
Introduction
The BCL-2 family can be divided into multidomain anti-apoptotic (e.g. BCL-2, BCL-XL, BCL-W, MCL-1, BFL-1) and pro-apoptotic (e.g. BAK, BAX) proteins. The functional interactions between these anti- and pro-apoptotic partners is controlled by a third group of proteins known as BH3-only proteins (e.g. BIM, BID, PUMA, BIK, BAD, NOXA, BMF) which contain one of four conserved BCL-2 homology (BH) domains. BH3-only proteins can directly bind and activate BAX/BAK or can insert their amphipathic BH3 α-helix into a groove on anti-apoptotic protein target(s) resulting in release and subsequent indirect BAX/BAK activation [1]. Cancer cells have long been known to evade cell death through overexpression of anti-apoptotic BCL-2 members or through down-regulation of BH3-only proteins [1]. To overcome these hurdles there is a great pharmacologic crusade to develop agents that directly engage BCL-2 family proteins to induce death regardless of the cell’s origin or genetic perturbations [2]. Despite early promise, many BH3-mimetics, have not effectively translated to the clinic or have been proven to work, at least in part, independent of the BCL-2 network [3–5].
Functional redundancy within the BCL-2 family can make it challenging to tailor effective therapeutic strategies without incurring resistance through upregulation of BCL-2 proteins that lie outside the mimetic’s binding profile [3, 6–9]. This is exemplified by diffuse large B-cell lymphoma (DLBCL) where MCL-1 contributes to intrinsic and acquired resistance to the rationally designed polyselective BCL-2, BCL-XL, and BCL-W inhibitor ABT-737 and the monoselective BCL-2 inhibitor ABT-199 [10, 11]. Despite the predominance of BCL-2 protein expression in DLBCL, either through the t(14;18) translocation and/or elevated BCL2 copy numbers, many BCL-2High DLBCL are resistant to direct BCL-2 inhibition and ultimately rely on MCL-1 for survival [11]. Additionally, although activated B-cell-like (ABC) DLBCL may rely on MCL-1 to a greater extent than germinal center B-cell-like (GCB) DLBCL, protein expression alone fails to predict reliance on BCL-2 or MCL-1 in either subtype. Rather, functional sequestration of pro-apoptotic BAK and BIM appear to define sensitivity to BH3-mimetic treatment [10, 12]. The importance of releasing BIM for cell death activation is exemplified by the treatment of BCL-2High DLBCL with ABT-199 or the BCL-XL-selective inhibitor A-1155463 which results in ejection of BIM from these proteins but subsequent sequestration by MCL-1 [11]. The significance of this paradigm is reflected in encouraging results using BCL-2/BCL-XL targeting BH3-mimetics in combination with agents that down-regulate MCL-1 in murine models of MYC-BCL2 double-hit lymphoma and human DLBCL [13, 14]. It is clear that release of endogenous BIM sequestered by multiple anti-apoptotics is key to overcoming cell death resistance in diseases such as DLBCL.
The physiologic dominance of BIM in regulating apoptosis in hematopoietic cells is reflected in the ability of its BH3 death domain to tightly engage the BH3-binding groove of all anti-apoptotic proteins and directly activate BAX and BAK [15]. To exploit BIM’s natural death-inducing functions we and others have shown that a hydrocarbon-stapled peptide modeled after the BIM BH3 α-helix (BIM SAHBA) broadly targets BCL-2 proteins with high affinity ex vivo, blocks inhibitory anti-apoptotic interactions, directly triggers BAX activation, dissociates BAK from MCL-1, and induces dose-responsive and BH3 sequence–specific cell death in hematologic cancers [16–18]. In the present study, we investigated the effect of BIM SAHBA on human DLBCL that differentially express and functionally depend on various BCL-2 anti-apoptotic proteins for survival [10]. We found that BIM SAHBA induced apoptosis in DLBCL regardless of anti-apoptotic protein expression but that it did so most effectively in DLBCL that were increasingly resistant to ABT-737 and ABT-199. These results led to the finding that BIM SAHBA preferentially displaced endogenous BIM from MCL-1 in these cells. Treatment with BIM SAHBA sensitized DLBCL to ABT-737 by preventing BIM relocation onto MCL-1 following displacement from BCL-2. BIM SAHBA’s functional affinity for MCL-1 and induction of apoptosis at the level of the mitochondria was confirmed in MCL-1 deficient mouse embryonic fibroblasts (MEFs). This work highlights the importance of displacement and sequestration of BIM by anti-apoptotic BCL-2 proteins and further implicates a functional role of MCL-1 in apoptotic resistance in DLBCL. This study also illuminates the use of peptide-based BH3 mimetics to uncover biologically relevant cell death mechanisms and confirms that functional intracellular BH3-mimetic affinities for anti-apoptotic proteins may only partially reflect their acellular binding profiles.
Results
Inverse correlation between DLBCL sensitivity to BIM SAHBA and ABT-737/ABT-199
A panel of 18 human DLBCL cell lines was treated with increasing concentrations of BIM SAHBA, ABT-737, and ABT-199 to determine cell death as a result of distinctive anti-apoptotic targeting (Figure 1 and Supplementary Figure 1A). The cell lines were divided into 3 groups based on their ABT-737 sensitivity: ABT-737 sensitive, ABT-737 moderately sensitive, and ABT-737 resistant (Figure 1A). Our ABT-737 sensitivity profiles were similar to those previously reported using other measures of cell death, namely Annexin V positivity and ATP content [10, 11]. BCL-2 dependence in these cell lines was reflected in similar results following treatment with ABT-199 (Supplementary Figure 1A). Together, these results support increased BCL-XL dependence in OCI-Ly10 and increased MCL-1 dependence in SU-DHL-10, HT, Pfeiffer, SU-DHL-5, SU-DHL-8, OCI-Ly7, and OCI-Ly4 cell lines as previously reported [10, 11, 17]. In contrast to treatment with ABT-737 and ABT-199, BIM SAHBA induced dose-responsive cell death in all DLBCL cell lines with EC50’s ranging from 2 μM to 18 μM (Figure 1B and Supplementary Table 1). Like treatment with ABT-737 and ABT-199, DLBCL could be divided into two groups based on their sensitivities to BIM SAHBA: BIM SAHBA ‘sensitive’ and BIM SAHBA ‘moderately sensitive’ (Figure 1B). No death was measured in any cell line treated with a hydrocarbon-stapled BH3 point mutant control (BIM SAHBA (R153D)) or vehicle alone indicating BIM-BH3 sequence-specific cell death induction (Supplementary Figure 1B and 1C) [16, 17, 19]. Based on our treatment analyses, there appeared to be an inverse correlation between DLBCL responses to ABT-737/ABT-199 and BIM SAHBA (Supplementary Table 1).
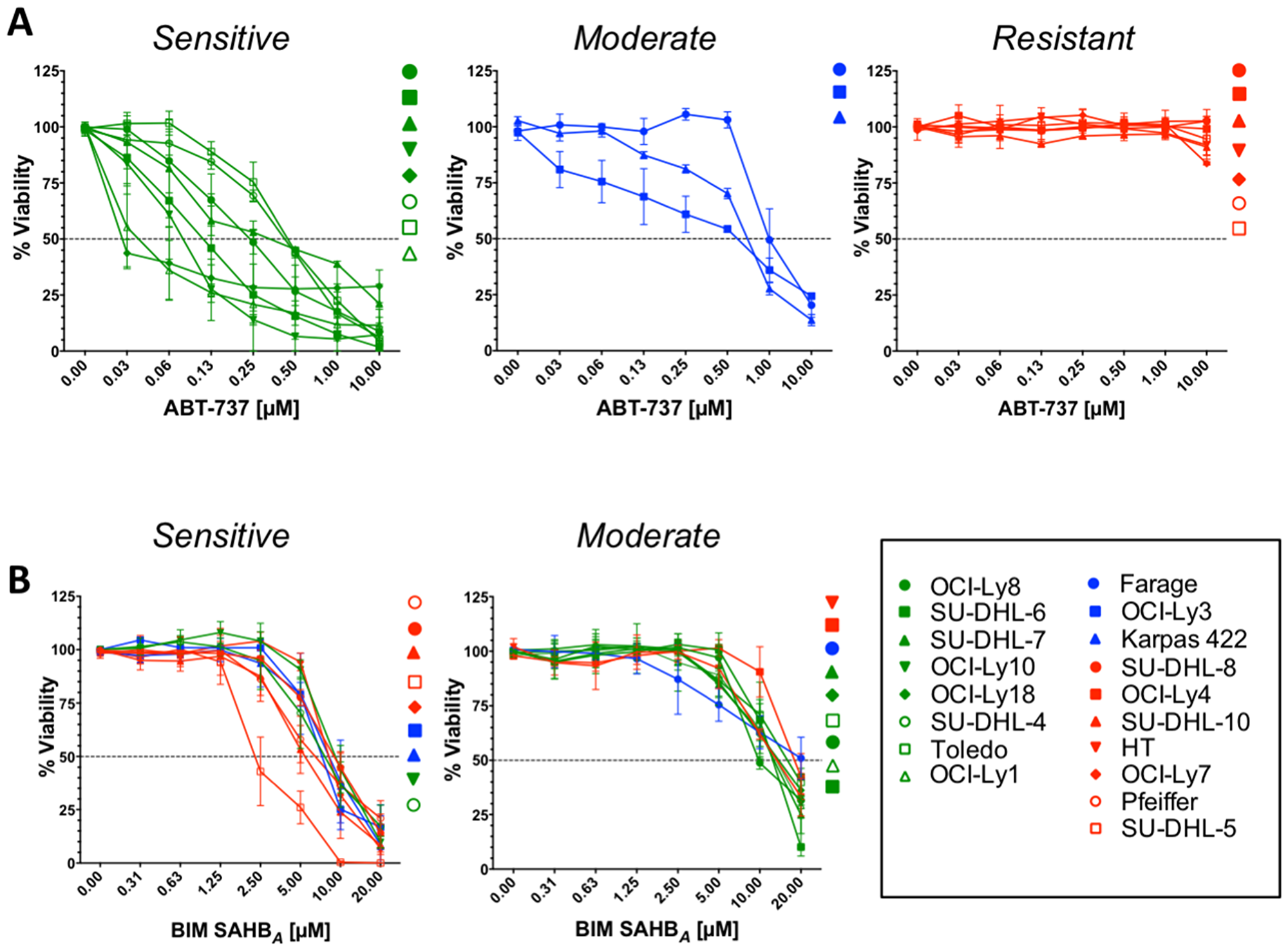
Figure 1: Sensitivity of DLBCLs to BIM SAHBA inversely correlates with their sensitivity to ABT-737. Cell viability in a panel of human DLBCL cell lines was measured after 24-hr incubation with increasing concentrations of (A) ABT-737 or (B) BIM SAHBA. Percent (%) viability was calculated as a percentage of control (DMSO) treated cells. Dose-response curves indicating highest sensitivity to ABT-737 are in green, those indicating moderate sensitivity to ABT-737 are in blue, and those indicating low to no sensitivity to ABT-737 are in red. Error bars are mean ± SEM for at least three independent preparations of cell and BH3 mimetic treatments.
BIM SAHBA induces caspase activation in DLBCL regardless of BCL-2 family protein expression
To confirm that BIM SAHBA treatment lead to the activation of the intrinsic apoptotic pathway and MOMP, activated caspase 3/7 was measured six hours following treatment of DLBCL with BIM SAHBA at their individual EC50 (Figure 2A). Cell death correlated with caspase 3/7 activation in all cell lines. The relative differences in caspase 3/7 activation between DLBCL may reflect differences in the kinetics of cell death in individual cell lines. Regardless, DLBCL partially or fully resistant to ABT-737 (shown in blue and red) had similar fold caspase activation compared to ABT-737 sensitive DLBCL (shown in green) (Figure 2A). Representative cell death morphology following treatment with BIM SAHBA displayed classical hallmarks of apoptosis such as cellular membrane blebbing, cell shrinkage, chromatin marginalization, and nuclear fragmentation consistent with previous findings (Figure 2B) [17]. No correlation existed between BIM SAHBA-induced killing and protein expression levels of BCL-2, BCL-XL, or MCL-1 (Figure 2C and Supplementary Figure 2A–2C). This is similar to other reports where no correlations were measured between protein expression of BCL-2 and BCL-XL and DLBCL responses to ABT-737 or ABT-199 [10, 11]. There was also no correlation in apoptosis and levels of other BCL-2 proteins known to be associated with cell death resistance in B-cell lymphomas including BIM, BID, PUMA, BAX, or BAK (Supplementary Figure 2D–2H) [10, 12, 20–23]. Given the importance of release of BIM for apoptosis induction in DLBCL, these results suggested that BIM SAHBA was able to block endogenous BIM binding to MCL-1, BCL-2 and/or BCL-XL expressing cells regardless of their sensitivity to ABT-737/ABT-199.
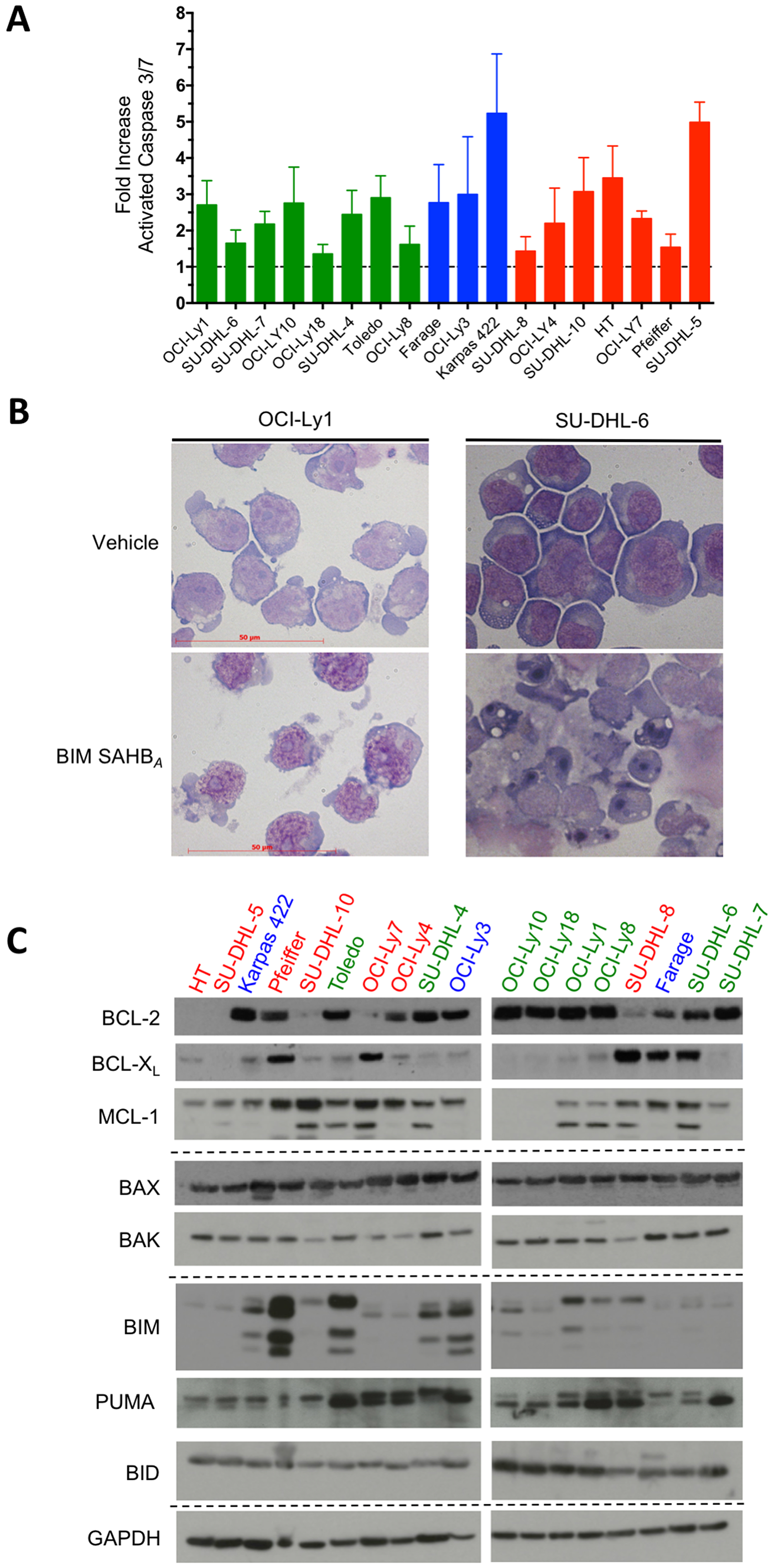
Figure 2: BIM SAHBA treatment results in caspase 3/7 activation and cellular hallmarks of apoptosis irrespective of BCL-2 family protein expression in DLBCL. (A) DLBCL cell lines were treated BIM SAHBA at their respective EC50 for 6-hr and activation of the intrinsic apoptotic pathway was assessed by monitoring the cleavage of pro-luminescent caspase 3/7 substrate. Caspase activity was calculated as fold change as compared to control (DMSO) treated cells. (B) BIM SAHBA-treatment results in hallmarks of apoptosis such as cellular membrane blebbing, cell shrinkage, chromatin marginalization, and nuclear fragmentation. (C) Western blot analysis of BCL-2 proteins in DLBCL cell lines. Colors of each cell line reflect their ABT-737 sensitivity patterns as depicted in Figure 1 and Supplementary Figure 1. Error bars are mean ± SEM for at least three independent preparations of cell and BIM SAHBA treatments.
BIM SAHBA dissociates BIM from MCL-1 to a greater extent than BCL-2
We next sought to investigate the ability of BIM SAHBA to release endogenous BIM from BCL-2 or MCL-1 in DLBCL that were sensitive (OCI-Ly1 > SU-DHL-6 > OCI-Ly8) or resistant (SU-DHL-5) to ABT-737 (Figure 1A, Supplementary Table 1) [10, 11]. The three ABT-737 sensitive cell lines were chosen as they represent an array of DLBCL phenotypes based on predominant anti-apoptotic BCL-2 family member expression (e.g. BCL-2, BCL-XL, and MCL-1). Importantly, these DLBCL depend, even if partially, on BCL-2, rather than BCL-XL for cell death control [10, 13]. Treatment of individual cell lines with the EC50 of either ABT-737 or BIM SAHBA alone was followed by immunoprecipitation of BCL-2 and MCL-1 and immunoblotting for endogenous BIM. As expected, treatment with ABT-737 resulted in dissociation of BIM from BCL-2 in BCL-2- expressing DLBCL (Figure 3). Once dissociated, BIM became sequestered by MCL-1, as measured particularly in OCI-Ly8 and SU-DHL-6 cells where the MCL-1:BIM complex following treatment with ABT-737 was greater than control treated cells (Figure 3A and 3C). In contrast, treatment with BIM SAHBA led to measurable, but minimal, displacement of endogenous BIM from BCL-2 in comparison to ABT-737. BIM SAHBA treatment resulted in much greater dissociation of BIM from MCL-1 with no detectable increased accumulation on BCL-2 in all three cell lines (Figure 3). Thus, although BIM SAHBA was better able to dissociate endogenous BIM from MCL-1, it was still able to prevent redistribution of BIM to BCL-2 [17]. Given these results, we were interested if step-wise treatment with ABT-737 and BIM SAHBA resulted in improved dissociation of BIM from either BCL-2 or MCL-1 and blockage of subsequent binding of released BIM on the alternate anti-apoptotic protein. To test this, cells were treated with either ABT-737 or BIM SAHBA for three hours followed by addition of the other compound (i.e. ABT-737 + BIM SAHBA or BIM SAHBA + ABT-737). BIM SAHBA was unable to release relocated BIM from MCL-1 that was dissociated from BCL-2 upon treatment first with ABT-737 (Figure 3). The increased BIM:MCL-1 association following treatment with ABT-737 overwhelmed the ability of BIM SAHBA to effectively target MCL-1 at the same doses used during monotherapy. However, treatment with BIM SAHBA before ABT-737 led to relocation of BIM from both BCL-2 and MCL-1. This phenomenon was greatest in OCI-Ly8 and SU-DHL-6 (Figure 3A and 3C) [10, 13]. In fact, although monotherapy of SU-DHL-5 and OCI-Ly1 with BIM SAHBA led to release of endogenous BIM from MCL-1, this effect was largely lost in both combination treatments (Figure 3B and Supplementary Figure 3A) [24, 25]. There were no gross differences in expression of BCL-2, MCL-1, or BIM during the six hour treatment timeframe in any cell line following individual or combination treatments (Supplementary Figure 4). Dissociation of BAK from MCL-1 and activation of BAX in these cell lines may also have played a role in cell death following BIM SAHBA treatment as has been reported previously [17, 26]. Dissociation of BAX from BCL-2 also occurs in these cell lines following treatment with both ABT-737 and BIM SAHBA (Supplementary Figure 5).
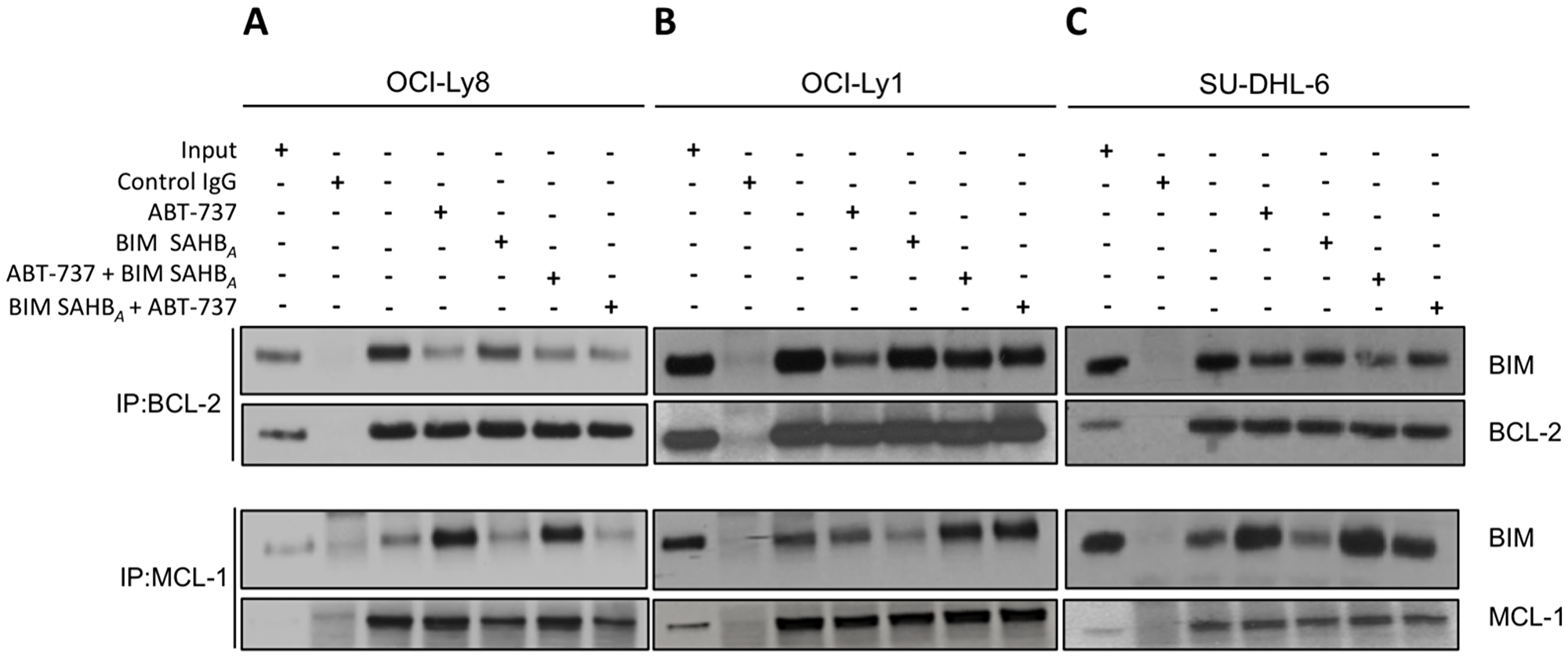
Figure 3: BIM SAHBA preferentially targets MCL-1 over BCL-2 in ABT-737-sensitive DLBCL. (A) OCI-Ly8, (B) OCI-Ly1, and (C) SU-DHL-6 were left untreated (third lane) or treated with ABT-737 (EC50; fourth lane), BIM SAHBA (EC50; fifth lane), ABT-737 for three hours followed by BIM SAHBA for three hours (sixth lane), or BIM SAHBA for three hours followed by ABT-737 for three hours (seventh lane). Lysates were immunoprecipitated with antibodies specific for BCL-2, MCL-1, or anti-Rabbit IgG (control), and immune complexes were resolved and immunoblotted for BIM, BCL-2 and MCL-1. Input lysate was loaded in the first lane and immunoprecipitates with a control IgG in the second lane. Treatments: OCI-Ly8: ABT-737 241 nM, BIM SAHBA 13.2 μM; OCI-Ly1: ABT-737 30.7 nM, BIM SAHBA 12 μM; SU-DHL-6: ABT-737 113 nM, BIM SAHBA 12 μM.
BIM SAHBA sensitizes DLBCL to ABT-737
To test if sequential treatment would lead to differential effects on cell viability, DLBCL were treated as above and cell death was measured after 24 hours. Consistent with the BIM relocation results (Figure 3 and Supplementary Figure 3A) treatment with BIM SAHBA prior to ABT-737 led to increased cell death when compared to the opposite treatment or monotherapy with either compound (Figure 4 and Supplementary Figure 3B). Equivalent results were measured when cells were treated with ABT-737 alone or with ABT-737 followed by BIM SAHBA as was predicted by our BIM relocation data. The largest effect was measured in OCI-Ly8 correlating with BIM SAHBA’s ability to fully block BIM sequestration on MCL-1 following treatment with ABT-737 (Figure 4C). Regardless of overall effect, treatment with BIM SAHBA prior to treatment with ABT-737 at a ratio of 10:1 universally led to increased cell death when compared to the reverse treatment in all cell lines (ABT-737 EC50: OCI-Ly8: 165 nM to 40.1 nM (76% decrease); OCI-Ly1: 93 nM to 66.4 nM (29% decrease); SU-DHL-6: 100 nM to 63.5 nM (37% decrease); and SU-DHL-5: >10,000 nM to 3,360 nM (~66% decrease) (Figure 4A–4C and Supplementary Figure 3B). In each case, synergy was observed when BIM SAHBA was given prior to ABT-737, as reflected by combination indices of less than 0.8 at 50%, 75%, and 90% effective doses. Apart from synergy in OCI-Ly1, ABT-737 treatment prior to BIM SAHBA, predominantly resulted in an additive cell killing effect with combination indices between 0.8 and 1.1 [27]. These effects were magnified in OCI-Ly8 at a BIM SAHBA:ABT-737 ratio of 20:1 where the EC50 decreased from 165 nM to 32.2 nM (80% decrease) and synergy was increased at all effective doses indicating that the amount of BH3 mimetic may play a role in sequential dosing but does not significantly alter synergy values (Figure 4D). The increased sensitivity to ABT-737, particularly in OCI-Ly8, correlated with the ability of MCL-1 to absorb redistributed BIM once released from BCL-2 in these cells. Our results align with previous work showing that normal lymphoid cell and human non-Hodgkin lymphoma resistance to ABT-737 is conferred by mutant BIM specific for BCL-2, BCL-XL, and BCL-W [28]. However, mutant BIM specific for MCL-1 led to ABT-737 sensitivity indicating that ABT-737-mediated killing only occurred when MCL-1 was neutralized [28]. Our data would suggest that BIM SAHBA is more selective for intracellular MCL-1 and effectively prevents BIM:MCL-1 association in a similar context.
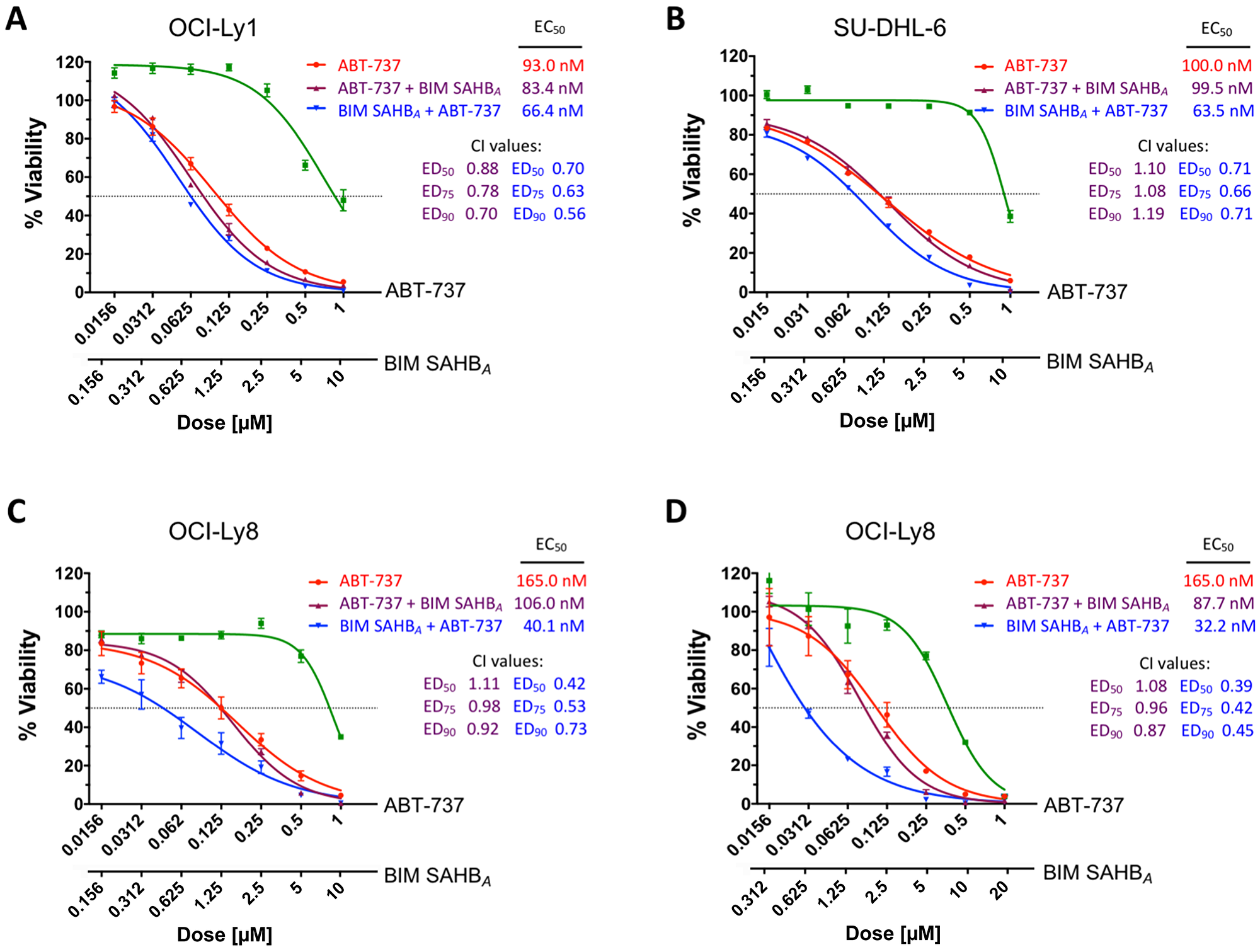
Figure 4: Treatment with BIM SAHBA increases DLBCL cell death when given before but not following treatment with ABT-737. Cell viability of (A) OCI-Ly1, (B) SU-DHL-6, and (C, D) OCI-Ly8 was measured after 24-hr treatment with increasing concentrations of ABT-737 (red), BIM SAHBA (green), ABT-737 for the first three hours followed by BIM SAHBA (purple), or BIM SAHBA for the first three hours followed by ABT-737 (blue). BIM SAHBA given prior to ABT-737 universally led to a greater synergistic decrease in cell viability, as reflected by CalcuSyn analysis (combination index [CI] <1). Error bars are mean ± SEM for at least three independent preparations of cells and BH3 mimetic treatments. EC50, 50% effective concentration; ED50, 50% effective dose; ED75, 75% effective dose; ED90, 90% effective dose.
To identify and correlate relevant BCL-2 pro-survival dependency in cells with either high (OCI-Ly8) or low (OCI-Ly1) MCL-1:BIM binding following ABT-737 treatment, we employed lentiviral expression of BIMS-derived BH3 variants [29]. These variants can determine biologically relevant BH3-only protein dependence within an intact cellular environment [30]. OCI-Ly8 and OCI-Ly1 were engineered to inducibly express: 1) BIMSWT able to target all pro-survival proteins, 2) BIMS2A able to target MCL-1 only, 3) BIMSBAD able to target BCL-2, BCL-XL and BCL-W; or 4) BIMS4E unable to bind any anti-apoptotic, serving as a negative control (Supplementary Figure 6) [29]. BIMSWT expression greatly reduced the viability of both OCI-Ly8 and OCI-Ly1. OCI-Ly8 cells were more sensitive to BIMSBAD compared to BIMS2A indicating more BCL-2 dependence. This anti-apoptotic dependency pattern supports the increased cell death in OCI-Ly8 when treated first with BIM SAHBA followed by ABT-737 where BIM, released from BCL-2 was unable to reoccupy itself on MCL-1 (Figure 4C). In contrast, targeting of MCL-1 by BIMS2A in OCI-Ly1 led to cell death equal to that of BIMSWT while the response to BIMSBAD was more moderate (Supplementary Figure 5B). Thus, OCI-Ly1 was overall more sensitive to BIM expression in this setting and more dependent on MCL-1 than BCL-2, supporting a different sequestration dynamic when endogenous BIM is dissociated from MCL-1 following BIM SAHBA treatment. This is despite both DLBCL having similar baseline expression of BCL-2 (Supplementary Figure 2) [10, 11]. Together, these results support that the preferential intracellular target of BIM SAHBA was MCL-1.
MCL-1 is required for BIM SAHBA-dependent defects in the outer mitochondrial membrane
Mouse embryonic fibroblasts (MEFs) are dependent upon MCL-1, and to a lesser extent, BCL-XL, for survival [31]. MCL-1 plays the more critical functional role in regulating apoptosis as reflected by the ability of ABT-737 to induce apoptosis only in MCL-1-deficient MEFs [9, 31]. We have previously shown that BIM SAHBA can induce apoptosis in MCL-1-dependent and ABT-737-resistant malignant hematopoietic cells lines and can dissociate BAK from MCL-1 [17]. We have also shown that BIM SAHBA can induce cell death in WT MEFs but not in BAX-/-BAK-/-MEFs [16, 17]. To investigate the general dependence on MCL-1 for BIM SAHBA-induced cell death we used MCL-1-/- MEFs and MEFs with inducible deletion of MCL-1 (Mcl-1f/f Rosa-ERCreT2 MEFs) to ensure there was no BCL-2 family compensation secondary to long-term MCL-1 loss [32]. WT and WT Rosa-ERCre T2 MEFs had equivalent expression of MCL-1 and MCL-1 protein was not detectable in either MCL-1-/- or Mcl-1f/f Rosa-ERCreT2 MEFs following treatment with tamoxifen (Figure 5A). Loss of MCL-1 in either MEF allowed for significant apoptosis in response to treatment with ABT-737 (Supplementary Figure 7). ABT-737 induced greater death in Mcl-1f/f Rosa-ERCreT2 MEFs compared to MCL-1-/- MEFs likely because MCL-1-/- MEFs have developed BCL-2 compensatory mechanisms through long-term culturing as has been previously documented in genetic knockout model systems [33, 34]. In contrast to ABT-737, BIM SAHBA induced death in both WT MEFs while either long-term or short-term absence of MCL-1 led to equivalent protection (Figure 5B). Only loss of MCL-1 was responsible for resistance to BIM treatment as indicated by equivalent cell death in MCL-1-/- or Mcl-1f/f Rosa-ERCreT2 MEFs.
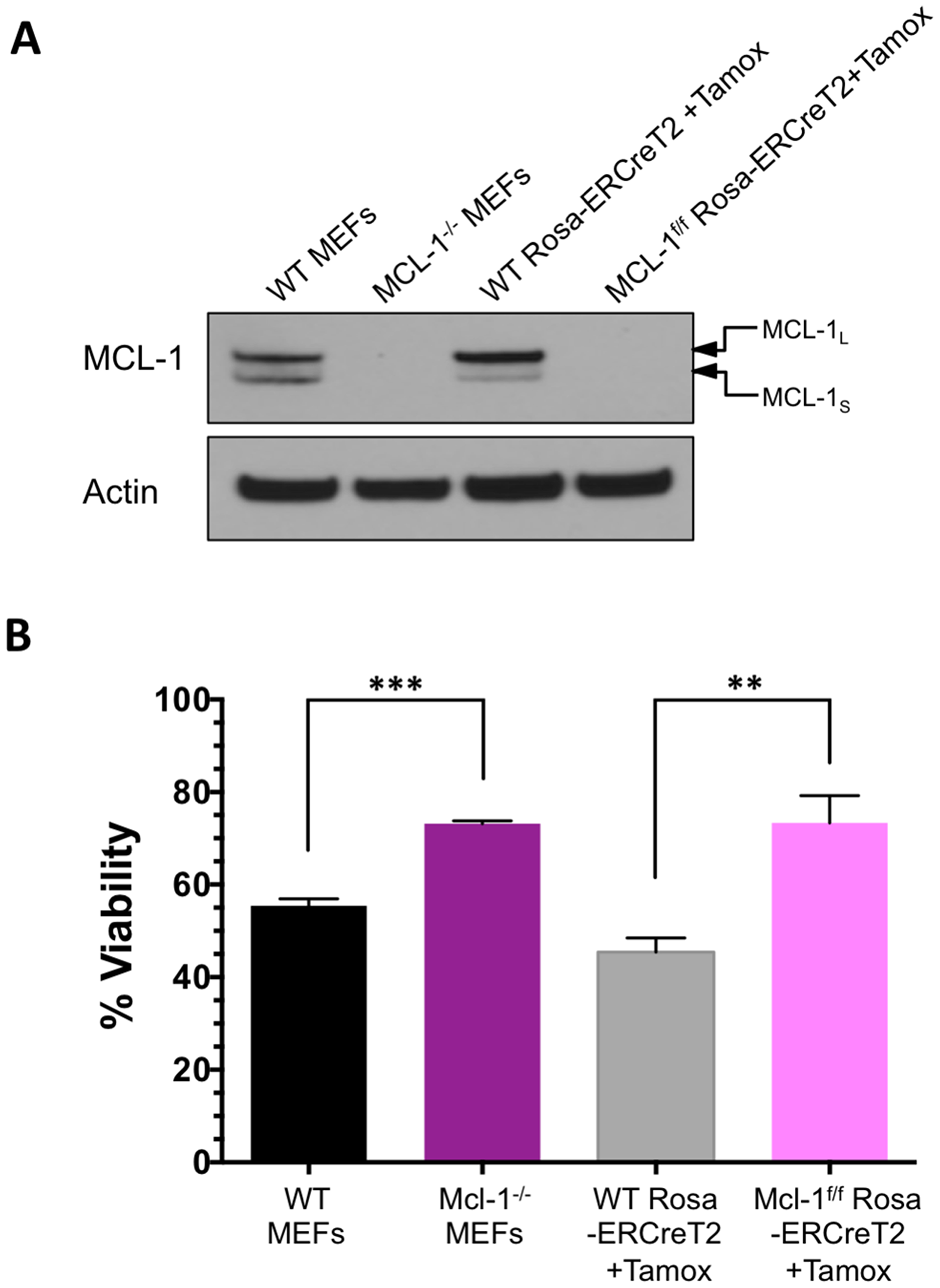
Figure 5: Absence of MCL-1 leads to resistance against BIM SAHBA-induced apoptosis. (A) Western blot analysis of MCL-1 expression in WT MEFs and MCL-1-/- MEFs at steady-state and Rosa-ERCreT2 control MEFs and Mcl-1f/f Rosa-ERCreT2 MEFs 72-hr following treatment with 100 nM of (4-hydroxy)-tamoxifen. Equivalent presence of MCL-1 was measured in the control MEFs and complete loss of MCL-1 was measured in MCL-1-/- and Mcl-1f/f Rosa-ERCreT2 MEFs. Short (S) and long (L) isoforms of MCL-1 are indicated by arrows. (B) Cell viability of these MEFs was measured following 24-hr treatment with 20 μM BIM SAHBA. Error bars are mean ± SEM for at least three independent preparations of cells and BH3 mimetic treatments.
We extended these results to investigate if treatment with BIM SAHBA caused MCL-1-dependent changes in mitochondria morphology. Treatment of WT MEFs with BIM SAHBA led to cristae disorganization and mitochondrial swelling, hallmarks of intrinsic apoptotic pathway activation (Figure 6A) [35, 36]. These phenotypic changes were not evident in treated MCL-1-/- MEFs indicating that BIM SAHBA preferentially targets MCL-1 in these cells (Figure 6B). These phenotypic data confirm BIM SAHBA’s ability to target intracellular MCL-1 and supports an MCL-1-dependent mechanism responsible for sensitivity to BIM SAHBA [17].
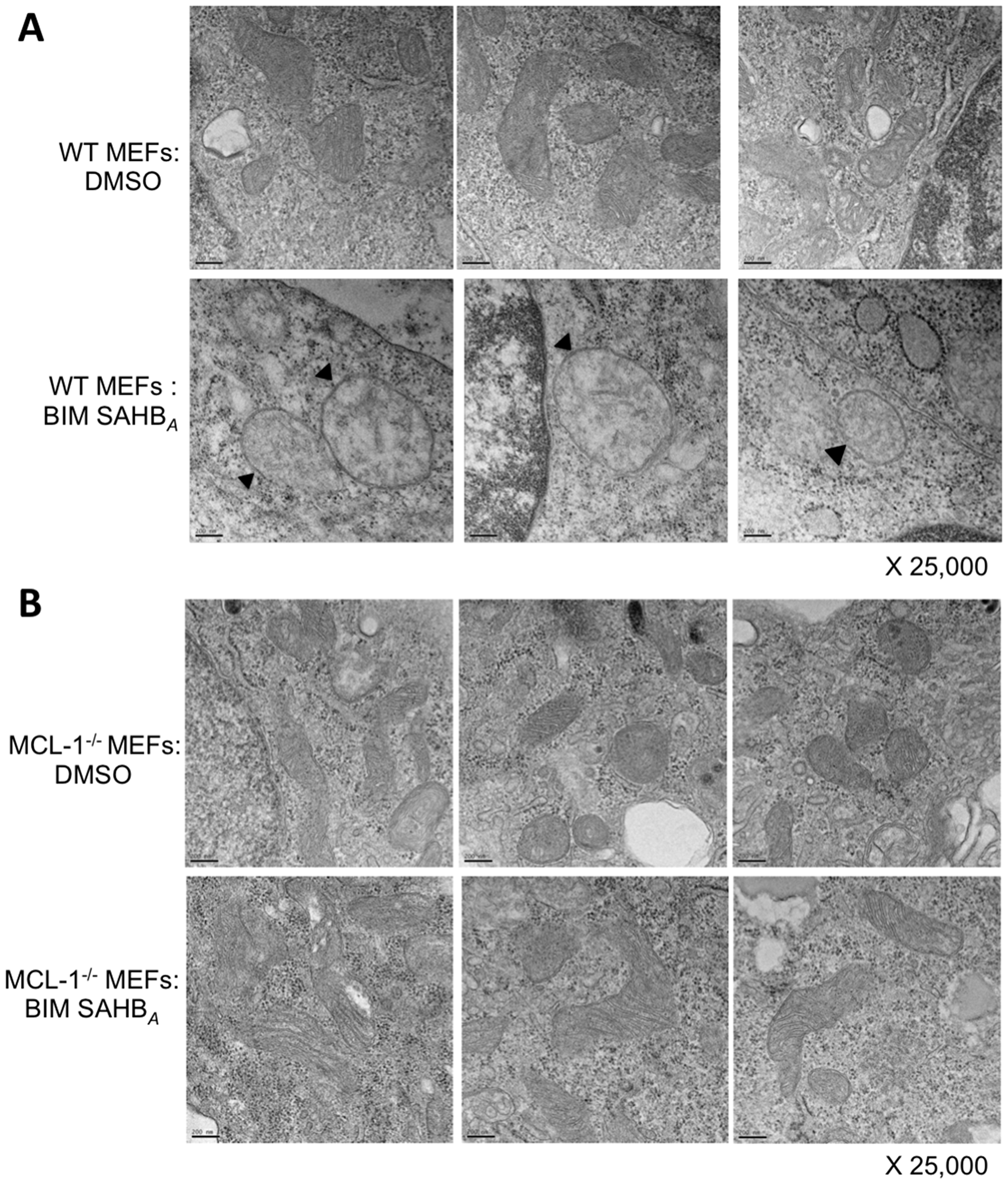
Figure 6: BIM SAHBA induces characteristic hallmarks of apoptosis at the level of the mitochondria in the presence of MCL-1. (A) WT MEFs treated for 6-hr with DMSO (control; upper panels) or 20 μM BIM SAHBA (lower panels) were monitored for mitochondrial apoptotic morphology using electron microscopy. (B) Identical treatment and imaging was performed on MCL-1-/- MEFs. Black arrowheads point to mitochondrial swelling and loss of cristae architecture in BIM SAHBA-treated WT MEFs but not in MCL-1-/- MEFS revealing a dependence on MCL-1 for BIM SAHBA-mediated apoptosis. x 25,000 magnification; scale bars, 200 nm.
DISCUSSION
The presence of BCL-2 family anti-apoptotic members dictates cellular sensitivity to BH3 mimetics. However, expression alone is not enough to predict apoptotic functional dependency patterns, especially as it relates to malignant cell death. This is particularly evident in cells where expression of BCL-2, BCL-XL, or BCL-W does not always result in sensitivity to ABT-737 [37–41]. Additionally, ex vivo sensitivity against these agents does not always correlate with long-term in vivo results either in pre-clinical human xenograft studies or in patients [25, 39]. A primary reason for these resistance patterns is the up-front presence or emergence of MCL-1 during the course of treatment confirming the dynamism of the BCL-2 family regulatory network [42–44]. Importantly, despite the ability of ABT-737, or its oral analogue ABT-263, to bind BCL-2, BCL-XL, and BCL-W, it appears to overwhelmingly bind BCL-2 even when the other anti-apoptotics are present [28, 45]. A recent example of this preferential intracellular attraction is that treatment with ABT-199 or a BCL-XL-specific mimetic induced more cell death in BCL-2 and BCL-XL expressing human small cell lung cancer (SCLC) and AML cell lines than treatment with ABT-263, indicating an intracellular preference for either BCL-2 or BCL-XL, but not both, by ABT-263 [46]. This phenomenon extends to in vivo testing in SCLC xenograft models where combination treatment with ABT-199 and the BCL-XL-specific mimetic resulted in better survival than treatment with ABT-263 alone [46]. Adding to the difficulty in navigating the BCL-2 targeting landscape, cancers derived from the same cell type can show different BCL-2 anti-apoptotic dependencies [10, 40, 41, 46, 47]. These differences often relate to levels of BIM expression and patterns of BIM:anti-apoptotic sequestration [47]. DLBCL is an example of such a disease where cell death resistance depends upon combinations of BIM bound to a number of antiapoptotics (BCL-2, BCL-XL, and MCL-1), and is therefore a useful model for testing the importance of both anti-apoptotic expression and BIM:anti-apoptotic sequestration differences relating to BH3-mimetic sensitivity [10–12, 25].
Like other BH3 mimetics, quantitative expression levels of BCL-2 proteins alone were not effective indicators of sensitivity to BIM SAHBA. However, given the natural affinity of the BIM BH3 death domain to engage all anti- and pro-apoptotic BCL-2 proteins, it was not surprising that the cell permeable hydrocarbon-stapled BIM BH3 therapeutic reactivated cell death in all DLBCL tested. The ability of BIM SAHBA to displace endogenous BIM from BCL-2 and MCL-1 supports the promise of the BIM BH3 domain as a therapeutic. Unexpectedly, like ABT-737/ABT-263 intracellular affinity for BCL-2 in many disease models, BIM SAHBA has a propensity for targeting intracellular MCL-1 over BCL-2. This was likely responsible for its inverse sensitivity profile against the DLBCL cell lines compared to that of ABT-737 and ABT-199. In this regard, a therapeutic with BIM SAHBA’s therapeutic profile could be of particular benefit against activated B-cell like (ABC) DLBCLs that express MCL1 at higher levels and have higher MCL-1:BIM binding than germinal center B-cell like (GCB) DLBCL [12]. However, despite the single agent efficacy of BIM SAHBA against DLBCL as shown here, combination BH3 mimetic therapy will likely be necessary for reactivation of cell death at clinically useful levels [47–49]. Our data would also suggest that dosing and the sequence of such therapies are critical.
We found that BIM SAHBA was unable to effectively dissociate a large amount of released endogenous BIM following treatment with ABT-737 when MCL-1 was present. However, prior to release from BCL-2, BIM SAHBA was able to efficiently bind MCL-1 and prevent relocation of BIM in the more BCL-2-dependent and lower MCL-1 expressing cell lines (Figure 3A and 3C). It should be noted that DLBCL were treated at their EC50 in our study so as to capture cells before they underwent complete apoptosis. Additional studies will be needed to determine if increased doses of BIM SAHBA, as used in Figure 4D, or various treatment times would adequately remove BIM from DLBCL with larger amounts of MCL-1 following treatment with ABT-737 or ultimately stabilize MCL-1 protein levels as do newly introduced high-affinity small molecule MCL-1 inhibitors [6, 50]. In addition to dosing amounts, the timing of death induced by such treatments, either alone or in combination, likely differ between cell lines. Such differences could also be reflected in the relative amount of caspase 3/7 activation (Figure 2A) or synergy calculations in these and other cell lines where such combination treatments have been tested [11, 13, 14, 51]. Additionally, this work does not rule out the possibility of simultaneous triggering of caspase-dependent (e.g. apoptosome/caspase 3 and endonuclease activity) and caspase-independent (e.g. AIF) pathways following BIM SAHBA treatment and caspase 3/7 activation. Despite this, the current study reinforces work indicating that the ability of BH3-mimetics to preferentially target intracellular BCL-2 anti-apoptotic members often does not completely reflect their in vitro binding profile(s) [4, 10, 28, 45]. We and others have shown that BIM SAHBA binds MCL-1, BCL-XL, BCL-2, and A1 with low nanamolar affinity and is able to dissociate BAK and BAX BH3 ligands from MCL-1 and BCL-XL respectively [17, 52]. Although able to bind all anti-apoptotics tested, the current study supports that the preferential intracellular target of BIM SAHBA is MCL-1 over BCL-2, at least in DLBCL. However, we cannot discount that direct activation of BAX or dissociation of other BH3-only proteins, such as BID, plays a role in the overall cell death effectiveness of BIM SAHBA in these same cells [16, 17, 26, 52].
Well-timed combination BH3 mimetic therapy may be a promising strategy against diseases like DLBCL where oncoproteins (e.g. MYC, BCL6, and BCL-2) play roles in blocking the intrinsic apoptotic network through direct upregulation of BCL-2 family members and other cell death-related proteins [53, 54]. Such complex perturbation in apoptotic regulation necessitates a combinatorial therapeutic approach involving multiple BH3 mimetics, which may include small molecule and peptide-based therapeutics, alone or with other conventional chemotherapies. A complete understanding of intracellular affinities and the therapeutic ramifications of targeted anti-apoptotic therapeutics on relocation of BIM will be critical as BH3-mimetic therapies are advanced.
Materials and Methods
Cell culture and therapeutic reagents
Human DLBCL cell lines (kind gift from Margaret A. Shipp, M.D., Dana-Farber Cancer Institute, Boston, MA 02115, USA and purchased from ATCC and DSMZ) were cultured in RPMI 1640 medium supplemented with 0.5 mg/ml penicillin/streptomycin, 2 mM L Glutamine, 1 mM HEPS, Non-Essential Amino Acids (all from Life technologies) and 10% heat inactivated fetal bovine serum (Denville Scientific). HEK293T were obtained from American Type Culture Collection. HEK293T cells and MEFs Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Life technologies) supplemented with same reagents as previously mentioned. For BIM SAHBA treatment, cells were cultured in advanced RPMI supplemented with 0.5 mg/ml penicillin/streptomycin, 2 mM L Glutamine, 1 mM HEPS, 1% NEAA. All cells were maintained at 37°C in 5% CO2. Wild-type ERCreT2 and Mcl-1f/f Rosa-ERCreT2 MEFs were a kind gift from Joseph T. Opferman Ph.D., St. Jude Children’s Research Hospital. BIM SAHBA and BIM SAHBA (R153D) were synthesized, purified, as previously reported [16, 17, 19]. SAHB constructs used in caspase assays and confocal cell death assays were a kind gift from Loren D. Walensky, M.D., Ph.D. and Gregory Bird, Ph.D., Dana-Farber Cancer Institute, Boston, MA. All other SAHBs used for cell treatments were synthesized at the University of Chicago. Doxycycline (Sigma), ABT-199 (Selleck Chemicals), ABT-737 (Selleck Chemicals), (4-hydroxy)-tamoxifen (Sigma).
Viability assays
DLBCL (2 × 104 cells) were treated with serial dilutions of BIM SAHBA, ABT-199, ABT-737 or DMSO for 2 h in serum free advanced RPMI media followed by addition of FBS to the media (to 10% [v/v]) as previously described [17]. Viability was assessed using Cell proliferation kit II XTT (Roche) according to the manufacturer’s instructions. Absorbance EC50s were calculated using GraphPad Prism 6 software. For combination treatments, DLBCL (2 × 104 cells) were treated with serial dilutions of BIM SAHBA (0–20 μM) or ABT-737 (0–1 or to 20 μM), 3 h in serum free advanced RPMI media followed by 3 h of treatment with either ABT-737, BIM SAHBA or DMSO (0.2%). FBS was then added to the media (to 10% [v/v]). Viability was assessed 24 h later using CellTiter-Glo according to the manufacturer’s instructions. (XTT) or luminescence (CellTiter-Glo) was detected by Synergy 2 microplate reader (BioTek). Synergy analyses were performed as previously described [17].
Caspase-3/7 activation assay
DLBCL (10,000 cells/well) were seeded in 96-well plates in advanced RPMI Growth Media and were treated with BIM SAHBA or DMSO for 6 hours. Caspase-Glo 3/7 chemiluminescence reagent (Promega) was used according to manufacturer’s directions. Luminescence was measured by a Synergy 2 microplate reader (BioTek) and was standardized to DMSO treated samples as previously described [17].
Western blot analysis
Following treatment of individual DLBCL with compounds, cells were washed with PBS and lysed in cell lysis buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 5 mM EDTA and 1 mM NaF supplemented with 1x protease inhibitor cocktail tablets [Roche]) as previously described [17]. Protein concentrations were determined by Pierce BCA Protein Assay Kit (Thermo Scientific). 30 μg of protein were electrophoretically separated on NuPAGE 12% Bis-Tris polyacrylamide gels (Invitrogen). Separated proteins were transferred to nitrocellulose membranes (BioRad). Blots were incubated with the following antibodies: MCL-1 (Santa Cruz, S-19), BCL-2 (Epitomics, 1017-1), BCL-XL (Santa Cruz, S18), BIM (Millipore AB17003), BAX (Santa Cruz, N20), BAK (Millipore 06-536), BID (Santa Cruz), GAPDH (Santa Cruz FL-335), PUMA (Santa Cruz). The immune complex was detected using anti-rabbit HRP-conjugated antibody (Envision Detection Kit, DakoCytomation) and the chemiluminescent detection kit according to the manufacturer’s specifications (Amersham). Proteins were visualized with CL-XPosure Film (Thermo Scientific) using AX200 X-Ray film processor (Aphatek).
Immunoprecipitation
DLBCL (9 × 106 cells) were lysed in 2 ml of Triton X-100 buffer (50 mM Tris-HCL [pH 7.4], 150 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 10% Glycerol, 1% Triton X-100 and 1x protease inhibitor cocktail tablets [Roche]) on ice for 30 min and then centrifuged at 18,000 × g for 20 min at 4°C. 400 μg proteins was pre-cleared using 30 μl of Pierce Protein A Magnetic Beads (Thermo Scientific) for 2 hours at 4°C then incubated overnight at 4°C with MCL-1 antibody (Protein Tech, 16225), BCL-2 antibody (Epitomics, 1017–1) or Rabbit IgG. 20 μl of Magnetic beads were added and incubated for 2 hours. Immunoprecipitates were then washed 5 times with 500 μl of Triton X-100 buffer and boiled in NuPAGE LDS Sample Buffer (Thermo Scientific) supplemented with 1 mM DTT and proteins were separated on NuPAGE 10% Bis-Tris polyacrylamide gels.
Wright-giemsa stain
DLBCL cells were treated with vehicle or BIM SAHBA in Opti-MEM media for 4 hours at 37°C. Cells stained as previously described [17]. Briefly, cells were collected and washed twice with PBS containing 2% FBS and resuspended at 2.5 × 106 cells/ml for mounting of the cellular suspension onto glass slides by cytospin. The slides were air dried and then stained with Accustain Modified Wright-Giemsa according to the manufacturer’s protocol (Sigma-Aldrich).
BIMs isoform lentivirus production and transduction
The TRIPZ vectors with inducible expression of the different variants of BIMs were a kind gift from Andreas Strasser, Ph.D. (The Walter and Eliza Hall Institute, Parkville, Melbourne) [29]. To generate cells stably expressing the inducible BIMs variants, DLBCL cells were infected with viral supernatants, produced by lipofectamine 2000 co-transfection of 293T cells with TRIPZ expression constructs and packaging plasmids according to the manufacture protocol and as previously described [29]. Cells were expanded then FACS-sorted for the expression of GFP using a BD FACSAria cell sorter. The expression of BIMs proteins was induced as previously described [29].
Electron microscopy
Indicated MEFs were plated in six-well format (2 × 105 cell/condition) were washed in serum free Advanced DMEM following overnight induction. The cells were then incubated with 20 μM BIM SAHBA or vehicle (0.05% [v/v] DMSO) for 2-hr, and then FBS was added for an additional 4-hr. The cells were washed twice with advanced DMEM media and fixed in the dish using a solution of 2% (w/v) glutaraldehyde, 4% (w/v) paraformaldehyde, in 0.1M sodium cacodylate buffer overnight at RT. The cells were then washed in 0.1 M sodium cacodylate buffer (pH 7.4) 3X5 min, post-fixed for 60 min in 1% (w/v) 1% Osmium Tetroxide in 0.1 M sodium cacodylate buffer, washed in sodium cacodylate buffer 2X5 min and Maleate buffer (pH5.1) once, 5 min, and then incubated in 1% (w/v) aqueous uranyl acetate in Maleate buffer for 60 min, followed wash in Maleate buffer 3X5 min. The cells were removed from the dish in 2:1 propylene oxide and incubated overnight in a 1:1 mixture of propylene oxide. The samples were subsequently embedded in TAAB Epon and polymerized at 60°C for 48 h. Ultrathin sections (~90 nm) were cut on a Reichert-Jung Ultracut E microtome, placed onto copper grids, stained with Uranyl acetate and Lead citrate, and then examined under 300KV at FEI Tecnai F30 and the Gatan CCD digital micrograph. Electron microscopic sample preparation and imaging was performed in conjunction with the Advanced Electron Microscopy facility at the University of Chicago.
Abbreviations
ABC: activated B-cell; CCD: charge-coupled device; DLBCL: diffuse large B-cell lymphoma; DMEM: Dulbecco’s modified Eagle medium; DMSO: dimethyl sulfoxide; DTT: dithiothreitol; EC50: half-maximal concentration of drug; EGTA: ethylene glycol-bis(β-aminoethyl)-N,N,N’,N’-tetraacetic acid; FACS: fluorescence-activated cell sorter; GCB: germinal center B-cell; GFP: green-fluorescent protein; HEPES: 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; Hr: hour; MEF: mouse embryonic fibroblast; Min: minute; mM: millimolar; MOMP: mitochondrial outer membrane permeabilization; NEAA: non-essential amino acids; nM: nanomolar; PAGE: polyacrylamide gel electrophoresis; PBS: phosphate buffered saline; RPMI: Roswell Park Memorial Institute medium; RT: room temperature; SDS: sodium dodecyl sulfate; SEM: standard error of the mean; μM: micromolar; WT: wild-type.
ACKNOWLEDGMENTS
The authors would like to thank the laboratories of Loren D. Walensky, Joseph T. Opferman, and Margaret A. Shipp for graciously providing the cell lines and initial SAHB constructs, the laboratory of Andreas Strasser for the lentiviral Bim vectors, and the Advanced Electron Microscopy facility at the University of Chicago for assistance in acquiring the TEM images used in this study.
CONFLICTS OF INTEREST
The authors report no conflicts of interest.
FUNDING
This work was supported by the National Institutes of Health (NIH) K08-CA151450 (JLL) and the Cancer Research Foundation (JLL). Additional support was provided by the National Center for Advancing Translational Sciences of the National Institutes of Health through Grant Number UL1 TR000430.
References
1. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15:49–63. https://doi.org/10.1038/nrm3722. [PubMed].
2. Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016; 16:99–109. https://doi.org/10.1038/nrc.2015.17. [PubMed].
3. Renault TT, Elkholi R, Bharti A, Chipuk JE. B cell lymphoma-2 (BCL-2) homology domain 3 (BH3) mimetics demonstrate differential activities dependent upon the functional repertoire of pro- and anti-apoptotic BCL-2 family proteins. J Biol Chem. 2014; 289:26481–91. https://doi.org/10.1074/jbc.M114.569632. [PubMed].
4. Varadarajan S, Vogler M, Butterworth M, Dinsdale D, Walensky LD, Cohen GM. Evaluation and critical assessment of putative MCL-1 inhibitors. Cell Death Differ. 2013; 20:1475–84. https://doi.org/10.1038/cdd.2013.79. [PubMed].
5. Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ, Cohen GM. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009; 16:1030–9. https://doi.org/10.1038/cdd.2009.48. [PubMed].
6. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015; 6:e1590. https://doi.org/10.1038/cddis.2014.561. [PubMed].
7. Pécot J, Maillet L, Le Pen J, Vuillier C, Trécesson SC, Fétiveau A, Sarosiek KA, Bock FJ, Braun F, Letai A, Tait SWG, Gautier F, Juin PP. Tight Sequestration of BH3 Proteins by BCL-xL at Subcellular Membranes Contributes to Apoptotic Resistance. Cell Rep. 2016; 17:3347–58. https://doi.org/10.1016/j.celrep.2016.11.064. [PubMed].
8. Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007; 67:1176–83. https://doi.org/10.1158/0008-5472.CAN-06-2203. [PubMed].
9. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006; 10:389–99. https://doi.org/10.1016/j.ccr.2006.08.027. [PubMed].
10. Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007; 12:171–85. https://doi.org/10.1016/j.ccr.2007.07.001. [PubMed].
11. Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, Leverson JD. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. 2015; 5:e368. https://doi.org/10.1038/bcj.2015.88. [PubMed].
12. Wenzel SS, Grau M, Mavis C, Hailfinger S, Wolf A, Madle H, Deeb G, Dörken B, Thome M, Lenz P, Dirnhofer S, Hernandez-Ilizaliturri FJ, Tzankov A, et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia. 2013; 27:1381–90. https://doi.org/10.1038/leu.2012.367. [PubMed].
13. Klanova M, Andera L, Brazina J, Svadlenka J, Benesova S, Soukup J, Prukova D, Vejmelkova D, Jaksa R, Helman K, Vockova P, Lateckova L, Molinsky J, et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin Cancer Res. 2016; 22:1138–49. https://doi.org/10.1158/1078-0432.CCR-15-1191. [PubMed].
14. Li L, Pongtornpipat P, Tiutan T, Kendrick SL, Park S, Persky DO, Rimsza LM, Puvvada SD, Schatz JH. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia. 2015; 29:1702–12. https://doi.org/10.1038/leu.2015.99. [PubMed].
15. Mérino D, Giam M, Hughes PD, Siggs OM, Heger K, O’Reilly LA, Adams JM, Strasser A, Lee EF, Fairlie WD, Bouillet P. The role of BH3-only protein Bim extends beyond inhibiting Bcl-2-like prosurvival proteins. J Cell Biol. 2009; 186:355–62. https://doi.org/10.1083/jcb.200905153. [PubMed].
16. Edwards AL, Wachter F, Lammert M, Huhn AJ, Luccarelli J, Bird GH, Walensky LD. Cellular Uptake and Ultrastructural Localization Underlie the Pro-apoptotic Activity of a Hydrocarbon-stapled BIM BH3 Peptide. ACS Chem Biol. 2015; 10:2149–57. https://doi.org/10.1021/acschembio.5b00214. [PubMed].
17. LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012; 122:2018–31. https://doi.org/10.1172/JCI46231. [PubMed].
18. Reynolds C, Roderick JE, LaBelle JL, Bird G, Mathieu R, Bodaar K, Colon D, Pyati U, Stevenson KE, Qi J, Harris M, Silverman LB, Sallan SE, et al. Repression of BIM mediates survival signaling by MYC and AKT in high-risk T-cell acute lymphoblastic leukemia. Leukemia. 2014; 28:1819–27. https://doi.org/10.1038/leu.2014.78. [PubMed].
19. Bird GH, Gavathiotis E, LaBelle JL, Katz SG, Walensky LD. Distinct BimBH3 (BimSAHB) stapled peptides for structural and cellular studies. ACS Chem Biol. 2014; 9:831–7. https://doi.org/10.1021/cb4003305. [PubMed].
20. Bai M, Skyrlas A, Agnantis NJ, Kamina S, Tsanou E, Grepi C, Galani V, Kanavaros P. Diffuse large B-cell lymphomas with germinal center B-cell-like differentiation immunophenotypic profile are associated with high apoptotic index, high expression of the proapoptotic proteins bax, bak and bid and low expression of the antiapoptotic protein bcl-xl. Mod Pathol. 2004; 17:847–56. https://doi.org/10.1038/modpathol.3800130. [PubMed].
21. Bosch R, Dieguez-Gonzalez R, Céspedes MV, Parreño M, Pavón MÁ, Grañena A, Sierra J, Mangues R, Casanova I. A novel inhibitor of focal adhesion signaling induces caspase-independent cell death in diffuse large B-cell lymphoma. Blood. 2011; 118:4411–20. https://doi.org/10.1182/blood-2011-04-345181. [PubMed].
22. Fitzsimmons L, Boyce AJ, Wei W, Chang C, Croom-Carter D, Tierney RJ, Herold MJ, Bell AI, Strasser A, Kelly GL, Rowe M. Coordinated repression of BIM and PUMA by Epstein-Barr virus latent genes maintains the survival of Burkitt lymphoma cells. Cell Death Differ. 2018; 25:241–254. https://doi.org/10.1038/cdd.2017.150. [PubMed].
23. Inomata M, Tagawa H, Guo YM, Kameoka Y, Takahashi N, Sawada K. MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood. 2009; 113:396–402. https://doi.org/10.1182/blood-2008-07-163907. [PubMed].
24. Adams CM, Mitra R, Gong JZ, Eischen CM. Non-Hodgkin and Hodgkin Lymphomas Select for Overexpression of BCLW. Clin Cancer Res. 2017; 23:7119–29. https://doi.org/10.1158/1078-0432.CCR-17-1144. [PubMed].
25. Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, McKeegan E, Salem AH, Zhu M, Ricker JL, Blum W, DiNardo CD, Kadia T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016; 6:1106–17. https://doi.org/10.1158/2159-8290.CD-16-0313. [PubMed].
26. Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, Walensky LD. BAX activation is initiated at a novel interaction site. Nature. 2008; 455:1076–81. https://doi.org/10.1038/nature07396. [PubMed].
27. Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011; 731:421–34. https://doi.org/10.1007/978-1-61779-080-5_34. [PubMed].
28. Mérino D, Khaw SL, Glaser SP, Anderson DJ, Belmont LD, Wong C, Yue P, Robati M, Phipson B, Fairlie WD, Lee EF, Campbell KJ, Vandenberg CJ, et al. Bcl-2, Bcl-x(L), and Bcl-w are not equivalent targets of ABT-737 and navitoclax (ABT-263) in lymphoid and leukemic cells. Blood. 2012; 119:5807–16. https://doi.org/10.1182/blood-2011-12-400929. [PubMed].
29. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012; 26:120–5. https://doi.org/10.1101/gad.182980.111. [PubMed].
30. Lopez J, Bessou M, Riley JS, Giampazolias E, Todt F, Rochegue T, Oberst A, Green DR, Edlich F, Ichim G, Tait SW. Mito-priming as a method to engineer Bcl-2 addiction. Nat Commun. 2016; 7:10538. https://doi.org/10.1038/ncomms10538. [PubMed].
31. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME, Huang DC, Kile BT. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014; 159:1549–62. https://doi.org/10.1016/j.cell.2014.11.036. [PubMed].
32. Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012; 14:575–83. https://doi.org/10.1038/ncb2488. [PubMed].
33. Eichhorn JM, Alford SE, Sakurikar N, Chambers TC. Molecular analysis of functional redundancy among anti-apoptotic Bcl-2 proteins and its role in cancer cell survival. Exp Cell Res. 2014; 322:415–24. https://doi.org/10.1016/j.yexcr.2014.02.010. [PubMed].
34. El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017; 13:e1006780. https://doi.org/10.1371/journal.pgen.1006780. [PubMed].
35. Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009; 11:1050–62. https://doi.org/10.1016/j.micinf.2009.08.013. [PubMed].
36. Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011; 21:92–101. https://doi.org/10.1016/j.devcel.2011.06.017. [PubMed].
37. Chonghaile TN, Roderick JE, Glenfield C, Ryan J, Sallan SE, Silverman LB, Loh ML, Hunger SP, Wood B, DeAngelo DJ, Stone R, Harris M, Gutierrez A, et al. Maturation stage of T-cell acute lymphoblastic leukemia determines BCL-2 versus BCL-XL dependence and sensitivity to ABT-199. Cancer Discov. 2014; 4:1074–87. https://doi.org/10.1158/2159-8290.CD-14-0353. [PubMed].
38. Anderson NM, Harrold I, Mansour MR, Sanda T, McKeown M, Nagykary N, Bradner JE, Lan Zhang G, Look AT, Feng H. BCL2-specific inhibitor ABT-199 synergizes strongly with cytarabine against the early immature LOUCY cell line but not more-differentiated T-ALL cell lines. Leukemia. 2014; 28:1145–8. https://doi.org/10.1038/leu.2013.377. [PubMed].
39. Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, Kunz J, Richter-Pechanska P, Marovca B, Pail O, Jenni S, Diaz-Flores E, Chang BH, Brown TJ, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017; 129:e26–e37. https://doi.org/10.1182/blood-2016-09-738070. [PubMed].
40. Khaw SL, Suryani S, Evans K, Richmond J, Robbins A, Kurmasheva RT, Billups CA, Erickson SW, Guo Y, Houghton PJ, Smith MA, Carol H, Roberts AW, et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood. 2016; 128:1382–95. https://doi.org/10.1182/blood-2016-03-707414. [PubMed].
41. Peirs S, Matthijssens F, Goossens S, Van de Walle I, Ruggero K, de Bock CE, Degryse S, Cante-Barrett K, Briot D, Clappier E, Lammens T, De Moerloose B, Benoit Y, et al. ABT-199 mediated inhibition of BCL-2 as a novel therapeutic strategy in T-cell acute lymphoblastic leukemia. Blood. 2014; 124:3738–47. https://doi.org/10.1182/blood-2014-05-574566. [PubMed].
42. Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, Anderson GR, Tingley JP, Wood KC. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci Rep. 2016; 6:27696. https://doi.org/10.1038/srep27696. [PubMed].
43. Luedtke DA, Niu X, Pan Y, Zhao J, Liu S, Edwards H, Chen K, Lin H, Taub JW, Ge Y. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther. 2017; 2:17012. https://doi.org/10.1038/sigtrans.2017.12. [PubMed].
44. Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010; 115:3304–13. https://doi.org/10.1182/blood-2009-07-233304. [PubMed].
45. Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 2012; 3:e366. https://doi.org/10.1038/cddis.2012.109. [PubMed].
46. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, Belmont LD, Nimmer P, Xiao Y, Ma XM, Lowes KN, Kovar P, Chen J, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015; 7:279ra40. https://doi.org/10.1126/scitranslmed.aaa4642. [PubMed].
47. Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, Pietanza MC, Hellmann MD, Kris MG, Hsieh JJ, et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nat Commun. 2017; 8:16078. https://doi.org/10.1038/ncomms16078. [PubMed].
48. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017; 16:273–84. https://doi.org/10.1038/nrd.2016.253. [PubMed].
49. Opferman JT. Attacking cancer’s Achilles heel: antagonism of anti-apoptotic BCL-2 family members. FEBS J. 2016; 283:2661–75. https://doi.org/10.1111/febs.13472. [PubMed].
50. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016; 538:477–82. https://doi.org/10.1038/nature19830. [PubMed].
51. Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006; 10:375–88. https://doi.org/10.1016/j.ccr.2006.10.006. [PubMed].
52. Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Molecular Cell. 2006; 24:199–210. https://doi.org/10.1016/j.molcel.2006.08.020. [PubMed].
53. Horn H, Ziepert M, Becher C, Barth TF, Bernd HW, Feller AC, Klapper W, Hummel M, Stein H, Hansmann ML, Schmelter C, Moller P, Cogliatti S, et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood. 2013; 121:2253–63. https://doi.org/10.1182/blood-2012-06-435842. [PubMed].
54. Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010; 362:1417–29. https://doi.org/10.1056/NEJMra0807082. [PubMed].