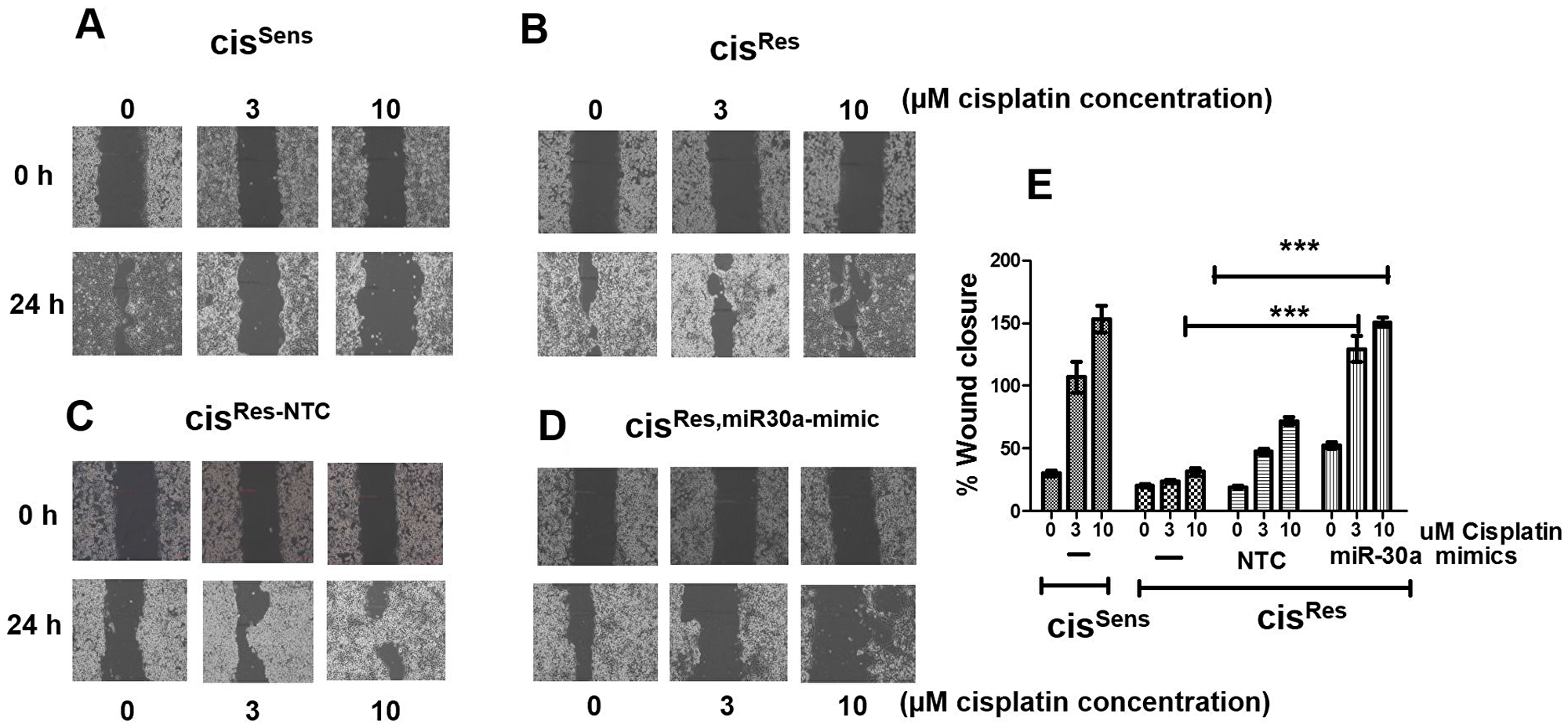
Figure 5: The effects of cisplatin treatment were analyzed on the migration of cisRes oral cancer cells transfected with either miR-30a mimics or NC (non-target control) by wound assay. cisSens cells were employed for the comparison. (A–D) Representative images of wound closure taken at 0 and 24 h after the scratch was made and cisplatin treatment was given. (E) quantification of wound closure as analyzed using Image J. Data are expressed as mean ± SD. ***p < 0.001.
Exosome-mediated miR-30a delivery in cisRes OSCC cells modulated their autophagic, apoptotic and chemo-sensitive response
Exosomes are known to play an essential role in cellular crosstalk within tumor-microenvironment [30]. In the present study, exosomes isolated from both cisRes OSCC cells and serum of OSCC patients showed a marked reduction in miR-30a expression levels (Figure 2). Having observed the essential role of miR-30a restoration on the induction of apoptosis and reversal of cisplatin sensitivity in the CisRes OSCC cells (Figure 4), we next intended to determine if there is any exosomal contribution in mediating these effects. To answer this, we employed a co-conditioning approach wherein, we first restored miR-30a levels in CisRes cells using mimics (CisRes, miR-30a-mimic), thereafter isolated exosomes from these cells and use the isolated exosomes to treat naïve CisRes cells (hereafter referred as cisRes; exomiR-30a-mimic) and analyzed the effects on various functional parameters. The quality of our exosomal preparation was checked and confirmed by a Western blot analysis of exosomal surface marker CD9 (Figure 6A). Notably, we found that expression of Beclin1 (a miR-30a target) is significantly decreased in the cisRes; exomiR-30a-mimic cells compared to cisRes cells. It is worth mentioning that this reduction in Beclin 1expression is comparable to the levels of Beclin1 measured in cisRes following miR-30a restoration and also in the parental cisSens (Figure 6B, 6C). Furthermore, cisplatin sensitivity was significantly enhanced in the cisRes; exomiR-30a-mimic cells when compared to the cisRes OSCC cells (Figure 6D) indicating that exosomal-mediated miR-30a transfer conferred a sensitive trait. Besides, exosomal-mediated miR-30a delivery led to the induction of apoptosis in the cisRes; exomiR-30a-mimic cells. There is an increment of Annexin V+PI+ population from 6.3 to 10.54% in cisRes vs cisRes; exomiR-30a-mimic cells (Figure 6E, 6F) and decreased Bcl2 expression (Figure 6G, 6H). Interestingly, miR-30a expression in the cisRes; exomiR-30a-mimic cells were significantly enhanced when compared to cisRes cells, though the extent of increase was less than that observed with the use of commercial mimics in CisRes, miR-30a-mimic (Figure 6I). These findings thus suggest that exosomes play an essential role in mediating miR-30a transfer and hence restoring cisplatin sensitivity, enhancing apoptosis and decreasing autophagic response in cisRes OSCC cells.
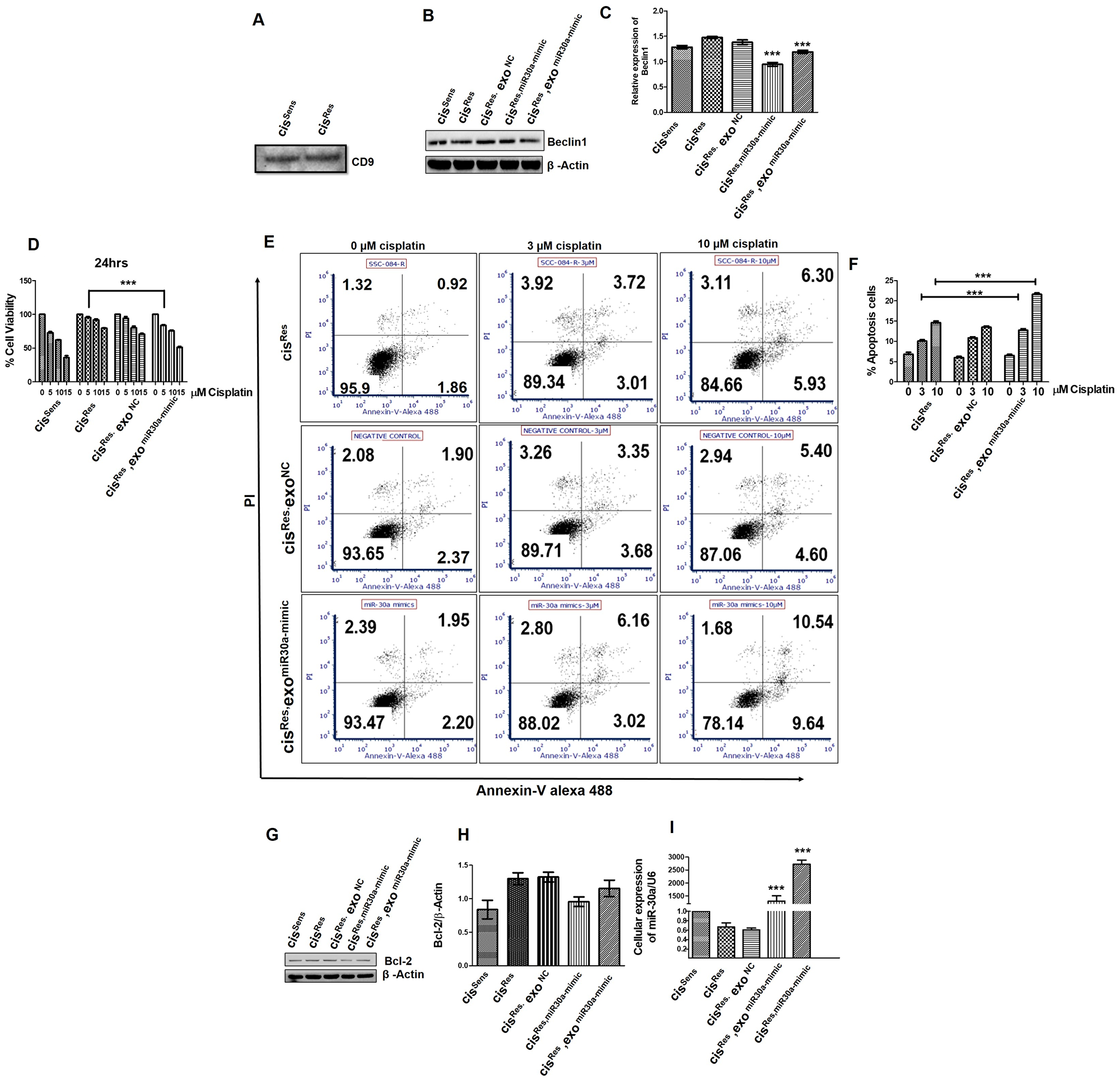
Figure 6: Exosomes were isolated from cisRes cells transfected with either NC or miR-30a mimics and were used to treat other naïve cisRes cells. cisSens, cisRes, and cisRes-miR-30a mimic cells were employed as controls and for comparison. (A) The expression of CD9 in isolated exosomes from different cells was determined by Western blotting. (B) Beclin1 protein expression as determined by western bot. (C) Densitometry analysis of Beclin1 Western blot normalized to β-actin as a loading control. (D) Following exosomes treatment, cells were exposed to different concentrations of cisplatin (μM), as indicated) for 24 h. Cell viability was measured by the MTT assay. (E) For determining apoptosis, after cisplatin treatment, cells were harvested, stained with FITC-conjugated annexin-V and PI and analyzed by flow cytometry. (F) Graphical representation of the percentage of apoptotic cells as analyzed by FACS analysis. (G) BCl2 protein expression as determined by Western blot. (H) Densitometry analysis of BCl2 Western blot normalized to β-actin as a loading control. (I) miR-30a expression levels as determined by real-time PCR. Error bars represent the mean ± SD of three independent experiments. ***p < 0.001.
DISCUSSION
Exosomes, the membrane-bound extracellular vesicles, play an essential role in transferring molecular and genetic information from one cell to another [24, 31]. Some of the miRNAs they cargo serve as a critical regulator of cell-cycle, metabolism, proliferation, migration, invasion, angiogenesis and hence act as a modulator of the tumor microenvironment and chemoresistance [32, 33]. Resistance to anticancer agents is a foremost obstacle to the successful treatment of OSCC, and therefore understanding of the underlying mechanisms is essential for overcoming this therapeutic barrier. In the present study, we provide evidence that exosomal-mediated delivery of miR30a plays an essential role in reversing the sensitivity of cisplatin-resistant OSCC cells, thereby offers a potential therapeutic strategy to overcome the drug-resistance hurdle in OSCC treatment.
Aberrant expression of miRNAs has been linked with tumor progression, metastasis and chemo-resistance observed in various cancer [34]. Pioneering studies have functionally validated many of these miRNAs and demonstrated their mode of action via specific gene-target [35, 36]. However, there is still much remains to be done: allocate a particular exosomal-miRNA with specific cancer, identify its functional gene-target, determine its relevance in drug-resistance and finally to validate these signatures as a prognostic biomarker.
Through a combinatorial approach involving comprehensive literature search and bioinformatics web-based tools, we identified miRNAs that were either up- or down-deregulated in OSCC namely (miR-572, miR-214, miR-200a, miR-7, miR-15a, miR-155a) and (miR-30a, miR-22, miR-21, miR-19a, miR-374, miR-122a, miR-195) respectively [37, 38]. A recent in silico comparative profiling study documented six miRNAs that may be associated with cisplatin resistance in oral cancer (miR130-b, miR-134, miR-149, miR-491, miR-181d, miR-30a) from a total of 36 miRNAs taken into consideration [26]. Studies in renal cell carcinoma, chronic myeloid leukemia, and ovarian cancer showed a diminution in miR-30a expression facilitated apoptosis through an autophagy-reliant pathway [28]. Besides, miR-30a overexpression in lung cancer cells has been reported to enhance their paclitaxel sensitivity [27]. In contrast, miR-30a was found to be overexpressed in cisplatin-resistant melanoma cells, and therefore treatment with miR-30a inhibitor enhanced their chemo-sensitivity [39]. Thus, miR-30a may be employed to reestablish the chemo-sensitivity of certain tumor cells, however, its implication in cisplatin-induced resistance in OSCC remains unknown thus far. Not only this, but the relevance of miR-30a downregulation on various functional attributes of OSCC also remains obscure. While this manuscript was in preparation, miR-30a downregulation in OSCC was shown to suppress cell proliferation and invasion via modulating fibroblast activation protein alpha [25]. Nevertheless, its association with cisplatin-resistance and related functional effects such as autophagy, apoptosis, and migration have not yet been studied. In the present study, we conclusively present evidence demonstrating the loss of miR-30a expression in the cisplatin-resistant oral cancer cells. It is important to mention that besides observation in cisplatin resistant cells, we report here for the first time, decreased miR-30 expression in exosomes isolated from the serum of oral cancer patients vs healthy controls; the effect being more pronounced in the patients with disease recurrence post cisplatin treatment, thus substantiating the association of miR-30a with cisplatin-resistance.
miRNA functions by binding to the 3′UTR of mRNA of the target gene, thereby interfering with its translation and expression. Hence, the identification of downstream targets of miRNA is indispensable for designing efficient treatment strategies as well as overcoming drug-related resistance. For example, HOXB8 (the developmental gene) and p27 (CDKN1B, cell cycle associated gene) have been identified as the direct targets of miR-196a, and HMGA2 as a miR-204 target [40]. In line with these results, we present here original evidence that Beclin1, an autophagy-related gene, acts as the miR-30a target in OSCC and more importantly, showed Beclin1 upregulation corresponded to the development of cisplatin resistance. This is of special relevance as autophagy has been recognized as a tumor cell survival mechanism triggered following various treatment strategies, and hence contributory towards chemoresistance. Our findings thus argue for considering miR-30a target, Beclin1, as a promising therapeutic approach to circumvent cisplatin-resistance in OSCC. Remarkably, our results highlight that miR-30a overexpression in cisplatin-resistant cells led to a decrease in Beclin1 and hence autophagic response, which in turn is reflected in enhancements in apoptosis via downregulation of Bcl2 and concomitant increase in caspase3 expression. Altogether these effects resulted in regaining cisplatin sensitivity in OSCC cells. It is worth mentioning that Beclin1 has recently been linked with OSCC progression [41, 42]. In corroboration with our findings, Zou et al. depicted the role of miR-30a in conferring chemosensitivity to the liver cancer cells via abating Beclin1-mediated autophagy [22]. The importance of our findings is strengthened by the fact that we show the major role of miR-30a in regulating two essential pathways: autophagy and apoptosis, ultimately resulting in sensitizing resistant oral cancer cells towards cisplatin, a commonly used chemotherapeutic drug.
Exosomes, the small extracellular vesicles and cargo of genetic material, proteins, and lipids, function as signaling aid between cancer and adjoining cells. Exosomes strongly influence tumor microenvironment as their uptake alter phenotypic and functional attributes of both the recipient and other cells in the neighborhood [43, 44]. Therefore, an in-depth understanding of exosomal contributions in moderating signaling networks associated with cancer progression, invasion, development of pre-metastatic niche, may identify potential targets to combat therapy resistance. A recent report revealed that exosomal-mediated shuttling of miR-155 from cancer-associated-tumor cells to neighboring cells in tumor microenvironment mediated chemoresistance in breast cancer cells [14]. Analogous to this, here we convincingly demonstrated that exosomal-delivered miR-30a is capable of transferring sensitive phenotype to the cisRes OSCC cells in unification with turning on the apoptotic machinery while disabling the autophagic response.
Taken together, we identified Beclin1 as the miR-30a target in oral cancer, and present here first evidence of the essential role of exosomal-miR-30a in reducing acquired chemo-resistance in OSCC via regulation of autophagic and apoptotic markers (Figure 7). Our study thus provides a strong rationale for employing exosomal-mediated miR-30a delivery as an effective therapeutic approach and also highlights its importance in retrieving sensitivity against cisplatin resistance, a major hurdle in conventional current approaches.
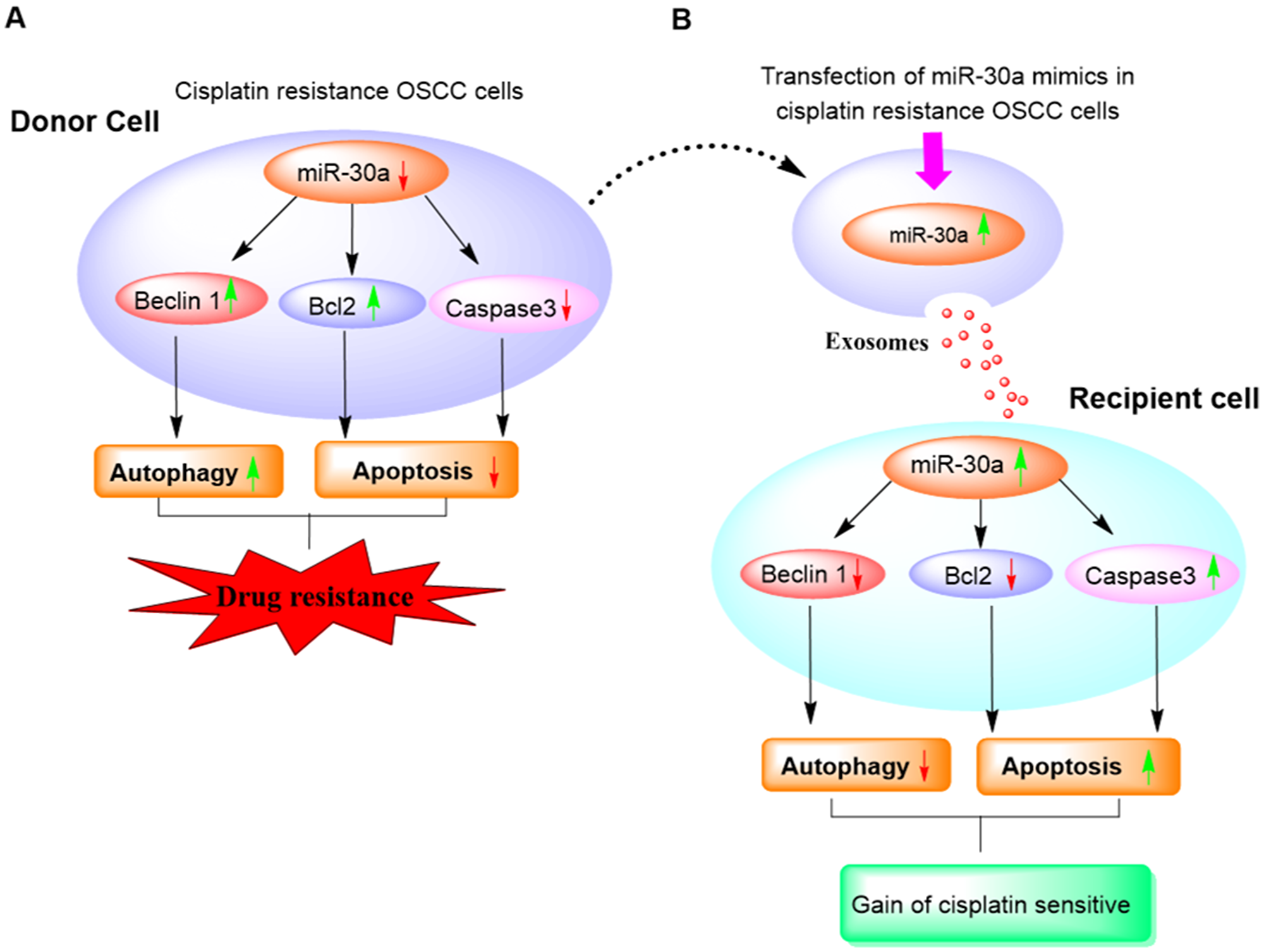
Figure 7: Diagram illustrating the role of exosome-mediated miR30a delivery in a reversal of cisplatin resistance in OSCC cells.
Materials and Methods
miR-30a target prediction
Prediction of miRNA targets implicated in OSCC chemoresistance was done using web-based tools: TargetScan [45], DIANA-micro-T-CDS [46] and RNA-hybrid [47]. The screening of miRNA targets using TargetScan is based on the mRNA’s 3′UTR seed-match and free-energy. RNA-hybrid utilizes seed-match, free-energy, and p-value for predicting potential miRNAs binding sites in the mRNA’s 3′UTRs, and determines the structure with minimum free-energy. DIANA-micro-T-CDS employs an mRNA reference sequence database to determine 3′UTR sequence; dynamic algorithm programming allows the identification of minimum free-energy. We first employed TargetScan and DIANA-micro-T-CDS to predict the most potential gene targets. From the screened potential gene targets, whose involvement in the chemoresistance have been reported were further considered. Final selection was done on the basis of scores from the two tools and also based on the evaluation of binding energy as analysed by RNAhybrid.
OSCC patients samples and cells
OSCC serum samples were provided by Dr. Rakesh Rawal, Department of Life Science, Gujarat University. The sample collection was done following the Institutional Ethics Committee of Gujarat University (GU/IEC/10/20118). Blood from healthy volunteers with or without tobacco history was collected (Table 1) and processed to obtain serum as described, with some modifications [48]. Briefly, blood samples collected in vacutainer tubes were allowed to stand at room temperature for 30–60 min to allow the clot formation. This was followed by centrifugation at 1200 g for 20 minutes at room temperature, subsequently, clear supernatant was collected in a fresh tube and stored at –80°C, until further use.
Table 1: Detail of clinical samples used in the study
| Variable Considered | Healthy Volunteers | Healthy Volunteers (with tobacco history) | OSCC patients | OSCC patients with tumor recurrence (post cisplatin treatment) | |
|---|---|---|---|---|---|
| Gender | Number of Male / Female (Mixed) | 7 | 7 | 7 | 6 |
| Tobacco history | Tobacco consumption/ chewing/smoking habit | No | Yes | Yes | Yes |
| Tumor site | No tumor is seen | No tumor is seen | Buccal Mucosa, Tongue, and Floor of Mouth | Buccal Mucosa, Tongue, and Floor of Mouth | |
| Tumor size | T1 & T2 | 0 | 0 | 5 | 6 |
| T3 & T4 | 0 | 0 | 2 | 0 | |
| Lymph node | Positive | No | No | 3 | |
| Metastasis | Positive | No | No | 0 | 0 |
Human OSCC cells UPCI: SCC084 and its cisplatin-resistant strain (referred to as cisSens and cisRes respectively) were received as a gift from Dr. Ruma Dey Ghosh (Tata-TCRC, Kolkata) and were cultured as previously described [26]. Briefly, both cisSens and cisRes cells were grown in DMEM medium with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37°C in a humidified 5% CO2 incubator. Besides, cisRes cells were maintained with 1 μg/ml cisplatin (Sigma-Aldrich) for 48 h, every alternate passage. However, for experimental purposes, cisRes cells were grown in the cisplatin-free culture medium.
Western blot
The total cell lysate was prepared using RIPA buffer as described [49] and Western blotting was carried out as detailed [50]. The following antibodies were used: rabbit polyclonal anti-Beclin1 (1:2000, Abcam), Anti-Bcl2 (1:500, Abcam), Anti-CD9 antibody (1:250 Abcam) and anti-rabbit or anti-mouse secondary antibodies conjugated to horse-radish-peroxidase (1:20,000, Abcam) and β-Actin (1:5000, Santacruz Inc.,). Following TBST washes, protein bands were visualized using ECL and ChemiDoc Imaging System and quantified with Image-J.
Exosomes isolation
Exosomes were isolated from both cell culture media (2 ml) and serum (200 μl) obtained from the clinical samples adopting the ultracentrifugation method as has been previously described with some modifications [51]. Briefly, an initial spin was performed at 300 g for 10 min at 4°C to remove the live cells. The supernatant was collected and subjected to a series of centrifugation: first at 2000 g for 10 min at 4°C to get rid of the dead cells and apoptotic bodies. The clear supernatant was collected and subjected to another round of centrifugation at 10,000 g for 30 min at 4°C. The supernatant was collected in a clean tube and ultracentrifuged for 1,00,000 g for 70 min at 4°C to obtain the exosomal pellet, which was then re-suspended in ice-cold PBS and subjected to another cycle of ultracentrifugation (as a washing step). The cleaned exosomal pellet was finally resuspended in 100 μl of PBS and stored at –80°C until further use [51].
RNA and miRNA isolation and real-time PCR (qPCR)
Total RNA and exosomal miRNA were isolated using Qiazol and miRNeasy Kit (QIAGEN) respectively [52]. 1 μg RNA was reverse-transcribed to cDNA using iscript-cDNA synthesis kit (BioRad) and subsequently, qPCR was done using Beclin1 syber-green probe and iQ SYBR® Green Supermix. 18S syber-green probe was employed as endogenous control (Supplementary Table 2). For miR-30a analysis, qPCR was performed using a TaqMan probe (AppliedBiosystems) with U6snRNA as an internal control. Results were evaluated by 2–ΔΔCt quantification method [52].
miRNA-mimics transfection
CisRes cells were transfected with either non-target control [NTC or Negative control (NC)] or miR-30a mimics using Lipofectamine RNAiMAX reagent as described [52]. 48 h later, cells were used in the respective experiments.
Cell viability
Cell viability was assessed by MTT assay as described [53]. Briefly, cells in 96-well plates (1 × 103 cells/well) were treated with different cisplatin concentrations. 48 h later MTT reagent was added and absorbance was read at 570 nm. The absorbance of untreated cells served as blank.
Annexin V-PI binding
For assessing apoptosis, 0.5 × 106 cells in 6-MW plates were treated with cisplatin (3 and 10 μM) for 72 h. Cells were then harvested with trypsin, washed with PBS and stained with Annexin V-Alexa 488 (Invitrogen) and PI (2 μg/ml, Sigma-Aldrich) for 15 min. Cells were analyzed using FACS S3e Cell Sorter.
Cell migration
Cell migration was evaluated by wound healing assay as described [53]. Briefly, 0.5 × 106 cells in 6-MW plates were transfected with NC or miR-30a mimics. 48 h later, a scratch was created using a sterile microtip. The wound area was washed gently with PBS and fresh media having different concentrations of cisplatin (0, 3 and 10 μM) was added. Photographs were taken at different times using a Zeiss inverted fluorescence microscope. The percentage of wound closure was calculated using Image J.
Exosomes co-conditioning
To study the possible role of exosomes in the transmission of chemoresistance, 0.5 × 106 cisRes cells in 6-MW plates were transfected with either NC or miR-30a mimics. After 48 h, cells were serum-starved for 24 h and subsequently, exosomes were isolated. Exosomal preparation was used to treat naïve cisRes cells for 48 h and then cellular miR-30a levels were analyzed.
Beclin1 cloning and luciferase reporter assay
Beclin1 3′UTR target sequence in miR-30a was synthesized from IDT (Supplementary Table 3). The reconstituted oligonucleotides were annealed and cloned in the pmirGLO dual-luciferase vector (Promega) at the restriction-endonuclease (RE) sites (XbaI, PmeI). Following ligation, clones were transformed into E. coli DH5α cells. The plasmid was isolated, purified and quantified using nanodrop. The cloned vector was verified both by RE digestion (Supplementary Figure 1) and sequencing (Supplementary Figure 2). 3′UTRs of Beclin1 were thus cloned downstream of the luciferase gene in the pmirGLO vector (pmirGLO-BECN1). 1 × 105 cisRes cells/well in the 24-MW plate were co-transfected with the pmirGLO-BECN1-vector and miR-30a or NC mimics using Lipofectamine-2000. 48 h later, luciferase reporter activity was assayed using a Dual-Luciferase Assay System (Promega, USA). The analysis was done following normalization with Renilla luciferase activity.
Statistical analysis
Statistical analysis was done using GraphPad Prism 5.01. Means of all data were compared by student t-test and ANOVA, followed by Bonferroni’s post-hoc test. Data are expressed as mean ± standard-deviation. p value < 0.05 was considered significant. In all cases, experiments have been done three times. Representative graph or figure has been shown as two other identical experiments gave similar results.
Abbreviations
miRNA: microRNA; OSCC: oral squamous cell carcinoma; NC: negative control; NTC: non-target control; Bcl2: B-cell lymphoma 2; UTR: untranslated region; ECL: enhanced chemo luminescence; TBST: tris buffer saline-Tween 20; BCA: bichinconic acid; PI: propidium iodide; RE: restriction enzyme; FACS: fluorescence associated cell sorting.
ACKNOWLEDGMENTS
Authors would like to acknowledge the Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, India, for supporting this research at NIPER- Ahmedabad. We would like to acknowledge the University of Pittsburgh Graduate School of Public Health, Pittsburgh, PA 15261, USA and Dr. Sussane Gollin for providing of SCC:084 and SCC:131 cell lines. We are thankful to Dr. Ruma Dey Ghosh (Tata translational cancer research institute, Kolkata, India) for providing us cell lines. We are also thankful to Dr. Rakesh Rawal, Department of life science Gujarat university, India) for providing oral cancer patients serum samples. AJ gratefully acknowledges the support of the Ramalingaswami Fellowship from the Department of Biotechnology, Govt. of India.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest.
FUNDING
The authors have received funding from the National Institute of Pharmaceutical Education and Research, Department of Pharmaceutics, Ministry of Chemicals and Fertilizers, Government of India. The funders had no role in study design, data collection, and analysis, decision to publish, or preparation of the manuscript.
References
1. Sharma S, Satyanarayana L, Asthana S, Shivalingesh KK, Goutham BS, Ramachandra S. Oral cancer statistics in India on the basis of first report of 29 population-based cancer registries. J Oral Maxillofac Pathol. 2018; 22:18–26. [PubMed].
2. Zhong LP, Zhang CP, Ren GX, Guo W, William WN Jr, Sun J, Zhu HG, Tu WY, Li J, Cai YL, Wang LZ, Fan XD, Wang ZH, et al. Randomized phase III trial of induction chemotherapy with docetaxel, cisplatin, and fluorouracil followed by surgery versus up-front surgery in locally advanced resectable oral squamous cell carcinoma. J Clin Oncol. 2013; 31:744–51. https://doi.org/10.1200/JCO.2012.43.8820. [PubMed].
3. White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, Gollin SM. The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines. Oral Oncol. 2007; 43:701–12. https://doi.org/10.1016/j.oraloncology.2006.09.001. [PubMed].
4. Roma-Rodrigues C, Fernandes AR, Baptista PV. Exosome in tumour microenvironment: overview of the crosstalk between normal and cancer cells. BioMed Res Int. 2014; 2014:179486. https://doi.org/10.1155/2014/179486. [PubMed].
5. Vautrot V, Chanteloup G, Elmallah M, Cordonnier M, Aubin F, Garrido C, Gobbo J. Exosomal miRNA: Small Molecules, Big Impact in Colorectal Cancer. J Oncol. 2019; 2019:8585276. https://doi.org/10.1155/2019/8585276. [PubMed].
6. Bae S, Brumbaugh J, Bonavida B. Exosomes derived from cancerous and non-cancerous cells regulate the anti-tumor response in the tumor microenvironment. Genes Cancer. 2018; 9:87–100. https://doi.org/10.18632/genesandcancer.172. [PubMed].
7. Kulkarni B, Kirave P, Gondaliya P, Jash K, Jain A, Tekade RK, Kalia K. Exosomal miRNA in chemoresistance, immune evasion, metastasis and progression of cancer. Drug Discov Today. 2019; 24:2058–67. https://doi.org/10.1016/j.drudis.2019.06.010. [PubMed].
8. Munson P, Shukla A. Exosomes: potential in cancer diagnosis and therapy. Medicines (Basel). 2015; 2:310–27. https://doi.org/10.3390/medicines2040310. [PubMed].
9. Acunzo M, Romano G, Wernicke D, Croce CM. MicroRNA and cancer—a brief overview. Adv Biol Regul. 2015; 57:1–9. https://doi.org/10.1016/j.jbior.2014.09.013. [PubMed].
10. Patel GK, Patton MC, Singh S, Khushman M, Singh AP. Pancreatic cancer exosomes: shedding off for a meaningful journey. Pancreat Disord Ther. 2016; 6:e148. https://doi.org/10.4172/2165-7092.1000e148. [PubMed].
11. Ding G, Zhou L, Qian Y, Fu M, Chen J, Chen J, Xiang J, Wu Z, Jiang G, Cao L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget. 2015; 6:29877–88. https://doi.org/10.18632/oncotarget.4924. [PubMed].
12. Charrier A, Chen R, Chen L, Kemper S, Hattori T, Takigawa M, Brigstock DR. Connective tissue growth factor (CCN2) and microRNA-21 are components of a positive feedback loop in pancreatic stellate cells (PSC) during chronic pancreatitis and are exported in PSC-derived exosomes. J Cell Commun Signal. 2014; 8:147–56. https://doi.org/10.1007/s12079-014-0220-3. [PubMed].
13. Zhou M, Chen J, Zhou L, Chen W, Ding G, Cao L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014; 292:65–69. https://doi.org/10.1016/j.cellimm.2014.09.004. [PubMed].
14. Santos JC, Lima ND, Sarian LO, Matheu A, Ribeiro ML, Derchain SF. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep. 2018; 8:829–40. https://doi.org/10.1038/s41598-018-19339-5. [PubMed].
15. Zeng A, Wei Z, Yan W, Yin J, Huang X, Zhou X, Li R, Shen F, Wu W, Wang X, You Y. Exosomal transfer of miR-151a enhances chemosensitivity to temozolomide in drug-resistant glioblastoma. Cancer Lett. 2018; 436:10–21. https://doi.org/10.1016/j.canlet.2018.08.004. [PubMed].
16. Liu T, Zhang X, Du L, Wang Y, Liu X, Tian H, Wang L, Li P, Zhao Y, Duan W, Xie Y, Sun Z, Wang C. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol Cancer. 2019; 18:43–60. https://doi.org/10.1186/s12943-019-0981-7. [PubMed].
17. Li Y, Zhang J, Liu Y, Zhang B, Zhong F, Wang S, Fang Z. MiR-30a-5p confers cisplatin resistance by regulating IGF1R expression in melanoma cells. BMC Cancer. 2018; 18:404. https://doi.org/10.1186/s12885-018-4233-9. [PubMed].
18. Wang T, Chen G, Ma X, Yang Y, Chen Y, Peng Y, Bai Z, Zhang Z, Pei H, Guo W. MiR-30a regulates cancer cell response to chemotherapy through SNAI1/IRS1/AKT pathway. Cell Death Dis. 2019; 10:153–68. https://doi.org/10.1038/s41419-019-1326-6. [PubMed].
19. Chen S, Wu J, Jiao K, Wu Q, Ma J, Chen D, Kang J, Zhao G, Shi Y, Fan D, Zhao G. MicroRNA-495-3p inhibits multidrug resistance by modulating autophagy through GRP78/mTOR axis in gastric cancer. Cell Death Dis. 2018; 9:1070–82. https://doi.org/10.1038/s41419-018-0950-x. [PubMed].
20. Gozuacik D, Akkoc Y, Ozturk DG, Kocak M. Autophagy-Regulating microRNAs and Cancer. Front Oncol. 2017; 7:65–87. https://doi.org/10.3389/fonc.2017.00065. [PubMed].
21. Sheng J, Qin H, Zhang K, Li B, Zhang X. Targeting autophagy in chemotherapy-resistant of hepatocellular carcinoma. Am J Cancer Res. 2018; 8:354–65. [PubMed].
22. Zou Z, Wu L, Ding H, Wang Y, Zhang Y, Chen X, Chen X, Zhang CY, Zhang Q, Zen K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J Biol Chem. 2012; 287:4148–56. https://doi.org/10.1074/jbc.M111.307405. [PubMed].
23. Yang X, Bai F, Xu Y, Chen Y, Chen L. Intensified Beclin-1 mediated by low expression of miR-30a-5p promotes chemoresistance in human small cell lung cancer. Cell Physiol Biochem. 2017; 43:1126–39. https://doi.org/10.1159/000481754. [PubMed].
24. Jiang LH, Zhang HD, Tang JH. MiR-30a: a novel biomarker and potential therapeutic target for cancer. J Oncol. 2018; 2018:5167829. https://doi.org/10.1155/2018/5167829. [PubMed].
25. Ruan P, Tao Z, Tan A. Low expression of miR-30a-5p induced the proliferation and invasion of oral cancer via promoting the expression of FAP. Biosci Rep. 2018; 38:BSR20171027. https://doi.org/10.1042/BSR20171027. [PubMed].
26. Ghosh RD, Ghuwalewala S, Das P, Mandloi S, Alam SK, Chakraborty J, Sarkar S, Chakrabarti S, Panda CK, Roychoudhury S. MicroRNA profiling of cisplatin-resistant oral squamous cell carcinoma cell lines enriched with cancer-stem-cell-like and epithelial-mesenchymal transition-type features. Sci Rep. 2016; 6:23932. https://doi.org/10.1038/srep23932. [PubMed].
27. Xu X, Jin S, Ma Y, Fan Z, Yan Z, Li W, Song Q, You W, Lyu Z, Song Y, Shi P, Liu Y, Han X, et al. miR-30a-5p enhances paclitaxel sensitivity in non-small cell lung cancer through targeting BCL-2 expression. J Mol Med (Berl). 2017; 95:861–71. https://doi.org/10.1007/s00109-017-1539-z. [PubMed].
28. Yang X, Chen Y, Chen L. The versatile role of microRNA-30a in human cancer. Cell Physiol Biochem. 2017; 41:1616–32. https://doi.org/10.1159/000471111. [PubMed].
29. Jo Y, Choi N, Kim K, Koo HJ, Choi J, Kim HN. Chemoresistance of Cancer Cells: Requirements of Tumor Microenvironment-mimicking In Vitro Models in Anti-Cancer Drug Development. Theranostics. 2018; 8:5259–75. https://doi.org/10.7150/thno.29098. [PubMed].
30. Whiteside TL. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin Immunol. 2018; 35:69–79. https://doi.org/10.1016/j.smim.2017.12.003. [PubMed].
31. Wan Z, Gao X, Dong Y, Zhao Y, Chen X, Yang G, Liu L. Exosome-mediated cell-cell communication in tumor progression. Am J Cancer Res. 2018; 8:1661–73. [PubMed].
32. Whiteside TL. Tumor-derived exosomes and their role in cancer progression. Adv Clin Chem. 2016; 74:103–41. https://doi.org/10.1016/bs.acc.2015.12.005. [PubMed].
33. Zhang C, Ji Q, Yang Y, Li Q, Wang Z. Exosome: function and role in cancer metastasis and drug resistance. Technol Cancer Res Treat. 2018; 17:1533033818763450. https://doi.org/10.1177/1533033818763450. [PubMed].
34. Frixa T, Donzelli S, Blandino G. Oncogenic microRNAs: key players in malignant transformation. Cancers (Basel). 2015; 7:2466–85. [PubMed].
35. Hong L, Han Y, Zhang Y, Zhang H, Zhao Q, Wu K, Fan D. MicroRNA-21: a therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets. 2013; 17:1073–80. https://doi.org/10.1517/14728222.2013.819853. [PubMed].
36. Weiner-Gorzel K, Dempsey E, Milewska M, McGoldrick A, Toh V, Walsh A, Lindsay S, Gubbins L, Cannon A, Sharpe D, O’Sullivan J, Murphy M, Madden SF, et al. Overexpression of the microRNA miR-433 promotes resistance to paclitaxel through the induction of cellular senescence in ovarian cancer cells. Cancer Med. 2015; 4:745–58. https://doi.org/10.1002/cam4.409. [PubMed].
37. Manikandan M, Deva Magendhra Rao AK, Arunkumar G, Manickavasagam M, Rajkumar KS, Rajaraman R, Munirajan AK. Oral squamous cell carcinoma: microRNA expression profiling and integrative analyses for elucidation of tumourigenesis mechanism. Mol Cancer. 2016; 15:28. https://doi.org/10.1186/s12943-016-0512-8. [PubMed].
38. Zeng Q, Tao X, Huang F, Wu T, Wang J, Jiang X, Kuang Z, Cheng B. Overexpression of miR-155 promotes the proliferation and invasion of oral squamous carcinoma cells by regulating BCL6/cyclin D2. Int J Mol Med. 2016; 37:1274–80. https://doi.org/10.3892/ijmm.2016.2529. [PubMed].
39. Cooper DR, Wang C, Patel R, Trujillo A, Patel NA, Prather J, Gould LJ, Wu MH. Human Adipose-Derived Stem Cell Conditioned Media and Exosomes Containing MALAT1 Promote Human Dermal Fibroblast Migration and Ischemic Wound Healing. Advances in Wound Care. 2018; 7:299–308. https://doi.org/10.1089/wound.2017.0775. [PubMed].
40. Tsai SC, Huang SF, Chiang JH, Chen YF, Huang CC, Tsai MH, Tsai FJ, Kao MC, Yang JS. The differential regulation of microRNAs is associated with oral cancer. Oncol Rep. 2017; 38:1613–20. https://doi.org/10.3892/or.2017.5811. [PubMed].
41. Liu JL, Chen FF, Chang SF, Chen CN, Lung J, Lo CH, Lee FH, Lu YC, Hung CH. Expression of Beclin family proteins is associated with tumor progression in oral cancer. PLoS One. 2015; 10:e0141308. https://doi.org/10.1371/journal.pone.0141308. [PubMed].
42. Kapoor V, Paliwal D, Baskar Singh S, Mohanti BK, Das SN. Deregulation of Beclin 1 in patients with tobacco-related oral squamous cell carcinoma. Biochem Biophys Res Commun. 2012; 422:764–69. https://doi.org/10.1016/j.bbrc.2012.05.079. [PubMed].
43. Barok M, Puhka M, Vereb G, Szollosi J, Isola J, Joensuu H. Cancer-derived exosomes from HER2-positive cancer cells carry trastuzumab-emtansine into cancer cells leading to growth inhibition and caspase activation. BMC Cancer. 2018; 18:504. https://doi.org/10.1186/s12885-018-4418-2. [PubMed].
44. Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. 2019; 18:32. https://doi.org/10.1186/s12943-019-0975-5. [PubMed].
45. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015; 4:e05005. https://doi.org/10.7554/eLife.05005. [PubMed].
46. Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, Filippidis C, Dalamagas T, Hatzigeorgiou AG. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013; 41:W169–73. https://doi.org/10.1093/nar/gkt393. [PubMed].
47. Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006; 34:W451–4. https://doi.org/10.1093/nar/gkl243. [PubMed].
48. Tuck MK, Chan DW, Chia D, Godwin AK, Grizzle WE, Krueger KE, Rom W, Sanda M, Sorbara L, Stass S, Wang W, Brenner DE. Standard operating procedures for serum and plasma collection: early detection research network consensus statement standard operating procedure integration working group. J Proteome Res. 2009; 8:113–17. https://doi.org/10.1021/pr800545q. [PubMed].
49. Garg R, Blando J, Perez CJ, Wang H, Benavides FJ, Kazanietz MG. Activation of nuclear factor κB (NF-κB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon). J Biol Chem. 2012; 287:37570–82. https://doi.org/10.1074/jbc.M112.398925. [PubMed].
50. Garg R, Gupta S, Maru GB. Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzo[a]pyrene-treated mice: mechanism of its anti-initiating action. Carcinogenesis. 2008; 29:1022–32. https://doi.org/10.1093/carcin/bgn064. [PubMed].
51. Li P, Kaslan M, Lee SH, Yao J, Gao Z. Progress in exosome isolation techniques. Theranostics. 2017; 7:789–804. https://doi.org/10.7150/thno.18133. [PubMed].
52. Gondaliya P, Dasare A, Srivastava A, Kalia K. miR29b regulates aberrant methylation in In-Vitro diabetic nephropathy model of renal proximal tubular cells. PLoS One. 2018; 13:e0208044. https://doi.org/10.1371/journal.pone.0208044. [PubMed].
53. Garg R, Blando JM, Perez CJ, Abba MC, Benavides F, Kazanietz MG. Protein kinase C epsilon cooperates with PTEN loss for prostate tumorigenesis through the CXCL13-CXCR5 pathway. Cell Rep. 2017; 19:375–88. https://doi.org/10.1016/j.celrep.2017.03.042. [PubMed].