
Introduction
Solid tumor treatment with CAR T cells is currently limited by the paucity of cell surface targets that are tumor restricted, broadly expressed throughout the tumor, and integrally associated with tumorigenicity. Designing CARs to minimize on-target off-tumor toxicity is an important consideration in CAR design given the potentially serious consequences of targeting normal tissue [1]. Central nervous system (CNS) tumors continue to pose an exceptional challenge in oncology due to the difficulty of delivering drugs through the blood-brain barrier, and the high morbidity of radiation and surgical approaches. The need for better therapies is particularly urgent for malignant gliomas, as well as other brain tumors that are refractory or recur after standard therapy. Currently, 5-year survival rates range from less than 5% for adult glioblastoma [2, 3] to 25% or less for recurrent or refractory medulloblastoma [4]. EGFR amplification, overexpression, or mutation is present in approximately half of glioblastomas and other malignant CNS tumors, including ependymoma and medulloblastoma, in both children and adults [5–12]. EGFR-targeted interventions such as tyrosine kinase inhibitors have had limited success in CNS tumors, likely due to difficulty accessing the tumors and tumor evasion of these pathway-based drugs [13, 14]. CAR T cells targeting EGFRvIII, a truncation mutant of EGFR that is common in glioblastoma, were safe but unable to eradicate the tumor, likely due to nonuniform expression of the target protein and antigen escape [15, 16].
To construct a CAR targeting EGFR-expressing CNS tumors, we chose the mAb806 scFv moiety due to its unique binding characteristics. mAb806 was initially raised against the EGFRvIII variant, but also binds to full-length EGFR that is expressed as a result of gene amplification [17, 18]. However, it does not recognize EGFR on normal cells [19]. This tumor specificity is likely due to changes in steric accessibility of the mAb806 epitope between tumor-expressed and wild type EGFR [20]. The mAb806 epitope is located on extracellular domain II of EGFR [21], and is inaccessible to the antibody during normal conformational states of the receptor, both the tethered, monomeric, unbound form and the untethered, dimerized, ligand bound form [22]. However, posttranslational modifications during overexpression of EGFR sterically expose the mAb806 binding site [21, 23]. In addition, multiple EGFR mutations found in glioblastoma, including EGFRvIII, produce a conformational change in the extracellular domain that may promote pro-growth signaling and also renders the mAb806 epitope accessible [20].
The mAb806 antibody has been tested in clinical trials for EGFR-overexpressing tumors, and showed specific binding to tumor tissue, including malignant glioma, with only mild gastrointestinal and dermatologic side effects [24]. A humanized version of the antibody is now in clinical trials as an antibody-drug-conjugate targeting recurrent glioblastoma [25]. We chose intracranial delivery of the CAR T cells as this approach effectively places the cells near their target, lowers the risk of side effects outside the CNS, and has shown promise in clinical trials for glioblastoma [26].
EGFR signaling plays an important role during brain development [27, 28], and has been shown in animal models to be involved in CNS injury, repair, and adult neurogenesis [29, 30]. Although no EGFR expression has been conclusively demonstrated in postnatal human brain [31–33], there remains theoretical concern that expression could be reactivated in the setting of cancer treatment, or that intracranially injected CAR T cells could emerge into the systemic circulation. Therefore, we tested tumor-specificity on physiologically EGFR-expressing fetal human astrocytes. In addition, we developed an in vivo teratoma assay to measure CAR T cell infiltration and activation in implanted human iPSCs that were allowed to differentiate into multiple tissues tissue types, including EGFR-expressing epithelia [34]. We show that second-generation EGFR806-CAR T cells with a short spacer can eradicate malignant glioma in a xenograft mouse model via intracranial delivery, and that CAR T cell activation is specific to tumor-expressed EGFR.
Results
EGFR806-CAR with extracellular short spacer shows efficient EGFR+ tumor lysis and cytokine production
To target EGFR-positive tumors, we engineered a 2nd generation CAR construct consisting of an extracellular binding domain derived from mAb806, a 4-1BB-z intracellular signaling domain, and truncated EGFR (EGFRt) to serve as a transduction marker and ablation target (Figure 1A). The EGFRt fragment does not contain the mAb806 binding site and thus is not recognized by the EGFR806-CAR [21]. Since the length of CAR extracellular spacer domains has been shown to affect CAR mediated cellular cytotoxicity [35], we addressed the functional impact of spacer lengths on EGFR806-CAR T cell activity by engineering CARs with modular spacer domains designated short (S, IgG4hinge alone), medium (M, IgG4hinge-CH3), and long (L, IgG4hinge-CH2-CH3) (Figure 1A). Thus, we purified CD8+CD45RO+CD62L+ central memory T cells (CD8+ TCM) [36] and transduced them with lentiviral vectors containing S-, M-, or L-spacer EGFR806-CARs. Initial transduction efficiencies based on EGFRt expression ranged from 74–90%, and transgene positive T cells were enriched to uniform purity (> 95%) by EGFRt selection (Figure 1B). Similar levels of surface and total CAR expression were confirmed by Protein-L staining (87–95%; Figure 1C) and a-CD3z western blot analysis (data not shown) respectively.
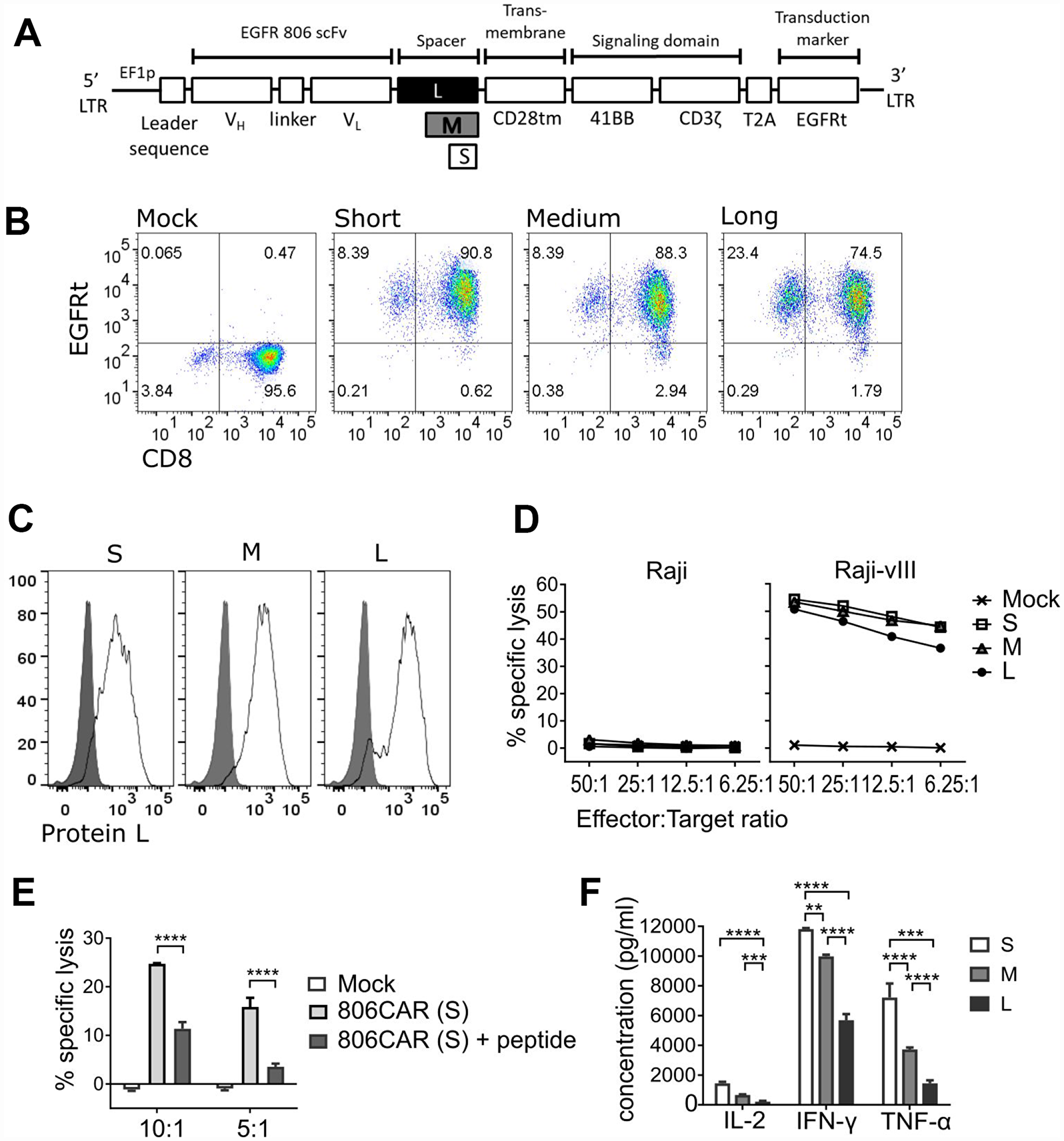
Figure 1: EGFR806-CAR T cells effectively target EGFR-expressing glioma cells in vitro. (A) Schematic of 2nd generation EGFR806-CAR extracellular domain variants. Spacer variants: Short (S) – IgG4-hinge; Medium (M) – IgG4-hinge-CH3; Long (L) – IgG4-hinge-CH2-CH3. (B) EGFRt expression by flow cytometry showing CAR expression in CD8+ TCM transduced with short, medium or long spacer 2nd generation EGFR806-CAR lentivirus and selected for EGFRt expression. Mock transduced cells were unselected. Representative data for one donor are shown, and all donors (N = 3) yielded >90% EGFRt positive T cells after the selection step. (C) Protein-L was used to label the scFv portion of the CAR, demonstrating surface expression. Representative data from one donor. (D) EGFR806-CAR T cells kill EGFRvIII-expressing Raji (Raji-vIII) cells but not untransduced Raji cells in a 4-hour chromium release assay. The x-axis shows the ratio of effector: target cells. (E) An EGFR aa. 287-302 peptide, which encompasses the mAb binding epitope, inhibits short-spacer EGFR806-CAR T cell lysis of Raji-vIII cells. The x-axis shows the ratio of effector: target cells. (F) Cytokine levels in supernatants obtained from 24hr co-cultures of EGFR806-CAR T cells expressing extracellular spacer variants and Raji-vIII cells at a 2:1 ratio. Cytokine data from three independent experiments was analyzed by Student t test. Error bars represent SEM. All panels: S, short spacer; M, medium spacer; L, long spacer. **P < 0.01; ***P < 0.01; ****P.001.
The in vitro lytic activity of the EGFR806-CAR T cells, as determined by chromium release assay, was analyzed against Raji cells transduced with an EGFRvIII construct (Figure 1D). Each EGFR806-CAR version conferred similar levels of specific lysis against EGFRvIII-expressing Raji cells, but did not recognize parental Raji (EGFR-negative) targets. EGFR806-CAR specificity was further verified by an inhibition study using an EGFR-derived peptide that contains the putative epitope of mAb806 (Figure 1E). At a concentration of 55 mM (100 mg/ml) [37], the soluble peptide inhibited the lytic capacity of short spacer EGFR806-CAR T cells by 54-78%, depending on effector to target (E: T) ratios. Although the tumor lytic capacity was similar for the three spacer variants, the short spacer CAR induced the most robust effector cytokine production upon tumor recognition, with 4.6- (P < 0.0001), 3.8- (P < 0.0001), and 3.1-fold (P=0.0001) higher IL-2, IFNg, and TNFa levels, respectively, compared to long spacer; and 2.2- (P > 0.05), 1.2- (P < 0.01) and 1.9-fold (P < 0.0001) higher IL-2, IFNg, and TNFa levels compared to medium spacer CAR (Figure 1F). All three cytokines were undetectable when CAR T cells were cultured in the absence of target, or with parental Raji cells (data not shown).
EGFR806-CAR T cells effectively lyse low-EGFR expressing glioblastoma cell lines independent of EGFRvIII expression
To validate EGFR as a suitable target for CAR based immunotherapy, we confirmed expression of EGFR on 35 separate malignant glioma samples via tumor tissue microarray. EGFR expression was high in 23% (H score >100), low (H score 5–100) in 20%, and absent in 57% of samples, and also absent in normal brain (Figure 2). We then used western blot and cell surface quantification of EGFR in three established glioblastoma cell lines (T98, U251T, and U87) to assess their ability to recapitulate tumor EGFR expression (Figure 3A, 3B). Epithelial carcinoma (A431) and Burkitt’s lymphoma (Raji) cell lines were used as positive and negative controls, respectively. U87 cells had the lowest level of EGFR surface expression, at 8% of the high-expressing A431 line (Figure 3B). EGFR was expressed on >99% of cells for all tumor cell lines (data not shown). We used RNA-Seq analysis to confirm the absence of EGFRvIII variant expression in the EGFR-positive cell lines, confirming that the activity of the EGFR806-CAR T cells in these cell lines is not mediated by binding to EGFRvIII (Table 1, Supplementary Figures 1 and 2). We then examined the in vitro lytic capacity of EGFR806-CAR T cells in the three human glioblastoma cell lines and A431 cells (Figure 3C). Similar to the Raji-vIII studies, we observed that the second generation EGFR806-CAR spacer variants mediated comparable levels of specific lysis at each effector: target ratio tested. Notably, cell targets with low surface EGFR expression relative to A431 were also efficiently lysed, although production of IL-2, TNFa and IFNg was lower when glioma cell lines were used as targets compared to A431 (Figure 3D). Based on these results, we selected the short spacer EGFR806 construct for further in-depth analysis. The short spacer CAR showed superior cytokine production against Raji-EGFRvIII cells, comparable activity against glioma cell lines, and minimizes lentiviral payload to allow the most efficient CAR transduction.
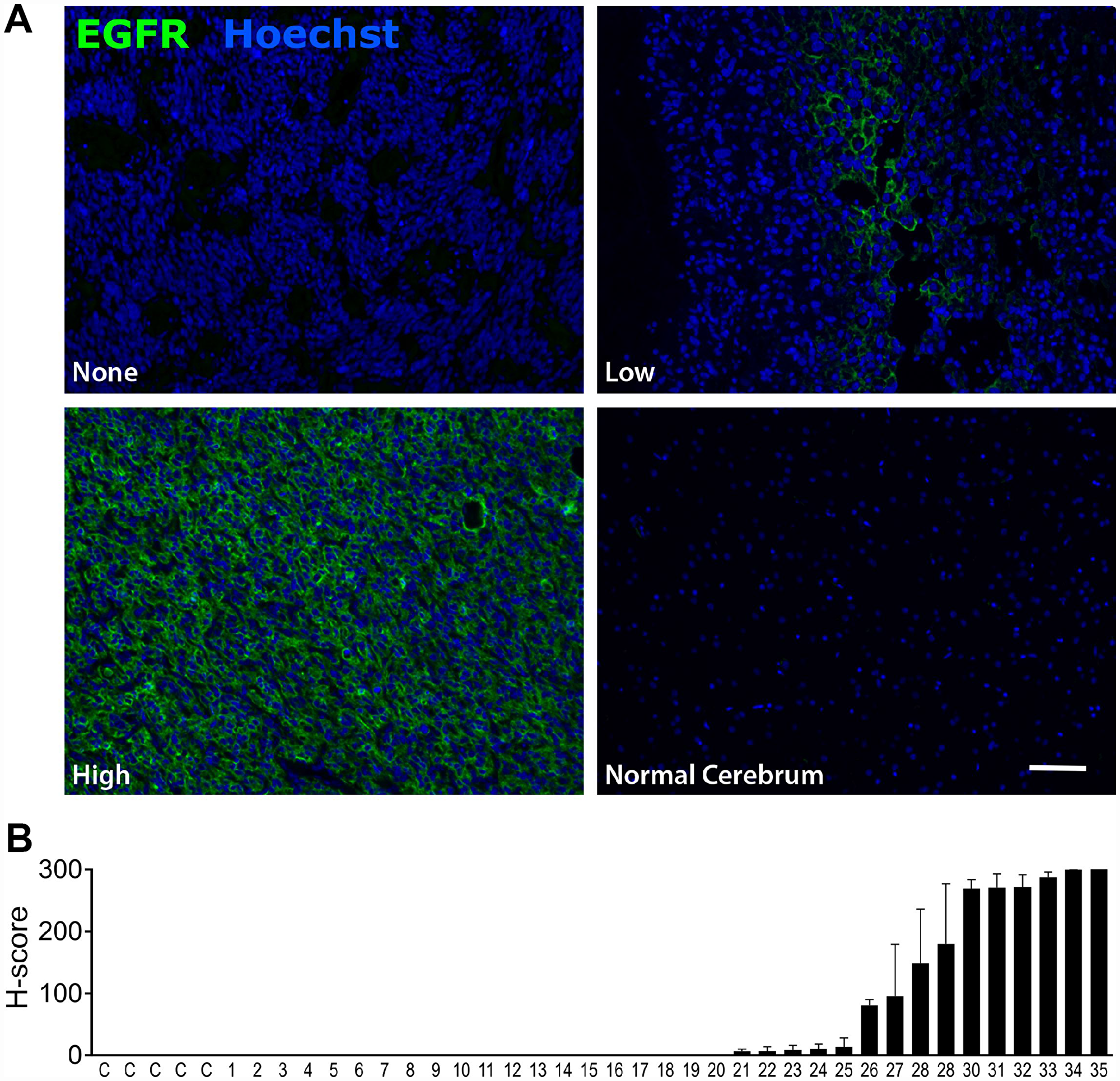
Figure 2: EGFR is expressed in glioblastoma but not in normal brain. (A) Representative immunofluorescent labeling for EGFR expression in human glioblastoma samples (top row and bottom left panels) and normal brain (bottom right panel). Scale bar 20μm. Images were acquired at 20 ×. (B) EGFR expression in normal brain (C=control) and 35 malignant glioma samples (1–35). Bars show mean EGFR immunofluorescence as quantified by H score. Error bars show SEM for 2 replicates from individual patients.
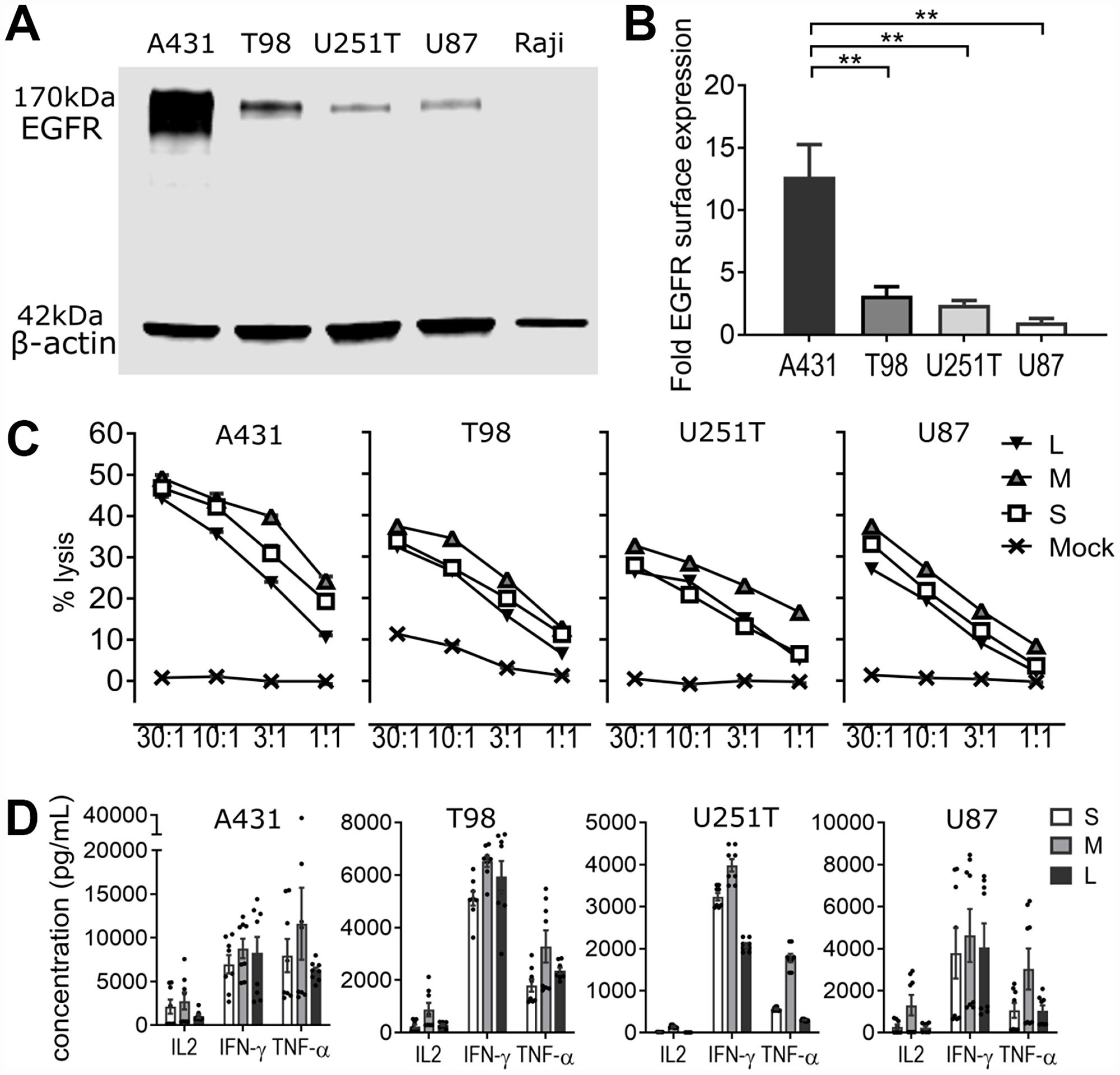
Figure 3: EGFR806-CAR T cells are selectively activated by tumor-specific EGFR expression in vitro. (A) EGFR quantification in tumor cell lysates via Western blot. Top: EGFR expression in tumor cell lines. Bottom: actin loading control. A431: EGFR+ squamous carcinoma cells (positive control); T98, U251T and U87: glioblastoma cell lines; Raji: EGFR- Burkitt’s lymphoma cells (negative control). (B) Quantification of EGFR surface antigen expression by flow cytometry. The y-axis shows normalized EGFR expression relative to the lowest-expressing U87 cells, ± SEM of three independent experiments. (C) Cytolytic capacity of EGFR806-CAR T cells expressing extracellular spacer variants against EGFR+ tumor cell targets in 4 hr chromium release assay. The x-axis shows the effector: target cell ratio. (D) Cytokine quantification of supernatants obtained from 24 hr co-cultures of EGFR806-CAR T cells and EGFR+ tumor targets at a 2:1 effector: target ratio. Each dot represents an individual replicate, bars show mean ± SEM. Data pooled from two separate experiments with CAR T cells from two different donors. S, short spacer; M, medium spacer; L, long spacer; Mock, untransduced CAR T cell control. **P < 0.01.
Table 1: EGFR and EGFRvIII expression in cell lines by RNASeq
| Cell line | Unique loci | Multiple loci | Total reads | Mapping (%) | EGFR | EGFRvIII | EGFR % |
|---|---|---|---|---|---|---|---|
| A431 | 32,728,176 | 3,077,612 | 35,805,788 | 95.32 | 92856 | 4 | 0.259 |
| T98 | 32,227,277 | 3,535,139 | 35,762,416 | 96.79 | 8465 | 5 | 0.024 |
| U251T | 25,629,699 | 2,680,718 | 28,310,417 | 92.20 | 2639 | 4 | 0.009 |
| U87 | 28,414,376 | 2,596,026 | 31,010,402 | 92.44 | 6088 | 6 | 0.020 |
| Raji-vIII | 24,459,634 | 3,252,848 | 27,712,482 | 94.97 | 11 | 2783 | 0.010 |
Lower on-target off-tumor activity of EGFR806-CAR relative to Erbitux-CAR
To test binding selectivity, we compared the EGFR806-CAR with an Erbitux-CAR, which binds EGFR expressed on both cancerous and normal tissues via a high-affinity binder derived from cetuximab [38]. Erbitux-CAR T cells have similar lysis capacity as EGFR806-CAR T cells against the glioma cell lines T98, U87, and U251T (data not shown). However, the 2nd generation short spacer EGFR806-CAR T cells demonstrated selective cytotoxicity and cytokine release relative to Erbitux-CAR T cells when co-cultured with primary fetal human astrocytes, which express wild-type EGFR (Figure 4A). Untransduced fetal human astrocytes were killed by Erbitux-CAR T cells, while EGFR806-CAR T cells showed only minimal lysis even at high effector: target ratios (Figure 4B). However, when the primary human astrocytes were transduced to express EGFRvIII ectopically, they were lysed at similar rates by EGFR806-CAR T cells and Erbitux-CAR T cells (Figure 4B). Cytokine production showed a concordant pattern. There was negligible release of IL-2, IFNg, and TNFa when EGFR806CAR T cells were incubated with untransduced fetal human astrocytes (Figure 4C), while high levels of the same cytokines were released during lysis of EGFRvIII-overexpressing astrocytes. Erbitux-CAR T cells, in contrast, showed robust cytokine production with both untransduced and EGFRvIII-expressing astrocytes (Figure 4C), although both target cell lysis and cytokine production were higher with EGFRvIII astrocytes (Figure 4B, 4C). The difference in cytokine production between Erbitux-CAR and EGFR806-CAR may be due to differences in effective antigen density, where the Erbitux-CAR recognizes both overexpressed EGFRvIII and native EGFR, while the EGFR806-CAR is only activated by the overexpressed EGFRvIII. These results show that the lower on-target toxicity observed with primary fetal human astrocytes is not due to an inherent functional deficiency of the EGFR806-CAR. Instead, the EGFR806-CAR has higher specificity for pathologically expressed EGFR and EGFRvIII compared to native EGFR, which may translate to a more favorable in vivo toxicity profile compared to the Erbitux-CAR.
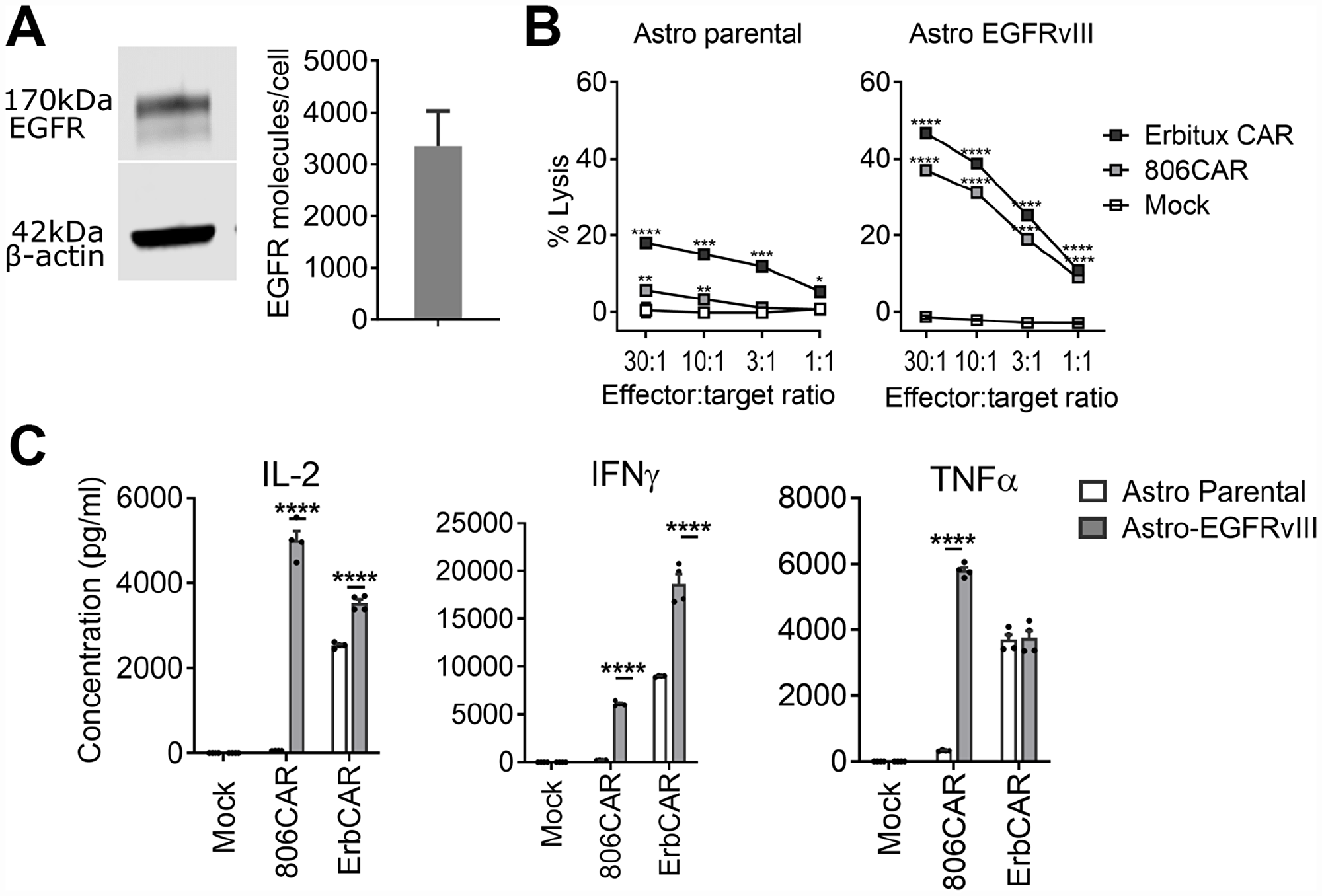
Figure 4: EGFR806-CAR T cells have lower activity against physiologically expressed EGFR compared to Erbitux-CAR. (A) Fetal human astrocytes express wild-type EGFR by Western blot (left) and surface expression is demonstrated by flow cytometry (right). (B) Lysis of fetal human astrocytes by EGFR806-CAR and Erbitux-CAR T cells in 4-hour chromium release assay. Left: untransduced fetal human astrocytes (Astro parental) are only minimally recognized by EGFR806-CAR T cells but are killed by Erbitux-CAR T cells. Right: after transduction with EGFRvIII (Astro EGFRvIII), fetal human astrocytes are killed efficiently by both effectors. Mock, untransduced CAR T cell control. Error bars show ± SEM for 3 replicate assays. (C) Cytokine production is stimulated by EGFRvIII-transduced, but not by wild-type EGFR expressing fetal human astrocytes when co-cultured with EGFR806-CAR T cells. Erbitux-CAR (ErbCAR) T cells show nonselective cytokine activation. Each dot represents an individual replicate, bars show the mean ± SEM of 4 replicates each. *P < 0.05; **P < 0.01; ***P < 0.01; ****P .001.
Short spacer EGFR806-CAR T cells eradicate glioblastoma xenografts
To confirm the in vitro anti-glioma activity of the EGFR806-CAR in vivo, we tested tumor killing capacity in a previously described orthotopic U87 glioma xenograft tumor model [39]. NSG mice were inoculated with the human U87 cell line by intracranial (i. c.) injection and seven days later treated with a single intratumoral dose of short spacer EGFR806-CAR T cells (Figure 5A, 5B). Control mice received mock transduced human donor T cells. EGFR806-CAR T cell treatment induced tumor regression in all animals (Figure 5C–5F). While all animals treated with control T cells had to be euthanized between day 28-30 (Figure 5G, 5H), 50% of EGFR806-CAR treated mice survived to the predetermined experimental end point of 90 days (P=0.0001, log rank test) (N = 8 for each group, pooled data from 2 separate experiments). This antitumor efficacy was markedly higher than in mice treated with T cells transduced with the medium or long spacer second-generation EGFR806-CAR or a third generation EGFR806-CAR with 4-1BB and CD28 costimulation domains (data not shown).
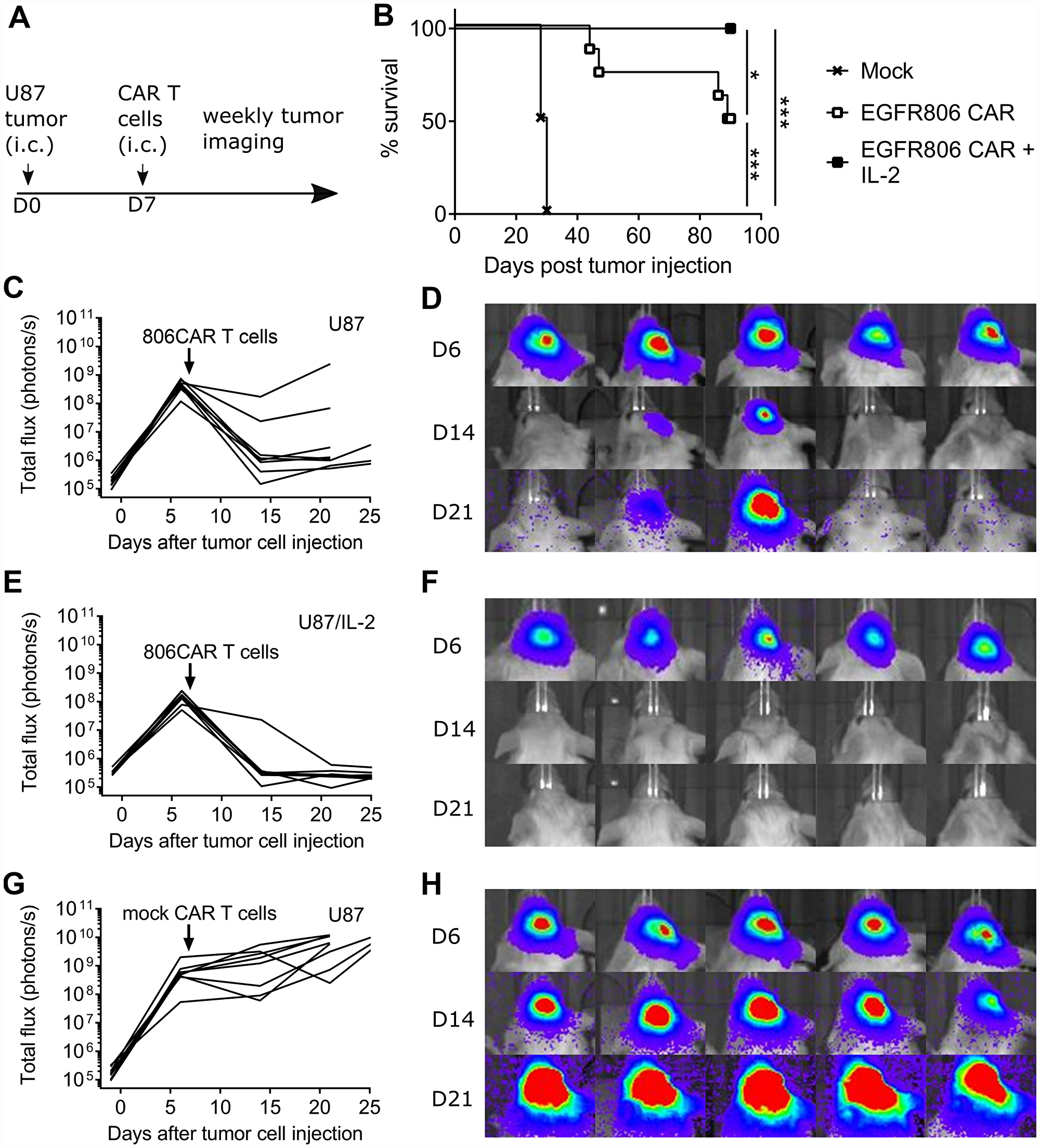
Figure 5: Short spacer second generation EGFR806-CAR T cells induce glioblastoma xenograft regression. (A) Experimental scheme. On day 0, NSG mice were intracranially (i. c.) injected with U87-ffluc cells with or without IL-2 overexpression. CAR T cells were delivered into the tumor bed on day 7, followed by weekly bioluminescence imaging. (B) Kaplan-Meier curve showing combined overall survival of mice treated with short spacer EGFR806 CAR T cells or mock transduced T cells. (C) Total tumor burden as expressed by total flux (photons/s) in individual mice bearing U87 intracranial tumors and treated with short spacer EGFR806-CAR T cells, with (D) showing representative bioluminescence images on days 6, 14, and 21 post treatment from the same group of animals. (E) Total tumor burden in individual mice bearing U87 intracranial tumors that overexpress IL-2, and treated with short-spacer EGFR806-CAR T cells, with (F) showing representative bioluminescence images from this group of animals. (G) Total tumor burden in individual mice bearing U87 intracranial tumors, treated with mock transduced T cells, with (H) showing representative images from this group. N = 8 for each group, pooled data from 2 experiments.
We then tested whether additional cytokine support could improve CAR T cell activity. Indeed, when mice were implanted with U87 tumors overexpressing IL-2, EGFR806-CAR T cells completely eradicated all tumors without recurrence in the 90-day experimental window. All animals survived until 90 days, which is significantly longer than in EGFR806-CAR treated mice bearing non-IL-2 tumors (P =0.0359, log rank test) (N = 8 for each group, pooled data from 2 separate experiments).
EGFR806-CAR T cells demonstrate a selective activation profile in a human iPSC teratoma model
To evaluate the potential cross-reactivity of the EGFR806-CAR to wild-type EGFR in a live tissue assay, we developed an in vivo human iPSC xenograft teratoma CAR T cell activation model. In this model, we also assessed whether T cell activation by concurrent glioma changes the likelihood of on-target off-tumor toxicity. NSG mice were injected intramuscularly (i. m.) with human iPSCs and maintained untreated until palpable teratomas were established (45–60 days) (Figure 6A). One cohort of mice was then subcutaneously injected in the other flank of the animal with U87 EGFR-positive tumor cells expressing truncated CD19, followed by i. v. CAR T cell administration 10 days later (Figure 6B). Mice were treated with 2nd generation short spacer EGFR806-CAR, Erbitux-CAR (positive control which is expected to be activated by both glioma and EGFR-expressing teratoma), and CD19-directed CAR T cells (CD19-CAR, negative control that is only activated by the CD19-overexpressing glioma, but not the teratoma). We used a defined ratio of 1:1 CD4: CD8 product for the EGFR806-CAR infusions to reflect the composition of CAR T products in clinical development. Established teratomas and glial tumors were harvested 3 days post CAR T cell injection and analyzed by immunohistochemistry for T cell infiltration and activation in EGFR-positive regions of teratoma. Histological analysis of established teratomas by H&E staining demonstrated that multiple tissue types from different germ layer origins were established (Supplementary Figure 3). Parallel immunofluorescence analysis confirmed that teratomas regionally expressed EGFR, but never CD19, while the glial tumors retained uniform EGFR and CD19 surface expression (Supplementary Figure 4).
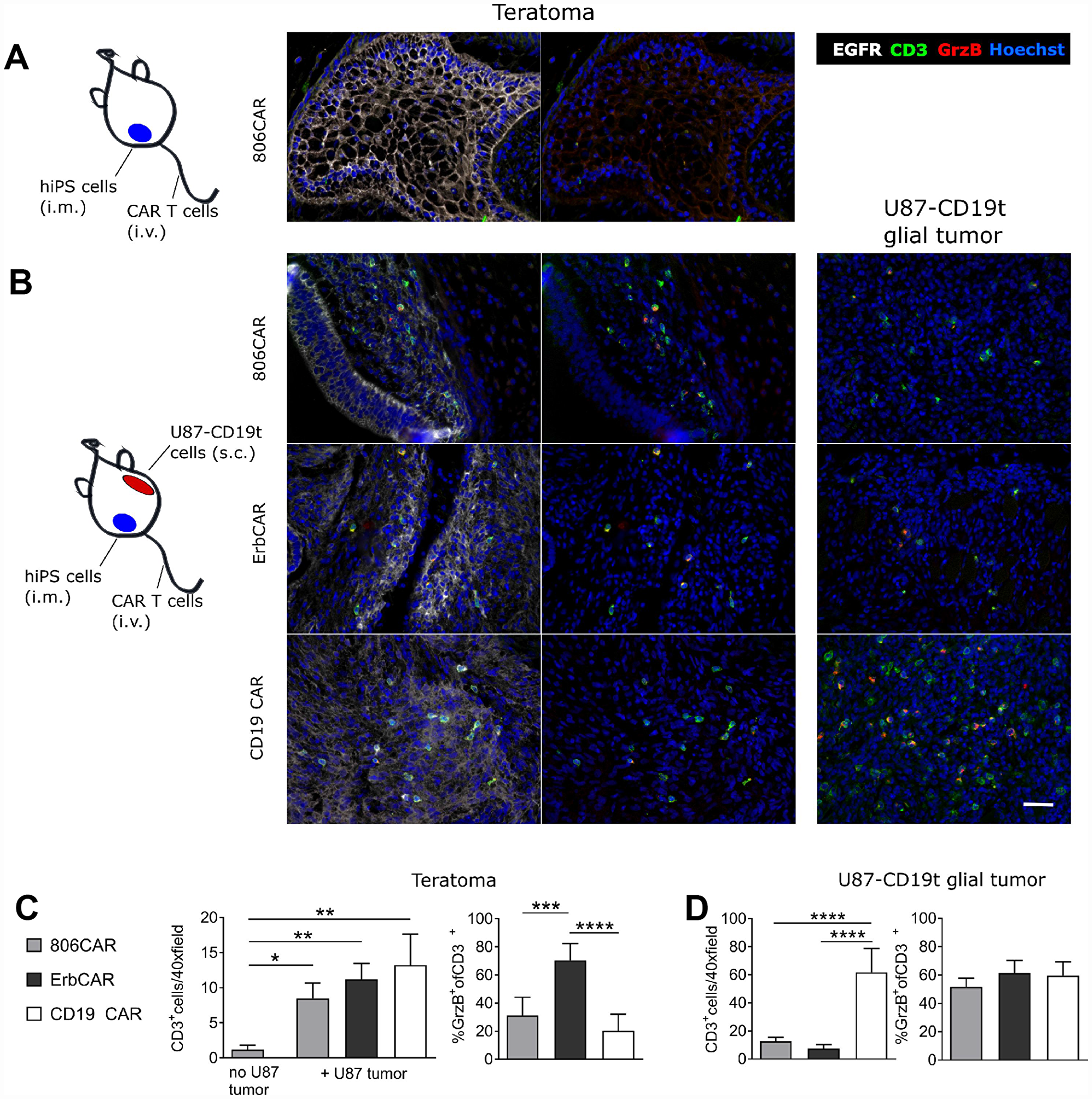
Figure 6: EGFR806-CAR T cells are selectively activated by tumor-specific EGFR expression in a human iPS cell teratoma model. (A) Teratoma-only model: mice were injected subcutaneously with human iPS (hiPS) cells in the left leg muscle, and EGFR806-CAR T cells were injected after teratoma has established. Immunolabeling of the teratoma (right 2 panels) shows minimal infiltration of CD3+ cells (green) into EGFR+ (white) regions of teratoma. (B) Teratoma-glioma model: mice were injected with hiPS cells in the left leg. After establishment of a palpable teratoma, U87 glioma cells expressing CD19t (U87-CD19t) were injected in the right flank. CAR T cells were infused 10 days after glioma cell injection. Representative images show immunolabeling demonstrating similar degrees of infiltration of CD3+ CAR T cells (green) into EGFR+ teratoma (left 2 columns) and EGFR+ U87-CD19t glioma (right column) after treatment with EGFR806 CAR (top), Erbitux CAR (middle) and CD19 CAR (bottom) T cells, with differing degrees of activation as indicated by Granzyme B (GrzB) labeling in red. (C) Left: infiltration of CAR T cells into teratoma is much lower in animals that did not receive concurrent U87-CD19t glioma grafts. Right: GrzB+ CAR T cells in EGFR+ teratoma as a percentage of total CD3+ teratoma resident T cells. Bars represent average cell number in 20 40× images from different EGFR+ tumor regions. Error bars, SEM. (D) Left: infiltration of CAR T cells into U87-CD19t glioma grafts on the other side of teratoma-bearing mice. Right: bars represent percentage GrzB+ CAR T cells of total CD3+ T cells in EGFR+ U87-CD19t glioma. Bars represent average cell number in 20 40× images from different glial tumor regions. Error bars, SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Scale bar, 20 μm. All images were acquired at 40× magnification.
EGFR806-CAR T cells showed negligible infiltration of the teratoma when no concurrent glioma was present (Figure 6A). In mice bearing both teratoma and glioma, the rate of teratoma infiltration increased, and was similar for EGFR806-CAR, Erbitux-CAR, and CD19-CAR T cells (Figure 6B, 6C). However, teratoma-resident Erbitux-CAR T cells significantly upregulated Granzyme B (GrzB) relative to EGFR806-CAR and CD19-CAR T cells (p < 0.0001; Figure 6B, 6C). In contrast, there was no statistically significant difference in T cell activation (%CD3+/GrzB+) between Erbitux-CAR, EGFR806-CAR, and CD19-CAR T cells that infiltrated the CD19+EGFR+ U87 glial tumor mass (Figure 6D). The number of glioma-infiltrating Erbitux-CAR and EGFR806-CAR T cells was similar, but lower than that of the CD19-CAR T cells (Figure 6D). This difference may be due to higher CD19t expression levels compared to the intrinsic EGFR expression in the glioma cells, and is less likely due to differential expansion of the different CAR T cells, given their similar density in the teratomas.
These results lend additional support to the conclusion that EGFR806-CAR T cells have lower on-target, off-tumor activity than nonselective Erbitux-CAR T cells, as indicated by their minimal activation in wild type EGFR-expressing teratoma compared to Erbitux-CAR T cells. The on-target, on-tumor response is comparable between both CAR constructs, indicating that the difference in activation in the teratoma is not due to overall decreased activity of the EGFR806-CAR T cells.
DISCUSSION
Designing an effective and safe CAR T cell therapy depends on choosing a tumor-specific target and tuning the signaling characteristics of the CAR. As the interaction of the extracellular domain with the target epitope is unique for each pairing, requirements for the extracellular spacer and intracellular signaling domains cannot be generalized across different CAR designs. We have developed a CAR based on the tumor-specific mAb806, which is selectively activated by tumor-expressed EGFR, be it full-length or the truncated EGFRvIII variant. Spacer length affected the efficacy of the EGFR806-CAR, with the short-spacer version showing the most robust in vivo tumor control.
We report consistent regression of orthotopic glioma in xenograft mouse models treated with EGFR806-CAR T cells, with an overall response comparable to other EGFR-directed CAR T cells (Erbitux-CAR, EGFRvIII-CAR, Nimotuzumab-CAR) on glioma xenografts [38, 40, 41]. We show that intracranial delivery is effective at controlling glioma growth in the brain, but the question of the optimal route of delivery is not yet settled in the field. In clinical trials of CD19-directed systemically delivered CAR T cells, small numbers of CAR T cells were detected in the cerebrospinal fluid [42, 43], however, it is unknown how efficiently they can traffic into the brain parenchyma. In clinical trials for glioblastoma, transient tumor regression has been achieved via intraventricular delivery [26], whereas systemic delivery, while safe, has not resulted in significant tumor response [15]. Certainly, many other factors, such as choice of epitope, CAR design, frequency of dosing, CAR T cell persistence, antigen expression, and antigen escape by the tumor are likely to be important for outcomes as well.
Affinity tuning of the CAR has been used as a strategy to reduce on-target off-tumor toxicity by using lower affinity CARs that spare normal tissue expressing low levels of the target [44–46]. For solid tumors, however, differences in CAR target expression levels throughout the tissue are the norm [47], which may decrease the efficacy of a low-affinity CAR. We thus did not rely on affinity tuning, and instead took advantage of the unique conformational properties of EGFR to ensure tumor specificity, as EGFR806-CAR binding is restricted to tumor-associated conformational states of the extracellular domains of EGFR. We ensured that EGFR806-CAR T cells are able to lyse tumor cells with a wide range of EGFR expression levels. We found that cytokine release and target cell killing by the EGFR806-CAR were similarly effective in all glioblastoma cell lines tested, and that it was active against EGFRvIII-transduced fetal human astrocytes, which expressed approximately 3000-4000 EGFR copies per cell. Other high affinity CARs require a minimum of 200-5000 cell-surface targets [48]. Glioblastoma expression of EGFRvIII is reported as several orders of magnitude higher [49], but decreased expression was reported upon treatment with EGFRvIII-directed CAR T cells [15]. Furthermore, we found that lower-level EGFR expression is common in glioblastoma. Therefore, we chose U87, the glioma cell line with the lowest EGFR expression level, for in vivo testing. Despite the EGFR806-CAR T cells’ robust in vivo activity, they did not show on-target off-tumor activation in EGFR-expressing teratomas, again confirming tumor specificity.
When used for CAR T cell targeting, the mAb806 scFv moiety remained highly specific for tumor-expressed EGFR, with no evidence of activity against native EGFR. In vitro, the EGFR806-CAR was minimally stimulated by normal fetal human astrocytes, but was activated by overexpressed EGFRvIII similarly to the nonselective Erbitux-CAR. With our novel human teratoma xenograft model, we have developed an efficient system for testing CAR T cell binding specificity in a wide variety of tissue types. EGFR806-CAR T cells only infiltrated EGFR-positive teratomas if the they were activated by a glioma at a different site in the same animal. However, the infiltrating EGFR806-CAR T cells did not become activated within the teratoma, unlike the nonselective Erbitux-CAR T cells. These findings suggest that teratoma infiltration may not be triggered by presence of local antigen, but rather by remote activation of the T cells. This is an intriguing observation given that lack of effector T cell infiltration into solid tumors has been a major obstacle in cell-based immunotherapy [50].
Important questions remain that cannot be easily answered in a xenograft mouse model. For example, how do the intracranially delivered CAR T cells traffic inside the CNS and what is the likelihood of escape into the systemic circulation? Human T cells are likely to behave very differently in the autologous host from which they were derived, and interaction with other tumor infiltrating immune cells will likely affect their performance. It is unknown whether CAR T cells can control infiltrative gliomas without injuring the surrounding brain parenchyma, and whether ablation of EGFR-expressing glioma cells will provide a selective survival advantage for EGFR-negative malignant cells.
Taken together, our data support the utility of targeting wild-type cell surface proteins on tumor cells that house a unique conformational epitope for selective CAR T cell targeting. The efficient and specific binding of the EGFR806-CAR supports its further development for clinical application.
Materials and Methods
Generation of CAR constructs and lentivirus
The chimeric monoclonal antibody (mAb) 806 has been previously described [18]. Variable short (IgG4-hinge), medium (IgG4-hinge-CH3) and long (IgG4-hinge-CH2-CH3) spacer 2nd generation 4-1BB-zeta CARs were constructed using the VL and VH segments of mAb 806 as previously described [44]. Each CAR sequence was linked to a T2A ribosomal skip sequence followed by a truncated EGFR (EGFRt) to facilitate selection of transduced cells. Second generation short spacer Erbitux-CAR T cells were produced by a similar mechanism, however the T2A sequence was followed by a truncated CD19 (CD19t). Lentiviral vector was produced in HEK 293T cells using the packaging vectors pCHGP-2, pCMV-Rev2, and pCMV-G [44].
Generation of central memory T cell lines expressing EGFR806-CARs
CD8+CD45RO+CD62L+ central memory T cells (TCM) were isolated from peripheral blood mononuclear cells (PBMCs) of healthy donors (Puget Sound Blood Center, Seattle, WA) by negative selection using CD8 isolation kits and CD45RA microbeads and positive selection with CD62L microbeads (Miltenyi Biotec). Following isolation, CD8+ TCM were stimulated with anti CD3/CD28 Dynabeads (Life Technologies) and transduced at a multiplicity of infection (MOI) of 3 on the third day of culture. EGFRt+ TCM subsets were enriched by immunomagnetic selection using biotinylated Erbitux and anti-biotin microbeads (Miltenyi Biotec), then expanded as previously described [44]. CD8+ TCM were maintained in RPMI medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 IU/mL recombinant human interleukin 2 (IL-2) and 1 ng/mL recombinant human interleukin 15 (IL-15).
Flow cytometry and immunophenotyping
Conjugated mAbs for CD3, CD4, CD8, CD45RO, and CD62L (Biolegend) were used for immunophenotyping. Tumor cell EGFR positivity and CAR construct expression, via the surrogate cell-surface marker EGFRt, was confirmed using biotinylated Erbitux (Cetuximab) and PE-conjugated streptavidin (SA-PE). CAR cell-surface and total expression was confirmed using biotinylated Protein-L (Genscript) and SA-PE or anti-CD247 (CD3z, BD Biosciences), respectively. Flow analysis was performed on an LSRFortessa (BD), sort-purifications on a FACSAriaII (BD) and data analyzed using FlowJo software (Treestar).
Tissue microarray staining and analysis
Tissue microarrays containing duplicates of 33 glioblastoma cases, 2 brain anaplastic astrocytoma, and 5 normal brain tissue samples were ordered from US Biomax, Inc. (Cat: GL806c). Arrays were stained with primary mouse anti-human EGFR antibody (Clone 31G7, Invitrogen) at a 1:100 dilution. Briefly, slides were baked for 30 minutes at 60°C and deparaffinized on the Leica Bond Automated Immunostainer, followed by antigen retrieval with Proteinase K for 10min at 37°C. Blocking was performed for 10min at RT using Normal Goat Serum (10% in TBS) followed by primary antibody in Leica primary antibody diluent for 30min. Secondary antibodies (Life Technologies) were incubated with TMAs for 2hr at RT and diluted 1:500 in PBS with 0.2% BSA.
TMAs were imaged using the Nuance Multispectral imaging system (Perkin Elmer) on a Nikon Eclipse Ci upright microscope at 20 ×. Images were captured every 20 nm wavelength of light from 420 nm-720 nm. Data were analyzed by InForm analysis software (Perkin Elmer) using a threshold of 0.07OD.
Cell line production and analysis
The A431, T98, U251T, U87 and Raji cell lines were obtained from the American Type Culture Collection (ATCC). Unless otherwise indicated, cell lines were maintained in DMEM or RPMI (Gibco) supplemented with 2 mM L-glutamine (Irvine Scientific; Santa Ana, CA), 25 mM HEPES (Irvine Scientific), and 10% heat-inactivated FCS (Hyclone). Total EGFR expression was analyzed by western blot analysis (Cat: sc-03, Santa Cruz Biotech) and antigen density was quantified by determining the anti-EGFR antibody (Clone EGFR.1, BD) binding capacity per cell using the BD QuantiBRITE system for fluorescence measurement [51]. Raji EGFR-vIII were generated by lentiviral transduction (EGFRvIII contains aa 1-29, 298-668 of EGFR) and enriched by immunomagnetic selection. U87 ffluc-IL2+ tumor cells were previously described [39].
In vitro cellular assays
Chromium release assay
CAR T cell cytotoxicity was determined by chromium release assay. Target cells were labeled with 51Cr (Perkin Elmer), washed and incubated in triplicate with CAR T cells at various effector to target (E: T) ratios. Supernatants were harvested for g-counting 4 hours later and specific lysis was calculated using the standard formula [52]. Soluble EGFR peptide (aa 287-302) was purchased from GenScript for peptide inhibition assays.
Cytokine release assay
To investigate cytokine secretion, CAR T cells and target cells were plated at a 2:1 ratio and incubated for 24 hours. Supernatant was then analyzed for IL-2, IFNg and TNFa production using the Bio-Plex multiplex bead array system (Bio-Rad).
Orthotopic xenograft model and exogenous T cell transplantation
The orthotopic xenograft model was performed as previously described [39]. Briefly, 8-12-week-old adult male NSG mice were injected intracranially (i. c.) on day 0 with 2 × 105 ffluc-IL2+ expressing U87 glioma cells 2 mm lateral, 0.5 mm anterior to the bregma and 2.5 mm deep from the dura. 7 days later, Mice were i. c. injected at the same site with a total of 2 × 106 CAR T cells placed 2.5, 2.35 and 2.25 mm deep from the dura. Bioluminescent imaging was performed weekly by intraperitoneal (i. p.) injection of 4.29mg/mouse D-luciferin (Xenogen), anesthesia by isoflurane and imaging 15 minutes post D-luciferin injection using the IVIS Spectrum Imaging System (Perkin Elmer). Luciferase activity was analyzed using Living Image Software Version 4.3 (Perkin Elmer) and photon flux was analyzed within regions of interest. All animal experiments were approved by the Institutional Animal Care and Use Committee.
iPSC teratoma model and IHC analysis
To generate teratomas in vivo, 6-8 week old NSG female mice (Jackson Laboratories or bred in-house) were injected with 0.5 × 106 human iPS cells prepared in a pro-survival cocktail [53] (generously provided by the Ellison Stem Cell Core of the institute for Stem Cell Regenerative Medicine at the University of Washington) via intramuscular (i. m.) injection into the inner left hind leg. Once teratomas became palpable (6-7 weeks), a subset of mice was subsequently engrafted with 5 × 106 CD19t-expressing U87 glioblastoma tumor cells in USP grade PBS (Ameresco) and 50% matrigel (Corning) via s. c. injection into the right flank. Ten days post glioma inoculation, all animals received a tail vein injection of either USP grade PBS (Ameresco) vehicle alone or 50 × 106 CAR T cells at a 1:1 ratio of CD4: CD8. All animals were euthanized 3 days after CAR T cell administration, perfused with PBS (Gibco) followed by 10% neutral buffered formalin (Thermo Scientific), and teratomas and subcutaneously engrafted tumors were collected for immunohistochemistry. IHC analysis was performed in EGFR+ regions.
EGFR teratoma immunohistochemistry
Slides were deparaffinized through a graded alcohol series, and antigen retrieval was performed using Digest-all 3 enzyme cocktail (Fisher, #3009) for 10 minutes at 37°C. Slides were blocked for 1 hour using 0.2% bovine serum albumin + 2% normal goat serum, incubated with primary antibody (clone 31G7, Fisher, #280005), diluted 1:100 for 1 hour in blocking buffer for 1 hour, RT. Slides were washed in PBS, and incubated in secondary antibody (Fisher #A-21240) diluted 1:500 in blocking buffer for 1 hour, RT. Slides were washed in ddH2O, and incubated with Hoechst dye (Fisher, #H3569) diluted to 2 μg/ml in ddH2O for 10 minutes. Slides were imaged on a Nuance Multispectral Imaging system on a Nikon Eclipse Ci microscope.
CD3 and Granzyme B teratoma immunohistochemistry
Slides were deparaffinized through a graded alcohol series, and antigen retrieval was performed using a decloaking chamber (Biocare medical, #DC2002) at 125°C for 3 minutes using Diva buffer (Biocare medical, #DV2004G1). Slides were blocked for 1 hour using 0.2% bovine serum albumin + 2% normal goat serum, incubated with primary antibodies (CD3 clone CD3-12, bio-rad #MCA1477, Granzyme B, rabbit polyclonal, LSBio # 34084), diluted 1:500 each for 1 hour in blocking buffer for 1 hour, RT. Slides were washed in PBS, and incubated in secondary antibody (Fisher #A-21240) diluted 1:500 in blocking buffer for 1 hour, RT. Slides were washed in ddH2O, and incubated with Hoechst dye (Fisher, #H3569) diluted to 2 μg/ml in ddH2O for 10 minutes. Slides were imaged on a Nuance Multispectral Imaging system on a Nikon Eclipse Ci microscope.
Statistical analyses
Statistical analyses were conducted using Prism Software (GraphPad). Data are presented as means ± SD or SEM as stated in the figure legends. Student’s t test was conducted as a two-sided unpaired test with a confidence interval of 95% and results with a P value less than 0.05 were considered significant. Statistical analyses of survival were conducted by log-rank testing and results with a P value less than 0.05 were considered significant.
Abbreviations
CAR: chimeric antigen receptor; CNS: central nervous system; EGFR: epidermal growth factor receptor; IFNg: interferon gamma; iPSC: induced pluripotent stem cell; TNFa: tumor necrosis factor alpha.
ACKNOWLEDGMENTS
We thank the Ellison Stem Cell Core of the Institute for Stem Cell and Regenerative Medicine at the University of Washington for providing human iPSC.
CONFLICTS OF INTEREST
Dr Orentas conducts consulting and sponsored research for Miltenyi Biotec, Inc. Dr Jensen has received grants and personal fees from Juno Therapeutics, a Celgene Company, during the conduct of the study but outside the submitted work. In addition, Dr Jensen holds a patent “Defined Composition Gene Modified T Cell Products”, with royalties paid to Juno Therapeutics. The remaining authors declare no conflicts of interest.
FUNDING
Stand up to Cancer and St. Baldrick’s Pediatric Dream Team Translational Research Grant, NIH R01 CA136551-05, Alex’s Lemonade Stand Phase I/II Infrastructure Grant, Washington State Life Sciences Discovery Fund, Ben Towne Foundation, William Lawrence and Blanche Hughes Foundation, Juno Therapeutics Inc, a Celgene Company.
References
1. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front Immunol. 2017; 8:1850. https://doi.org/10.3389/fimmu.2017.01850. [PubMed].
2. McLendon RE, Halperin EC. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer. 2003; 98:1745–1748. https://doi.org/10.1002/cncr.11666. [PubMed].
3. Ostrom QT, Gittleman H, Xu J, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016; 18:v1–v75. https://doi.org/10.1093/neuonc/now207. [PubMed].
4. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, Schüller U, Gururangan S, McLendon R, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013; 14:1200–1207. https://doi.org/10.1016/S1470-2045(13)70449-2. [PubMed].
5. Alexiou GA, Moschovi M, Stefanaki K, Panagopoulos D, Tsotra M, Siozos G, Sfakianos G, Prodromou N. Supratentorial ependymomas in children: Analysis of nine cases. J Pediatr Neurosci. 2013; 8:15–18. https://doi.org/10.4103/1817-1745.111415. [PubMed].
6. Bax DA, Gaspar N, Little SE, Marshall L, Perryman L, Regairaz M, Viana-Pereira M, Vuononvirta R, Sharp SY, Reis-Filho JS, Stávale JN, Al-Sarraj S, Reis RM, et al. EGFRvIII deletion mutations in pediatric high-grade glioma and response to targeted therapy in pediatric glioma cell lines. Clin Cancer Res. 2009; 15:5753–5761. https://doi.org/10.1158/1078-0432.CCR-08-3210. [PubMed].
7. Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015; 15:302–310. https://doi.org/10.1038/nrc3918. [PubMed].
8. Li G, Mitra SS, Monje M, Henrich KN, Bangs CD, Nitta RT, Wong AJ. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. J Neurooncol. 2012; 108:395–402. https://doi.org/10.1007/s11060-012-0842-3. [PubMed].
9. Mendrzyk F, Korshunov A, Benner A, Toedt G, Pfister S, Radlwimmer B, Lichter P. Identification of Gains on 1q and Epidermal Growth Factor Receptor Overexpression as Independent Prognostic Markers in Intracranial Ependymoma. Clin Cancer Res. 2006; 12:2070–2079. https://doi.org/10.1158/1078-0432.CCR-05-2363. [PubMed].
10. Patereli A, Alexiou GA, Stefanaki K, Moschovi M, Doussis-Anagnostopoulou I, Prodromou N, Karentzou O. Expression of epidermal growth factor receptor and HER-2 in pediatric embryonal brain tumors. Pediatr Neurosurg. 2010; 46:188–192. https://doi.org/10.1159/000316640. [PubMed].
11. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014; 46:444–450. https://doi.org/10.1038/ng.2938. [PubMed].
12. Binder ZA, Thorne AH, Bakas S, Wileyto EP, Bilello M, Akbari H, Rathore S, Ha SM, Zhang L, Ferguson CJ, Dahiya S, Bi WL, Reardon DA, et al. Epidermal Growth Factor Receptor Extracellular Domain Mutations in Glioblastoma Present Opportunities for Clinical Imaging and Therapeutic Development. Cancer Cell. 2018; 34:163–177.e7. https://doi.org/10.1016/j.ccell.2018.06.006. [PubMed].
13. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, Drappatz J, O’Rourke DM, Wong M, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017; 18:1373–1385. https://doi.org/10.1016/S1470-2045(17)30517-X. [PubMed].
14. Westphal M, Maire CL, Lamszus K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs. 2017; 31:723–735. https://doi.org/10.1007/s40263-017-0456-6. [PubMed].
15. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017; 9. https://doi.org/10.1126/scitranslmed.aaa0984. [PubMed].
16. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018; 15:422–442. https://doi.org/10.1038/s41571-018-0003-5. [PubMed].
17. Luwor RB, Johns TG, Murone C, Huang HJ, Cavenee WK, Ritter G, Old LJ, Burgess AW, Scott AM. Monoclonal antibody 806 inhibits the growth of tumor xenografts expressing either the de2-7 or amplified epidermal growth factor receptor (EGFR) but not wild-type EGFR. Cancer Res. 2001; 61:5355–5361. https://doi.org/10.1038/sj.onc.1207870. [PubMed].
18. Panousis C, Rayzman VM, Johns TG, Renner C, Liu Z, Cartwright G, Lee FT, Wang D, Gan H, Cao D, Kypridis A, Smyth FE, Brechbiel MW, et al. Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR. Br J Cancer. 2005; 92:1069–1077. https://doi.org/10.1038/sj.bjc.6602470. [PubMed].
19. Jungbluth AA, Stockert E, Huang HJS, Collins VP, Coplan K, Iversen K, Kolb D, Johns TJ, Scott AM, Gullick WJ, Ritter G, Cohen L, Scanlan MJ, et al. A monoclonal antibody recognizing human cancers with amplification/overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003; 100:639–644. https://doi.org/10.1073/pnas.232686499. [PubMed].
20. Orellana L, Thorne AH, Lema R, Gustavsson J, Parisian AD, Hospital A, Cordeiro TN, Bernadó P, Scott AM, Brun-Heath I, Lindahl E, Cavenee WK, Furnari FB, et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc Natl Acad Sci U S A. 2019; 116:10009–10018. https://doi.org/10.1073/pnas.1821442116. [PubMed].
21. Gan HK, Burgess AW, Clayton AH, Scott AM. Targeting of a Conformationally Exposed, Tumor-Specific Epitope of EGFR as a Strategy for Cancer Therapy. Cancer Res. 2012; 72:2924–2930. https://doi.org/10.1158/0008-5472.CAN-11-3898. [PubMed].
22. Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol. 2014; 6:a020768. https://doi.org/10.1101/cshperspect.a020768. [PubMed].
23. Gan HK, Walker F, Burgess AW, Rigopoulos A, Scott AM, Johns TG. The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J Biol Chem. 2007; 282:2840–2850. https://doi.org/10.1074/jbc.M605136200. [PubMed].
24. Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, Papenfuss AT, Poon AM, Hopkins W, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007; 104:4071–4076. https://doi.org/10.1073/pnas.0611693104. [PubMed].
25. van den Bent M, Gan HK, Lassman AB, Kumthekar P, Merrell R, Butowski N, Lwin Z, Mikkelsen T, Nabors LB, Papadopoulos KP, Penas-Prado M, Simes J, Wheeler H, et al. Efficacy of depatuxizumab mafodotin (ABT-414) monotherapy in patients with EGFR-amplified, recurrent glioblastoma: results from a multi-center, international study. Cancer Chemother Pharmacol. 2017; 80:1209–1217. https://doi.org/10.1007/s00280-017-3451-1. [PubMed].
26. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, Kurien A, Priceman SJ, Wang X, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016; 375:2561–2569. https://doi.org/10.1056/NEJMoa1610497. [PubMed].
27. Sibilia M, Kroismayr R, Lichtenberger BM, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation. 2007; 75:770–787. https://doi.org/10.1111/j.1432-0436.2007.00238.x. [PubMed].
28. Wagner B, Natarajan A, Grünaug S, Kroismayr R, Wagner EF, Sibilia M. Neuronal survival depends on EGFR signaling in cortical but not midbrain astrocytes. EMBO J. 2006; 25:752–762. https://doi.org/10.1038/sj.emboj.7600988. [PubMed].
29. Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature. 2010; 467:323–327. https://doi.org/10.1038/nature09347. [PubMed].
30. Galvez-Contreras AY, Quinones-Hinojosa A, Gonzalez-Perez O. The role of EGFR and ErbB family related proteins in the oligodendrocyte specification in germinal niches of the adult mammalian brain. Front Cell Neurosci. 2013; 7:258. https://doi.org/10.3389/fncel.2013.00258. [PubMed].
31. Liu TF, Tatter SB, Willingham MC, Yang M, Hu JJ, Frankel AE. Growth factor receptor expression varies among high-grade gliomas and normal brain: epidermal growth factor receptor has excellent properties for interstitial fusion protein therapy. Mol Cancer Ther. 2003; 2:783–787. [PubMed].
32. Tissue expression of EGFR - Summary - The Human Protein Atlas. Available 2018 Apr 4, from https://www.proteinatlas.org/ENSG00000146648-EGFR/tissue.
33. Vinukonda G, Hu F, Mehdizadeh R, Dohare P, Kidwai A, Juneja A, Naran V, Kierstead M, Chawla R, Kayton R, Ballabh P. Epidermal Growth Factor Preserves Myelin and Promotes Astrogliosis after Intraventricular Hemorrhage. Glia. 2016; 64:1987–2004. https://doi.org/10.1002/glia.23037. [PubMed].
34. Bulic-Jakus F, Katusic Bojanac A, Juric-Lekic G, Vlahovic M, Sincic N. Teratoma: from spontaneous tumors to the pluripotency/malignancy assay. Wiley Interdiscip Rev Dev Biol. 2016; 5:186–209. https://doi.org/10.1002/wdev.219. [PubMed].
35. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015; 3:125–135. https://doi.org/10.1158/2326-6066.CIR-14-0127. [PubMed].
36. Wang X, Wong CW, Urak R, Taus E, Aguilar B, Chang WC, Mardiros A, Budde LE, Brown CE, Berger C, Forman SJ, Jensen MC. Comparison of naïve and central memory derived CD8+ effector cell engraftment fitness and function following adoptive transfer. Oncoimmunology. 2016; 5:e1072671. https://doi.org/10.1080/2162402X.2015.1072671. [PubMed].
37. Johns TG, Adams TE, Cochran JR, Hall NE, Hoyne PA, Olsen MJ, Kim YS, Rothacker J, Nice EC, Walker F, Ritter G, Jungbluth AA, Old LJ, et al. Identification of the epitope for the epidermal growth factor receptor-specific monoclonal antibody 806 reveals that it preferentially recognizes an untethered form of the receptor. J Biol Chem. 2004; 279:30375–30384. https://doi.org/10.1074/jbc.M401218200. [PubMed].
38. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H, Lee DA, Heimberger AB, Champlin RE, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent anti-tumor activity. Cancer Res. 2015; 75:3505–3518. https://doi.org/10.1158/0008-5472.CAN-15-0139. [PubMed].
39. Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004; 64:9160–9166. https://doi.org/10.1158/0008-5472.CAN-04-0454. [PubMed].
40. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, Boesteanu AC, Cogdill AP, Chen T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015; 7:275ra22. https://doi.org/10.1126/scitranslmed.aaa4963. [PubMed].
41. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, Sayour EJ, McLendon R, Herndon JE 2nd, Healy P, Archer GE, Bigner DD, Johnson LA, et al. EGFRvIII-Specific Chimeric Antigen Receptor T Cells Migrate to and Kill Tumor Deposits Infiltrating the Brain Parenchyma in an Invasive Xenograft Model of Glioblastoma. PLoS One. 2014; 9:e94281. https://doi.org/10.1371/journal.pone.0094281. [PubMed].
42. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, Yeung C, Liles WC, Wurfel M, Lopez JA, Chen J, Chung D, Harju-Baker S, et al. Endothelial Activation and Blood–Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017; 7:1404–1419. https://doi.org/10.1158/2159-8290.CD-17-0698. [PubMed].
43. Rheingold SR, Chen LN, Maude SL, Aplenc R, Barker C, Barrett DM, Callahan C, Cebry K, Kulikovskaya I, Lacey SF, Levine BL, Melenhorst JJ, Shaw PA, et al. Efficient Trafficking of Chimeric Antigen Receptor (CAR)-Modified T Cells to CSF and Induction of Durable CNS Remissions in Children with CNS/Combined Relapsed/Refractory ALL. Blood. 2015; 126:3769–3769. https://doi.org/10.1182/blood.V126.23.3769.3769.
44. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T-cells. Clin Cancer Res. 2013; 19:3153–3164. https://doi.org/10.1158/1078-0432.CCR-13-0330. [PubMed].
45. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B, Zhou L, Loew A, Young RM, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015; 75:3596–3607. https://doi.org/10.1158/0008-5472.CAN-15-0159. [PubMed].
46. Abate-Daga D, Davila ML. CAR models: next-generation CAR modifications for enhanced T-cell function. Mol Ther Oncolytics. 2016; 3:16014. https://doi.org/10.1038/mto.2016.14. [PubMed].
47. Schmidts A, Maus MV. Making CAR T Cells a Solid Option for Solid Tumors. Front Immunol. 2018; 9:2593. https://doi.org/10.3389/fimmu.2018.02593. [PubMed].
48. Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol Sci. 2016; 37:220–230. https://doi.org/10.1016/j.tips.2015.11.004. [PubMed].
49. Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997; 57:4130–4140. [PubMed].
50. Sackstein R, Schatton T, Barthel SR. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Invest. 2017; 97:669–697. https://doi.org/10.1038/labinvest.2017.25. [PubMed].
51. Schwartz A, Marti GE, Poon R, Gratama JW, Fernández-Repollet E. Standardizing flow cytometry: a classification system of fluorescence standards used for flow cytometry. Cytometry. 1998; 33:106–114. https://doi.org/10.1002/(SICI)1097-0320(19981001)33:2<106::AID-CYTO4>3.0.CO;2-H. [PubMed].
52. Gonzalez S, Naranjo A, Serrano LM, Chang WC, Wright CL, Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J Gene Med. 2004; 6:704–711. https://doi.org/10.1002/jgm.489. [PubMed].
53. Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, Palpant NJ, Gantz J, Moyes KW, Reinecke H, Van Biber B, Dardas T, Mignone JL, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012; 489:322–325. https://doi.org/10.1038/nature11317. [PubMed].