
Introduction
During gene transcription, a pre-mature messenger RNA (pre-mRNA) molecule is generated that requires further processing to a mature in a mRNA molecule that will be translated into a protein. In this maturation step, the introns are usually removed and the exons are ligated. This process, called splicing, is one of the post-transcriptional processes essential for RNA translation into functional proteins and requires the activity of the splice factors. Besides simple intron removal and exon coupling, the activity of those splice factors enable that multiple protein isoforms can be translated out of one pre-mRNA transcript by selective incorporation of pre-mRNA parts in the mature mRNA transcript [1–4]. This is called alternative splicing (AS) and provides an essential layer of post-transcriptional regulation that only recently received much attention from the research community. In particular, it is becoming clear that tumor cells benefit greatly from this flexible regulatory process since many specific isoforms have been identified as promoting and supporting neoplastic transformation, tumor growth and progression. Many reports have linked AS to up-regulation of proto-oncogenes, deregulated cell division, increased survival, altered metabolism, onset of angiogenesis, increased invasion and metastasis in different cancer types including breast cancer [5–8].
Splicing is a complex multistep process catalyzed by the spliceosome, a large, dynamic, multicomponent complex consisting of five small nuclear ribonucleoproteins (snRNPs) U1, U2, U4, U5 and U6 and many associated proteins. In the human spliceosome, the 141 core factors are highly abundant and/or are specifically associated with the U1, U2, U5, U4/U6 snRNPs, or the U4/U6. U5 tri-snRNP [9, 10]. The auxiliary splice factors that are not part of the core spliceosome regulate AS and are less abundant when co-purified with the core spliceosome members [9, 10]. Splice factors are highly diverse considering both function and structure. For example, hnRNPs are characterized by a RNA Recognition Motif (RRM) domain that accommodates site-specific binding to the target RNA typically resulting in splicing inhibition by suppressing assembly of the spliceosome [11] or attraction of snRNPs [12, 13]. SR splice factors contain a domain consisting of arginine/serine repeats (RS domain) and at least one RRM domain [14–16] and facilitate recruitment of the snRNPs to the splice sites [2]. Additionally, their activity is regulated through phosphorylation by SR protein kinases (SRPKs). Currently almost 250 splice factors distributed over different classes have been identified, all playing a specific role at a specific stage of the splicing process [9, 17, 18]).
Breast cancer is the most frequent type of cancer in women with an estimation of 268,670 new cases and 41,400 deaths in the United States in 2018 [19]. In order to develop and progress, (breast) cancer cells move through various steps to fulfill their requirements for certain oncogenic properties. These processes – the so-called ‘hallmarks of cancer’ – have been summarized by Hanahan and Weinberg in 2000 and 2011 [20, 21] and currently include ten processes essential for tumor development and progression. In this review, we will discuss the spliceosomal changes across the different hallmarks of breast cancer. Since many of the already known spliceosome target genes have already been reviewed extensively elsewhere [5–8], we will focus on the role of splice factors as potential oncogenes or tumor suppressors in breast cancer. We will highlight newly identified splice factors of which abnormal regulation is linked to the different hallmarks of breast cancer [21]. For the hallmarks that have not yet been linked to splice factors expression, we identified factors strongly related to hallmark-specific oncogenic processes using publicly available RNA sequencing data. Finally, we discuss the clinical relevance of using splice factors as biomarkers and potential targets in breast cancer therapy.
SPLICE FACTOR DYSREGULATION IN BREAST CANCER
Cancer-specific splicing events are established via different routes: 1) changes in expression levels, activity and localization of splice factors and/or 2) mutations in functional domains of splicing related proteins and/or mutations in regulatory sequences, such as enhancer/silencer sequences and branch points [22, 23]. Both processes can result in differential splice factor activity leading to differential splice site usage or increased or suppressed intron or exon inclusion. Those deregulatory events in splicing have been shown to play a prominent role in breast cancer.
Altered expression, activity and localization of splice factors
Altered expression
In two independent studies, the comparison of the transcriptome of human breast tumors versus matched healthy tissue revealed that 10%–50% of the protein-coding genes have altered transcript variant expression levels [24, 25]. These patient data are in line with recent in vitro findings that show a significant switch in splicing pattern during epithelial-to-mesenchymal transition (EMT) accompanied with a specific EMT splicing signature [26]. Interestingly, this shift in splicing pattern was correlated to the expression levels of specific splice factors; all three studies revealed splice factor RBFOX2 as one of the most differentially expressed between the epithelial and mesenchymal cell state [24–26]. Moreover, expression levels of MBNL1, QKI, PTBP1, ELAV1, HNRNPC, KHDRBS1, SRSF2 and TIAR were also linked to the mesenchymal state [24]. By applying a splicing motif analysis in EMT regulated alternative transcripts, Shapiro et al. concluded that the MBNL, CELF, hnRNP, or ESRP splice factors were most likely involved in the EMT splicing patterns [26]. Furthermore, depletion of the mesenchymal splice factor RBFOX2 or overexpression of the epithelial factor ESRP in mesenchymal cells induced a more epithelial morphology and reduced cell motility [26]. Altogether, these data clearly suggest that splice factors can be in control of EMT and breast cancer progression.
Post-translational modifications and chromatin structure
Next to changes in expression levels, activity of certain splice factors can also be regulated by post-translational modifications (PTMs) such as acetylation, phosphorylation and ubiquitination. Strong interactions between ubiquitination and the spliceosome have been demonstrated and SR proteins are widely known to regulate the activation of other splicing factors by phosphorylation [27–29]. For example, acetylation and ubiquitination of the splicing factor SRSF5 has been shown to control tumor growth [30]. The phosphorylation status of SRSF1 and SRSF7 controls their function as only non-phosphorylated SR proteins were shown to facilitate the recruitment of mRNA to nuclear export receptors [31–34].
Moreover, the intracellular distribution can be crucial for downstream signaling events. Although most splice factors reside in the nucleus, cytoplasmic splicing has been recently shown to take place in mammalian cells implying that splice factors might have differential activities depending on their intracellular location [35]. Splice factor dynamics is also highly dependent on the chromatin structure that is often disturbed in cancer cells [36]. Non-coding RNAs (ncRNAs) and in particular long ncRNAs (lncRNAs) can alter the chromatin environment preventing the recruitment of a repressive chromatin-splicing adapter complex and consequently regulate AS of the FGFR2 [37]. Moreover, histone hyper acetylation has been shown to affect the distribution of several splicing factors such as SRSF1, SRSF2 SRSF3 and U2AF65, causing accumulation in the nuclear speckles [38] and decreased spliceosomal assembly at 3′ splice sites, while calcium-mediated histone hyperacetylation regulates AS of genes important in heart development [39]. Finally, splicing can be regulated by miRNAs within the supraspliceosome that can target different RNAs via alternative base pairing, thereby regulating gene expression and quality control of AS [40].
Mutations in splice factors or regulatory sites
Next to altered splice factor expression levels and activity, abnormal splicing can be caused by mutations in the genes that encode these factors. Few studies and our own analysis (Supplementary Table 1) demonstrate that the splice factors U2AF1, SRSF2 and SF3B1 are often mutated in myelodysplastic syndromes [41], but also in solid cancers amongst which breast cancer [42]. These mutations mainly caused haematopoiesis due to impaired 3′-splice site recognition followed by RNA splicing deficiencies [41]. U2AF1 mutations specifically affected AS of genes in various pathways pivotal for cancer development, such as apoptosis via CASP8, DNA damage response via ATR and FANCA and DNA methylation via DNMT3B [43].
For the luminal breast cancer subtype, mutations in SF3B1 were found to be possible driver mutations [44–46]. These mutations result in a change-of-function and have been associated with hundreds of atypical splice sites at the 3′ end of the intron, thereby inducing AS of SF3B1 downstream target genes [47, 48]. Accordingly, our splice factor mutation analysis of breast cancer tumors from the COSMIC database revealed frequently mutated spliceosome genes amongst which SF3B1 (Supplementary Table 1). Interestingly, 10 splicing factors were classified as driver genes of which mutations are selected during tumor development by the Intogen database (Supplementary Table 1), 5.4% of all driver genes in breast cancer were regulating splicing, suggesting a major role for these proteins in breast cancer oncogenesis.
Next to mutations that could affect the functionality of splice factors, mutations in 5′- or 3′ splice site or branch point can disrupt or create splice sites [49] and thereby cause AS [50, 51]. Furthermore, specificity of AS is controlled by cis-regulatory elements that regulate the recruitment of trans-acting splicing factors to the splice site; exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs) [2]. Mutations can modulate activity of these elements thereby affecting AS. For instance, a point mutation in exon 18 of important tumor-suppressor gene BRCA1 disrupts an ESE resulting in exon skipping [52], while mutations in the ESEs and branchpoint recognized by SRSF2 dysregulates spliceosome assembly and result in AS in myelodysplasia [53].
ROLE OF SPLICE FACTORS IN THE HALLMARKS OF BREAST CANCER
In order to develop and progress, (breast) cancer cells move through various oncogenic processes. These hallmarks of cancer were summarized by Hanahan and Weinberg in 2000 and 2011 and now contain ten processes essential for tumor development and progression [20, 21]. Although the splice factor research in relation to breast cancer emerged only recently, there are already five hallmarks of cancer known to be affected by splice factors: sustaining proliferation, activation of invasion and metastasis, resisting cell death, deregulating cellular energetics and angiogenesis. Here we will discuss splice factors and up –and downstream pathways important in these five hallmarks (Figure 1, Supplementary Table 2). Moreover, we could relate expression of representative genes of four other hallmarks to splice factor levels in human breast cancer tumors (Figure 2, Supplementary Table 3), suggesting that also these hallmarks might be modulated by splice factors. However, the causal relationship between splice factor levels and these hallmarks of cancer remains to be elucidated.
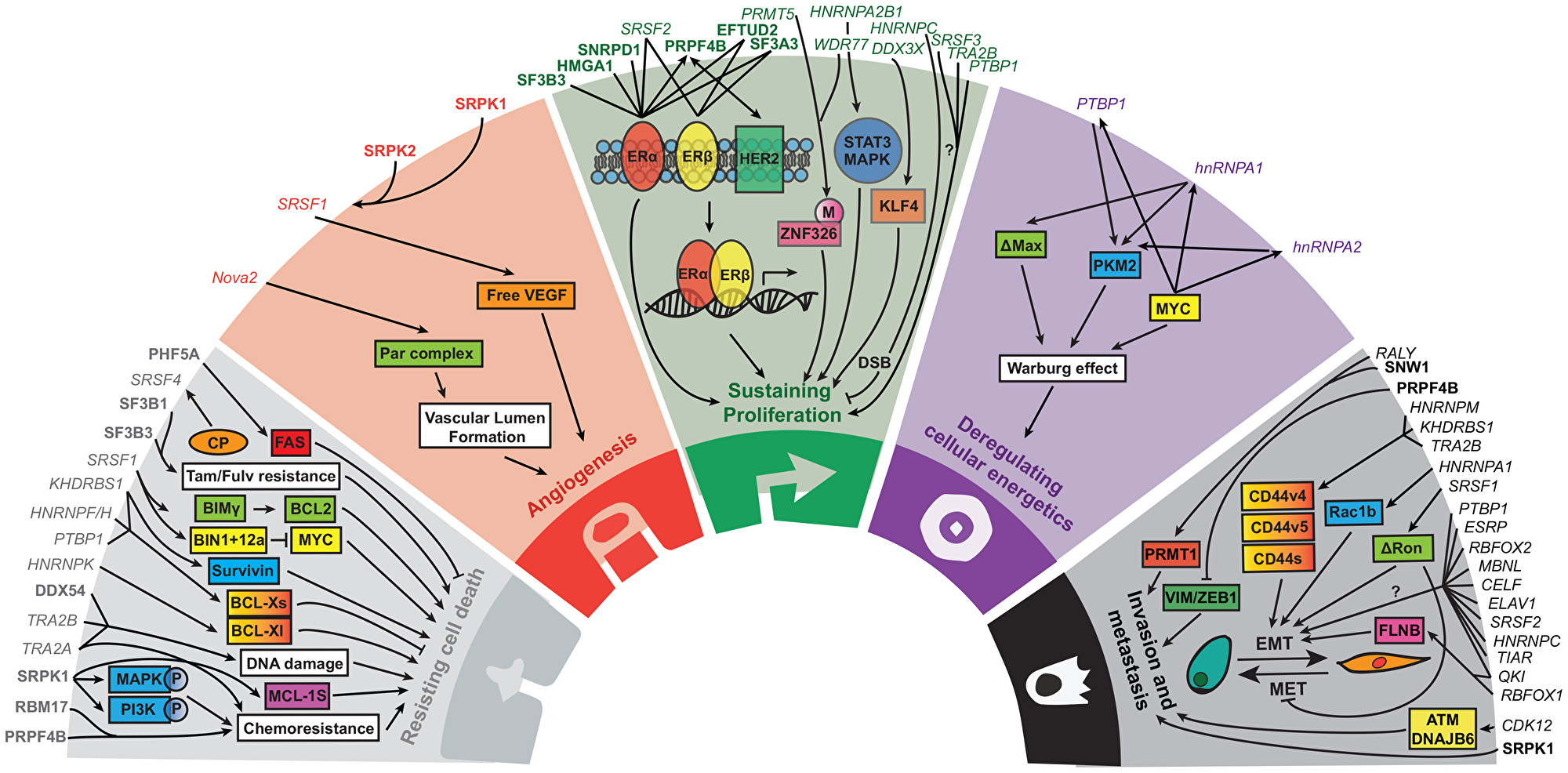
Figure 1: The role of splice factors and their associated pathways in the five hallmarks of cancer. Core splicing factors are listed in bold. Non-core splicing factors are listed in italic. Adapted from Hanahan and Weinberg, 2011 [21].
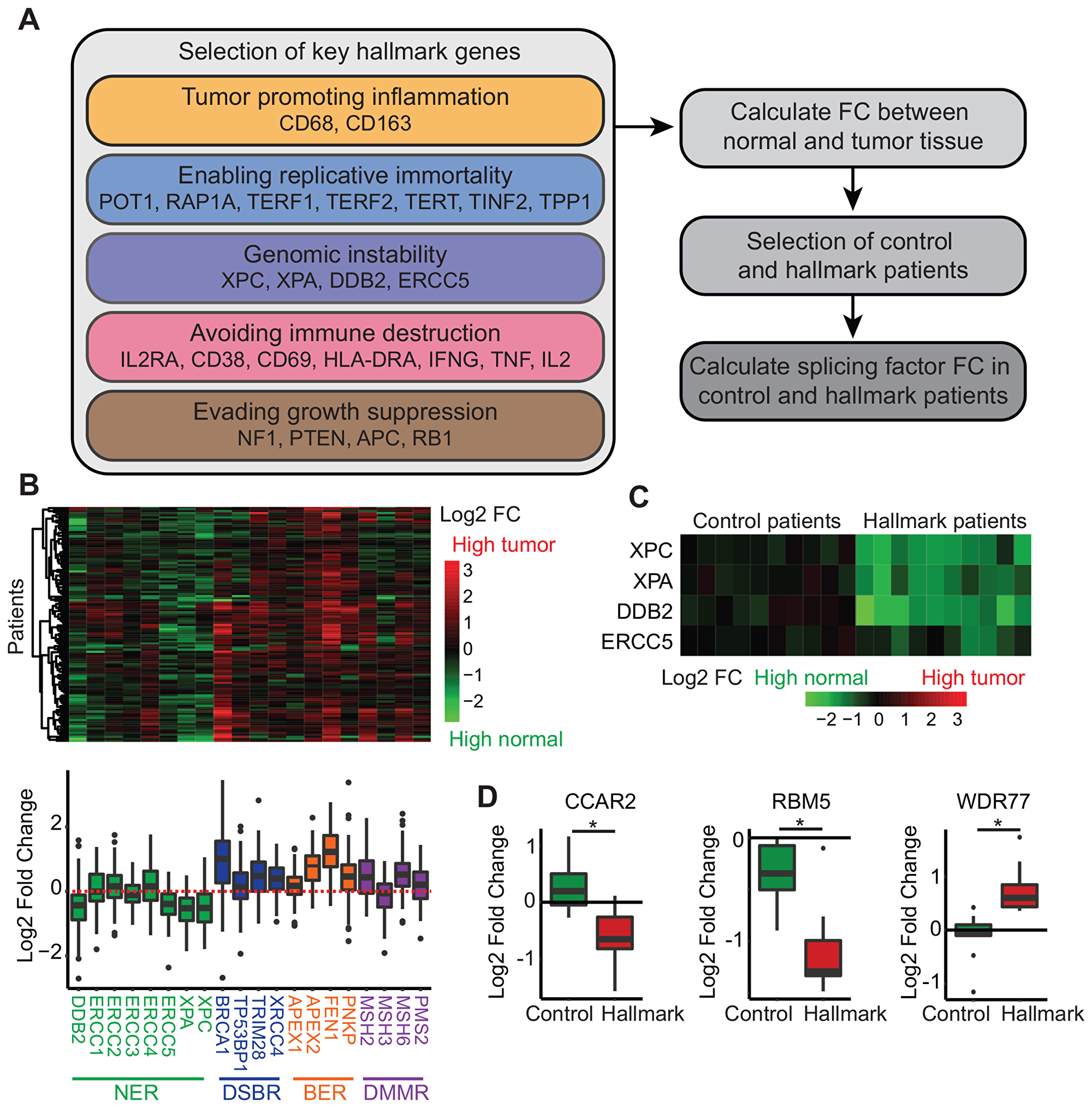
Figure 2: Relation of splice factor expression levels to other hallmarks of cancer. (A) Steps used to link splice factor RNA expression levels to the other hallmarks of cancer. (B) Heatmap displaying the log2 fold change of genome instability markers comparing primary tumor tissue to normal tissue. NER = nucleotide excision repair, DSBR = double strand break repair, BER = base excision repair, DMMR = DNA mismatch repair. (C) Heatmap of log2 fold change of genome instability markers in ten control and hallmark patients comparing normal to primary tumor tissue. (D) Log2 fold change of splice factors in control and hallmark patients comparing normal to primary tumor tissue.
Sustaining proliferation
Hormone-receptor dependent pathways
Sustained proliferation is probably the most critical and studied cancer hallmark. Normal tissues precisely control cell number through many signaling pathways, amongst which the well-known MAPK and PI3K cascades [20, 54, 55]. In transformed cells, these pathways are upregulated resulting in uncontrolled growth. In breast cancer this mainly occurs via mutations resulting in overexpression of hormone receptors including the estrogen receptor (ER), androgen receptor (AR), epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2) and their corresponding ligands [20, 56, 57].
The ER consists of two subtypes: ERα and ERβ. Upon ligand binding ERα and/or ERβ will form homo- or heterodimers, leading to nuclear translocation and possibly transcriptional activation [58]. ERα stimulates cell proliferation and survival by regulating the transcription of hundreds of genes [59, 60]. The role of ERβ in cancer has yet to be defined as several studies report contradictory correlations between transcript level and patient prognosis [61–63]. Emerging evidence suggests a role for splice factors in ER cancer signaling either by direct or indirect interactions with the receptors. For instance, RNA levels of SF3B3 correlate significantly with the overall survival of patients bearing ERα-positive tumors [64], PRPF4B protein expression levels are directly regulated by ERα activation [65] and HMGA1 is directly involved in ERα splicing [66]. Nassa et al. discovered that the interactome of both ERα and ERβ contains multiple splice factors of which some are common for both receptor subtypes (e. g. EFTUD2, SRSF2), while others only interact with a single receptor subtype, such as SNRPD1 for ERα and SF3A1 for ERβ [67]. Introduction of ERβ expression diminished 72% of ERα induced splicing events, but also introduced distinct splicing events in 28 genes that were functionally involved in many cellular processes including DNA replication and repair, DNA transcription, cell cycle and apoptosis [68]. Whether these changes are counteracting the proliferative effects of ERα transcriptional activity is yet unknown.
In addition to being regulated by ER signaling, PRPF4B levels are also controlled by HER2 signaling. Knockdown of HER2 results in downregulation of PRPF4B, whereas HER2 upregulation increases the levels of this splice factor in breast cancer cell lines [69]. Moreover, HER2 itself can be alternatively spliced resulting in different variants exhibiting either pro- or anti-tumorigenic functions. Skipping exon 16 results in the variant HER2D16, that is linked to increased resistance to HER2-targeting therapy and associated with cancer cell dissemination [70, 71]. Inclusion of intron 8 results in a truncated version of HER2 named Herstatin. Herstatin binds to the extracellular domain of HER2, preventing transfer to the cell membrane and receptor dimerization and phosphorylation, thereby exhibiting anti-tumor activities [72–74]. Although HER2 and ER expression patterns are inversely correlated because of negative feedback loops, they are interconnected via downstream pathways, such as the MAPK pathway. This crosstalk between ER and HER2 seems to be important in endocrine resistance [75] and therefore treatment might be improved by identifying more pathways or proteins regulated by both ER and HER2. Since splice factors like PRPF4B fit this double activation pattern, it might be worth investigating the role of PRPF4B in hormone receptor growth resistance conditions.
Hormone receptor independent pathways
Next to involvement in known proliferation pathways, splice factors are also implicated in tumor growth via partly unknown mechanisms that might be independent of these hormone cascades. Examples are PTBP1 which inhibits cell growth in breast cancer cell lines [76], Tra2β, a target of transcription factor and oncogene ETS-1 that is upregulated in breast cancer and is associated with cancer cell survival [77], SRSF3 that upon inhibition decreased breast cancer cell proliferation [78], the loss of PRMT5 or WDR77 resulting in AS and loss of proliferative genes [79] and HNRNPC which is highly expressed in breast tumors and of which knockdown results in double strand breaks and reduced proliferation [80]. Furthermore, DDX3× modulates the cell cycle by affecting splicing and expression of the cell cycle repressor KLF4, resulting in G1 arrest [81], while hnRNPA2B1 knockdown affects MAPK and STAT3 signaling resulting in prolonged S-phase [82]. Altogether, we can conclude that splice factors play an important role in breast cancer sustained proliferation by either directly or indirectly activating hormone receptors and other growth associated pathways.
Furthermore, in line with the existing concept of breast cancer cells being addicted to oncogenes such as MYC for their proliferative and survival capacity, few recent studies have reported the dependence of breast cancer on spliceosomal components [83, 84]. In general, the overall increase in gene expression in cancer cell implies their aggravated dependence on splicing factors which open up a new strategic window for targeting BC. Indeed, several splice factors including BUD31, SF3B1 and SRSF1 [85] are known to be gene targets of the oncoprotein MYC and inhibition or knockdown of those in MYC hyperactivated breast cancer cells impairs tumorigenesis [86].
Activation of invasion and metastasis formation
Because the human breasts are non-vital organs, primary tumors in breast tissue can be surgically removed without major consequences. Ultimately, it is the formation of metastatic lesions in secondary organs that causes breast cancer mortality. Metastasis formation is often described as a multi-step process, also called the invasion-metastasis cascade [87, 88]. Primary breast cancer cells start to locally invade into the surrounding tissue and often those cells undergo a phenotypic switch characterized by epithelial-to-mesenchymal transition (EMT). In this process, epithelial cells bearing strong adhesion structures can switch to a migratory mesenchymal phenotype with loss of cell polarity and cell-cell contacts [89]. Those features acquired by mesenchymal cells allow not only infiltration into adjacent tissue, but also escape into the blood or lymphatic vessels.
Many splice factors have been linked to this EMT process. A genome-wide screen for EMT inducers identified many RNA-binding proteins, with splice factors QKI and RBFOX1 as main candidates. These factors regulated splicing of the actin binding protein FLNB, followed by release of FOXC1 leading to an intermediate mesenchymal cell state [90]. Moreover, the ratio of splice factors ESRP1/RBFOX2 was decreased during EMT and related to cancer progression and metastatic potential [91]. Next, the splice factor ESPR regulates fibroblast growth factor receptor 2 (FGFR2) splicing and thereby affects ligand binding, favoring FGFR2-IIIb which is specific to epithelial cells [92]. Genome-wide analysis of the ESRP splicing network uncovered hundreds of alternatively spliced genes that are involved in EMT related processes such as cellular adhesion and migration including ITGA6 and RALGPS2 [92].
Another important class of splice factors involved in metastasis formation are the heterogeneous nuclear ribonucleoproteins (hnRNPs), which can control splice site selection by either directly antagonizing the recognition of splice sites or interfere with proteins bound to enhancers [93]. For instance, cytoplasmic localization of hnRNPA1 is associated with metastatic relapse and activates RON translation that is known to affect cell migration and dissemination [94]. Conversely, nuclear hnRNPA1 acts as a tumor suppressor and inhibits exon 3b inclusion of the small GTPase Rac1, thereby repression formation of the Rac1b isoform [95]. Rac1b has a constitutively activated GTPase domain [96, 97] and is overexpressed in breast cancer [95, 98]. Matrix metalloproteinase-3 treatment interferes with hnRNPA1-Rac1 interactions, resulting in increased Rac1b levels and EMT [95, 99]. Moreover, breast cancer patients express high hnRNPA1 and low Rac1b levels in normal breast tissue, but low hnRNPA1 and high Rac1b levels in cancer tissue, suggesting that splicing of the Rho GTPase is also in vivo regulated by hnRNPA1 [95].
hnRNPM competes with the pro-epithelial splice factor ESRP1 for guanine-uridine rich motifs to regulate splicing of exons in genes involved in EMT-related pathways [100]. Furthermore, hnRNPM controls EMT by modulating CD44 isoform expression, which in turn increases TGFβ signaling. Elimination of hnRNPM prevents TGFβ induced breast cancer metastasis in mice by decreasing the mesenchymal-related standard CD44 isoform. hnRNPM mRNA levels were shown to correlate with aggressive breast cancer subtypes (basal and ER negative) and increased CD44 standard levels in breast cancer patients [101, 102]. Interestingly, the adhesion molecule CD44 that regulates the aggressive phenotype of breast cancer cells seem to be a common target of AS. KHDRBS1 is a factor involved in a dynamic protein complex variable in size and sensitive to EGF stimulation. EGF activation favors the smaller KHDRBS1 complex, which induces CD44 exon v5 inclusion resulting in enhanced cell migration [103]. Furthermore, SR splice factor TRA2β is overexpressed in invasive breast cancer and induces exon v4 and v5 inclusion [104], suggesting that besides the standard CD44 isoform also the v4 and v5 isoforms are related to increased invasion and metastasis formation.
The third hnRNP, PTBP1 is upregulated in progressively transformed human mammary epithelial cells (HMECs). Knockdown of PTBP1 impairs tumor cell growth, colony formation, in vitro invasiveness of breast cancer cell lines and transformation state of HMECs [76].
Next to the hnRNPs, the splice factor SRSF protein kinase 1 (SRPK1) was shown to be highly expressed in more aggressive basal breast cancer, correlating to less metastasis-free survival and specifically increased number of lung and brain metastases in patients. Stable knockdown of this kinase reduced metastasis to distant organs in a mouse model and inhibited focal adhesion reorganization, which were surprisingly not correlated to a downstream decrease in serine/arginine-rich (SR) splice factor phosphorylation [105]. However, an important role for these SR splice factors – in particular serine and arginine splice factor 1 (SRSF1) – cannot be excluded. SRSF1 is amplified and upregulated in breast cancer and transforms immortal cells when overexpressed [106, 107]. This transformation is mediated by SRSF1 collaboration with transcription factor MYC thereby amplifying eIF4E activation. This potential mechanism is further supported by patient data that reveal a significant co-expression of MYC and SRSF1 in human breast tumors [107]. Furthermore, SRSF1 mutants prevent tumorigenesis and soft agar colony formation by inhibiting activation of the B-Raf-MEK-ERK pathway [106]. Finally, SRSF1 activates EMT and cell migration by induction of DRon, a constitutively active isoform of the Ron tyrosine kinase receptor that is causally connected to EMT [108]. Interestingly, hnRNPA1 has been shown to antagonize SRSF1-mediated EMT activation: through the inhibition of DRon production, hnRNPA1 activates the MET program at distant sites, thereby enhancing metastasis formation [108].
Next to its oncogenic roles in sustaining proliferation, PRPF4B demonstrated an anti-oncogenic role in relation to EMT: loss of PRPF4B resulted in reduced EGFR degradation, increased expression of mesenchymal markers vimentin and ZEB1, detachment from the extracellular matrix and anoikis resistance [109]. Besides EMT, some splice factors have also been linked to the metastatic cascade in general, and their role in a specific step of the metastatic cascade remains to be elucidated. For example, RALY and SNW1 stimulate exon 2 inclusion in PRMT1, promoting breast cancer invasiveness [110] and CDK12 promote alternative last exon splicing of DNA damage genes ATM and DNAJB6 thereby increasing migration and invasiveness of breast cancer cells [111].
Resisting cell death
Resisting physiological stresses
During tumorigenesis or anticancer therapy, cancer cells are exposed to numerous physiological stresses. In normal cells, these cellular stresses will cause apoptosis. However, cancer cells adapt in such environments and rewire their apoptotic program to survive. The SR related splice factor SRSF1 appears to play an important role in this process by promoting AS of crucial regulators of apoptosis BIM γ1 and γ2. Both isoforms lack the BH3 domain necessary to bind the anti-apoptotic Bcl-2 family members. Moreover, SRSF1 stimulates AS of a BIN1 isoform that is not able to bind MYC anymore, thereby losing its tumor suppressor activity leading to decreased levels of apoptosis [107]. This is in agreement with the observed upregulation of SRPK1 that contributes to the cytoplasmic accumulation of RNA-binding motif protein 4 (RBM4). This leads to the production of anti-apoptotic isoforms IR-A and MCL-1L and decreased sensitivity to apoptotic signals in breast cancer cells [112]. Furthermore, depletion of splice factor PHF5A increased apoptotic signaling by promoting expression of short truncated FAS-activated serine/threonine kinase enabling Fas-mediated apoptosis [113], while KHDRBS1 regulates exon 3 inclusion of the anti-apoptotic protein survivin that is higher expressed in advanced breast cancers [114]. The relation between splice factor levels and the ratio between the pro-apoptotic Bcl-Xs and anti-apoptotic Bcl-Xl splice variants is less evident. The activity of the splice factors PTBP1 [115], hnRNPF/H [116] and KHDRBS1 [117] increase the expression of Bcl-Xs, whereas hnRNPK favors Bcl-Xl expression [118]. In the end, the overall altered splicing in transformed cells is likely defined by different key factors which most probably changes during the different stages of cancer progression.
Transformation of normal cells into cancer cells almost invariably results in reduced genome stability. Tumor cells adapt to the load of mutations by activation of the DNA damage response (DDR) which prevents further proliferation and requires extra time to repair the lesions and might even result in apoptosis. Interestingly, splice factors TRA2α and TRA2β are clearly upregulated in breast cancer and those oncogene-like factors limit the amount of DNA damage thereby preventing cell death before entering the G2 phase. Indeed, dual knockdown of these factors results in a decreased expression of full length of CHEK1 (G2 checkpoint protein), leading to increased levels of the DNA damage marker γH2AX and decreased cell viability [77]. Moreover, upon DNA damage DDX54 interacts with pre-mRNAs containing introns with weak acceptor splice sites, leading to lower intron retention and increased survival [119].
Drug resistance
In addition to resisting cell death due to physiological stresses, cancer cells might also gain properties resulting in resistance to cytotoxic agents. For example, overexpression of RNA-binding protein and splice factor RBM17 occurs in many cancer types and is associated with drug resistance to general chemotherapeutic agents such as doxorubicin and vincristine [120]. Next, SRPK1 inhibition increased apoptotic potential and cell killing when combined with gemcitabine and cisplatin treatments through impaired MAPK1, MAPK3 and PI3K pathways [121]. Furthermore, TRA2A overexpression results in AS of RSRC2 and decreased protein expression, contributing to paclitaxel resistance in triple-negative breast cancer patients [122]. Low levels of PRPF4B correlate to patient acquired resistance to microtubule targeting chemotherapeutics, presumably by regulating the spindle assembly checkpoint [69]. Moreover, the subunits of the SF3B complex SF3B1 and SF3B3 are upregulated in ER-α positive cells with acquired tamoxifen and fulvestrant resistance, with SF3B3 overexpression relating to a decrease in overall survival [64]. Opposite to the previous factors, spliceosome component SRSF4 induces splicing events followed by apoptosis in cancer cells when combined with the cytotoxic agent cisplatin. Knockdown of this factor reverses these splicing events and as a result significantly reduces cisplatin induced apoptosis [123]. Interestingly, this confirms that there is a dual role for different splice factors in apoptosis regulation.
Deregulating cellular energetics
Closely related to uncontrolled cell proliferation is the deregulation of cellular energetics, which is necessary to feed cells during growth and division. In aerobic conditions, healthy cells fuel their energy by processing glucose through glycolysis in the cytoplasm and oxidative phosphorylation in the mitochondria. Because the mitochondria consume high amounts of oxygen, energy production in anaerobic conditions relies only on glycolysis. However, cancer cells can reprogram their glucose metabolism using mainly glycolysis even in the presence of oxygen, named the Warburg effect. Splice factors that are suggested to control the Warburg effect are multiplayers PTBP1, hnRNPA1 and hnRNPA2 which not surprisingly are also involved in breast cancer growth and invasion. All three factors favor pyruvate kinase exon 10 inclusion causing higher levels of the M2 isoform (PKM2) compared to M1 (PKM1) resulting in decreased oxygen consumption contributing to the Warburg effect [76, 124]. PTBP1, hnRNPA1, hnRNPA2 levels are regulated by MYC [124]. Interestingly, hnRNPA1 also regulates MYC by regulating AS of the MYC-interacting protein Max, resulting in increased Delta Max levels in glioblastoma. Delta Max but not Max stimulates the expression of glycolytic genes and is required for tumor growth in vivo [125].
PTPBP1, hnRNPA1 and hnRNPA2 are currently the only splice factors that have been related to the Warburg effect. However, since hypoxia is driving AS in breast cancer cells [126] and other splicing events of key metabolic genes such as PFKFB4 – which is responsible for retaining fructose-2,6-biphosphate, a key regulator of glycolysis – are altered in tumor tissue [127], we hypothesize that more spliceosome components are involved in cancerous cell metabolism.
Angiogenesis
New blood vessel formation or angiogenesis is critical for tumor progression since it i) provides the tumor with nutrients needed for growth and ii) brings the tumor cells in close proximity to blood circulation facilitating invasion and metastasis formation. Vascular endothelial growth factor (VEGF) is a key component in both physiological and pathological angiogenesis. Breast cancer patients with elevated VEGF levels have a higher risk to develop metastases or death compared to other patients [128] and therefore inhibition of this factor is a promising therapeutic strategy [129]. VEGF can be alternatively spliced by using a distal splice site selection in exon 8, resulting in the anti-angiogenic isoform VEGFxxxb bearing a different C-terminus [130–132]. Splice site selection is dependent on SRPK1/2 phosphorylation of the RNA-binding splice factor SRSF1. Furthermore, SRPK1 regulates VEGF splicing and activity in prostate cancer: SRPK1 knockdown results in up-regulation of the anti-angiogenic isoform VEGFxxxb and decreased angiogenesis in a xenograft model [133]. Accordingly, mutations in the tumor suppressor gene WT1 lead to increased SRPK1 levels and hyper phosphorylated SRSF1, reducing anti-angiogenic VEGFxxxb levels [134]. Treatment with SRPK1/2 inhibitors results in reduced angiogenesis, suggesting that AS regulation might provide a promising strategy to inhibit angiogenesis through depletion of pro-angiogenic components such as VEGF [135]. This is confirmed by the prediction that targeting of specific VEGF isoforms might be the best strategy to reduce free VEGF in tumors [136]. Another example is the splicing regulator Nova2 that is involved in vascular lumen formation, an essential step in angiogenesis. Nova2 targets exons implicated in the partitioning-defective (Par) complex and its regulators including Par3, Arhgef6 and Rapgap1. The Par complex interacts with tight junctions and cadherins and is important for lumen formation by endothelial cells, thereby being essential for cellular and tissue homeostasis [137–140]. Nova2 knockdown interferes with vascular lumen development in vivo and impairs endothelial cell polarity [137]. Although Nova2 has not been linked to tumorigenesis yet, it might be a potential target to inhibit angiogenesis.
Splice factors in the other hallmarks
In the last decade, splice factors levels have extensively been related to five hallmarks (Figure 1), suggesting a strong relation between splicing regulation and cancer development and progression. However, the remaining five hallmarks (genomic instability, tumor promoting inflammation, enabling replicative immortality, avoiding immune destruction and evading growth suppression) are still unaddressed. Here, we used RNA sequencing data from primary breast tumors from The Cancer Genome Atlas to investigate the potential role of splice factors in these hallmarks. For all of these hallmarks, we selected representative genes based on literature (Figure 2A, Supplementary Table 4) and calculated their log2 fold change (FC) between normal and tumor tissue. Key representative genes were selected based on their differential expression between normal and tumor tissue. For example, genome instability is characterized by a loss of repair mechanisms [21]. Comparing normal and primary tumor expression levels for genes involved in repair mechanisms, we identified four genes involved in nucleotide excision repair to be significantly downregulated in tumor tissue (Figure 2B). Next, we selected ten patients that were not affected (control patients) and ten patients that were heavily affected (hallmark patients) by the hallmark of interest (Figure 2C for genome instability). Finally, splice factor expression levels were compared between control and hallmark patients using a student’s t-test and after correction for multiple testing, splice factors significantly related to the specific hallmark could be identified (Supplementary Table 3). Interestingly, we could detect splice factors related to all remaining hallmarks, except for tumor promoting inflammation. Some of these splicing factors have already been associated with other hallmarks. EFTUD2 expression levels are linked to markers of replicative immortality while it was previously shown to interact with ER and affect breast cancer proliferation [67]. Loss of WDR77 resulted in loss of proliferative genes and expression levels are linked to genome instability [79]. Interestingly, we also identified splice factors that have not been linked to other hallmarks in breast cancer before, such as CRNKL1, RALY and JUP. Future functional studies can use our analysis as a starting point to unravel the causal relationship between splice factor levels and these hallmarks of cancer.
Future perspectives and conclusion
As splice factors are frequently overexpressed in cancer compared to normal tissue, but also in highly invasive compared to less invasive tumors, splice factors might be a new promising therapeutic avenue in preventing breast cancer metastasis thereby lowering mortality in women. A possible drawback of inhibiting these factors could be the generation of adverse side effects when considering their critical function in intron removal in normal tissue. However, recent studies demonstrate that splice factor inhibition might certainly be applicable to the clinical situation. Both tumor-bearing mice and pre-diagnostic human samples of ER positive and triple negative breast cancer demonstrate autoantibody reactivity against spliceosomal proteins suggesting that at least partial inhibition for some of these factors should be possible [141, 142]. Furthermore, screens with natural products with antitumor characteristics resulted in the identification of spliceosome targeting drugs exhibiting cytostatic effects in multiple tumor cell lines by causing cell cycle arrest in G1 and G2/M phase [143–145]. Antitumor activities were confirmed in animal models and remarkably, general cytotoxicity was not observed. Moreover, these potential drugs seem to be more effective in cancer cells with some of them even targeting multidrug-resistant cells [146–148]. Recently, the natural compound resveratrol demonstrated to inhibit the oncogenic splice factor hnRNPA1 by inducing tumor suppressive miRNAs miR-424 and miR-503 via p53 thereby controlling tumor growth [149]. Also treatment with the CLK inhibitor T-025 reduced SR protein phosphorylation, which resulted in general effects on exon skipping and reduced cancer cell growth in vitro and in vivo [150]. Of note, most of the drugs described in the literature target the SF3B complex, a five-polypeptide subcomplex of the spliceosomal U2 snRNP. Small molecules affecting spliceosomal function by inhibiting different splicing complexes are known [151], but their potential role in combatting (breast) cancer has to be investigated. Other interesting targets would be the SR proteins or its upstream kinases like SRPK1 that has been demonstrated to be critical in multiple steps of breast cancer progression and for which inhibitors have been developed [105, 133, 152, 153]. Finally, pharmacological inhibition of the spliceosome would be a promising therapeutic strategy for MYC-addicted breast cancer tumors.
Next to the use of small molecules, the potential of using splice-switching antisense oligonucleotides (SSOs) to modulate AS is of great therapeutic interest. SSOs are single-stranded oligonucleotides consisting of 20-30 nucleotides that bind to pre-mRNA and sterically prevent splicing factor binding, resulting in splice site switching. In contrast to normal antisense oligonucleotides (ASOs), SSOs are chemically modified to prevent breakdown of the targeted transcript to specifically target splicing without affecting total transcript levels [154]. Furthermore, SSOs are easy to synthesize and deliver, are relatively stable and can enter many different cell types [155, 156]. Although the use of SSOs was initially mainly studied in neuromuscular diseases such as Duchenne muscular dystrophy and spinal muscular atrophy [157], the therapeutic potential of SSOs as anti-cancer therapy is currently widely exploited [158]. For example, treatment with SSO stimulating exon 11 skipping caused better response to PARP inhibitors by inducing DNA double strand breaks [159]. Additionally, treatment with SSOs inducing exon 15 HER2 skipping caused downregulation of full length HER2 resulting in decreased downstream signaling, reduced cell proliferation and induction of apoptosis [160]. As a next step, clinical studies have to prove the use of these SSOs as anti-cancer therapy in breast cancer patients.
Since 94% of human genes are alternatively spliced [161, 162], splicing can be a very powerful new layer of oncogenic control. As discussed in detail above, splice factors demonstrated to play major roles in different hallmarks of cancer during tumorigenesis (Figure 1 and Supplementary Table 2) and some of these factors were already described as new oncogenes or tumor suppressors. Since the role of many splice factors is not limited to a specific step or cancer hallmark they might provide a new approach to combat (breast) cancer progression. However, there are still many AS events that have been associated with cancer progression which cannot be attributed to specific splice factors yet. The introduction of the RNAi libraries and more recently the CRISPR Cas9 technology together with the development of high-throughput screening technologies [105, 163–165] would allow systematic evaluation of spliceosomal components in multiple aspects of breast cancer progression, such as proliferation and migration. Future studies should apply these technologies to uncover the complete signaling landscape of splice factors in breast cancer progression that then can be used to develop specific splice factor inhibitors preventing metastasis formation and patient deaths.
Abbreviations
AR: androgen receptor; AS alternative splicing; DDR: DNA damage response; EGFR: epidermal growth factor receptor; EMT: epithelial-to-mesenchymal transition; ER: estrogen receptor; FC: fold change; HER2: human epidermal growth factor receptor 2; HMEC: human mammary epithelial cell; Par: partitioning-defective; PK: pyruvate kinase; RBM4: RNA-binding motif protein 4; snRNP: small nuclear ribonucleoprotein; SR: serine/arginine-rich; SRPK1: splicing factor SRSF protein kinase 1; SRSF1: serine and arginine splicing factor 1; VEGF: vascular endothelial growth factor.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
This work was supported by the ERC Advanced grant Triple-BC (grant no. 322737) and the Dutch Cancer Society project (grant nr 2011-5124).
References
1. Black DL. Protein diversity from alternative splicing: a challenge for bioinformatics and post-genome biology. Cell. 2000; 103:367–70. https://doi.org/10.1016/S0092-8674(00)00128-8. [PubMed].
2. Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003; 72:291–336. https://doi.org/10.1146/annurev.biochem.72.121801.161720. [PubMed].
3. Smith CW, Valcárcel J. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem Sci. 2000; 25:381–88. https://doi.org/10.1016/S0968-0004(00)01604-2. [PubMed].
4. Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, Spirohn K, Begg BE, Duran-Frigola M, et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell. 2016; 164:805–17. https://doi.org/10.1016/j.cell.2016.01.029. [PubMed].
5. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev. 2010; 24:2343–64. https://doi.org/10.1101/gad.1973010. [PubMed].
6. Ghigna C, Valacca C, Biamonti G. Alternative splicing and tumor progression. Curr Genomics. 2008; 9:556–70. https://doi.org/10.2174/138920208786847971. [PubMed].
7. Fackenthal JD, Godley LA. Aberrant RNA splicing and its functional consequences in cancer cells. Dis Model Mech. 2008; 1:37–42. https://doi.org/10.1242/dmm.000331. [PubMed].
8. Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014; 33:5311–18. https://doi.org/10.1038/onc.2013.533. [PubMed].
9. Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012; 45:567–80. https://doi.org/10.1016/j.molcel.2011.12.034. [PubMed].
10. Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, Lührmann R. Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol Cell Biol. 2011; 31:2667–82. https://doi.org/10.1128/MCB.05266-11. [PubMed].
11. Zhu J, Mayeda A, Krainer AR. Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell. 2001; 8:1351–61. https://doi.org/10.1016/S1097-2765(01)00409-9. [PubMed].
12. Tange TO, Damgaard CK, Guth S, Valcárcel J, Kjems J. The hnRNP A1 protein regulates HIV-1 tat splicing via a novel intron silencer element. EMBO J. 2001; 20:5748–58. https://doi.org/10.1093/emboj/20.20.5748. [PubMed].
13. House AE, Lynch KW. An exonic splicing silencer represses spliceosome assembly after ATP-dependent exon recognition. Nat Struct Mol Biol. 2006; 13:937–44. https://doi.org/10.1038/nsmb1149. [PubMed].
14. Ge H, Manley JL. A protein factor, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell. 1990; 62:25–34. https://doi.org/10.1016/0092-8674(90)90236-8. [PubMed].
15. Krainer AR, Conway GC, Kozak D. Purification and characterization of pre-mRNA splicing factor SF2 from HeLa cells. Genes Dev. 1990; 4:1158–71. https://doi.org/10.1101/gad.4.7.1158. [PubMed].
16. Fu XD, Maniatis T. The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3′ splice site. Proc Natl Acad Sci U S A. 1992; 89:1725–29. https://doi.org/10.1073/pnas.89.5.1725. [PubMed].
17. Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003; 12:5–14. https://doi.org/10.1016/S1097-2765(03)00270-3. [PubMed].
18. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009; 136:701–18. https://doi.org/10.1016/j.cell.2009.02.009. [PubMed].
19. American Cancer Society. Cancer Facts & Figures 2018. American Cancer Society. 2018; 28–43. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2018/cancer-facts-and-figures-2018.pdf.
20. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57–70. https://doi.org/10.1016/S0092-8674(00)81683-9. [PubMed].
21. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013. [PubMed].
22. Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003; 17:419–37. https://doi.org/10.1101/gad.1048803. [PubMed].
23. Stoilov P, Meshorer E, Gencheva M, Glick D, Soreq H, Stamm S. Defects in pre-mRNA processing as causes of and predisposition to diseases. DNA Cell Biol. 2002; 21:803–18. https://doi.org/10.1089/104454902320908450. [PubMed].
24. Wen J, Toomer KH, Chen Z, Cai X. Genome-wide analysis of alternative transcripts in human breast cancer. Breast Cancer Res Treat. 2015; 151:295–307. https://doi.org/10.1007/s10549-015-3395-2. [PubMed].
25. Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, Lucier JF, Thibault P, Rancourt C, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009; 16:670–76. https://doi.org/10.1038/nsmb.1608. [PubMed].
26. Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011; 7:e1002218. https://doi.org/10.1371/journal.pgen.1002218. [PubMed].
27. Valcárcel J, Green MR. The SR protein family: pleiotropic functions in pre-mRNA splicing. Trends Biochem Sci. 1996; 21:296–301. https://doi.org/10.1016/S0968-0004(96)10039-6. [PubMed].
28. Hagiwara M. Alternative splicing: a new drug target of the post-genome era. Biochim Biophys Acta. 2005; 1754:324–31. https://doi.org/10.1016/j.bbapap.2005.09.010. [PubMed].
29. Tenenbaum SA, Aguirre-Ghiso J. Dephosphorylation shows SR proteins the way out. Mol Cell. 2005; 20:499–501. https://doi.org/10.1016/j.molcel.2005.11.005. [PubMed].
30. Chen Y, Huang Q, Liu W, Zhu Q, Cui CP, Xu L, Guo X, Wang P, Liu J, Dong G, Wei W, Liu CH, Feng Z, et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun. 2018; 9:2464. https://doi.org/10.1038/s41467-018-04815-3. [PubMed].
31. Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci U S A. 2004; 101:9666–70. https://doi.org/10.1073/pnas.0403533101. [PubMed].
32. Sanford JR, Ellis JD, Cazalla D, Cáceres JF. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc Natl Acad Sci U S A. 2005; 102:15042–47. https://doi.org/10.1073/pnas.0507827102. [PubMed].
33. Cáceres JF, Screaton GR, Krainer AR. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998; 12:55–66. https://doi.org/10.1101/gad.12.1.55. [PubMed].
34. Huang Y, Gattoni R, Stévenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003; 11:837–43. https://doi.org/10.1016/S1097-2765(03)00089-3. [PubMed].
35. Buckley PT, Khaladkar M, Kim J, Eberwine J. Cytoplasmic intron retention, function, splicing, and the sentinel RNA hypothesis. Wiley Interdiscip Rev RNA. 2014; 5:223–30. https://doi.org/10.1002/wrna.1203. [PubMed].
36. Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999; 9:171–74. https://doi.org/10.1016/S0959-437X(99)80026-4. [PubMed].
37. Gonzalez I, Munita R, Agirre E, Dittmer TA, Gysling K, Misteli T, Luco RF. A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat Struct Mol Biol. 2015; 22:370–76. https://doi.org/10.1038/nsmb.3005. [PubMed].
38. Schor IE, Llères D, Risso GJ, Pawellek A, Ule J, Lamond AI, Kornblihtt AR. Perturbation of chromatin structure globally affects localization and recruitment of splicing factors. PLoS One. 2012; 7:e48084. https://doi.org/10.1371/journal.pone.0048084. [PubMed].
39. Sharma A, Nguyen H, Geng C, Hinman MN, Luo G, Lou H. Calcium-mediated histone modifications regulate alternative splicing in cardiomyocytes. Proc Natl Acad Sci U S A. 2014; 111:E4920–28. https://doi.org/10.1073/pnas.1408964111. [PubMed].
40. Sperling R. Small non-coding RNA within the endogenous spliceosome and alternative splicing regulation. Biochim Biophys Acta Gene Regul Mech. 2019 Jul 16. https://doi.org/10.1016/j.bbagrm.2019.07.007. [Epub ahead of print]. [PubMed].
41. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–69. https://doi.org/10.1038/nature10496. [PubMed].
42. Yoshida K, Ogawa S. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA. 2014; 5:445–59. https://doi.org/10.1002/wrna.1222. [PubMed].
43. Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015; 25:14–26. https://doi.org/10.1101/gr.181016.114. [PubMed].
44. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012; 486:353–60. https://doi.org/10.1038/nature11143. [PubMed].
45. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D, et al, and Oslo Breast Cancer Consortium (OSBREAC). The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012; 486:400–04. https://doi.org/10.1038/nature11017. [PubMed].
46. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534:47–54. https://doi.org/10.1038/nature17676. [PubMed]. Erratum in: Author Correction: Landscape of somatic mutations in 560 breast cancer whole-genome sequences. [Nature. 2019]. https://doi.org/10.1038/s41586-019-0883-2. [PubMed].
47. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3’ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015; 11:e1004105. https://doi.org/10.1371/journal.pcbi.1004105. [PubMed].
48. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, Dutertre M, Stern MH. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016; 7:10615. https://doi.org/10.1038/ncomms10615. [PubMed].
49. Shiraishi Y, Kataoka K, Chiba K, Okada A, Kogure Y, Tanaka H, Ogawa S, Miyano S. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 2018; 28:1111–25. https://doi.org/10.1101/gr.231951.117. [PubMed].
50. Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015; 84:165–98. https://doi.org/10.1146/annurev-biochem-060614-034242. [PubMed].
51. Wang Y, Xiao X, Zhang J, Choudhury R, Robertson A, Li K, Ma M, Burge CB, Wang Z. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nat Struct Mol Biol. 2013; 20:36–45. https://doi.org/10.1038/nsmb.2459. [PubMed].
52. Mazoyer S, Puget N, Perrin-Vidoz L, Lynch HT, Serova-Sinilnikova OM, Lenoir GM. A BRCA1 nonsense mutation causes exon skipping. Am J Hum Genet. 1998; 62:713–15. https://doi.org/10.1086/301768. [PubMed].
53. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, Cho H, Kim MK, Zebari AS, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015; 27:617–30. https://doi.org/10.1016/j.ccell.2015.04.006. [PubMed].
54. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13:140–56. https://doi.org/10.1038/nrd4204. [PubMed].
55. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007; 26:3279–90. https://doi.org/10.1038/sj.onc.1210421. [PubMed].
56. Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014; 40:862–71. https://doi.org/10.1016/j.ctrv.2014.03.004. [PubMed].
57. Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, Yue W. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002; 80:239–56. https://doi.org/10.1016/s0960-0760(01)00189-3. [PubMed].
58. Pace P, Taylor J, Suntharalingam S, Coombes RC, Ali S. Human estrogen receptor beta binds DNA in a manner similar to and dimerizes with estrogen receptor alpha. J Biol Chem. 1997; 272:25832–38. https://doi.org/10.1074/jbc.272.41.25832. [PubMed].
59. Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell. 2011; 145:622–34. https://doi.org/10.1016/j.cell.2011.03.042. [PubMed].
60. Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003; 144:4562–74. https://doi.org/10.1210/en.2003-0567. [PubMed].
61. Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, Matsuura M, Ito Y, Akiyama F, Sakamoto G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008; 26:3727–34. https://doi.org/10.1200/JCO.2007.14.2968. [PubMed].
62. Guo L, Meng J, Yilamu D, Jakulin A, Fu M, Wang B, Abulajiang G. Significance of ERβ expression in different molecular subtypes of breast cancer. Diagn Pathol. 2014; 9:20. https://doi.org/10.1186/1746-1596-9-20. [PubMed].
63. Chantzi NI, Tiniakos DG, Palaiologou M, Goutas N, Filippidis T, Vassilaros SD, Dhimolea E, Mitsiou DJ, Alexis MN. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J Cancer Res Clin Oncol. 2013; 139:1489–98. https://doi.org/10.1007/s00432-013-1467-4. [PubMed].
64. Gökmen-Polar Y, Neelamraju Y, Goswami CP, Gu X, Nallamothu G, Janga SC, Badve S. Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor-positive breast cancer. Mod Pathol. 2015; 28:677–85. https://doi.org/10.1038/modpathol.2014.146. [PubMed].
65. Lahsaee S, Corkery DP, Anthes LE, Holly A, Dellaire G. Estrogen receptor alpha (ESR1)-signaling regulates the expression of the taxane-response biomarker PRP4K. Exp Cell Res. 2016; 340:125–31. https://doi.org/10.1016/j.yexcr.2015.12.013. [PubMed].
66. Ohe K, Miyajima S, Tanaka T, Hamaguchi Y, Harada Y, Horita Y, Beppu Y, Ito F, Yamasaki T, Terai H, Mori M, Murata Y, Tanabe M, et al. HMGA1a Induces Alternative Splicing of the Estrogen Receptor-αlpha Gene by Trapping U1 snRNP to an Upstream Pseudo-5’ Splice Site. Front Mol Biosci. 2018; 5:52. https://doi.org/10.3389/fmolb.2018.00052. [PubMed].
67. Nassa G, Tarallo R, Guzzi PH, Ferraro L, Cirillo F, Ravo M, Nola E, Baumann M, Nyman TA, Cannataro M, Ambrosino C, Weisz A. Comparative analysis of nuclear estrogen receptor alpha and beta interactomes in breast cancer cells. Mol Biosyst. 2011; 7:667–76. https://doi.org/10.1039/C0MB00145G. [PubMed].
68. Dago DN, Scafoglio C, Rinaldi A, Memoli D, Giurato G, Nassa G, Ravo M, Rizzo F, Tarallo R, Weisz A. Estrogen receptor beta impacts hormone-induced alternative mRNA splicing in breast cancer cells. BMC Genomics. 2015; 16:367. https://doi.org/10.1186/s12864-015-1541-1. [PubMed].
69. Corkery DP, Le Page C, Meunier L, Provencher D, Mes-Masson AM, Dellaire G. PRP4K is a HER2-regulated modifier of taxane sensitivity. Cell Cycle. 2015; 14:1059–69. https://doi.org/10.1080/15384101.2015.1007775. [PubMed].
70. Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Ménard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006; 13:221–32. https://doi.org/10.1677/erc.1.01047. [PubMed].
71. Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, Lesko SM, Brogi E, Jones FE. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009; 8:2152–62. https://doi.org/10.1158/1535-7163.MCT-09-0295. [PubMed].
72. Azios NG, Romero FJ, Denton MC, Doherty JK, Clinton GM. Expression of herstatin, an autoinhibitor of HER-2/neu, inhibits transactivation of HER-3 by HER-2 and blocks EGF activation of the EGF receptor. Oncogene. 2001; 20:5199–209. https://doi.org/10.1038/sj.onc.1204555. [PubMed].
73. Hu P, Zhou T, Qian L, Wang J, Shi M, Yu M, Yang Y, Zhang X, Shen B, Guo N. Sequestering ErbB2 in endoplasmic reticulum by its autoinhibitor from translocation to cell surface: an autoinhibition mechanism of ErbB2 expression. Biochem Biophys Res Commun. 2006; 342:19–27. https://doi.org/10.1016/j.bbrc.2006.01.115. [PubMed].
74. Hu P, Feng J, Zhou T, Wang J, Jing B, Yu M, Hu M, Zhang X, Shen B, Guo N. In vivo identification of the interaction site of ErbB2 extracellular domain with its autoinhibitor. J Cell Physiol. 2005; 205:335–43. https://doi.org/10.1002/jcp.20409. [PubMed].
75. Todorović-Raković N, Nešković-Konstantinović Z, Nikolić-Vukosavljević D. Cross-talk between ER and HER2 in breast carcinoma. Arch Oncol. 2006; 14:146–50. https://doi.org/10.2298/AOO0604146T.
76. He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis. 2014; 3:e84. https://doi.org/10.1038/oncsis.2013.47. [PubMed].
77. Best A, Dagliesh C, Ehrmann I, Kheirollahi-Kouhestani M, Tyson-Capper A, Elliott DJ. Expression of Tra2 β in Cancer Cells as a Potential Contributory Factor to Neoplasia and Metastasis. Int J Cell Biol. 2013; 2013:843781. https://doi.org/10.1155/2013/843781. [PubMed].
78. Chang YL, Hsu YJ, Chen Y, Wang YW, Huang SM. Theophylline exhibits anti-cancer activity via suppressing SRSF3 in cervical and breast cancer cell lines. Oncotarget. 2017; 8:101461–74. https://doi.org/10.18632/oncotarget.21464. [PubMed].
79. Rengasamy M, Zhang F, Vashisht A, Song WM, Aguilo F, Sun Y, Li S, Zhang W, Zhang B, Wohlschlegel JA, Walsh MJ. The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 2017; 45:11106–20. https://doi.org/10.1093/nar/gkx727. [PubMed].
80. Wu Y, Zhao W, Liu Y, Tan X, Li X, Zou Q, Xiao Z, Xu H, Wang Y, Yang X. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 2018; 37:1–19. https://doi.org/10.15252/embj.201899017. [PubMed].
81. Cannizzaro E, Bannister AJ, Han N, Alendar A, Kouzarides T. DDX3X RNA helicase affects breast cancer cell cycle progression by regulating expression of KLF4. FEBS Lett. 2018; 592:2308–22. https://doi.org/10.1002/1873-3468.13106. [PubMed].
82. Hu Y, Sun Z, Deng J, Hu B, Yan W, Wei H, Jiang J. Splicing factor hnRNPA2B1 contributes to tumorigenic potential of breast cancer cells through STAT3 and ERK1/2 signaling pathway. Tumour Biol. 2017; 39:1010428317694318. https://doi.org/10.1177/1010428317694318. [PubMed].
83. Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, Karlin KL, Dominguez-Vidaña R, Hartman JD, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015; 525:384–88. https://doi.org/10.1038/nature14985. [PubMed].
84. Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabò A, Amati B, Wee KB, Guccione E. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015; 523:96–100. https://doi.org/10.1038/nature14351. [PubMed].
85. Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, Allain FH, Krainer AR. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell. 2015; 60:105–17. https://doi.org/10.1016/j.molcel.2015.09.005. [PubMed].
86. Gao Y, Koide K. Chemical perturbation of Mcl-1 pre-mRNA splicing to induce apoptosis in cancer cells. ACS Chem Biol. 2013; 8:895–900. https://doi.org/10.1021/cb300602j. [PubMed].
87. Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010; 70:5649–69. https://doi.org/10.1158/0008-5472.CAN-10-1040. [PubMed].
88. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003; 3:453–58. https://doi.org/10.1038/nrc1098. [PubMed].
89. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014; 7:re8. https://doi.org/10.1126/scisignal.2005189. [PubMed].
90. Li J, Choi PS, Chaffer CL, Labella K, Hwang JH, Giacomelli AO, Kim JW, Ilic N, Doench JG, Ly SH, Dai C, Hagel K, Hong AL, et al. An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. Elife. 2018; 7:1–28. https://doi.org/10.7554/eLife.37184. [PubMed].
91. Fici P, Gallerani G, Morel AP, Mercatali L, Ibrahim T, Scarpi E, Amadori D, Puisieux A, Rigaud M, Fabbri F. Splicing factor ratio as an index of epithelial-mesenchymal transition and tumor aggressiveness in breast cancer. Oncotarget. 2017; 8:2423–36. https://doi.org/10.18632/oncotarget.13682. [PubMed].
92. Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009; 33:591–601. https://doi.org/10.1016/j.molcel.2009.01.025. [PubMed].
93. Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Adv Exp Med Biol. 2007; 623:123–47. https://doi.org/10.1007/978-0-387-77374-2_8. [PubMed].
94. Cammas A, Lacroix-Triki M, Pierredon S, Le Bras M, Iacovoni JS, Teulade-Fichou MP, Favre G, Roché H, Filleron T, Millevoi S, Vagner S. hnRNP A1-mediated translational regulation of the G quadruplex-containing RON receptor tyrosine kinase mRNA linked to tumor progression. Oncotarget. 2016; 7:16793–805. https://doi.org/10.18632/oncotarget.7589. [PubMed].
95. Pelisch F, Khauv D, Risso G, Stallings-Mann M, Blaustein M, Quadrana L, Radisky DC, Srebrow A. Involvement of hnRNP A1 in the matrix metalloprotease-3-dependent regulation of Rac1 pre-mRNA splicing. J Cell Biochem. 2012; 113:2319–29. https://doi.org/10.1002/jcb.24103. [PubMed].
96. Fiegen D, Haeusler LC, Blumenstein L, Herbrand U, Dvorsky R, Vetter IR, Ahmadian MR. Alternative splicing of Rac1 generates Rac1b, a self-activating GTPase. J Biol Chem. 2004; 279:4743–49. https://doi.org/10.1074/jbc.M310281200. [PubMed].
97. Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem. 2003; 278:50442–48. https://doi.org/10.1074/jbc.M308215200. [PubMed].
98. Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, Harbeck N, Schmitt M, Lengyel E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000; 19:3013–20. https://doi.org/10.1038/sj.onc.1203621. [PubMed].
99. Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005; 436:123–27. https://doi.org/10.1038/nature03688. [PubMed].
100. Harvey SE, Xu Y, Lin X, Gao XD, Qiu Y, Ahn J, Xiao X, Cheng C. Coregulation of alternative splicing by hnRNPM and ESRP1 during EMT. RNA. 2018; 24:1326–38. https://doi.org/10.1261/rna.066712.118. [PubMed].
101. Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, Siziopikou KP, Peng J, Xiao X, Cheng C. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014; 28:1191–203. https://doi.org/10.1101/gad.241968.114. [PubMed].
102. Sun H, Liu T, Zhu D, Dong X, Liu F, Liang X, Chen C, Shao B, Wang M, Wang Y. HnRNPM and CD44s expression affects tumor aggressiveness and predicts poor prognosis in breast cancer with axillary lymph node metastases. Genes Chromosomes Cancer. 2017; 56:598–607. https://doi.org/10.1002/gcc.22463. [PubMed].
103. Huot MÉ, Vogel G, Richard S. Identification of a Sam68 ribonucleoprotein complex regulated by epidermal growth factor. J Biol Chem. 2009; 284:31903–13. https://doi.org/10.1074/jbc.M109.018465. [PubMed].
104. Watermann DO, Tang Y, Zur Hausen A, Jäger M, Stamm S, Stickeler E. Splicing factor Tra2-beta1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006; 66:4774–80. https://doi.org/10.1158/0008-5472.CAN-04-3294. [PubMed].
105. van Roosmalen W, Le Dévédec SE, Golani O, Smid M, Pulyakhina I, Timmermans AM, Look MP, Zi D, Pont C, de Graauw M, Naffar-Abu-Amara S, Kirsanova C, Rustici G, et al. Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J Clin Invest. 2015; 125:1648–64. https://doi.org/10.1172/JCI74440. [PubMed].
106. Shimoni-Sebag A, Lebenthal-Loinger I, Zender L, Karni R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1-MAPK-ERK activation and cellular transformation. Carcinogenesis. 2013; 34:2498–504. https://doi.org/10.1093/carcin/bgt247. [PubMed].
107. Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012; 19:220–28. https://doi.org/10.1038/nsmb.2207. [PubMed].
108. Bonomi S, di Matteo A, Buratti E, Cabianca DS, Baralle FE, Ghigna C, Biamonti G. HnRNP A1 controls a splicing regulatory circuit promoting mesenchymal-to-epithelial transition. Nucleic Acids Res. 2013; 41:8665–79. https://doi.org/10.1093/nar/gkt579. [PubMed].
109. Corkery DP, Clarke LE, Gebremeskel S, Salsman J, Pinder J, Le Page C, Meunier L, Xu Z, Mes-Masson AM, Berman JN, Johnston B, Dellaire G. Loss of PRP4K drives anoikis resistance in part by dysregulation of epidermal growth factor receptor endosomal trafficking. Oncogene. 2018; 37:174–84. https://doi.org/10.1038/onc.2017.318. [PubMed].
110. Bondy-Chorney E, Baldwin RM, Didillon A, Chabot B, Jasmin BJ, Côté J. RNA binding protein RALY promotes Protein Arginine Methyltransferase 1 alternatively spliced isoform v2 relative expression and metastatic potential in breast cancer cells. Int J Biochem Cell Biol. 2017; 91:124–35. https://doi.org/10.1016/j.biocel.2017.07.008. [PubMed].
111. Tien JF, Mazloomian A, Cheng SG, Hughes CS, Chow CC, Canapi LT, Oloumi A, Trigo-Gonzalez G, Bashashati A, Xu J, Chang VC, Shah SP, Aparicio S, Morin GB. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017; 45:6698–716. https://doi.org/10.1093/nar/gkx187. [PubMed].
112. Lin JC, Lin CY, Tarn WY, Li FY. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. RNA. 2014; 20:1621–31. https://doi.org/10.1261/rna.045583.114. [PubMed].
113. Zheng YZ, Xue MZ, Shen HJ, Li XG, Ma D, Gong Y, Liu YR, Qiao F, Xie HY, Lian B, Sun WL, Zhao HY, Yao L, et al. PHF5A Epigenetically Inhibits Apoptosis to Promote Breast Cancer Progression. Cancer Res. 2018; 78:3190–206. https://doi.org/10.1158/0008-5472.CAN-17-3514. [PubMed].
114. Gaytan-Cervantes J, Gonzalez-Torres C, Maldonado V, Zampedri C, Ceballos-Cancino G, Melendez-Zajgla J. Protein Sam68 regulates the alternative splicing of survivin DEx3. J Biol Chem. 2017; 292:13745–13757. https://doi.org/10.1074/jbc.M117.800318. [PubMed].
115. Bielli P, Bordi M, Di Biasio V, Sette C. Regulation of BCL-X splicing reveals a role for the polypyrimidine tract binding protein (PTBP1/hnRNP I) in alternative 5′ splice site selection. Nucleic Acids Res. 2014; 42:12070–81. https://doi.org/10.1093/nar/gku922. [PubMed].
116. Koumbadinga GA, Mahmood N, Lei L, Kan Y, Cao W, Lobo VG, Yao X, Zhang S, Xie J. Increased stability of heterogeneous ribonucleoproteins by a deacetylase inhibitor. Biochim Biophys Acta. 2015; 1849:1095–103. https://doi.org/10.1016/j.bbagrm.2015.05.001. [PubMed].
117. Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol. 2007; 176:929–39. https://doi.org/10.1083/jcb.200701005. [PubMed].
118. Revil T, Pelletier J, Toutant J, Cloutier A, Chabot B. Heterogeneous nuclear ribonucleoprotein K represses the production of pro-apoptotic Bcl-xS splice isoform. J Biol Chem. 2009; 284:21458–67. https://doi.org/10.1074/jbc.M109.019711. [PubMed].
119. Milek M, Imami K, Mukherjee N, Bortoli F, Zinnall U, Hazapis O, Trahan C, Oeffinger M, Heyd F, Ohler U, Selbach M, Landthaler M. DDX54 regulates transcriptome dynamics during DNA damage response. Genome Res. 2017; 27:1344–59. https://doi.org/10.1101/gr.218438.116. [PubMed].
120. Sampath J, Long PR, Shepard RL, Xia X, Devanarayan V, Sandusky GE, Perry WL 3rd, Dantzig AH, Williamson M, Rolfe M, Moore RE. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am J Pathol. 2003; 163:1781–90. https://doi.org/10.1016/S0002-9440(10)63538-9. [PubMed].
121. Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007; 67:2072–80. https://doi.org/10.1158/0008-5472.CAN-06-2969. [PubMed].
122. Liu T, Sun H, Zhu D, Dong X, Liu F, Liang X, Chen C, Shao B, Wang M, Wang Y, Sun B. TRA2A Promoted Paclitaxel Resistance and Tumor Progression in Triple-Negative Breast Cancers via Regulating Alternative Splicing. Mol Cancer Ther. 2017; 16:1377–88. https://doi.org/10.1158/1535-7163.MCT-17-0026. [PubMed].
123. Gabriel M, Delforge Y, Deward A, Habraken Y, Hennuy B, Piette J, Klinck R, Chabot B, Colige A, Lambert C. Role of the splicing factor SRSF4 in cisplatin-induced modifications of pre-mRNA splicing and apoptosis. BMC Cancer. 2015; 15:227. https://doi.org/10.1186/s12885-015-1259-0. [PubMed].
124. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010; 463:364–68. https://doi.org/10.1038/nature08697. [PubMed].
125. Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, Zhu S, Gu Y, Villa GR, Akhavan D, Nathanson D, Gini B, Mareninov S, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013; 17:1000–08. https://doi.org/10.1016/j.cmet.2013.04.013. [PubMed].
126. Han J, Li J, Ho JC, Chia GS, Kato H, Jha S, Yang H, Poellinger L, Lee KL. Hypoxia is a Key Driver of Alternative Splicing in Human Breast Cancer Cells. Sci Rep. 2017; 7:4108. https://doi.org/10.1038/s41598-017-04333-0. [PubMed].
127. Minchenko OH, Ogura T, Opentanova IL, Minchenko DO, Esumi H. Splice isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-4: expression and hypoxic regulation. Mol Cell Biochem. 2005; 280:227–34. https://doi.org/10.1007/s11010-005-8009-6. [PubMed].
128. Gasparini G. Prognostic value of vascular endothelial growth factor in breast cancer. Oncologist. 2000; 5:37–44. https://doi.org/10.1634/theoncologist.5-suppl_1-37. [PubMed].
129. Moreira IS, Fernandes PA, Ramos MJ. Vascular endothelial growth factor (VEGF) inhibition--a critical review. Anticancer Agents Med Chem. 2007; 7:223–45. https://doi.org/10.2174/187152007780058687. [PubMed].
130. Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008; 8:880–87. https://doi.org/10.1038/nrc2505. [PubMed].
131. Bates DO, Cui TG, Doughty JM, Winkler M, Sugiono M, Shields JD, Peat D, Gillatt D, Harper SJ. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002; 62:4123–31. [PubMed].
132. Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R, Foster R, Digby-Bell J, Shields JD, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004; 64:7822–35. https://doi.org/10.1158/0008-5472.CAN-04-0934. [PubMed].
133. Mavrou A, Brakspear K, Hamdollah-Zadeh M, Damodaran G, Babaei-Jadidi R, Oxley J, Gillatt DA, Ladomery MR, Harper SJ, Bates DO, Oltean S. Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene. 2015; 34:4311–19. https://doi.org/10.1038/onc.2014.360. [PubMed].
134. Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, Symonds KE, Nowak DG, Royer-Pokora B, et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell. 2011; 20:768–80. https://doi.org/10.1016/j.ccr.2011.10.016. [PubMed].
135. Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G, Hagiwara M, Harper SJ, Woolard J, Ladomery MR, Bates DO. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010; 285:5532–40. https://doi.org/10.1074/jbc.M109.074930. [PubMed].
136. Finley SD, Popel AS. Predicting the effects of anti-angiogenic agents targeting specific VEGF isoforms. AAPS J. 2012; 14:500–09. https://doi.org/10.1208/s12248-012-9363-4. [PubMed].
137. Giampietro C, Deflorian G, Gallo S, Di Matteo A, Pradella D, Bonomi S, Belloni E, Nyqvist D, Quaranta V, Confalonieri S, Bertalot G, Orsenigo F, Pisati F, et al. The alternative splicing factor Nova2 regulates vascular development and lumen formation. Nat Commun. 2015; 6:8479. https://doi.org/10.1038/ncomms9479. [PubMed].
138. Macara IG. Par proteins: partners in polarization. Curr Biol. 2004; 14:R160–62. https://doi.org/10.1016/j.cub.2004.01.048. [PubMed].
139. Koh W, Mahan RD, Davis GE. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J Cell Sci. 2008; 121:989–1001. https://doi.org/10.1242/jcs.020693. [PubMed].
140. Iden S, Rehder D, August B, Suzuki A, Wolburg-Buchholz K, Wolburg H, Ohno S, Behrens J, Vestweber D, Ebnet K. A distinct PAR complex associates physically with VE-cadherin in vertebrate endothelial cells. EMBO Rep. 2006; 7:1239–46. https://doi.org/10.1038/sj.embor.7400819. [PubMed].
141. Ladd JJ, Chao T, Johnson MM, Qiu J, Chin A, Israel R, Pitteri SJ, Mao J, Wu M, Amon LM, McIntosh M, Li C, Prentice R, et al. Autoantibody signatures involving glycolysis and splicesome proteins precede a diagnosis of breast cancer among postmenopausal women. Cancer Res. 2013; 73:1502–13. https://doi.org/10.1158/0008-5472.CAN-12-2560. [PubMed].
142. Katayama H, Boldt C, Ladd JJ, Johnson MM, Chao T, Capello M, Suo J, Mao J, Manson JE, Prentice R, Esteva F, Wang H, Disis ML, Hanash S. An Autoimmune Response Signature Associated with the Development of Triple-Negative Breast Cancer Reflects Disease Pathogenesis. Cancer Res. 2015; 75:3246–54. https://doi.org/10.1158/0008-5472.CAN-15-0248. [PubMed].
143. Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo). 1996; 49:1204–11. https://doi.org/10.7164/antibiotics.49.1204. [PubMed].
144. Mizui Y, Sakai T, Iwata M, Uenaka T, Okamoto K, Shimizu H, Yamori T, Yoshimatsu K, Asada M. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo). 2004; 57:188–96. https://doi.org/10.7164/antibiotics.57.188. [PubMed].
145. Sakai Y, Tsujita T, Akiyama T, Yoshida T, Mizukami T, Akinaga S, Horinouchi S, Yoshida M, Yoshida T. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J Antibiot (Tokyo). 2002; 55:863–72. https://doi.org/10.7164/antibiotics.55.863. [PubMed].
146. Albert BJ, McPherson PA, O’Brien K, Czaicki NL, Destefino V, Osman S, Li M, Day BW, Grabowski PJ, Moore MJ, Vogt A, Koide K. Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol Cancer Ther. 2009; 8:2308–18. https://doi.org/10.1158/1535-7163.MCT-09-0051. [PubMed].
147. Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, Tani T, Horinouchi S, Yoshida M. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007; 3:576–83. https://doi.org/10.1038/nchembio.2007.18. [PubMed].
148. Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007; 3:570–75. https://doi.org/10.1038/nchembio.2007.16. [PubMed].
149. Otsuka K, Yamamoto Y, Ochiya T. Regulatory role of resveratrol, a microRNA-controlling compound, in HNRNPA1 expression, which is associated with poor prognosis in breast cancer. Oncotarget. 2018; 9:24718–30. https://doi.org/10.18632/oncotarget.25339. [PubMed].
150. Iwai K, Yaguchi M, Nishimura K, Yamamoto Y, Tamura T, Nakata D, Dairiki R, Kawakita Y, Mizojiri R, Ito Y, Asano M, Maezaki H, Nakayama Y, et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol Med. 2018; 10. https://doi.org/10.15252/emmm.201708289. [PubMed].
151. Zaharieva E, Chipman JK, Soller M. Alternative splicing interference by xenobiotics. Toxicology. 2012; 296:1–12. https://doi.org/10.1016/j.tox.2012.01.014. [PubMed].
152. Batson J, Toop HD, Redondo C, Babaei-Jadidi R, Chaikuad A, Wearmouth SF, Gibbons B, Allen C, Tallant C, Zhang J, Du C, Hancox JC, Hawtrey T, et al. Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease. ACS Chem Biol. 2017; 12:825–32. https://doi.org/10.1021/acschembio.6b01048. [PubMed].
153. Hatcher JM, Wu G, Zeng C, Zhu J, Meng F, Patel S, Wang W, Ficarro SB, Leggett AL, Powell CE, Marto JA, Zhang K, Ki Ngo JC, et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem Biol. 2018; 25:460–470.e6. https://doi.org/10.1016/j.chembiol.2018.01.013. [PubMed].
154. Summerton J. Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta. 1999; 1489:141–58. https://doi.org/10.1016/S0167-4781(99)00150-5. [PubMed].
155. Geary RS. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2009; 5:381–91. https://doi.org/10.1517/17425250902877680. [PubMed].
156. Moulton HM, Moulton JD. Morpholinos and their peptide conjugates: therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim Biophys Acta. 2010; 1798:2296–303. https://doi.org/10.1016/j.bbamem.2010.02.012. [PubMed].
157. Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016; 44:6549–63. https://doi.org/10.1093/nar/gkw533. [PubMed].
158. Denichenko P, Mogilevsky M, Cléry A, Welte T, Biran J, Shimshon O, Barnabas GD, Danan-Gotthold M, Kumar S, Yavin E, Levanon EY, Allain FH, Geiger T, et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat Commun. 2019; 10:1590. https://doi.org/10.1038/s41467-019-09523-0. [PubMed].
159. Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, Tirosh I, Narayan R, Root DE, Golub TR, Subramanian A, Doench JG. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017; 15:e2003213. https://doi.org/10.1371/journal.pbio.2003213. [PubMed].
160. Wan J, Sazani P, Kole R. Modification of HER2 pre-mRNA alternative splicing and its effects on breast cancer cells. Int J Cancer. 2009; 124:772–77. https://doi.org/10.1002/ijc.24052. [PubMed].
161. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008; 40:1413–15. https://doi.org/10.1038/ng.259. [PubMed]. Erratum in: Nat Genet. 2009 Jun;41(6):762. https://doi.org/10.1038/ng0609-762d.
162. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008; 456:470–76. https://doi.org/10.1038/nature07509. [PubMed].
163. Fokkelman M, Roosmalen W, Rogkoti VM, Le Dévédec S, Geiger B, Water B. PhagoKinetic Track Assay: Imaging and Analysis of Single Cell Migration. Bio Protoc. 2016; 6:e1699. https://doi.org/10.21769/BioProtoc.1699.
164. McDonald ER 3rd, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, Golji J, Kao I, Li L, et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell. 2017; 170:577–592.e10. https://doi.org/10.1016/j.cell.2017.07.005. [PubMed].
165. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, Meyers RM, Ali L, Goodale A, et al. Defining a Cancer Dependency Map. Cell. 2017; 170:564–576.e16. https://doi.org/10.1016/j.cell.2017.06.010. [PubMed].