
Introduction
Despite recent advances in the management of the disease, esophageal cancer (EC) is the sixth most lethal malignant disease with nearly half a million novel cases and over 400,000 deaths worldwide [1]. Multimodal therapy including neoadjuvant chemo-radiotherapy followed by surgical resection of the cancer and regional lymph nodes represents the standard of care in this tumor entity [2]. Esophageal adenocarcinoma (AC) and squamous cell carcinoma (SCC) account for >90% of all malignant neoplasms of the esophagus. In recent years, the incidence for both tumor entities changed in Western countries with an increase of AC and a decrease of SCC, which is likely caused by changes in lifestyle habits [3]. Today, therapeutic decisions as to whether a patient receives neoadjuvant treatment, operation or palliative treatment are made according to the preoperative TNM staging [4]. However, esophageal cancer is highly heterogeneous and tumors with identical TNM stage demonstrate marked differences in clinical course and treatment response. Thus, the identification of markers predicting malignant potential and prognosis are of great importance.
The p16 tumor suppressor has been reported to play a pivotal role in cancer, since it inhibits cyclin-dependent kinases (CDKs) 4 and 6 at the G1 to S-phase transition of the cell cycle and thus prevents phosphorylation of the retinoblastoma (RB1) protein [5]. Maintaining hypophosphorylation of RB family members promotes binding to E2F1 and leads to G1 cell cycle arrest [6]. p16 is encoded by the CDKN2A gene localized on chromosome 9p21 within the INK4/ARF locus (reviewed in [7]). p16 plays an important role during carcinogenesis and tumor progression in numerous tumor entities including cancers of the colon, liver, gall bladder, and skin (reviewed in [8]). Its expression is associated with unfavorable or favorable tumor phenotype depending on the analyzed tumor entity (reviewed in [6, 8]). In EC, different types of p16 inactivation have been described, such as homozygous and heterozygous CDKN2A deletions, deleterious point mutations and p16 promoter methylation [9–13]. Previous studies additionally suggest, that alterations of p16 occur early during tumorigenesis as they are commonly seen in Barret’s dysplasia and peritumoral mucosa [14].
To elucidate the potential role of both p16 expression and CDKN2A deletion as prognostic biomarkers we examined our preexisting EC tissue microarray (TMA) built from tumor samples of more than 690 individual EC patients. The database attached to this TMA contains comprehensive molecular, pathological and clinical follow up data.
Results
p16 and Ki67 immunohistochemistry (IHC)
p16 immunostaining was interpretable in 351 AC and 280 SCC. Non-informative cases were due to lack of tissue samples or absence of unequivocal cancer tissue in the TMA spot. 30.2% (N = 106) of all AC and 13.9% (N = 39) of SCC showed positive staining for p16. Representative images of p16 immunostaining in AC and SCC are given in Figure 1. p16 positivity was not associated with any clinical parameters in AC (Table 1) whereas in SCC positive p16 immunostaining correlated with gender (P = 0.032) and low tumor stage (P = 0.014, Table 2).
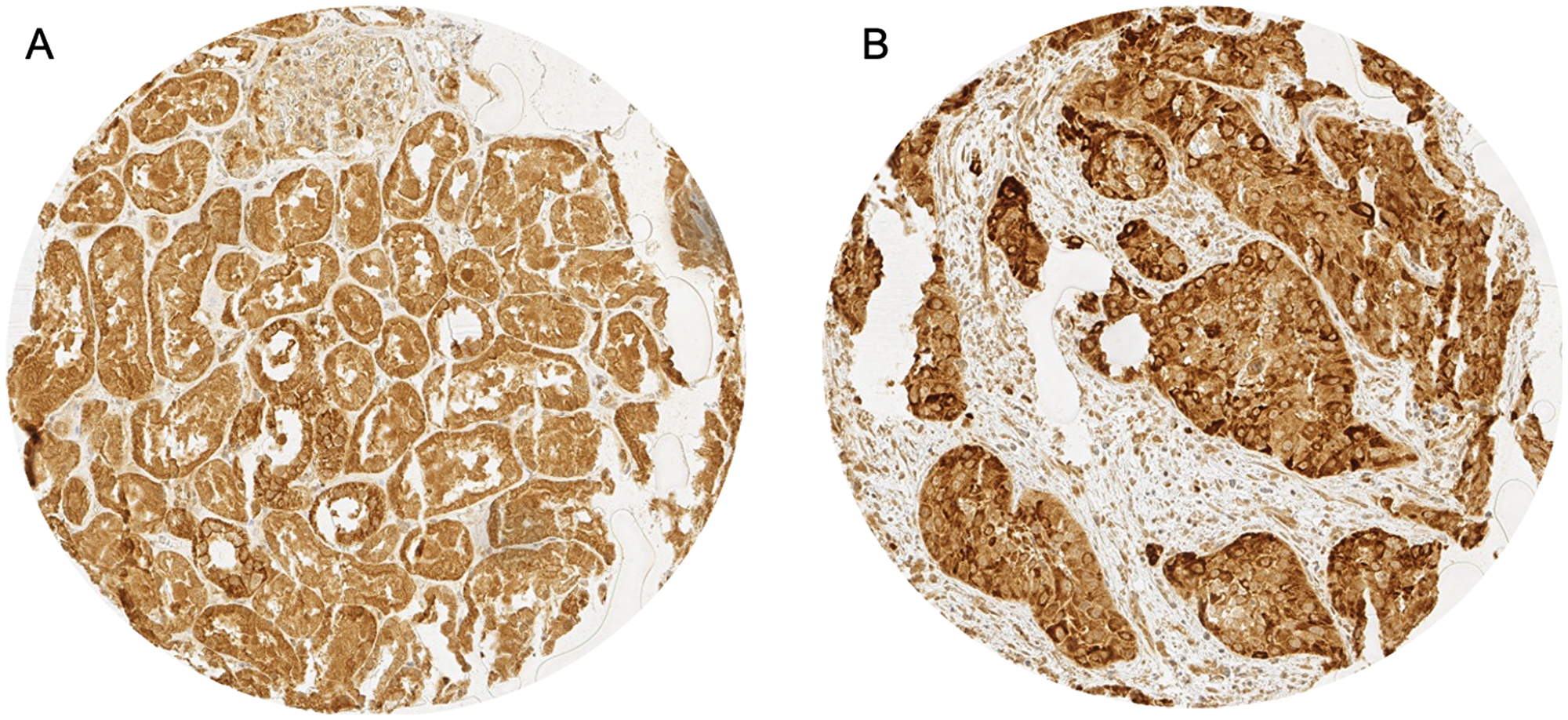
Figure 1: Representative images of p16 immunostaining in (A) p16 cytoplasmatic (red arrow) and nuclear (blue arrow) staining in adenocarcinoma and (B) p16 cytoplasmatic staining in squamous cell carcinoma.
Table 1: Association of p16 immunostaining, CDKN2A FISH and Ki67LI with clinico-pathological parameters in adenocarcinoma
| p16 IHC | CDKN2A FISH | Ki67 IHC | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | neg. | pos. | p | n | no del | het del | hom del | p | n | <10% | 10–80% | >80% | p | ||
| All tumors | 351 | 69.8 | 30.2 | 202 | 67.9 | 25.7 | 6.4 | 312 | 54.5 | 40.4 | 5.1 | ||||
| Age group | <65 yrs | 120 | 69.2 | 30.8 | 0.852 | 63 | 58.8 | 31.7 | 9.5 | 0.153 | 105 | 54.3 | 41.0 | 4.8 | 0.973 |
| >65 yrs | 231 | 70.1 | 29.9 | 139 | 72.0 | 23.0 | 5.0 | 207 | 54.6 | 40.1 | 5.3 | ||||
| Sex | male | 293 | 71.0 | 29.0 | 0.316 | 170 | 68.8 | 25.3 | 5.9 | 0.623 | 261 | 54.8 | 39.8 | 5.4 | 0.198 |
| female | 56 | 64.3 | 35.7 | 31 | 61.3 | 29.0 | 9.7 | 50 | 54.0 | 44.0 | 2.0 | ||||
| Tumor stage | pT1 | 75 | 66.7 | 33.3 | 0.426 | 30 | 80.0 | 16.7 | 3.3 | 0.281 | 62 | 77.4 | 22.6 | 0.0 | 0.001 |
| pT2 | 37 | 62.2 | 37.8 | 20 | 55.0 | 30.0 | 15.0 | 33 | 48.5 | 51.5 | 0.0 | ||||
| pT3 | 213 | 71.8 | 28.2 | 133 | 67.6 | 25.6 | 6.8 | 192 | 47.4 | 44.8 | 7.8 | ||||
| pT4 | 24 | 79.2 | 20.8 | 18 | 61.1 | 38.9 | 0.0 | 23 | 56.5 | 39.1 | 4.3 | ||||
| Lymph node metastasis | pN0 | 115 | 69.6 | 30.4 | 0.298 | 56 | 66.0 | 28.6 | 5.4 | 0.734 | 95 | 66.3 | 30.5 | 3.2 | 0.009 |
| pN1 | 58 | 63.8 | 36.2 | 30 | 76.7 | 13.3 | 10.0 | 53 | 60.4 | 37.7 | 1.9 | ||||
| pN2 | 87 | 67.8 | 32.2 | 51 | 68.6 | 25.5 | 5.9 | 78 | 52.6 | 39.7 | 7.7 | ||||
| pN3 | 89 | 77.5 | 22.5 | 64 | 35.9 | 29.7 | 6.2 | 84 | 38.1 | 54.8 | 7.1 | ||||
| UICC Stage | I | 77 | 68.8 | 31.2 | 0.887 | 34 | 73.6 | 17.6 | 8.8 | 0.535 | 62 | 79.0 | 21.0 | 0.0 | 0.001 |
| II | 46 | 67.4 | 32.6 | 23 | 60.9 | 39.1 | 0.0 | 39 | 43.6 | 51.3 | 5.1 | ||||
| III | 188 | 72.3 | 27.7 | 118 | 67.0 | 25.4 | 7.6 | 173 | 50.3 | 43.9 | 5.8 | ||||
| IV | 36 | 69.4 | 30.6 | 24 | 66.6 | 29.2 | 4.2 | 34 | 41.2 | 47.1 | 11.8 | ||||
| Distant metastasis | M0 | 312 | 69.6 | 30.4 | 0.774 | 176 | 68.8 | 25.0 | 6.2 | 0.763 | 276 | 56.5 | 39.1 | 4.3 | 0.061 |
| M1 | 39 | 71.8 | 28.2 | 26 | 61.5 | 30.8 | 7.7 | 36 | 38.9 | 50.0 | 11.1 | ||||
| Surgical resection margin | R0 | 259 | 68.0 | 32.0 | 0.075 | 144 | 68.0 | 26.4 | 5.6 | 0.737 | 223 | 57.8 | 38.1 | 4.0 | 0.063 |
| R1 | 83 | 78.3 | 21.7 | 52 | 65.4 | 25.0 | 9.6 | 81 | 45.7 | 46.9 | 7.4 | ||||
| R2 | 3 | 33.3 | 66.7 | 2 | 100.0 | 0.0 | 0.0 | 3 | 33.3 | 33.3 | 33.3 | ||||
| Grading | G1 | 20 | 65.0 | 35.0 | 0.491 | 5 | 80.0 | 20.0 | 0.0 | 0.261 | 17 | 94.1 | 5.9 | 0.0 | 0.005 |
| G2 | 128 | 65.6 | 34.4 | 67 | 55.3 | 34.3 | 10.4 | 113 | 61.1 | 35.4 | 3.5 | ||||
| G3 | 191 | 73.3 | 26.7 | 126 | 73.0 | 22.2 | 4.8 | 173 | 45.7 | 48.0 | 6.4 | ||||
| G4 | 6 | 66.7 | 33.3 | 1 | 100.0 | 0.0 | 0.0 | 4 | 50.0 | 50.0 | 0.0 | ||||
Table 2: Association of p16 immunostaining, CDKN2A FISH and Ki67LI with clinico-pathological parameters in squamous cell carcinomas
| P16 IHC | CDKN2A FISH | Ki67 IHC | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All tumors | n | negative | positive | p | n | no del | het del | hom del | p | n | <10% | 10–80% | >80% | P | |
| 280 | 86.1 | 13.9 | 161 | 66.5 | 30.4 | 3.1 | 261 | 42.1 | 51.7 | 6.2 | |||||
| Age group | <65 yrs | 108 | 86.1 | 13.9 | 0.988 | 64 | 76.6 | 23.4 | 0.0 | 0.033 | 101 | 43.6 | 52.5 | 4.0 | 0.506 |
| >65 yrs | 172 | 86.0 | 14.0 | 96 | 59.4 | 35.4 | 5.2 | 160 | 41.2 | 51.2 | 7.5 | ||||
| Sex | male | 206 | 88.8 | 11.2 | 0.032 | 118 | 65.2 | 31.4 | 3.4 | 0.884 | 191 | 41.4 | 53.9 | 4.7 | 0.211 |
| female | 74 | 78.4 | 21.6 | 42 | 69.0 | 28.6 | 2.4 | 70 | 44.3 | 45.7 | 10.0 | ||||
| Tumor stage | pT1 | 52 | 73.1 | 26.9 | 0.014 | 27 | 77.8 | 22.2 | 0.0 | 0.019 | 43 | 51.2 | 44.2 | 4.7 | 0.611 |
| pT2 | 56 | 83.9 | 16.1 | 34 | 64.7 | 23.5 | 11.8 | 54 | 42.6 | 53.7 | 3.7 | ||||
| pT3 | 153 | 90.8 | 9.2 | 86 | 64.0 | 36.0 | 0.0 | 147 | 39.5 | 52.4 | 8.2 | ||||
| pT4 | 18 | 88.9 | 11.1 | 13 | 61.5 | 30.8 | 7.7 | 17 | 41.2 | 58.8 | 0.0 | ||||
| Lymph node metastasis | pN0 | 137 | 83.9 | 16.1 | 0.407 | 77 | 64.9 | 31.2 | 3.9 | 0.421 | 126 | 43.7 | 50.8 | 5.6 | 0.897 |
| pN1 | 60 | 86.7 | 13.3 | 32 | 78.1 | 21.9 | 0.0 | 58 | 34.5 | 56.9 | 8.6 | ||||
| pN2 | 55 | 85.5 | 14.5 | 35 | 54.3 | 40.0 | 5.7 | 51 | 45.1 | 49.0 | 5.9 | ||||
| pN3 | 27 | 96.3 | 3.7 | 16 | 75.0 | 25.0 | 0.0 | 25 | 44.0 | 52.0 | 4.0 | ||||
| UICC stage | I | 69 | 76.8 | 23.2 | 0.059 | 41 | 70.7 | 22.0 | 7.3 | 0.493 | 60 | 46.7 | 50.0 | 3.3 | 0.587 |
| II | 65 | 87.7 | 12.3 | 32 | 62.5 | 37.5 | 0.0 | 63 | 42.9 | 49.2 | 7.9 | ||||
| III | 95 | 91.6 | 8.4 | 56 | 66.1 | 32.1 | 1.8 | 90 | 40.0 | 51.1 | 8.9 | ||||
| IV | 49 | 85.7 | 14.3 | 31 | 64.5 | 32.3 | 3.2 | 47 | 38.3 | 59.6 | 2.1 | ||||
| Distant metastasis | M0 | 230 | 86.5 | 13.5 | 0.654 | 127 | 67.0 | 29.9 | 3.1 | 0.931 | 213 | 43.2 | 49.8 | 7.0 | 0.255 |
| M1 | 50 | 84.0 | 16.0 | 33 | 63.7 | 33.3 | 3.0 | 48 | 37.5 | 60.4 | 2.1 | ||||
| Surgical resection margin | R0 | 209 | 84.7 | 15.3 | 0.284 | 118 | 67.8 | 28.8 | 3.4 | 0.525 | 195 | 43.1 | 50.3 | 6.7 | 0.228 |
| R1 | 56 | 87.5 | 12.5 | 32 | 59.4 | 40.6 | 0.0 | 50 | 32.0 | 62.0 | 6.0 | ||||
| R2 | 13 | 100.0 | 0.0 | 9 | 77.8 | 22.2 | 0.0 | 14 | 64.3 | 35.7 | 0.0 | ||||
| Grading | G1 | 5 | 100.0 | 0.0 | 0.615 | 1 | 0.0 | 100.0 | 0.0 | 0.651 | 3 | 0.0 | 100.0 | 0.0 | 0.002 |
| G2 | 176 | 86.4 | 13.6 | 106 | 67.0 | 30.2 | 2.8 | 164 | 48.2 | 49.4 | 2.4 | ||||
| G3 | 98 | 84.7 | 15.3 | 52 | 67.4 | 28.8 | 3.8 | 93 | 33.3 | 53.8 | 12.9 | ||||
Ki67LI was evaluable in 312 AC and 261 SCC. Immunostaining was low in 54.5% (N = 170) of AC, moderate Ki67LI was seen in 40.4% (N = 126) and strong KI67LI in 5.1% (N = 16). In SCC, low Ki67LI was present in 42.1% (N = 110), moderate in 51.7% (N = 135) and strong in 6.2% (N = 16). Association with clinical data was found in AC between high-level Ki67 staining and high tumor stage (P = 0.001), presence of lymph node metastasis (P = 0.009), high UICC stage (P = 0.001) and poor grading (P = 0.005, Table 1). For SCC, merely a link between Ki67 immunostaining and poor grading (P = 0.002, Table 2) was revealed.
CDKN2A fluorescence in-situ hybridization (FISH)
CDKN2A FISH analysis was interpretable in 202 (50.8%) samples of AC and 161 (54.8%) samples of SCC. Non-informative cases were caused by inefficient hybridization, missing tissue spots or absence of representative tumor tissue on the TMA spot. Representative images are shown in Figure 2. Homozygous CDKN2A deletions were detectable in 13 samples (6.4%) and heterozygous deletions in 52 samples (25.7%) of AC. In SCC, homozygous deletions were detectable in 5 patients (3.1%) and heterozygous deletions in 49 tissue spots (30.4%).
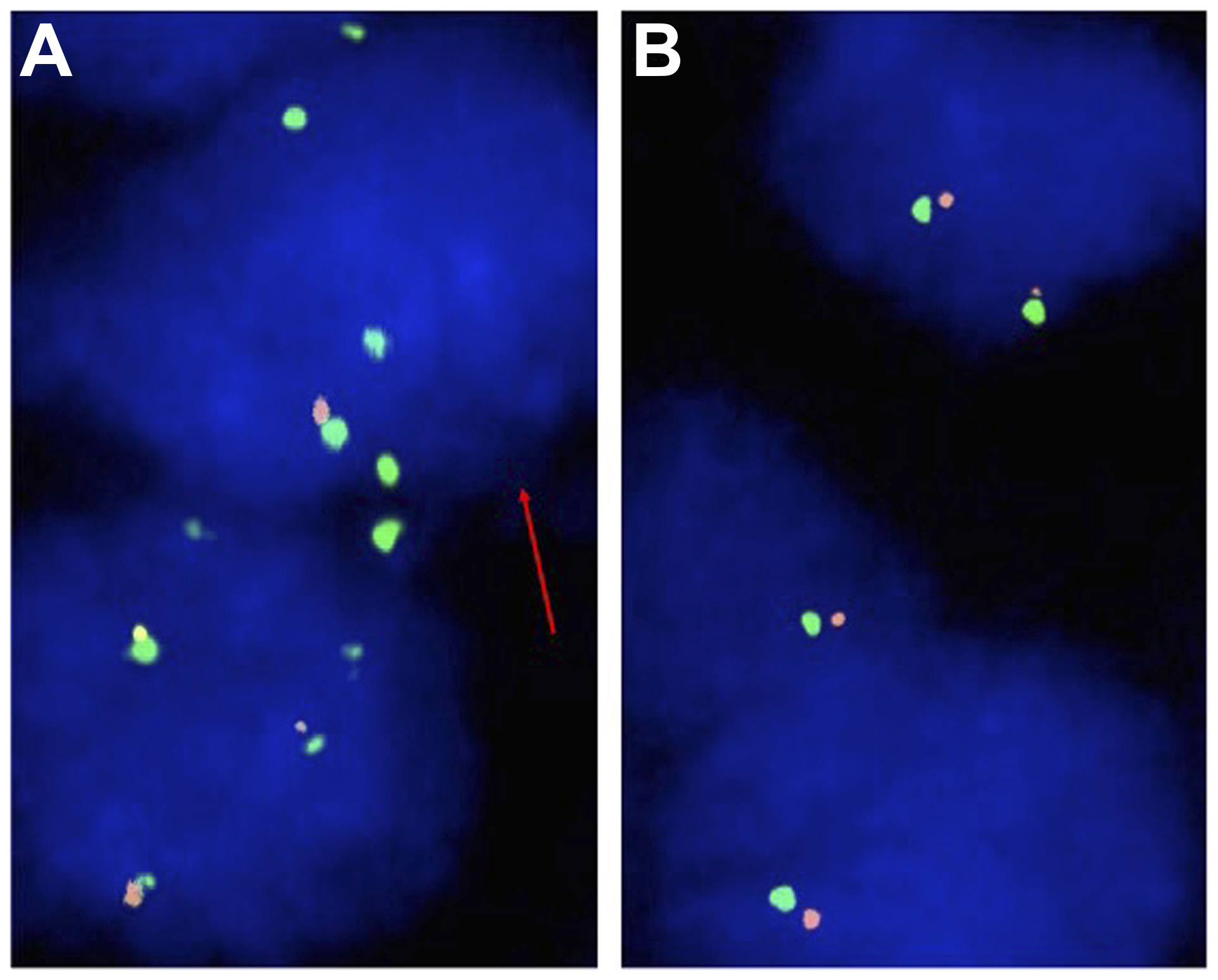
Figure 2: Representative FISH images of CDKN2A (A) Heterozygous CDKN2A deletion indicated by the lack of one orange CDKN2A signal and two green centromere 9 signals in the tumor cell nucleus (red arrow) and (B) Normal CDKN2A copy number indicated by two orange CDKN2A signals and two green centromere 9 signals.
In AC no links were evident between deletion rates and clinico-pathological parameters (Table 1), while in SCC CDKN2A deletions were associated with patients’ age (P = 0.033) and tumor stage (P = 0.024, Table 2).
A correlation between p16 immunostaining and CDKN2A deletion was found for AC (P = 0.039) but not for SCC (P = 0.610). However, in both histological tumor types, all cases with homozygous gene deletion were negative for p16 immunostaining (data not shown).
Combination of p16 and Ki67 IHC with CDKN2A deletions
Since a correlation between p16 immunostaining and CDKN2A deletion was found for AC a combined analysis of IHC and FISH was performed.
Data on both p16 immunostaining and CDKN2A deletion were available from 172 AC and 142 SCC. In AC, 37 samples (21.5%) were immunopositive for p16 and showed no CDKN2A deletion, while in SCC, combined p16 positivity and absence of CDKN2A deletion was seen in 16 samples (9.2%). There was no correlation detectable between clinical parameters and the combination of p16 expression with CDKN2A deletion.
Furthermore, no association was found between p16 and Ki67 immunostaining for either histological type (AC: P = 0.400 and SCC: P = 0.764). In addition, links between Ki67 immunostaining and CDKN2A deletion status were also not detectable (AC: P = 0.172; SCC: P = 0.712).
Survival analysis
Kaplan-Meier survival analysis for OS in AC showed shortened survival rates for patients with high Ki67 labeling index (P = 0.009, Figure 3A). Negative p16 immunostaining was also associated with a shortened overall survival (OS) compared to cancers showing p16 staining (P = 0.026, Figure 3B). CDKN2A deletions had no influence on the OS in AC (P = 0.679, Figure 3C).
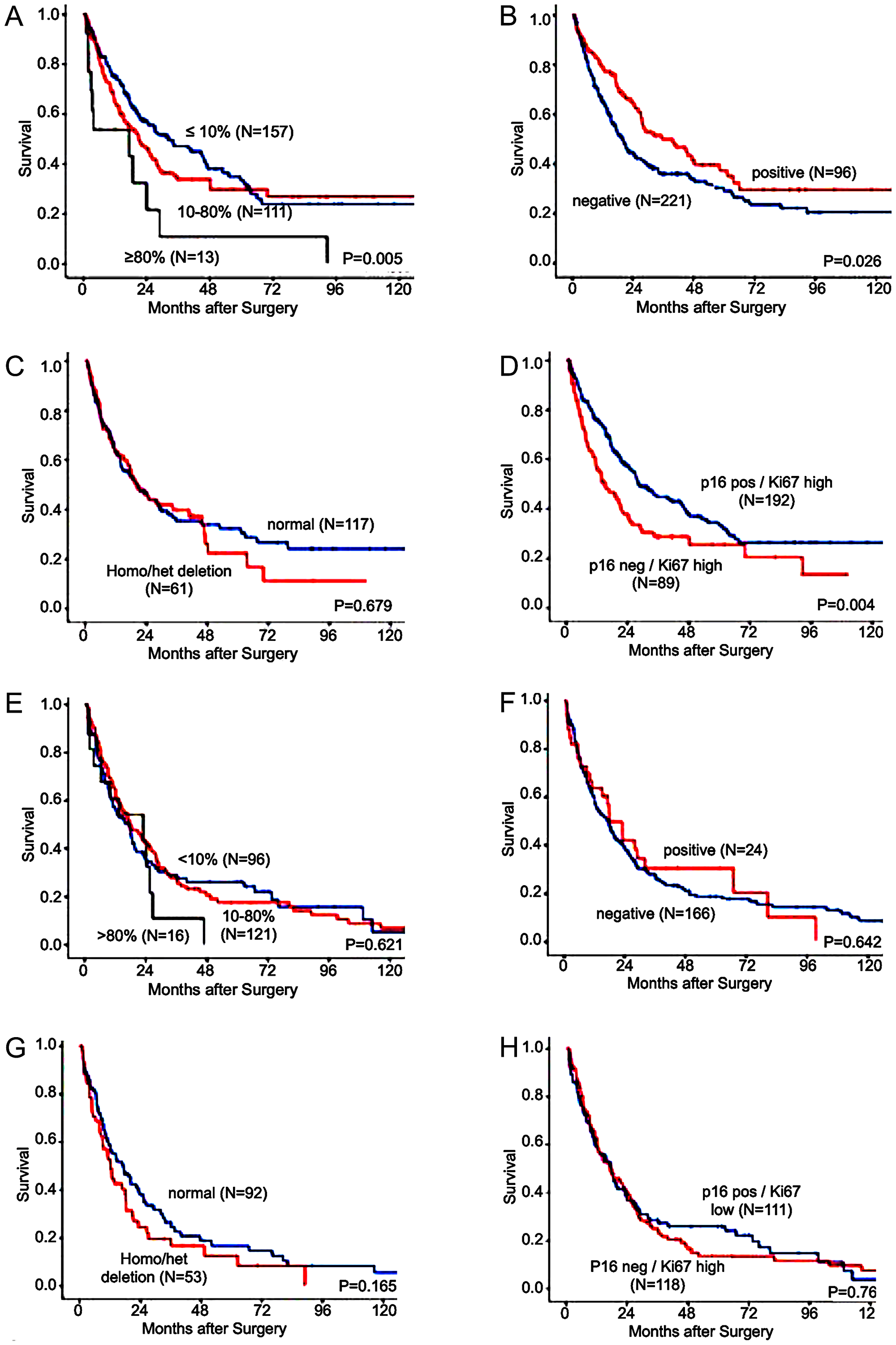
Figure 3: Association of immunohistochemistry (IHC) and fluorescence in-situ hybridization (FISH) results with the median overall-survival in patients with esophageal cancer. (A) KI67 immunostaining divided into ≤10%, 10–80%, and ≥80% Ki67 stained tumor cells and overall survival in adenocarcinoma, (B) p16 immunostaining divided in negative and positive and overall survival in adenocarcinoma, (C) CDKN2A FISH analysis divided in deletion (homozygous and heterozygous) and overall survival in adenocarcinoma, (D) combined p16 staining and Ki67LI and overall survival in adenocarcinoma, (E): KI67 immunostaining divided into ≤10%, 10–80%, and ≥80% Ki67 stained tumor cells and overall survival in squamous cell carcinoma, (F) p16 immunostaining divided in negative and positive and overall survival in squamous cell carcinoma (G): CDKN2A FISH analysis divided in deletion (homozygous and heterozygous) and overall survival in squamous cell carcinoma, (H): combined p16 staining and Ki67LI and overall survival in squamous cell carcinoma.
Combined analysis of Ki67 and p16 IHC suggested a superior prognostic value as compared to analysis of Ki67 and p16 expression alone, revealing favorable prognosis for patients with a combination of positive p16 immunostaining and low Ki67LI (P = 0.004, Figure 3D).
For SCC, neither the Ki67 labeling index (P = 0.621, Figure 3E) nor the p16 immunostatus (P = 0.682, Figure 3F) nor the CDKN2A deletion status (P = 0.165, Figure 3G) nor the combined analysis of Ki67LI and p16 (P = 0.7610, Figure 3H) reached statistical significance as a prognostic marker.
Multivariate Cox-regression analysis
Multivariate Analyses were performed evaluation the prognostic relevance of Ki67LI, p16 expression and CDKN2A deletion in relationship to patients’ age, sex and clinic-pathological parameters (pT, pN, pM, UICC stage, Grade, resection margin) in AC and SCC. For AC, the proportional cox-regression model revealed higher patients’ age (P = 0.0050), lymph node metastasis (P = 0.0070), resection margin (P = 0.0010) and the Ki67LI (P = 0.0040) as independent prognostic markers. In SCC, only tumor stage (P = 0.0260) proved to be independent prognosticators (Table 3).
Table 3: Multivariate Cox-regression model for esophageal adenocarcinoma and squamous cell carcinoma
| Hazard ratio | Adenocarcinoma 95% confidence interval | p value | Hazard Ratio | Squamous cell carcinoma 95% confidence interval | p value | |||
|---|---|---|---|---|---|---|---|---|
| lower | upper | lower | upper | |||||
| Age group | 2.008 | 1.235 | 3.264 | 0.005 | 1.104 | 0.733 | 1.664 | 0.635 |
| Sex (male vs. female) | 0.675 | 0.371 | 1.225 | 0.196 | 0.871 | 0.547 | 1.384 | 0.558 |
| Tumor stage (pT) | 1.410 | 0.992 | 2.004 | 0.055 | 1.504 | 1.050 | 2.156 | 0.026 |
| Lymph node metastsis (pN) | 1.501 | 1.119 | 2.013 | 0.007 | 1.038 | 0.776 | 1.388 | 0.803 |
| UICC stage | 1.030 | 0.586 | 1.813 | 0.918 | 1.017 | 0.648 | 1.595 | 0.943 |
| Distant metastasis (M) | 0.961 | 0.426 | 2.169 | 0.924 | 1.377 | 0.632 | 2.998 | 0.421 |
| Resection margin (R) | 2.161 | 1.385 | 3.373 | 0.001 | 1.097 | 0.757 | 1.590 | 0.626 |
| Grading (G) | 0.920 | 0.587 | 1.440 | 0.715 | 1.163 | 0.755 | 1.792 | 0.494 |
| Ki67 labeling index | 1.714 | 1.183 | 2.481 | 0.004 | 0.871 | 0.604 | 1.257 | 0.462 |
| p16 immunostaining | 1.078 | 0.680 | 1.708 | 0.750 | 0.954 | 0.541 | 1.682 | 0.869 |
| CDKN2A deletion | 1.118 | 0.826 | 1.514 | 0.469 | 1.184 | 0.813 | 1.722 | 0.378 |
DISCUSSION
The results of our study show that loss of p16 expression – but not 9p21 deletion - and high Ki67LI are prognosticators of poor survival in AC of the esophagus.
Aim of the present study was to assess whether p16 alterations are associated with adverse clinical outcome in patients with EC. Therefore, in our analysis, 351 AC and 280 SCC were analyzed by IHC (p16 expression) and FISH (CDKN2A deletion). Under the selected experimental conditions, our immunohistochemical analysis revealed 30% of AC and 14% of SCC positive for p16. Immunohistochemical analysis of p16 expression in EC is available in two studies only with expression rates varying between 15–60% and also depending on the underlying histological subtype [9, 15]. Discrepancy in expression frequencies is rather common when protein detection is performed using IHC. This is likely caused by variable immunohistochemistry conditions. It is well known that varying antibody conditions lead to significant changes in the rate of positive cases [16]. This is all the more expected in the case of ubiquitously expressed proteins, such as p16. Therefore, in some cases it may be difficult to differentiate between truly p16 negative cancers and lacking sensitivity.
Deletion rates differed between AC and SCC from about 25% in AC to almost 50% heterozygous deletions in SCC. Our deletion rate in AC is somewhat lower than reported in other studies [13, 14, 17–19], which is partially caused by more stringent criteria for defining CDKN2A deletions. These were applied to avoid false deletion calling due to truncation of the nuclei during tissue sectioning. Loss of heterozygosity (LOH) studies revealed LOH frequencies in SCC ranging from 65–79% [17–19]. However, LOH assays are influenced by ploidy changes, which are frequent in EC. These inevitably impact the assay sensitivity and vary markedly between individual tumors. In contrast, FISH allows for precise gene copy number determination in individual cells, rendering it independent of cancer tissue purity or aneuploidy. FISH is, thus, considered the gold standard for gene copy number analysis.
p16 is known to be a major tumor suppressor protein and its alteration has been associated with tumor progression in different entities [8]. p16 loss was linked to shortened overall survival and positive nodal stage in AC in our study. Other parameters, such as tumor stage and grade barely failed to reach statistical significance, which was probably first of all due to the low number of samples in some subgroups. p16 loss or downregulation but also its clear overexpression has been evaluated as negative prognosticators in several tumor types. Almost 50% of all tumors show p16 inactivation as one of the main drivers during carcinogenesis including pancreatic and biliary, head and neck, lung, bladder and colon carcinoma [20, 21]. Different mechanisms of p16 inactivation have been described earlier including promoter hypermethylation, point and missense mutations, loss of heterozygosity (LOH) and genetic deletions [13].
As p16 is considered a tumor suppressor and negative regulator for cell proliferation we correlated p16 expression with the cell proliferation marker Ki67 to analyze whether the loss of p16 was associated with increased cell proliferation. Ki67 is a known index marker for aggressive tumor behavior, including dedifferentiation. In line with the results of our analysis, high Ki67 indices have been linked to advanced tumor stages in EC [22, 23]. That Ki67 had independent prognostic value in our multivariate analysis, but not p16, suggests that Ki67 may be more promising candidate for clinical testing than p16. Interestingly, an association between loss of p16 and high Ki67LI was not seen, therefore regulative functions mediated by p16 protein expression are more complex than solely explained by increased cell proliferation. Although, possibly, the high rate of cancers with low Ki67LI experienced in our study may be misleading in this context rendering some results insignificant. Thus, the link may be masked by a slightly less sensitive Ki67 staining protocol than in the above mentioned study by Takeuchi [22, 23].
Another aim was to assess the relationship between CDKN2A deletions and p16 expression. A correlation was found for AC but not for SCC, which may be explained by different molecular mechanisms for inactivation of CDKN2A in the two histological tumor types. Inactivation of CDKN2A involves four types of genetic alterations: homozygous deletion, promoter hypermethylation, loss of heterozygosity and point mutation. Homozygous deletion and promoter hypermethylation constitute the majority of p16 alterations, but some cancers are known to prefer specific types of alterations. Promoter hypermethylation has been described as a main pathway for inactivation of CDKN2A for SCC of the esophagus [6, 24], while deletion appears to happen early in the development of Barrett’s mucosa [13], a recognized precursor lesion for esophageal AC.
Deletions are a common mechanism of gene inactivation for numerous tumor suppressor genes. As expected, all cancers with bi-allelic (homozygous) CDKN2A deletion completely lacked p16 expression, which indirectly validates our experimental approaches both for FISH and IHC. Differences in p16 expression levels in samples with or without heterozygous CDKN2A deletion demonstrate that EC cells have the ability to compensate the loss of one p16 (CDKN2A) allele, either by increased transcriptional activation of the remaining allele or by increased stabilization of p16 protein or mRNA (reviewed in [6, 24]). This also explained the absent of significant associations between CDKN2A deletions and clinic-pathological parameters as well as overall survival in our study. However, rare (5–6% depending on the histological subtype) biallelic CDKN2A deletions lead to catastrophic events for the cell with total loss of p16 expression.
It is a limitation of our study that the numbers of samples interpretable for p16 and Ki67 or p16 and CDKN2A are different and that combined analyses of these markers could only be made in subsets of the cancers. However, we do not consider this as a serious issue given that the total numbers of patients in these subsets are still comparatively high.
In summary, the results of our study show that loss of p16 expression - but not CDKN2A deletion - is linked to shortened overall survival in patients with esophageal AC. Furthermore, strong Ki67LI is an independent prognosticator of poor survival in AC. Rare homozygous 9p21 deletions can be considered as catastrophic events leading to complete loss of p16 expression.
Materials and Methods
Patients
For this study, specimens from patients that had undergone tumor resection in curative intent between 1992 and 2014 at the University Medical Center, Hamburg-Eppendorf were included. Tissue samples from 691 patients were analyzed including 398 AC and 293 SCC. All data including sex, tumor histology, size, lymph node metastasis and disease stage (UICC 7th edition) were obtained by reviewing a combination of clinical and pathological records, outpatient clinic medical records, epidemiological cancer surveillance data bases and by communication with the patients and their attending physicians. Overall (raw) survival was used as the clinical endpoint in this study. Clinical follow-up data were available for 635 patients with a median follow-up of 13.4 months (1 to 208.3 months). All resections were performed as en-bloc esophagectomies with radical two field lymph node dissection. Fifty patients underwent neoadjuvant therapy (AC n = 30, SCC n = 20) but therapy was unknown for the remaining patients. Patients who died within 30 days due to postoperative complications were not considered for survival analysis. The study was approved of by the Ethics Committee of the Chamber of Physicians of Hamburg, Germany (WF-035/14).
TMA construction
The TMA was constructed as previously described [25]. In brief, tissue cores were obtained from formalin-fixed paraffin-embedded (FFPE) tissue blocks from patients with pathologically proven EC. Representative areas of the tumor were selected based on hematoxylin–eosin staining. 691 tissue cylinders with a diameter of 0.6 mm were punched from the ‘‘donor’’ tissue blocks using a custom-made semi-automatic robotic precision instrument and placed into one empty recipient paraffin block. The resulting TMA blocks were used to produce 4 µm sections that were transferred to an adhesive-coated slide system (Instrumedics Inc., Hackensack, NJ).
IHC
Freshly cut TMA sections were immunostained on one day and in one experiment. Slides were deparaffinized, rehydrated, washed in DAKO buffer (K8002) and transferred to a DAKO Link 48 autostainer device. The immunohistochemical staining of p16 was performed with the commercially available CINtec p16 Histology Kit (Dilution 1:150, Cat# 725-4713, Ventana Medical Systems Inc., Arizona, USA) according to the manufacturer’s instructions. Staining was evaluated according to the following scoring system: The staining intensity (0, 1+, 2+, and 3+) and the fraction of positive tumor cells were recorded for each tissue spot. A final score was built from these 2 parameters according to the following score as previously described [26]: Negative stainings showed complete absence of staining. Weak scores had staining intensities of 1+ in ≤70% of tumor cells or of 2+ in ≤30% of tumor cells. Moderate scores had staining intensities of 1+ in >70% of tumor cells, staining intensities of 2+ in >30% but in >70% of tumor cells or staining intensities of 3+ in ≤30% of tumor cells. Strong scores had staining intensities of 2+ in ≤70% of tumor cells or staining intensities of 3+ in ≤30% of tumor cells. For statistical analysis all cancers with weak, moderate and strong staining was grouped as p16 positive. For Ki67 immunostaining standard indirect immunoperoxidase procedures were used for the detection of Ki67 (abcam, clone SPM171, dilution 1:150). Sections were heated in an autoclave at 121°C for 10 minutes in citrate puffer (pH 9.0). Diaminobenzidine was used as a chromogen, and sections were counterstained with Mayer’s haematoxylin. Ki67 staining was evaluated as follows: the number of invasive cancer cell nuclei that were positive for Ki67 immunostaining was divided by the total number of invasive cancer cell nuclei present in a histological sample resulting in the Ki67 labeling index (Ki67LI). For each spot, the procedure was repeated three times and the mean value was calculated. Three groups were stratified with scores ranging from 0 to 2 (low, moderate, strong). 0 represented tissue spots with Ki67LI <10%, 1 stood for Ki 67LI between 10 and 80% and 2 for Ki67LI >80%.
FISH
Four micrometer TMA sections were also used for FISH. For proteolytic slide pretreatment, a commercial kit was used (paraffin pretreatment reagent kit; Abbott, Wiesbaden, Germany). TMA sections were deparaffinized, air-dried, and dehydrated in 70%, 85%, and 100% ethanol, followed by denaturation for 5 min at 74°C in 70% formamide 2× SSC solution. The commercial Vysis CDKN2A/CEP 9 FISH probe kit (#04N61-020; Abbott, Wiesbaden, Germany) was used for detection of the 9p21 status. Hybridization was performed overnight at 37°C in a humidified chamber. Slides were subsequently washed and counterstained with 0.2 µmol/L 4′-6-diamidino-2-phenylindole in antifade solution. Stained slides were manually interpreted with an epifluorescence microscope, and the predominant FISH signal numbers were recorded in each tissue spot. Presence of fewer CDKN2A signals than centromere 9 probe signals in at least 60% of the tumor nuclei was considered to inidicate heterozygous deletion. Complete absence of CDKN2A signals in the tumor cells, but presence of centromere 9 and CDKN2A signals in adjacent normal cells, was considered to be a homozygous deletion. Tissue spots lacking any detectable CDKN2A signals in all (tumor and normal cells) or lack of any normal cells as an internal control for successful hybridization of the CDKN2A probe were excluded from analysis. These thresholds are based on our previous study analyzing PTEN deletions on a prostate cancer TMA where our approach resulted in a 100% concordance with aCGH data [27].
Statistical analysis
SPSS Statistics for Mac (Version 17, SPSS) was used for statistical analysis. Contingency tables and the chi2-test were performed to search for associations between molecular parameters and tumor phenotype. Survival curves were calculated according to Kaplan-Meier. The Log-Rank test was applied to detect significant differences between groups. Cox proportional hazards regression analysis was performed to test the statistical independence and significance between pathological, molecular and clinical variables. Separate analyses were performed using different sets of parameters available either before or after prostatectomy. All tests were two-sided. P values <0.05 were considered statistically significant.
Abbreviations
AC: adenocarcinomas; EC: esophageal cancer; FFPE: formalin-fixed paraffin-embedded; FISH: fluorescence in-situ hybridization; IHC: immunohistochemistry; Ki67LI: Ki67 labeling index; OS: overall survival; SSC: squamous cell carcinomas; TMA: tissue microarray.
ACKNOWLEDGMENTS
We thank Christina Möller-Koop, Janett Lütgens, Sünje Seekamp, and Inge Brandt for excellent technical assistance.
CONFLICTS OF INTEREST
We certify that there is no actual or potential conflicts of interest in relation to this article.
FUNDING
We certify that there are no sources of any support for the work, received in the form of grants, equipment, and/or drugs in relation to this article.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108. https://doi.org/10.3322/caac.21262. [PubMed].
2. Cartwright E, Keane FK, Enzinger P, Hong T, Chau I. Is There a Precise Adjuvant Therapy for Esophagogastric Carcinoma? Am Soc Clin Oncol Educ Book. 2018; 38:280–291. https://doi.org/10.1200/EDBK_200785. [PubMed].
3. Castro C, Bosetti C, Malvezzi M, Bertuccio P, Levi F, Negri E, La Vecchia C, Lunet N. Patterns and trends in esophageal cancer mortality and incidence in Europe (1980–2011) and predictions to 2015. Ann Oncol. 2014; 25:283–290. https://doi.org/10.1093/annonc/mdt486. [PubMed].
4. van Hagen P, Hulshof MC, van Lanschot JJ, Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, Cuesta MA, Blaisse RJ, Busch OR, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012; 366:2074–2084. https://doi.org/10.1056/NEJMoa1112088. [PubMed].
5. Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004; 18:2699–2711. https://doi.org/10.1101/gad.1256504. [PubMed].
6. Li J, Poi MJ, Tsai MD. Regulatory mechanisms of tumor suppressor P16(INK4A) and their relevance to cancer. Biochemistry. 2011; 50:5566–5582. https://doi.org/10.1021/bi200642e. [PubMed].
7. Serra S, Chetty R. p16. J Clin Pathol. 2018; 71:853–858. https://doi.org/10.1136/jclinpath-2018-205216. [PubMed].
8. Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, Ramon y Cajal S. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011; 30:2087–2097. https://doi.org/10.1038/onc.2010.614. [PubMed].
9. Cao F, Zhang W, Zhang F, Han H, Xu J, Cheng Y. Prognostic significance of high-risk human papillomavirus and p16(INK4A) in patients with esophageal squamous cell carcinoma. Int J Clin Exp Med. 2014; 7:3430–3438. [PubMed].
10. Esteve A, Martel-Planche G, Sylla B, Hollstein M, Hainaut P, Montesano R. Low frequency of p16/CDKN2 gene mutations in esophageal carcinomas. Int J Cancer. 1996; 66:301–304. https://doi.org/10.1002/(SICI)1097-0215(19960503)66:3<301::AID-IJC5>3.0.CO;2-2. [PubMed].
11. Fujiwara S, Noguchi T, Takeno S, Kimura Y, Fumoto S, Kawahara K. Hypermethylation of p16 gene promoter correlates with loss of p16 expression that results in poorer prognosis in esophageal squamous cell carcinomas. Dis Esophagus. 2008; 21:125–131. https://doi.org/10.1111/j.1442-2050.2007.00735.x. [PubMed].
12. Suzuki H, Fujioka Y, Nagashima K. Cyclin D1 gene amplification and p16 gene deletion in patients with esophageal carcinosarcoma. Diagn Mol Pathol. 1998; 7:253–259. https://doi.org/10.1097/00019606-199810000-00004. [PubMed].
13. Tokugawa T, Sugihara H, Tani T, Hattori T. Modes of silencing of p16 in development of esophageal squamous cell carcinoma. Cancer Res. 2002; 62:4938–4944. [PubMed].
14. Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett’s esophagus. Oncogene. 1996; 13:1867–1873. [PubMed].
15. Kato H, Yoshikawa M, Fukai Y, Tajima K, Masuda N, Tsukada K, Kuwano H, Nakajima T. An immunohistochemical study of p16, pRb, p21 and p53 proteins in human esophageal cancers. Anticancer Res. 2000; 20:345–349. [PubMed].
16. Schlomm T, Iwers L, Kirstein P, Jessen B, Köllermann J, Minner S, Passow-Drolet A, Mirlacher M, Milde-Langosch K, Graefen M, Haese A, Steuber T, Simon R, et al. Clinical significance of p53 alterations in surgically treated prostate cancers. Mod Pathol. 2008; 21:1371–1378. https://doi.org/10.1038/modpathol.2008.104. [PubMed].
17. Giroux MA, Audrezet MP, Metges JP, Lozac’h P, Volant A, Nousbaum JB, Labat JP, Gouerou H, Ferec C, Robaszkiewicz M. Infrequent p16/CDKN2 alterations in squamous cell carcinoma of the oesophagus. Eur J Gastroenterol Hepatol. 2002; 14:15–18. https://doi.org/10.1097/00042737-200201000-00004. [PubMed].
18. Hu N, Wang C, Su H, Li W, Emmert-Buck MR, Li G, Roth MJ, Tang Z, Lu N, Giffen C, Albert PS, Taylor PR, Goldstein AM. High frequency of CDKN2A alterations in esophageal squamous cell carcinoma from a high-risk Chinese population. Genes Chromosomes Cancer. 2004; 39:205–216. https://doi.org/10.1002/gcc.10315. [PubMed].
19. Suzuki H, Zhou X, Yin J, Lei J, Jiang HY, Suzuki Y, Chan T, Hannon GJ, Mergner WJ, Abraham JM, Melzer SJ. Intragenic mutations of CDKN2B and CDKN2A in primary human esophageal cancers. Hum Mol Genet. 1995; 4:1883–1887. https://doi.org/10.1093/hmg/4.10.1883. [PubMed].
20. Ueki T, Hsing AW, Gao Y, Wang B, Shen M, Cheng J, Deng J, Fraumeni JF, Rashid A. Alterations of p16 and prognosis in biliary tract cancers from a population-based study in China. Clin Cancer Res. 2004; 10:1717–1725. https://doi.org/10.1158/1078-0432.CCR-1137-3. [PubMed].
21. Serrano M. The tumor suppressor protein p16INK4a. Exp Cell Res. 1997; 237:7–13. https://doi.org/10.1006/excr.1997.3824. [PubMed].
22. Takeuchi H, Ozawa S, Ando N, Kitagawa Y, Ueda M, Kitajima M. Cell-cycle regulators and the Ki-67 labeling index can predict the response to chemoradiotherapy and the survival of patients with locally advanced squamous cell carcinoma of the esophagus. Ann Surg Oncol. 2003; 10:792–800. https://doi.org/10.1245/ASO.2003.10.014. [PubMed].
23. Vallböhmer D, Lenz HJ. Predictive and prognostic molecular markers in outcome of esophageal cancer. Dis Esophagus. 2006; 19:425–432. https://doi.org/10.1111/j.1442-2050.2006.00622.x. [PubMed].
24. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998; 1378:F115–F177. https://doi.org/10.1016/s0304-419x(98)00017-1. [PubMed].
25. Mirlacher M, Simon R. Recipient block TMA technique. Methods Mol Biol. 2010; 664:37–44. https://doi.org/10.1007/978-1-60761-806-5_4. [PubMed].
26. Dancau AM, Simon R, Mirlacher M, Sauter G. Tissue microarrays. Methods Mol Biol. 2010; 576:49–60. https://doi.org/10.1007/978-1-59745-545-9_4. [PubMed].
27. Krohn A, Diedler T, Burkhardt L, Mayer P, De Silva C, Meyer-Kornblum M, Kötschau D, Tennstedt P, Huang J, Gerhäuser C, Mader M, Kurtz S, Sirma H, et al. Genomic deletion of PTEN is associated with tumor progression and early PSA recurrence in ERG fusion-positive and fusion-negative prostate cancer. Am J Pathol. 2012; 181:401–412. https://doi.org/10.1016/j.ajpath.2012.04.026. [PubMed].