
Introduction
The phosphoinositide-3-kinase (PI3K) signaling pathway regulates cell survival, proliferation, metabolism and various other processes linked to cell growth. Human cancers frequently acquire mutations resulting in aberrant activation of PI3K signaling which is often associated with increased tumour progression and resistance to cancer therapies. The PI3K pathway is activated by growth-factor mediated activation of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) which transduce signals through class I PI3K via phosphorylation of the 3-hydroxyl group of the inositol ring of phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] to generate phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3 [PIP3]) [1, 2]. PIP3 is subsequently dephosphorylated by either PTEN to form phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] or by SHIP phosphatases to form phosphatidylinositol-3,4-bisphosphate [PtdIns(3,4)P2]. The 3’phosphorylated phosphatidylinositols like PIP3 and PtdIns(3,4)P2 recruit cytosolic proteins containing pleckstrin homology (PH) domains, such as the serine/threonine kinase Akt. Once recruited to membranes, the kinase activity of Akt is activated by phosphorylation to promote downstream signaling to cell survival, growth, proliferation, cell migration and angiogenesis, through phosphorylation of specific intracellular protein substrates [3].
Inositol Polyphosphate 4-Phosphatase, Type II (INPP4B), a lipid phosphatase that preferentially hydrolyzes PtdIns(3,4)P2 to generate phosphatidylinositol-3-monophosphate [PtdIns(3)P], was characterized as a gene with a key role in PI3K pathway signaling [4]. Since both PtdIns(3,4)P2 and PIP3 promote Akt recruitment to the plasma membrane, INPP4B was predicted to act as a tumour suppressor by inhibiting Akt recruitment, activation and thus downstream PI3K pathway signaling [5–8]. The first evidence characterizing INPP4B as a gene with importance in cancer was from a RNA interference screen in immortalized human mammary epithelial cells (HMEC) designed to identify candidate tumour suppressors [9]. Several studies have subsequently validated a tumour suppressor role for INPP4B in breast, ovarian, skin, and prostate cancer among others [10, 4, 8, 11–23]. In these studies, INPP4B loss resulted in elevated Akt activation, increased cell survival and a more aggressive growth phenotypes associated with poor outcomes for cancer patients [13, 16, 24]. These findings for INPP4B contribute to the increasing role of phosphoinositide phosphatases other than PTEN in cancer; these include the INPP5-family members such as INPP5J/PIPP, INPP5D/SHIP1, INPPL1/SHIP2, and INPP5E [25–29]. Notably, despite the abundance of clinical data supporting a tumour suppressor role for INPP4B, there is no evidence that Inpp4b deletion alone in mouse models leads to tumour formation [17, 19, 30]. However when Inpp4b loss was combined with Pten heterozygosity, it altered the penetrance of the Pten-spectrum of tumours, and notably malignant thyroid cancer was observed [17, 19, 30]. Thus it has been suggested that INPP4B may be a tumour suppressor in the context of PTEN loss, and may have weak tumour suppressive function otherwise [31].
Conversely, emerging findings in malignancies including acute myeloid leukemia (AML), colon cancer, melanoma and breast cancer among others suggest that overexpression of INPP4B is also associated with promoting aggressive cancer phenotypes [32–36]. Signaling downstream of PtdIns(3)P has been explored as a possible mechanism. For instance, PtdIns(3)P mediated activation of Serum/Glucocorticoid Regulated Kinase Family Member 3 (SGK-3) was observed downstream of INPP4B overexpression in some cancers [34, 36–39]. Moreover, PtdIns(3)P has very important cellular roles, which include endosomal trafficking and autophagy which are currently unexplored in the context of INPP4B overexpression [40]. Moreover, INPP4B was reported to have both tumour promoting and tumour suppressing features in different subsets of the same cancer. For instance in melanoma and breast cancer, both INPP4B loss and INPP4B overexpression were associated with downstream oncogenic signaling through Akt and SGK3, respectively [8, 37, 38, 41]. Altogether, these findings point to a putative contextual role for INPP4B in cancer [42, 43]. Nevertheless, mechanisms underlying the context-dependent cancer functions of INPP4B remain to be elucidated.
A growing body of evidence links altered levels of INPP4B expression to the progression of cancer. However, a role for INPP4B in the transformation of primary cells remains unexplored. Herein, we sought to investigate the consequences of Inpp4b-deficiency and INPP4B-overexpression on the cellular transformation of primary MEF in combination with oncogenic drivers including H-RasV12, E1A or SV40 T-Large. Exploring a role for Inpp4b in MEF transformation may provide insight on whether co-operating driver mutational signaling will alter Inpp4b context dependent outcome in tumourigenesis.
Results
Characterization of primary Inpp4b+/+, Inpp4b+/- and Inpp4b-/- MEF
To investigate the role of Inpp4b loss on cellular transformation we generated E13.5 MEF from a constitutive Inpp4b exon 10 knockout (Inpp4b-/-) mouse model [30]. PCR genotyping of cultured MEFs derived from timed-matings of Inpp4b+/- mice was performed to determine Inpp4b+/+, Inpp4b+/- or Inpp4b-/- genotypes (Figure 1A). RT-QPCR and immunoblots were performed to validate loss of both transcript and protein levels of Inpp4b in Inpp4b-/- MEF (Figure 1B). Growth characteristics of primary MEF was evaluated in short term growth assays where we observed no significant differences in the mean growth rates of Inpp4b+/+, Inpp4b+/- and Inpp4b-/- MEF (Figure 1C). Similarly, long term clonogenic growth potential was tested in primary Inpp4b+/+ and Inpp4b-/- MEF. After 11 days of growth, only sparse spontaneous clone formation was observed in both Inpp4b+/+ and Inpp4b-/- MEF, with no measurable difference between genotypes (Figure 1D). Finally, neither Inpp4b+/+ nor Inpp4b-/- MEF were observed to grow as anchorage independent colonies in soft agar (Figure 1E).
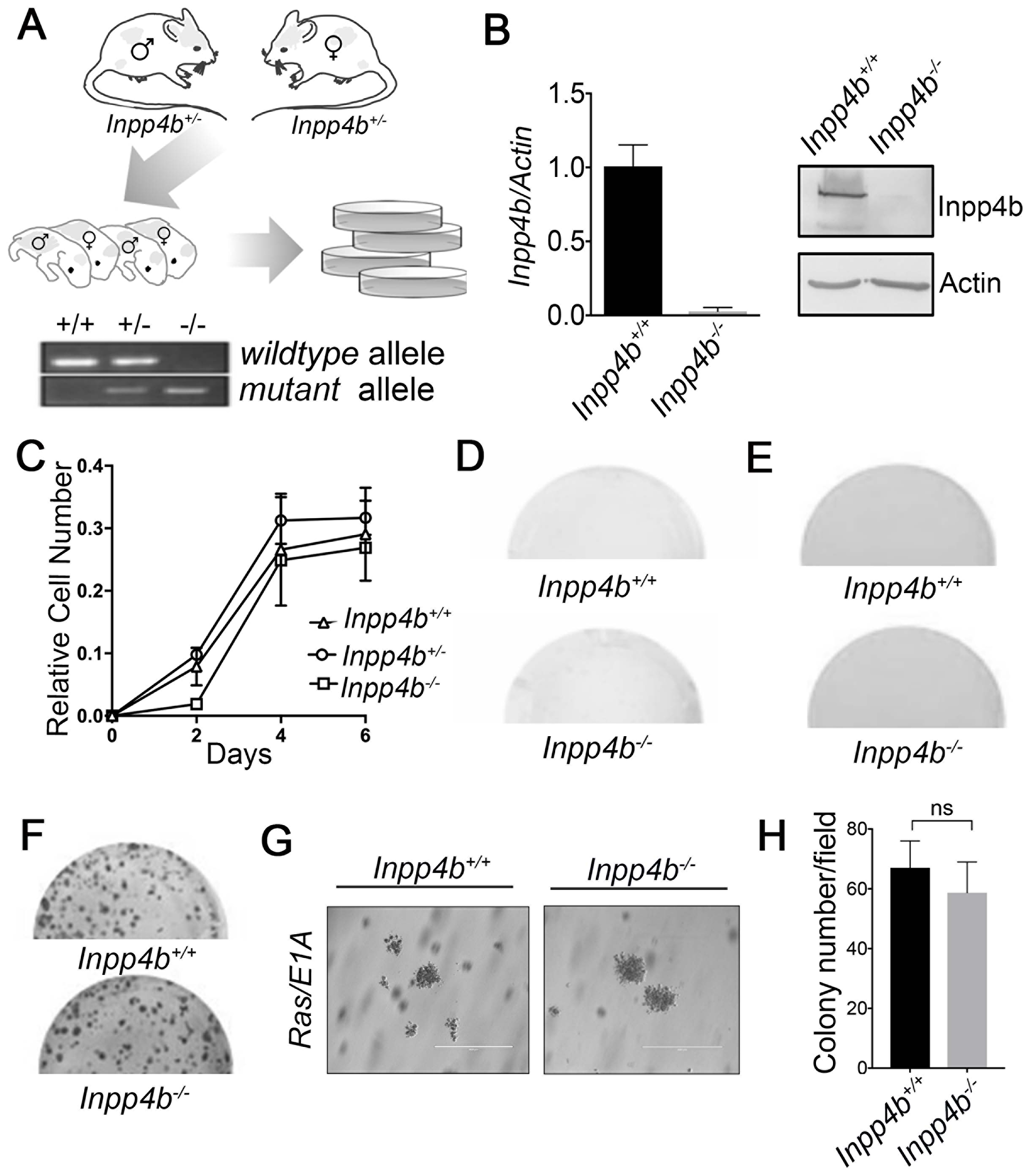
Figure 1: Generation and characterization of primary Inpp4b+/+, Inpp4b+/- and Inpp4b-/- MEF. (A) Schematic illustrating breeding strategy for generation of primary MEF and a typical Inpp4b genotyping PCR result is illustrated. (B) RT-qPCR and Immunoblot demonstrating Inpp4b expression levels in Inpp4b+/+ and Inpp4b-/- MEF. (C) Growth curve, (D) colony formation (E) and soft agar assay of primary Inpp4b+/+ and Inpp4b-/- MEF. (F) Colony formation assay with Inpp4b+/+ and Inpp4b-/- MEF empty vector and 12SLRC H-RasV12/E1A infection. (G) Representative soft agar assay of 12SLRC H-RasV12 /E1A infected Inpp4b+/+ and Inpp4b-/- MEF. Photos of representative colonies at 20X. (H) Quantitation of quantitation of foci formation assay.
To investigate the necessity for Inpp4b in cellular transformation, we examined the consequences of Inpp4b deficiency on H-RasV12/E1A mediated MEF transformation. For these experiments we infected early passage Inpp4b+/+ and Inpp4b-/- MEF with retrovirus expressing both H-RasV12 and E1A (12SLRC). Upon infection of cells, we observed no difference in the morphology in all H-RasV12/E1A transformed cells of either genotype. Moreover, we observed no difference between the transformed MEF from Inpp4b+/+ and Inpp4b-/- backgrounds on foci formation in clonogenic assays (Figure 1F) and anchorage independent colonies in soft agar (Figure 1G, 1H). These data suggest that Inpp4b-deficiency does not lead to spontaneous transformation of MEF and that Inpp4b expression is dispensable for H-RasV12/E1A mediated MEF transformation.
Neither loss nor overexpression of INPP4B cooperate with H-RasV12 in MEF transformation
To characterize the cooperativity of Inpp4b-deficiency with H-RasV12 overexpression in cellular transformation, early passage Inpp4b+/+ and Inpp4b-/- MEF were infected with retroviral particles generated from the pBabe-H-RasV12-puro vector. Morphologically, both H-RasV12; Inpp4b+/+ H-RasV12; Inpp4b-/- MEF demonstrated the expected features of oncogene-induced senescence (OIS) including complete growth inhibition (Figure 2A) and large multinucleated senescent-like cells (Figure 2B). Neither H-RasV12; Inpp4b+/+ nor H-RasV12; Inpp4b-/- MEF were able to form colonies in clonogenic assays (Figure 2C). Similarly, no anchorage-independent colonies were observed in soft agar (Figure 2D). Conversely, positive control MEF infected with H-RasV12 and E1A retroviruses generated rapidly proliferating cells (Figure 2A), abundant foci (Figure 2E, top) and numerous anchorage independent colonies (Figure 2E, bottom).
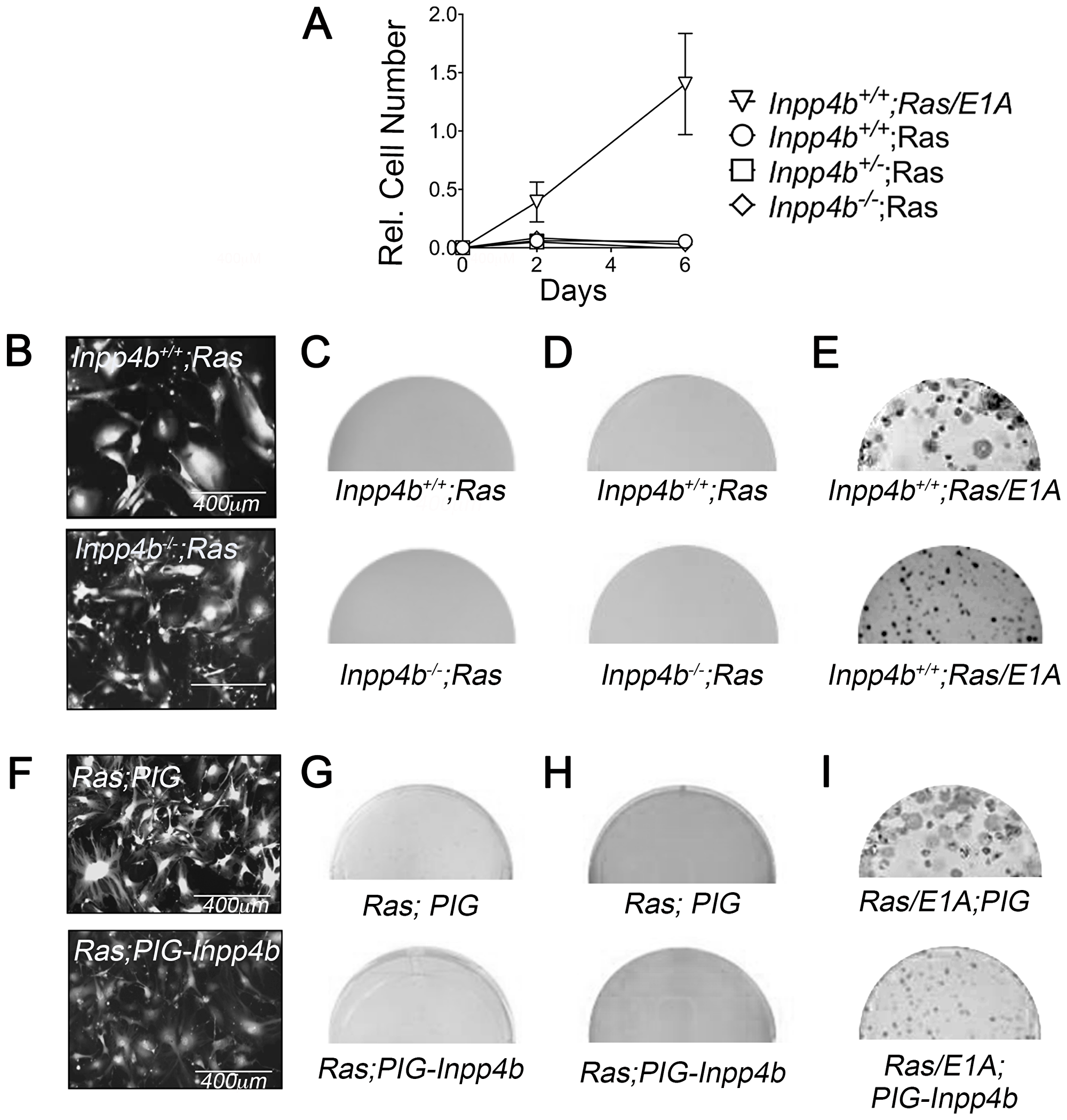
Figure 2: Inpp4b loss or overexpression do not cooperate with H-RasV12 in MEF transformation. (A) 6-day growth curve of Inpp4b+/+, Inpp4b+/- and Inpp4b-/- MEF after HRasV12 infection. 12SLRC H-RasV12 /E1A infected Inpp4b+/+ MEF used as control. (B) Morphology of eGFP-expressing Inpp4b+/+ and Inpp4b-/- MEF after H-RasV12 infection. (C) Colony formation and (D) Soft agar assay of primary Inpp4b+/+ and Inpp4b-/- MEF after H-RasV12 infection. (E) 12SLRC H-RasV12/E1A infected colony assay (top) and soft agar (bottom) controls. Full wells are depicted. (F) Morphology of eGFP-expressing Inpp4b+/+ MEF after H-RasV12 and PIG or PIG-Inpp4b infection. (G) Colony formation and; (H) Soft agar assay of primary Inpp4b+/+ MEF after H-RasV12 and PIG or PIG-Inpp4b infection (I) 12SLRC H-RasV12/E1A and PIG or PIG-Inpp4b infected colony assay (top) and soft agar (bottom) controls.
Next, to examine the potential of INPP4B as an oncogene in MEF, we investigated whether INPP4B-overexpression could cooperate with H-RasV12 in cellular transformation. Retrovirus generated from pWZL-H-RasV12-hygro vector was combined with either MSCV-Puro-IRES-GFP (PIG)-empty or PIG-INPP4B retrovirus to co-infect wild-type MEF followed by selection with hygromycin B and puromycin. Morphologically both H-RasV12; PIG or H-RasV12; PIG-INPP4B infected MEF appeared large and multinucleated (Figure 2F) as expected with H-RasV12 infection. Foci were not observed in clonogenic assays (Figure 2G) and similarly, no anchorage-independent colonies were observed in soft agar assays (Figure 2H). Inpp4b+/+ MEF infected with H-RasV12 and E1A retroviruses which generated abundant foci (Figure 2I, top) and anchorage independent colonies (Figure 2I, bottom) were used as positive controls. Overall, neither deficiency nor overexpression of INPP4B demonstrated cooperativity with H-RasV12-overexpression in cellular transformation.
Neither loss nor overexpression of INPP4B cooperate with E1A in MEF transformation
To investigate whether loss of Inpp4b is sufficient to cooperate with E1A in promoting cellular transformation, early passage Inpp4b+/+ and Inpp4b-/- MEF were infected with pWZL-E1A-hygro retrovirus and selected with hygromycin B. Immediately after selection, MEF were plated for growth analysis, clonogenic assay and soft agar assays. Inpp4b+/+, Inpp4b+/- and Inpp4b-/- MEF infected with E1A alone were growth inhibited compared to H-RasV12; E1A controls (Figure 3A). Morphologically both Inpp4b+/+ and Inpp4b-/- MEF appeared smaller and more dispersed compared to primary MEF (Figure 3B); clonal outgrowth on plastic observed after 21 days was similarly very minimal in both Inpp4b+/+ and Inpp4b-/- MEF (Figure 3C) and no anchorage independent growth colonies formed in soft agar (Figure 3D). Conversely, Inpp4b+/+ and Inpp4b-/- MEF overexpressing both H-RasV12 and E1A rapidly proliferated in 6-day growth curves (Figure 3A), grew many more foci in the clonogenic assay (Figure 3E, top) and formed large robust colonies in soft agar (Figure 3E, Bottom).
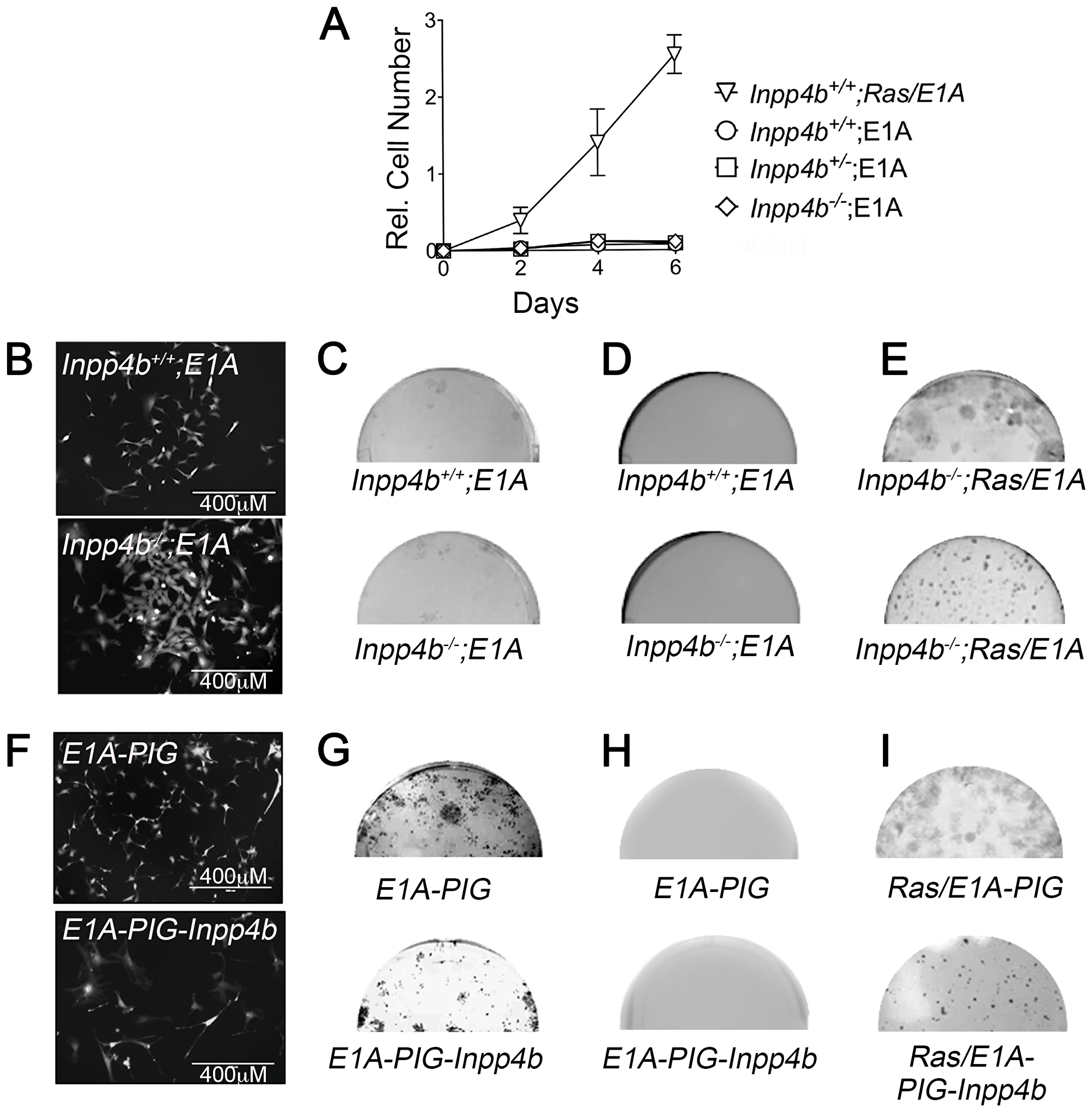
Figure 3: Inpp4b loss or overexpression do not cooperate with E1A in MEF. (A) 6-day growth curve of MEF after E1A infection. 12SLRC H-RasV12/E1A infected Inpp4b+/+ MEF used as control. (B) Morphology of eGFP-expressing Inpp4b+/+ and Inpp4b-/- MEF after E1A infection. (C) Colony formation and; (D) Soft agar assay of primary Inpp4b+/+ and Inpp4b-/- MEF after E1A infection and (E) 12SLRC H-RasV12/E1A infected colony assay (left) and soft agar (right) controls. Half wells are depicted. (F) Morphology of eGFP-expressing Inpp4b+/+ MEF after E1A and PIG or PIG-Inpp4b infection. (G) Colony formation and; (H) Soft agar assay of primary Inpp4b+/+ MEF after E1A and PIG or PIG-Inpp4b infection (I) 12SLRC H-RasV12/E1A and PIG or PIG-Inpp4b infected colony assay (left) and soft agar (right) controls.
Next, INPP4B overexpression was tested for its ability to cooperate with E1A to induce cellular transformation in MEF. For these experiments, wild-type MEF were retrovirally infected with pWZL-E1A-hygro vector with either PIG or PIG-INPP4B and selected with hygromycin B and puromycin for 48 hours. Morphologically, E1A; PIG MEF appeared smaller than primary cells (Figure 3F, top) whereas E1A; PIG-INPP4B MEF appeared larger, more elongated with long spindle-like protrusions and multinucleated (Figure 3F, bottom). No significant differences in foci counts were observed between E1A; PIG and E1A; PIG-INPP4B in clonogenic assays (Figure 3G). Anchorage independent growth was not observed in either E1A; PIG and E1A; PIG-INPP4B MEF (Figure 3H), but both clones and foci were clearly observed in the control cells infected with H-RasV12 and E1A retroviruses (Figure 3I). Overall, as with H-RasV12-overexpression, neither Inpp4b-deficiency nor INPP4B-overexpression were able to cooperate with E1A in cellular transformation.
Inpp4b deficiency increases colony forming potential in SV40 T-Large MEF
We next investigated cooperativity of Inpp4b-deficiency with SV40 T-large overexpression. As before, SV40 T-Large retrovirus was used to infect early passage Inpp4b+/+ and Inpp4b-/- MEF. Morphologically, no differences were observed between Inpp4b+/+ and Inpp4b-/- MEF lines (Figure 4A). Immunoblot analysis of SV40 T-Large Inpp4b+/+ and Inpp4b-/- demonstrated similar levels of p53 protein and no changes in PTEN expression (Figure 4C). Notably, unlike H-RasV12 and E1A, SV40 T-Large infected Inpp4b-/- MEF demonstrated a significant (P = 0.025) increase in foci formation (Figure 4C) and a significant (P = 0.0011) increase in anchorage independent colonies in soft agar (Figure 4D). These findings demonstrate cooperativity between SV40 T-Large overexpression and Inpp4b deficiency in MEF and suggest that Inpp4b may function as a tumour suppressor in the context of SV40 T-Large transduction.
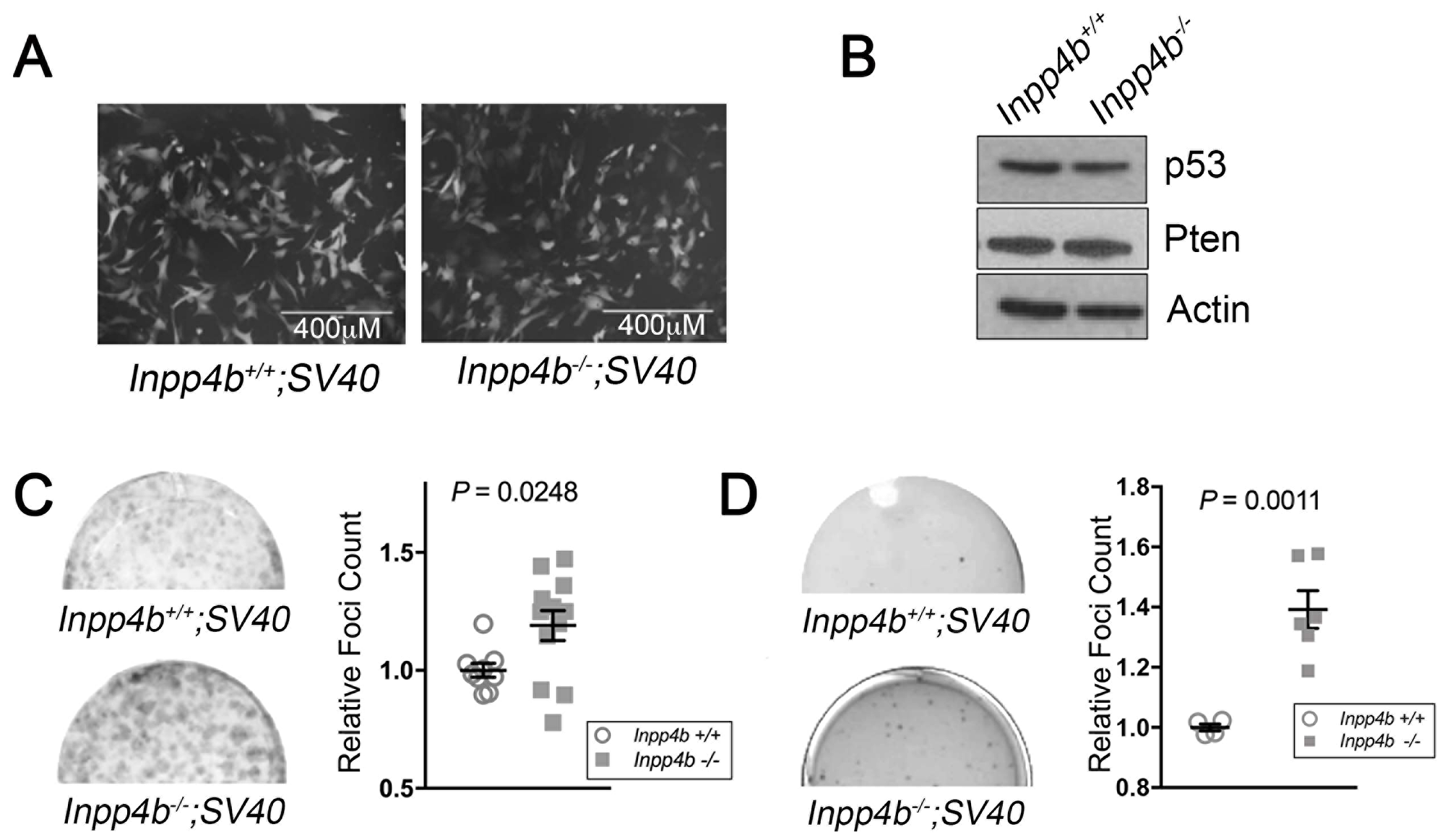
Figure 4: Inpp4b deficiency increases colony forming potential of SV40 T-Large in MEF. (A) Morphology of eGFP-expressing Inpp4b+/+ and Inpp4b-/- MEF after SV40 T-Large infection. (B) Immunoblot of Inpp4b+/+ and Inpp4b-/- SV40 T-Large MEF. (C) Representative clonogenic and (D) Soft agar assay plates with quantitation below. P-values were determined using the Student’s t-test.
Given this increase in SV40 T-Large transformation potential observed in Inpp4b-/- MEF, we reasoned that INPP4B overexpression would limit anchorage independent growth of SV40 T-Large MEF. To investigate this, early passage SV40 T-Large MEF were transduced with PIG and PIG-INPP4B retrovirus (Figure 5A). Although there were no gross morphological differences between PIG and PIG-INPP4B transduced SV40 T-Large MEF (Figure 5B), we did observe that INPP4B overexpression in MEF SV40 T-Large MEF displayed a significant reduction (P = 0.0018) in colony number as compared to control (Figure 5C). Immunoblot analysis of SV40 T-Large Inpp4b+/+ and Inpp4b-/- demonstrated no changes Pten protein levels upon INPP4B overexpression (Figure 5A).
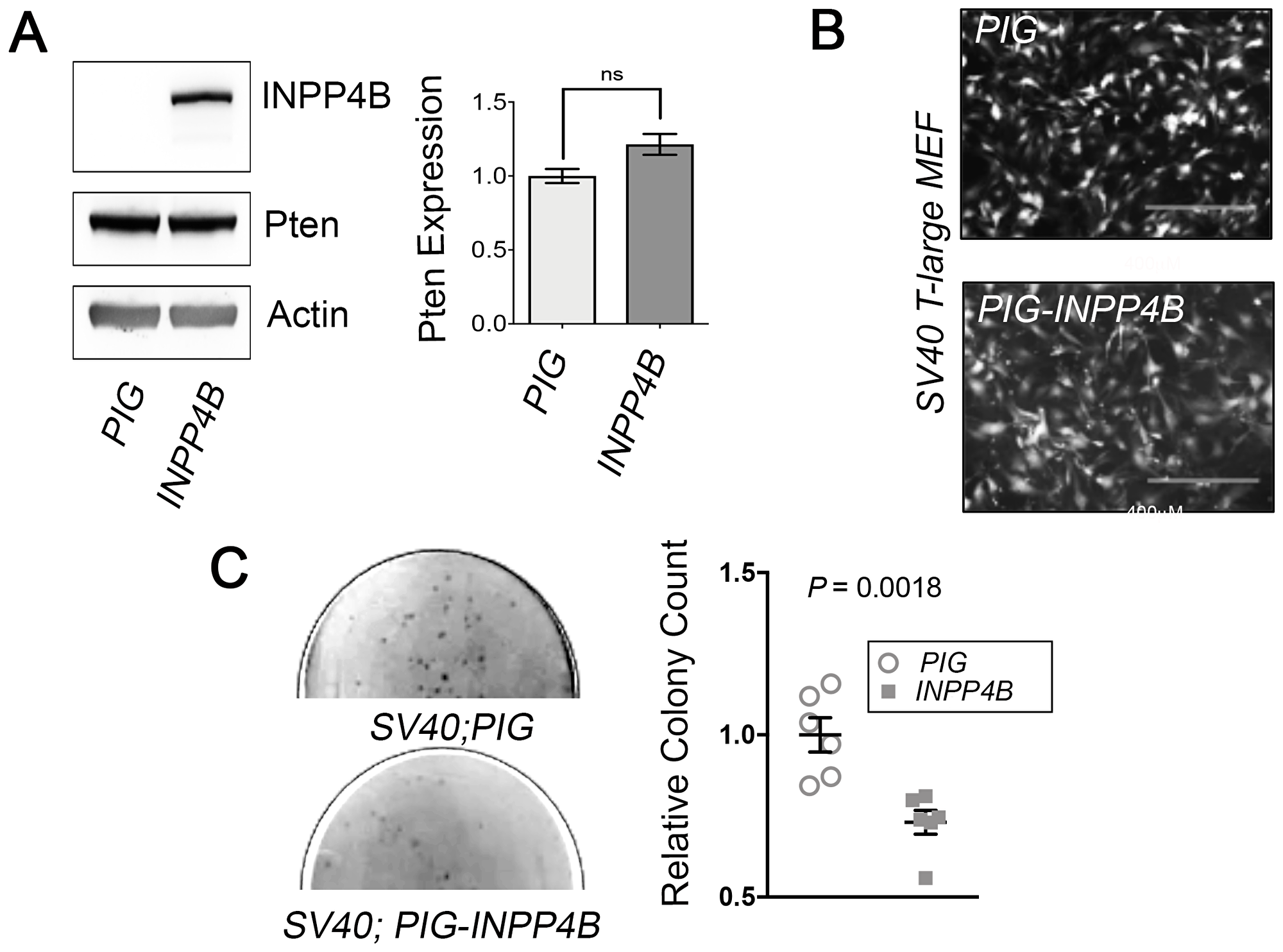
Figure 5: Inpp4b overexpression decreases transformation potential in SV40 T-Large MEF. (A) Immunoblot measuring INPP4B overexpression and Pten levels in SV40T-Large MEF (n = 5). (B) Morphology of eGFP-expressing Inpp4b+/+ SV40T-Large MEF with PIG or PIG-Inpp4b transduction. (C) Representative soft agar assay plates and quantitation of Inpp4b+/+ SV40T-Large MEF with PIG or PIG-Inpp4b colonies. P-values were determined using the Student’s t-test.
Inpp4b modulates EGF stimulated PI3K signaling in SV40 T-Large MEF
To explore the relationship of Inpp4b deficiency on PI3K signaling in MEF, low-passage Inpp4b+/+ and Inpp4b-/- SV40T-Large MEF were serum starved overnight and stimulated with epidermal growth factor (EGF). We consistently observed that Inpp4b-/- MEF achieved higher peak levels of pSer473-Akt activation at 5–10 minutes compared to timepoints in Inpp4b+/+ MEF. Inpp4b-/- MEF maintained prolonged peak levels of pSer473-Akt beyond 10 minutes of EGF and remained higher than Inpp4b+/+ MEF up to 30 min (Figure 6A). Conversely, INPP4B overexpression in SV40 T-Large MEF led to markedly decreased levels of pSer473-Akt after EGF stimulation. This decrease was observed at the peak pSer473-Akt concentrations of 5 and 10 minutes (Figure 6B). Together these results indicate that INPP4B regulates Akt activation dynamics in stimulated SV40T-Large MEF.
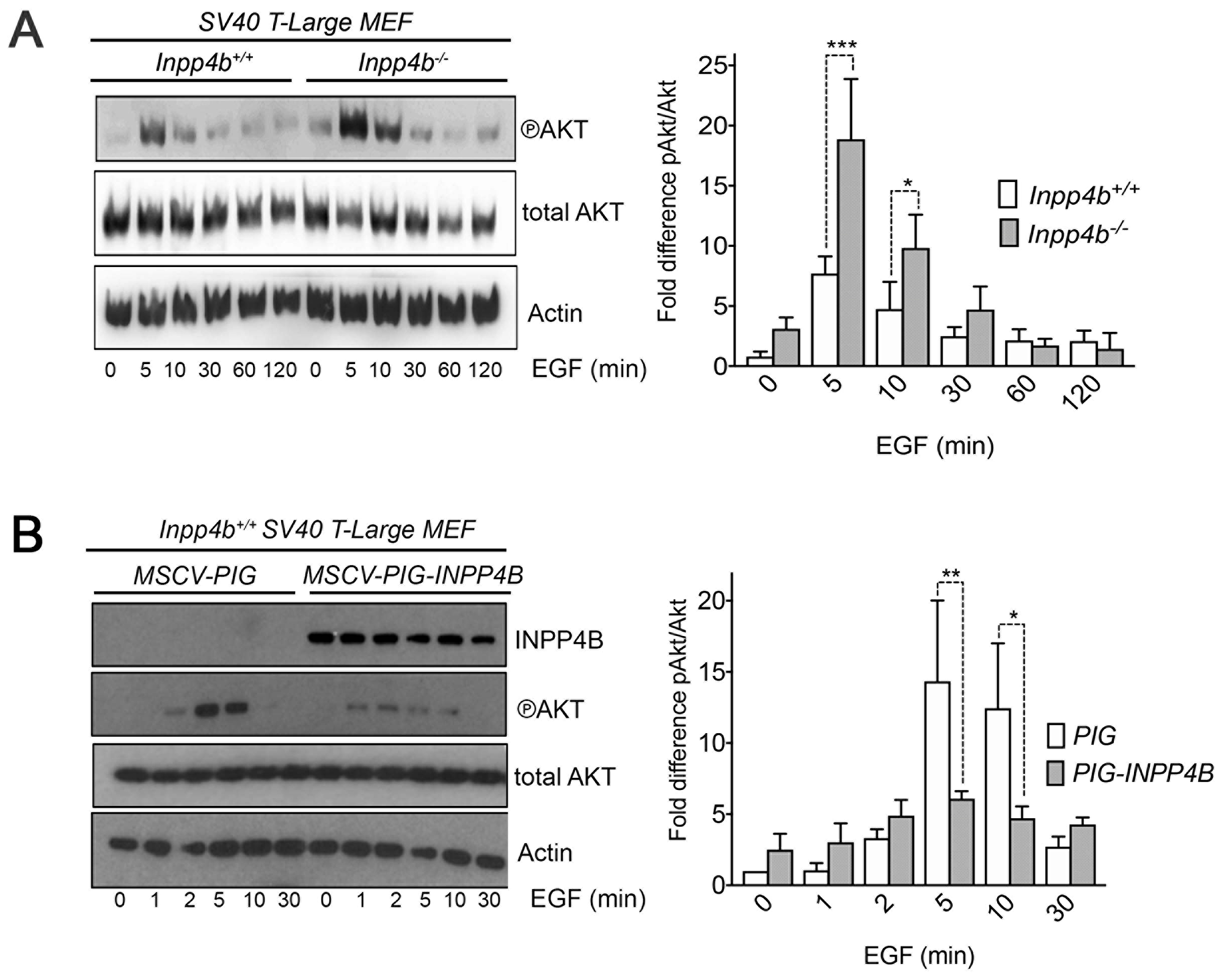
Figure 6: Inpp4b modulates EGF stimulated Akt activation in SV40 T-Large MEF. SV40T-Large MEF were serum starved and stimulated with 100 ng/mL of EGF and immunoblotted with pSer473-Akt. (A) Immunoblot of pSer473-Akt of SV40 T-Large Inpp4b+/+ and Inpp4b-/- MEF with quantitation of pSer473-Akt activation status (n = 4) (B) Immunoblot of pSer473-Akt SV40T-Large; PIG or SV40T-Large; PIG -INPP4B MEF with quantitation plot of pSer473-Akt activation status (n = 3). P-values were determined using ANOVA. *P < 0.05, **P < 0.01, ***P < 0.005.
DISCUSSION
Altered levels of INPP4B expression have been linked with cancer progression in various different human tumour types [42]. Previous works have demonstrated that INPP4B knockdown induced anchorage-independent cell growth, increased invasion and migration, and augmented Akt activation in immortalized HMECs [8, 9]. In contrast, other reports show that INPP4B overexpression enhanced proliferation and promotes anchorage-independent growth of HEMn-MP melanocytes and FHC normal colon epithelial cells [34, 38]. Herein, we present results which indicate that deficiency of Inpp4b in MEF can cooperate with very specific oncogenic alterations to induce cellular transformation. In our study, we observed that neither heterozygosity nor deficiency of Inpp4b were sufficient to alter growth characteristics of MEF on their own (Figure 1C). Spontaneous immortalization of MEF was also not observed in colony formation assays (Figure 1D, 1E). Similarity, Inpp4b deficiency was inconsequential for H-RasV12/E1A induced cellular transformation of MEF (Figure 1F, 1G). Inpp4b deficiency did not inhibit H-RasV12/E1A-induced cellular transformation as both Inpp4b-/- and Inpp4b+/+ MEF displayed long-term growth potential consistent with immortalization and in an anchorage independent manner in the soft agar assay (Figure 1G, 1H). When co-expressed in MEF, H-RasV12 and E1A potently induce cellular transformation due to the strong proliferative signaling through H-RasV12 concomitant with inhibition of the Rb tumour suppressor pathway by E1A which bypasses Ras-induced senescence [44, 45]. Inpp4b deficiency did not cooperate with H-RasV12 nor E1A oncogene overexpression to promote MEF transformation suggesting that Inpp4b may not play a role in E1A or Ras signaling pathways in MEF (Figures 2–3).
However, Inpp4b deficiency did promote SV40 T-Large mediated transformation of MEF, as demonstrated by an increase in anchorage independent growth and colony formation as compared to controls (Figure 4C–4D). Increased SV40 T-Large mediated cellular transformation was associated with elevated Akt activation as demonstrated by elevated pSer473-Akt after stimulation of starved MEF with EGF (Figure 6). Together, these findings are consistent with studies by Westbrook et al. [9] and Gewinner et al. [8] which demonstrated that shRNA knock-down of INPP4B in HMEC cells immortalized with SV40 T-Large and hTERT promoted cellular transformation. In addition, that Inpp4a-/- MEF also displayed elevated pAkt levels compared to wild type after SV40 T-Large immortalization [46] support the notion that 4-phosphatase function is tumour suppressive in part through regulation of Akt signaling. Other mechanisms which have been linked to INPP4B loss include loss of ATM and ATR, both upstream regulators of the p53 pathway [22, 47] and INPP4B was reported to downregulate PTEN protein through its protein phosphatase activity [34]. Our efforts in this study demonstrated that p53 and Pten expression levels were unchanged between Inpp4b+/+ and Inpp4b-/- SV40 T-Large MEF (Figure 4B) indicating that these mechanisms may not be associated with the phenotypes we observed in MEF.
Conversely, emerging evidence suggests that INPP4B overexpression may also promote tumourigenesis and cancer progression [32–36]. Thus, we tested whether INPP4B overexpression in MEF drives or cooperates with other transforming oncogenes in cellular transformation. We observed that INPP4B overexpression was unable to promote transformation in combination with H-RasV12, E1A nor SV40 T-Large MEF. In fact, overexpression of INPP4B in SV40 T-Large MEF significantly decreased cellular transformation and concomitantly decreased pSer473-Akt levels (Figures 5, 6). These findings were reminiscent of those reported by Fedele et al. [10] in serum starved and EGF stimulated MCF7 at similar time points, where peak pAkt levels were lower after INPP4B knockdown, but at 30 minutes INPP4B overexpressing cells displayed slightly increased pAkt levels compared to the control [10]. Taken together, these findings highlight a tumour suppressive role for Inpp4b in the context of SV40 T-Large that coincides with elevated Akt activation and suggest that a functional alteration conferred by SV40 T-Large transduction cooperates with Inpp4b loss to promote cellular transformation in vitro.
In summary, our study aimed to address the role for Inpp4b in cellular transformation. Given the numerous contrasting models for INPP4B function in cancer, we investigated both deficiency and overexpression to elucidate predominant tumour suppressor or oncogenic roles, respectively in MEF. With respect to MEF transformation, we observed that INPP4B overexpression did not promote oncogenesis when combined H-RasV12, E1A or SV40-T-large transduction. Although Inpp4b deficiency did not cooperate with overexpression H-RasV12, nor E1A, we did observe that Inpp4b deficiency can increase the transformation potential of SV40-T-large transduction suggesting that Inpp4b may function in a tumour suppressive manner in this context, at least in part through its control of Akt activation. This suggests that an event specifically associated with suppressing SV40-T-large transformation is relieved by Inpp4b deficiency. Further investigation is required to elucidate the specific mechanisms at play.
Materials and Methods
MEF preparation and culture conditions
Timed breedings were performed with C57BL/6J Inpp4b+/- pairs and embryos were dissected from euthanized mothers at 13.5 days post-coitum. The skin fibroblasts were separated from the head and viscera and incubated in 2 mL of 0.25% trypsin for 30 minutes. The solution was then mixed vigorously by pipetting, followed by the addition of another 2 mL of trypsin and incubated at 37°C and 5% CO2. After 30 minutes, the trypsin was neutralized by adding 1 mL of fetal bovine serum (FBS), and re-suspended and grown in complete growth media (Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin) for 12–16 hours. Once the MEF had grown to confluency, the cells were frozen down and designated as Passage 1. Primary MEF were passaged every 3 days to not exceed 1.5 × 106 cells per 10 cm dish. MEF were considered primary from Passage 1-Passage 4. All experiments were performed with primary MEF unless otherwise indicated.
Genotyping
DNA was extracted by lysing the heads of the mouse embryos in DNA extraction buffer (1M Tris (pH8), 20% SDS, 0.5M EDTA, 5M NaCl made up in water) containing 10% proteinase K at 55°C shaking for approximately 16 hours. An equal volume of 100% ethanol was added and centrifuged at 21.9 × g for 10 minutes. The supernatant was discarded, and the DNA pellet was washed with 70% ethanol, dried completely, and re-suspended in 50 uL of nuclease-free water. PCR was performed using 2X FroggaMix (FroggaBio), to distinguish Inpp4b+/+, Inpp4b+/-, and Inpp4b-/-. Genotyping primer pairs: wild type forward: 5′ GCTTCTGATAAAACATGGG 3′, wild type reverse: 5′ TGGGCACATTTATAAGCCTTC 3′, mutant forward: 5′ GCTTCTGATAAAACATGGG 3′, and mutant reverse: 5′ TGTTTTAAAAGCCTTGCTTGCTAAGTGTC 3′.
Retroviral constructs
The following plasmids were purchased from AddGene: pWZL hygro H-RasV12 (#18749), pBabe-puro H-RasV12 (#1768), pWZL hygro 12S E1A(#18748), and MSCV-puro-IRES-GFP (#21654). pWZL hygro SV40T-large was a kind gift from Pier Paolo Pandolfi, and 12SLRC H-RasV12 /E1A was a kind gift from Marisol Soengas. The MSCV-PIG-FLAG-Inpp4b was cloned by adding FLAG-Inpp4b to the MSCV-puro-IRES-GFP plasmid through Gibson Assembly (NEB).
Retroviral transduction
For transfection of retroviral constructs, 3.0 × 106 293T cells were seeded on a 10 cm dish 24 hours prior to calcium phosphate transfection. Media was changed 3–4 hours prior to transfection. For each 10 cm dish, 10 ug of retroviral plasmid and 5 ug of retroviral packaging vector, pCL-Eco, were combined with 2M CaCl2 and made up to a final volume of 300 uL with sterile water. This DNA/CaCl2 mixture was vortexed while 300 uL of 2X HEPES-Buffered saline (HBS; 140 mM NaCl, 1.5 mM Na2HPO4) was slowly added to mixture. 600 ul of DNA/CaCl2/HBS mixture was gently pipetted onto 293T cells. Media was changed 24 hours after transfection and supernatants was collected at 48- and 72-hours post-infection. MEF were seeded at 8.0 × 105 cells/10 cm dish 24 hours prior to infection. Fresh virus-rich media was collected and filtered using a 0.45-micron filter and added to respective plate along with 8 μg/ml protamine sulfate. Infections were repeated every 8 hours to increase infection efficiency. PWZL-hygro and PWZL- H-RasV12 -hygro were used as negative controls, and coinfection with PWZL-E1A-hygro and pBABE- H-RasV12 -puro were used as positive controls. Wild Type MEF cells were transfected with MSCV-PIG-Inpp4b and PWZL- H-RasV12-hygro to investigate INPP4B overexpression in cooperation with H-RasV12 overexpression. Inpp4b-/- MEF cells were transfected with pBABE- H-RasV12 -puro to investigate the effects of Inpp4b deficiency in cooperation with H-RasV12 overexpression on cellular transformation. Cells were selected for 4 days with hygromycin B at 75 μg/ml or 2 days of puromycin at 2 μg/ml selection, then cells recovered for 24 hours before plating for experiments. MEF infected with both puromycin and hygromycin B resistance plasmids were first selected for 4 days with hygromycin B at 75 ug/mL, allowed to recover for 24 hours and subsequently selected with 2 μg/ml puromycin; The cells were then allowed to recover for 24–48 hours before plating experiments.
Growth assays
MEF were seeded at a density of 2.5 × 104 cells per well in a 12-well plate for a 6-day growth curve. At each time point, cells were fixed with 10% formalin for 10 minutes and stored in phosphate-buffered saline (PBS) at 4°C. Once all time points were collected, cells were stained with 0.1% crystal violet, 20% methanol solution, then washed with water and dried for at least 4 hours. The crystal violet stain was solubilized by 10% acetic acid for 20 minutes and the absorbance was measured at 590 nm with Spectramax M3.
Clonogenic assays
MEF were plated at 1000 cells per well in a 6-well plate and allowed to grow for 10–20 days without media change. Cells were then washed with PBS, fixed with 10% formalin and stained with 0.1% crystal violet, 20% methanol solution, washed with water and allowed to dry. Colonies were imaged and counted using Image J. Cell counts were normalized to the control group.
Anchorage independence assays
200 mL of 6% Noble Agarose in sterile water was heated until liquified and brought to 42°C. Soft agar plates were prepared by mixing 2 mL of melted 6% Noble Agarose in MilliQ water in 18 ml of complete DMEM. 3 mL of 0.06% Noble Agar was plated per well of a 6 well dish and placed at 4°C to solidify. 1 hour prior to plating the dish was preheated in a 37°C cell incubator. 20,000 infected MEF were placed in 7.2 ul of media at 37°C then mixed with 0.8 ml of 42°C, 3% Noble Agarose in MilliQ water to obtain a final concentration of 5000 cells per well in triplicate of a 6 well dish in 0.3% Agar in DMEM. Colonies were stained using 0.05% crystal violet in 40% methanol. Colonies were counted using OpenCFU colony counting software (http://opencfu.sourceforge.net).
qPCR
RNA was isolated using the Qiagen RNeasy Mini Kit, and subsequently treated with DNase. Reverse transcription was performed using the SuperScript IV VILO Master Mix (Invitrogen, 11766050). TaqMan Fast Advanced Master Mix (appliedbiosystems, 4444557). TaqMan probes for Inpp4b (Mm01247230_m1) and actin (Mm02619580_g1) were purchased from Thermo Fisher Scientific. All of the above conducted according to manufacturers’ instructions.
Akt signaling assay
Passage 3–5 SV40 T-Large MEFs were seeded at 200 000 cells per well in 6 wells of a 6 well dish. After the MEF adhered to the bottom of the dish (4–6 hours after plating) the cells were washed twice with serum free DMEM then plated in starvation media (DMEM + 0.01% FBS). 16 hours later each well was allocated to the indicated time point (minutes). 100 ng/mL EGF was used to activate PI3K signaling. Activation was terminated at the indicated time points with ice cold PBS, followed by drying of wells, and subsequently flash frozen with liquid nitrogen and placed at –80°C prior to immunoblot.
Western blots
Immunoblots were performed using standard conditions as previously published [32]. The following antibodies were used in this study. Anti-phospho-Ser473-Akt (NEB 14543); anti-pan Akt (NEB 29020); anti-β-Actin (NEB 4967); anti-FLAG (Abgent AP1013a-ev). Anti-INPP4B antibody detecting murine Inpp4b was a kind gift from Jean Vacher [30].
Statistics
To compare means of 2 groups p-values were calculated using unpaired two-tailed Student’s t-test. For colony counts of the clonogenic assay and soft agar assay two-way parametric analysis of variance (ANOVA) was used to determine the significance value. A P-value < 0.05 was considered significant.
Abbreviations
AML: Acute myeloid leukemia; E1A: Adenovirus early region 1A; EGF: Epidermal growth factor; H-Ras: Harvey rat sarcoma viral oncogene homolog; HMEC: Human mammary epithelial cells; INPP4B: Inositol Polyphosphate 4-Phosphatase, Type II; MEF: Mouse embryonic fibroblast; PIG: MSCV-Puro-IRES-GFP; OIS: Oncogene-induced senescence; PtdIns: Phosphatidylinositol; pAKT: Phosphorylated Akt; Ras: Rat Sarcoma; SGK-3: Serum/Glucocorticoid Regulated Kinase Family Member 3; SV40: Simian Vacuolating Virus 40.
ACKNOWLEDGMENTS
We sincerely thank Dr. Lina Penn and Dr. Mark Minden for reagents and invaluable advice, and all members of the Salmena lab for critical discussions. We also thank Bell Wu and Aobo He for experimental assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
LS is the recipient of a Tier II Canada Research Chair. EMM is the recipient of a student incentive award from the Centre for Collaborative Drug Research. This work is funded in part by awards from CIHR (123343) to JV; and NSERC (RGPIN-2015-03984) and CIHR (201604PJT) to LS.
References
1. Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature. 1988; 332:644–646. https://doi.org/10.1038/332644a0. [PubMed].
2. Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC. PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell. 1989; 57:167–175. https://doi.org/10.1016/0092-8674(89)90182-7. [PubMed].
3. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017; 169:381–405. https://doi.org/10.1016/j.cell.2017.04.001. [PubMed].
4. Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011; 2:321–328. https://doi.org/10.18632/oncotarget.260. [PubMed].
5. Scheid MP, Huber M, Damen JE, Hughes M, Kang V, Neilsen P, Prestwich GD, Krystal G, Duronio V. Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for protein kinase B (PKB) activation; phosphatidylinositol (3,4)P2 is required for PKB phosphorylation at Ser-473: studies using cells from SH2-containing inositol-5-phosphatase knockout mice. J Biol Chem. 2002; 277:9027–9035. https://doi.org/10.1074/jbc.M106755200. [PubMed].
6. Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997; 272:8474–8481. https://doi.org/10.1074/jbc.272.13.8474. [PubMed].
7. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997; 275:665–668. https://doi.org/10.1126/science.275.5300.665. [PubMed].
8. Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, Cantley LC. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009; 16:115–125. https://doi.org/10.1016/j.ccr.2009.06.006. [PubMed].
9. Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, Depinho RA, Chin L, Elledge SJ. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005; 121:837–848. https://doi.org/10.1016/j.cell.2005.03.033. [PubMed].
10. Fedele CG, Ooms LM, Ho M, Vieusseux J, O'Toole SA, Millar EK, Lopez-Knowles E, Sriratana A, Gurung R, Baglietto L, Giles GG, Bailey CG, Rasko JE, et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc Natl Acad Sci U S A. 2010; 107:22231–22236. https://doi.org/10.1073/pnas.1015245107. [PubMed].
11. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. https://doi.org/10.1038/nature11412. [PubMed].
12. Hodgson MC, Shao L, Frolov A, Li R, Peterson LE, Ayala G, Ittmann MM, Weigel NL, Agoulnik IU. Decreased expression and androgen regulation of the tumor suppressor gene INPP4B in prostate cancer. Cancer Res. 2011; 71:572–582. https://doi.org/10.1158/0008-5472.CAN-10-2314. [PubMed].
13. Hodgson MC, Deryugina EI, Suarez E, Lopez SM, Lin D, Xue H, Gorlov IP, Wang Y, Agoulnik IU. INPP4B suppresses prostate cancer cell invasion. Cell Commun Signal. 2014; 12:61. https://doi.org/10.1186/s12964-014-0061-y. [PubMed].
14. Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty JD, Fulton LL, Fulton R, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013; 368:2059–2074. https://doi.org/10.1056/NEJMoa1301689. [PubMed].
15. Salmena L, Shaw P, Fans I, McLaughlin, Rosen B, Risch H, Mitchell C, Sun P, Narod SA, Kotsopoulos J. Prognostic value of INPP4B protein immunohistochemistry in ovarian cancer. Eur J Gynaecol Oncol. 2015; 36:260–267. [PubMed].
16. Rynkiewicz NK, Fedele CG, Chiam K, Gupta R, Kench JG, Ooms LM, McLean CA, Giles GG, Horvath LG, Mitchell CA. INPP4B is highly expressed in prostate intermediate cells and its loss of expression in prostate carcinoma predicts for recurrence and poor long term survival. Prostate. 2015; 75:92–102. https://doi.org/10.1002/pros.22895. [PubMed].
17. Li Chew C, Lunardi A, Gulluni F, Ruan DT, Chen M, Salmena L, Nishino M, Papa A, Ng C, Fung J, Clohessy JG, Sasaki J, Sasaki T, et al. In Vivo Role of INPP4B in Tumor and Metastasis Suppression through Regulation of PI3K-AKT Signaling at Endosomes. Cancer Discov. 2015; 5:740–751. https://doi.org/10.1158/2159-8290.CD-14-1347. [PubMed].
18. Chen M, Nowak DG, Trotman LC. Molecular pathways: PI3K pathway phosphatases as biomarkers for cancer prognosis and therapy. Clin Cancer Res. 2014; 20:3057–3063. https://doi.org/10.1158/1078-0432.CCR-12-3680. [PubMed].
19. Kofuji S, Kimura H, Nakanishi H, Nanjo H, Takasuga S, Liu H, Eguchi S, Nakamura R, Itoh R, Ueno N, Asanuma K, Huang M, Koizumi A, et al. INPP4B is a PtdIns(3,4,5)P3 phosphatase that can act as a tumor suppressor. Cancer Discov. 2015; 5:730–739. https://doi.org/10.1158/2159-8290.CD-14-1329. [PubMed].
20. Yuen JW, Chung GT, Lun SW, Cheung CC, To KF, Lo KW. Epigenetic inactivation of inositol polyphosphate 4-phosphatase B (INPP4B), a regulator of PI3K/AKT signaling pathway in EBV-associated nasopharyngeal carcinoma. PLoS One. 2014; 9:e105163. https://doi.org/10.1371/journal.pone.0105163. [PubMed].
21. Zhang L, Zeng D, Chen Y, Li N, Lv Y, Li Y, Xu X, Xu G. miR-937 contributes to the lung cancer cell proliferation by targeting INPP4B. Life Sci. 2016; 155:110–115. https://doi.org/10.1016/j.lfs.2016.05.014. [PubMed].
22. Ip LRH, Poulogiannis G, Viciano FC, Sasaki J, Kofuji S, Spanswick VJ, Hochhauser D, Hartley JA, Sasaki T, Gewinner CA. Loss of INPP4B causes a DNA repair defect through loss of BRCA1, ATM and ATR and can be targeted with PARP inhibitor treatment. Oncotarget. 2015; 6:10548–10562. https://doi.org/10.18632/oncotarget.3307. [PubMed].
23. Hsu I, Yeh CR, Slavin S, Miyamoto H, Netto GJ, Tsai YC, Muyan M, Wu XR, Messing EM, Guancial EA, Yeh S. Estrogen receptor alpha prevents bladder cancer via INPP4B inhibited akt pathway in vitro and in vivo. Oncotarget. 2014; 5:7917–7935. https://doi.org/10.18632/oncotarget.1421. [PubMed].
24. Tokunaga E, Yamashita N, Kitao H, Tanaka K, Taketani K, Inoue Y, Saeki H, Oki E, Oda Y, Maehara Y. Biological and clinical significance of loss of heterozygosity at the INPP4B gene locus in Japanese breast cancer. Breast. 2016; 25:62–68. https://doi.org/10.1016/j.breast.2015.10.006. [PubMed].
25. Ooms LM, Binge LC, Davies EM, Rahman P, Conway JR, Gurung R, Ferguson DT, Papa A, Fedele CG, Vieusseux JL, Chai RC, Koentgen F, Price JT, et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell. 2015; 28:155–169. https://doi.org/10.1016/j.ccell.2015.07.003. [PubMed].
26. Gilby DC, Goodeve AC, Winship PR, Valk PJ, Delwel R, Reilly JT. Gene structure, expression profiling and mutation analysis of the tumour suppressor SHIP1 in Caucasian acute myeloid leukaemia. Leukemia. 2007; 21:2390–2393. https://doi.org/10.1038/sj.leu.2404864. [PubMed].
27. Prasad NK, Tandon M, Badve S, Snyder PW, Nakshatri H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis. 2008; 29:25–34. https://doi.org/10.1093/carcin/bgm213. [PubMed].
28. Rudge SA, Wakelam MJ. Phosphatidylinositolphosphate phosphatase activities and cancer. J Lipid Res. 2016; 57:176–192. https://doi.org/10.1194/jlr.R059154. [PubMed].
29. Rodgers SJ, Ferguson DT, Mitchell CA, Ooms LM. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci Rep. 2017; 37. https://doi.org/10.1042/BSR20160432. [PubMed].
30. Ferron M, Boudiffa M, Arsenault M, Rached M, Pata M, Giroux S, Elfassihi L, Kisseleva M, Majerus PW, Rousseau F, Vacher J. Inositol polyphosphate 4-phosphatase B as a regulator of bone mass in mice and humans. Cell Metab. 2011; 14:466–477. https://doi.org/10.1016/j.cmet.2011.08.013. [PubMed].
31. Vo TT, Fruman DA. INPP4B is a tumor suppressor in the context of PTEN deficiency. Cancer Discov. 2015; 5:697–700. https://doi.org/10.1158/2159-8290.CD-15-0609. [PubMed].
32. Dzneladze I, He R, Woolley JF, Son MH, Sharobim MH, Greenberg SA, Gabra M, Langlois C, Rashid A, Hakem A, Ibrahimova N, Arruda A, Löwenberg B, et al. INPP4B overexpression is associated with poor clinical outcome and therapy resistance in acute myeloid leukemia. Leukemia. 2015; 29:1485–1495. https://doi.org/10.1038/leu.2015.51. [PubMed].
33. Rijal S, Fleming S, Cummings N, Rynkiewicz NK, Ooms LM, Nguyen NY, Teh TC, Avery S, McManus JF, Papenfuss AT, McLean C, Guthridge MA, Mitchell CA, et al. Inositol polyphosphate 4-phosphatase II (INPP4B) is associated with chemoresistance and poor outcome in AML. Blood. 2015; 125:2815–2824. https://doi.org/10.1182/blood-2014-09-603555. [PubMed].
34. Guo ST, Chi MN, Yang RH, Guo XY, Zan LK, Wang CY, Xi YF, Jin L, Croft A, Tseng HY, Yan XG, Farrelly M, Wang FH, et al. INPP4B is an oncogenic regulator in human colon cancer. Oncogene. 2016; 35:3049–3061. https://doi.org/10.1038/onc.2015.361. [PubMed].
35. Chen Y, Sun Z, Qi M, Wang X, Zhang W, Chen C, Liu J, Zhao W. INPP4B restrains cell proliferation and metastasis via regulation of the PI3K/AKT/SGK pathway. J Cell Mol Med. 2018; 22:2935–2943. https://doi.org/10.1111/jcmm.13595. [PubMed].
36. Jin H, Yang L, Wang L, Yang Z, Zhan Q, Tao Y, Zou Q, Tang Y, Xian J, Zhang S, Jing Y, Zhang L. INPP4B promotes cell survival via SGK3 activation in NPM1-mutated leukemia. J Exp Clin Cancer Res. 2018; 37:8. https://doi.org/10.1186/s13046-018-0675-9. [PubMed].
37. Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell. 2014; 56:595–607. https://doi.org/10.1016/j.molcel.2014.09.023. [PubMed].
38. Chi MN, Guo ST, Wilmott JS, Guo XY, Yan XG, Wang CY, Liu XY, Jin L, Tseng HY, Liu T, Croft A, Hondermarck H, Scolyer R, et al. INPP4B is upregulated and functions as an oncogenic driver through SGK3 in a subset of melanomas. Oncotarget. 2015; 6:39891–39907. https://doi.org/10.18632/oncotarget.5359. [PubMed].
39. Chen L, Cao Y, Rong D, Wang Y, Cao Y. MicroRNA-605 functions as a tumor suppressor by targeting INPP4B in melanoma. Oncol Rep. 2017; 38:1276–1286. https://doi.org/10.3892/or.2017.5740. [PubMed].
40. Noda T, Matsunaga K, Taguchi-Atarashi N, Yoshimori T. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin Cell Dev Biol. 2010; 21:671–676. https://doi.org/10.1016/j.semcdb.2010.04.002. [PubMed].
41. Perez-Lorenzo R, Gill KZ, Shen CH, Zhao FX, Zheng B, Schulze HJ, Silvers DN, Brunner G, Horst BA. A tumor suppressor function for the lipid phosphatase INPP4B in melanocytic neoplasms. J Invest Dermatol. 2014; 134:1359–1368. https://doi.org/10.1038/jid.2013.511. [PubMed].
42. Dzneladze I, Woolley JF, Rossell C, Han Y, Rashid A, Jain M, Reimand J, Minden MD, Salmena L. SubID, a non-median dichotomization tool for heterogeneous populations, reveals the pan-cancer significance of INPP4B and its regulation by EVI1 in AML. PLoS One. 2018; 13:e0191510. https://doi.org/10.1371/journal.pone.0191510. [PubMed].
43. Woolley JF, Dzneladze I, Salmena L. Phosphoinositide signaling in cancer: INPP4B Akt(s) out. Trends Mol Med. 2015; 21:530–532. https://doi.org/10.1016/j.molmed.2015.06.006. [PubMed].
44. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88:593–602. https://doi.org/10.1016/s0092-8674(00)81902-9. [PubMed].
45. de Stanchina E, McCurrach ME, Zindy F, Shieh SY, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19(ARF) tumor suppressor. Genes Dev. 1998; 12:2434–2442. https://doi.org/10.1101/gad.12.15.2434. [PubMed].
46. Ivetac I, Gurung R, Hakim S, Horan KA, Sheffield DA, Binge LC, Majerus PW, Tiganis T, Mitchell CA. Regulation of PI(3)K/Akt signalling and cellular transformation by inositol polyphosphate 4-phosphatase-1. EMBO Rep. 2009; 10:487–493. https://doi.org/10.1038/embor.2009.28. [PubMed].
47. Wang P, Ma D, Wang J, Fang Q, Gao R, Wu W, Cao L, Hu X, Zhao J, Li Y. INPP4B-mediated DNA repair pathway confers resistance to chemotherapy in acute myeloid leukemia. Tumour Biol. 2016; 37:12513–12523. https://doi.org/10.1007/s13277-016-5111-1. [PubMed].