
INTRODUCTION
Molecular chaperones are involved in protein folding and homeostasis [1]. Hsp90 is an essential, evolutionarily-conserved molecular chaperone that is ubiquitously expressed both in normal and cancer cells [2-5]. While Hsp90 has been shown to bind to and prevent the aggregation of a wide range of proteins [6], the list of proteins that require active chaperoning by Hsp90 is more restricted (www.picard.ch/downloads/Hsp90interactors.pdf), and is comprised primarily of key components of numerous signal transduction pathways. In cancer cells, Hsp90 plays a vital role in protecting selected mutated, over-expressed and/or deregulated oncoproteins from misfolding and degradation [7, 8]. Therefore it is not surprising that multiple Hsp90 inhibitors are being actively evaluated in the clinic (Table 1) [9].
Table 1: Current Hsp90 inhibitors in clinical trials. * non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC), chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), or B-cell prolymphocytic leukemia (B-PLL), acute myelogenous leukemia (AML), myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), gastrointestinal stromal tumor (GIST), metastatic breast cancer (MBC), multiple myeloma (MM), hormone-resistant prostate cancer (HRPC).
Inhibitor |
Phase |
Route |
Indication* |
Structure |
Imaging |
Combination |
Source |
Retaspimycin |
II |
i.v. |
Advanced dedifferentiated liposarcoma, NSCLC patients with ALK translocations |
|
- |
- |
Infinity |
IPI-493 |
I |
oral |
Advanced solid tumors, hematologic malignancies |
- |
- |
- |
Infinity |
AUY922 |
I/II |
i.v. |
Advanced solid malignancies, MBC, stage IIIB-IV NSCLC, advanced gastric cancer, metastatic colorectal cancer |
|
89Zr-trastuzumab, VEGF-89ZR-bevacizumab PET imaging |
Trastuzumab, erlotinib, capecitabine, cetuximab |
Novartis |
Ganetespib |
I/II |
i.v. |
AML, MDS, CML, or myeloproliferative disorders, stage IIIB or IV NSCLC, GIST, colorectal cancer, SCLC, esophagogastric cancer, metastatic ocular melanoma, metastatic pancreatic cancer, metastatic HRPC, MBC, solid tumors |
- |
- |
Docetaxel |
Synta |
KW-2478 |
I/II |
i.v. |
MM |
|
- |
Bortezomib |
Kyowa Hakko Kirin |
AT13387 |
I/II |
oral |
GIST, metastatic solid tumors |
|
- |
Imatinib |
Astex Therapeutics |
HSP990 |
I |
oral |
advanced solid tumors |
- |
- |
- |
Novartis |
MPC-3100 |
I |
oral |
Cancer patients who have failed other treatments |
- |
- |
- |
Myrexis, Inc. |
DS-2248 |
I |
oral |
Advanced solid tumors |
- |
- |
- |
Daiichi Sankyo Inc. |
Debio 0932 |
I |
oral |
Advanced solid tumors or lymphoma |
|
- |
- |
Debiopharm/Curis |
PU-H71 |
- |
i.v. |
Breast cancer, prostate cancer or lymphoma |
|
[124I]-PU-H71 PET imaging |
- |
Memorial Sloan-Kettering Cancer Center |
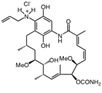
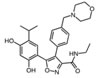
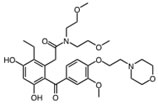
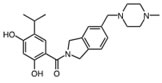
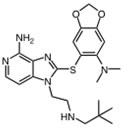
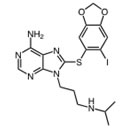
Hsp90 is dimeric and each protomer can be divided into three domains (Figure 1) [10, 11]: i) an N-terminal domain, containing nucleotide, co-chaperone (proteins that assist the chaperone activity of Hsp90), and drug binding sites; ii) a middle (M) domain, which provides binding sites for client proteins and co-chaperones; iii) a C-terminal domain containing a dimerization motif, a second inhibitor binding region and binding sites for additional co-chaperones [12-14]. N and M domains are connected by an unstructured charged-linker region of significant but variable length, which provides conformational flexibility to the protein [15, 16].
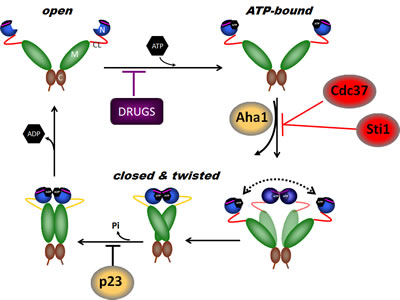
Figure 1: The Hsp90 chaperone cycle. Hsp90 is a dimer of two monomers, each containing an N-domain (“N”, blue); a charged linker (“CL”, red); an M-domain (“M”, green); and a C-domain (“C”, brown). The ATP lid (purple) in the N-domain alters conformation upon ATP binding to the N-terminal domains of the “open” conformation, promoting “lid” closure followed by transient dimerization of the N-domains. Subsequent structural changes result in the “closed and twisted” conformation that is competent for ATP hydrolysis. The co-chaperones Sti1 and Cdc37, and pharmacologic inhibitors such as those discussed in the text, slow or block, respectively, the chaperone cycle at an early stage, while the co-chaperone Aha1 enhances the ATPase activity of Hsp90 by assisting the extensive and energy-intensive conformational changes necessary to achieve competence for ATP hydrolysis. The co-chaperone p23 stabilizes a late Hsp90 conformation and slows, but does not block, the rate of ATP hydrolysis. For more details, please see [4].
Hsp90 chaperone function is coupled to its ability to bind and hydrolyze ATP, which in turn promotes an ordered series of conformational changes known as the chaperone cycle that is necessary for Hsp90’s chaperone function [17, 18]. Hsp90 inhibitors currently in clinical evaluation all share the property of preventing the chaperone cycle by occupying Hsp90’s N-domain ATP binding pocket [19, 20]. The regulation of eukaryotic Hsp90 function is complex and depends on several factors, including the regulated interaction of specific co-chaperones (e.g., Aha1 stimulates Hsp90 ATPase activity, whereas p23/Sba1 and Hop/Sti1 inhibit ATP hydrolysis [21, 22]), and various post-translational modifications [23, 24]. A more detailed understanding of the mechanics of Hsp90 regulation in normal and cancer cells may provide additional therapeutic strategies to effectively inhibit this protein.
HSP90 PHOSPHORYLATION REGULATES CHAPERONE FUNCTION
Hsp90 is subject to several post-translational modifications, including phosphorylation, acetylation, and S-nitrosylation, that contribute to Hsp90 regulation, although regulation of these processes within the cell is not well understood [3, 4]. While Hsp90 acetylation and S-nitrosylation have been identified more recently [23, 24], Hsp90 phosphorylation was first reported in the early 1980s [25].
Hsp90 is a substrate for several serine/threonine and tyrosine kinases, including double-stranded DNA-dependent protein kinase, Akt, B-Raf, Protein Kinase A (PKA), Casein Kinase 2 (CK2), c-Src and Wee1 [3, 4, 26] (Table 2). These kinases are also clients of Hsp90, suggesting the possible existence of complex feedback loops whereby these kinases may modulate their own chaperoning and functional activity. Indeed, others have suggested that client binding to Hsp90 may directly influence its chaperone activity [2, 27]. Previous work has shown that brief exposure of cells to okadaic acid, a serine/threonine phosphatase inhibitor, leads to Hsp90 threonine hyperphosphorylation and is accompanied by decreased Hsp90 association with its client kinase v-Src [28]. These data suggest that Hsp90 threonine phosphorylation is tightly regulated and therefore is likely to impact chaperone function.
We reported recently that Wee1Swe1 phosphorylates a conserved tyrosine residue (Y24 in yeast Hsp90 and Y38 in human Hsp90α) in the N-domain of Hsp90 in a cell cycle-dependent manner [29]. Interestingly, deletion of Swe1 in yeast or pharmacologic inhibition of Wee1 in cancer cells confers hypersensitivity to Hsp90 inhibition [29].
Table 2: Hsp90 phosphorylation sites and identified kinases. Yeast (Hsc82, Hsp82) and human (Hsp90α, Hsp90β) phosphorylation residues are marked by an asterisk (*); orthologue residue is also shown when present. These phosphorylation sites were taken from published literature and the websites PhosphoSitePlus® (http://www.phosphosite.org/) and Saccharomyces Genome Database (SGD) (http://www.yeastgenome.org/). Marked phosphorylation residues (†) were identified in rat and (§) mouse Hsp90.
Residues |
|||||
yHsp90 |
yHsc90 |
hHsp90a |
hHsp90b |
Protein Kinase |
Reference |
N/A |
N/A |
T5* |
N/A |
Ds-DNA activated protein kinase |
[43] |
N/A |
N/A |
T7* |
N/A |
Ds-DNA activated protein kinase |
[43] |
T22* |
T22 |
Y36 |
Y31 |
Casein kinase II |
[33] |
Y24* |
Y24 |
Y38* |
Y33 |
Swe1/Wee1 |
[29] |
S36 |
S36 |
S50 |
S45* |
UNKNOWN |
[44, 45] |
N/A |
N/A |
S52* |
N/A |
UNKNOWN |
[46] |
S39 |
S39 |
S53 |
S48* |
UNKNOWN |
[44, 45] |
Y47 |
Y47 |
Y61 |
Y56* |
UNKNOWN |
[47-50] |
S49 |
N/A |
S63*§ |
S58* |
Casein kinase II |
[49, 51] |
S51 |
S51 |
T65* |
T60 |
Casein kinase II |
[51] |
N/A |
N/A |
S68* |
S63 |
Casein kinase II |
[51] |
T58 |
T58 |
S72* |
S67 |
Casein kinase II |
[51] |
N/A |
N/A |
T88* |
T83 |
PKA |
[52] |
N/A |
N/A |
T90* |
T85 |
UNKNOWN |
[53] |
Y184 |
Y184 |
Y197 |
Y192* |
UNKNOWN |
[48] |
S198 |
S198 |
S211† |
S206 |
PKA , PKG |
[54] |
N/A |
N/A |
S231* |
S226* |
Casein kinase II |
[31, 55, 56] |
N/A |
N/A |
S252* |
N/A |
UNKNOWN |
[57] |
N/A |
N/A |
S263* |
S255* |
Casein kinase II, B-Raf |
[31, 55, 56, 58] |
N/A |
N/A |
N/A |
S261*†§ |
UNKNOWN |
[49, 57, 59-64] |
N/A |
N/A |
Y284* |
Y276* |
UNKNOWN |
[50, 65] |
S282* |
S278 |
N/A |
N/A |
UNKNOWN |
SGD |
T273 |
T269 |
T293* |
T285* |
UNKNOWN |
[66] |
T285 |
T281 |
T305 |
T297* |
UNKNOWN |
[67] |
S297* |
S293 |
N/A |
N/A |
UNKNOWN |
SGD |
Y289 |
Y285 |
Y309 |
Y301* |
c-Src (does not exist in yeast) |
[68] |
Y293 |
Y289 |
Y313§ |
Y305* |
UNKNOWN |
[69] |
S295 |
S291 |
S315 |
S307* |
UNKNOWN |
[63] |
S297 |
S293 |
T317* |
T309* |
UNKNOWN |
[66] |
S334* |
S330 |
N/A |
N/A |
UNKNOWN |
SGD |
S379* |
S375 |
S399 |
S391* |
UNKNOWN |
SGD, [49, 59, 65] |
T443* |
T429 |
S453§ |
S445* |
UNKNOWN |
SGD, [59] |
N/A |
N/A |
N/A |
T446§ |
UNKNOWN |
[49, 59] |
N/A |
N/A |
S460 |
S452* |
PKA |
[54] |
N/A |
N/A |
Y466* |
N/A |
UNKNOWN |
[50], PhosphoSitePlus® |
S456 |
S452 |
S476§ |
S468 |
UNKNOWN |
[49] |
N/A |
N/A |
N/A |
S482§ |
UNKNOWN |
[46] |
Y472 |
Y468 |
Y492* |
Y484* |
UNKNOWN |
[45, 70], PhosphoSitePlus® |
Y473 |
Y469 |
Y493* |
Y485* |
UNKNOWN |
PhosphoSitePlus® |
S478 |
S474 |
T495§ |
S490 |
UNKNOWN |
[45] |
T502 |
T498 |
N/A |
T514§ |
UNKNOWN |
[45] |
Y508 |
Y504 |
Y498 |
Y520§ |
UNKNOWN |
[45] |
T520 |
T516 |
T540 |
S532* |
UNKNOWN |
[63] |
N/A |
N/A |
T566§ |
N/A |
UNKNOWN |
[49] |
S568 |
S564 |
S589* |
S581 |
UNKNOWN |
[66] |
N/A |
N/A |
Y604*§ |
Y596* |
UNKNOWN |
[71] PhosphoSitePlus® |
Y606 |
Y602 |
Y627*§ |
Y619* |
UNKNOWN |
PhosphoSitePlus® |
S616* |
S612 |
N/A |
N/A |
UNKNOWN |
SGD |
S619* |
S615 |
N/A |
N/A |
UNKNOWN |
SGD |
N/A |
N/A |
S641§ |
N/A |
UNKNOWN |
[49] |
S657* |
S653 |
S677 |
S668 |
UNKNOWN |
[72] |
S663* |
S659 |
N/A |
N/A |
UNKNOWN |
[72] |
N/A |
N/A |
Y689* |
Y681 |
UNKNOWN |
[50], PhosphoSitePlus® |
N/A |
N/A |
T725* |
N/A |
UNKNOWN |
[63] |
N/A |
N/A |
S726* |
S718* |
UNKNOWN |
[63] |
CK2 DEPENDENT PHOSPHORYLATION OF HSP90
CK2 is a serine/threonine protein kinase and an Hsp90 client [30] that phosphorylates multiple serine and threonine residues in human Hsp90α (hHsp90α) and yeast Hsp82 (yHsp90) [31, 32]. Recently, we used S. cerevisiae to show that CK2 phosphorylates a single conserved threonine residue (T22) in the N-domain of yHsp90 both in vitro and in vivo [33]. T22 resides in helix-1 of the Hsp90 N-domain that, together with other adjacent amino acids, is involved in an important hydrophobic interaction with the ATPase catalytic loop in the M-domain that helps to establish Hsp90’s ATP hydrolysis-competent state.
The functional importance of T22 was uncovered initially in a genetic screen, where its mutation to isoleucine affected the chaperoning of v-Src and glucocorticoid receptor (GR) in yeast [34]. Subsequent work demonstrated that the T22I mutant has 6-fold higher ATPase activity compared to WT yHsp90 [35]. In contrast, we have shown that mutation of T22 to non-phosphorylatable alanine (T22A) did not affect its ATPase activity, while phospho-mimetic mutation of this residue to glutamic acid (T22E) reduced ATPase activity by 60% compared to WT yHsp90. Nevertheless, both mutants in yeast and the equivalent mutations in hHsp90α (T36A and T36E) affected Hsp90-dependent chaperoning of kinase (v-Src, Mpk1/Slt2, Raf-1, ErbB2 and CDK4) and non-kinase (cystic fibrosis transmembrane conductance regulator protein and GR) clients [33].
To explore further whether ATP binding is a prerequisite for T22 phosphorylation, we examined the ability of CK2 to phosphorylate two conformationally distinct Hsp90 N-domain mutants. CK2 was able to efficiently phosphorylate yHsp90-E33A, which binds ATP equivalently to wild-type but, upon ATP binding, favors a “closed” (N-domain dimerized) conformation (i.e., is unable to hydrolyze ATP). However, CK2 was unable to phosphorylate yHsp90-D79N, which cannot bind ATP and thus favors an “open” (N-domain undimerized) conformation (Figure 2A). Since T22 is not accessible to solvent once ATP-dependent N-domain dimerization has occurred [18], these data suggest that ATP binding to the open conformation of Hsp90 sets in motion rapid conformational change within and adjacent to helix-1 that is a prerequisite for CK2-mediated phosphorylation. Indeed, a recent study of the bacterial ortholog of Hsp90, HtpG, confirms that this region of the N-domain undergoes very rapid conformational change upon ATP binding that significantly precedes ATP-induced N-domain dimerization [35, 36] Since T22 phosphorylation slows the rate of ATP hydrolysis without affecting ATP binding, it is possible that eukaryotic cells utilize this post-translational modification to adjust the rate of the Hsp90 cycle to meet the optimal chaperone requirements of individual client proteins.
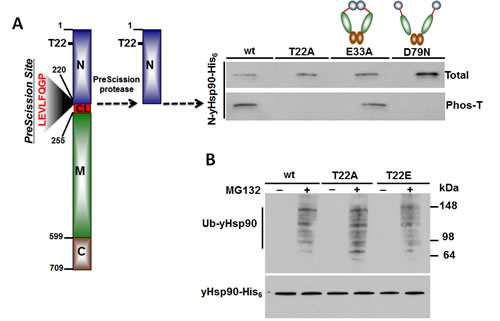
Figure 2. A) CK2-mediated threonine phosphorylation of the N-domain of WT, T22A, E33A and D79N yHsp90-His6 in vitro. Threonine phosphorylation was detected with a pan anti-phosphothreonine antibody. For more details, please see [33]. B) WT, T22A, and T22E yHsp90-His6-expressing yeast were treated with the proteasome inhibitor MG132 (50 mM for 1 h), and yHsp90 ubiquitination was assessed using anti-ubiquitin antibody to probe salt-stripped (0.5 M NaCl) His6 pull-downs.
We showed previously that phosphorylation of Y24 is a signal for Hsp90 polyubiquitination and degradation by cytoplasmic proteasomes [33]. We explored the possibility that T22 phosphorylation of yHsp90 may serve a similar purpose. Yeast expressing WT Hsp90 as well as the phospho mutants (T22A and T22E) were treated with the proteasome inhibitor MG132 (50 µM for 1h). This resulted in equivalent accumulation of polyubiquitinated yHsp90 in each case (Figure 2B). Therefore, unlike Y24, T22 phosphorylation is not likely to be a determinant of Hsp90 degradation. Instead, one can speculate that T22 phosphorylation occurs dynamically to allow for the fine-tuning of chaperone activity in response to environmental cues.
CK2 PHOSPHORYLATION OF THREONINE 22 IMPACTS CHAPERONING OF HEAT SHOCK FACTOR (HSF)
The phospho-mimetic T22E mutant, like T22I, is temperature sensitive whereas T22A-expressing yeast grow like wild-type cells at elevated temperature (Figure 3A). This phenotype may reflect the different ATPase activities of these mutants, since it is generally accepted that Hsp90 binding serves to down-regulate HSF transcriptional activity [37]. Mutations of Hsp90 that compromise its chaperone function, or Hsp90 inhibitor administration, lead to strong induction of Hsf1 activity in yeast, even in the absence of heat shock [38].
To examine the importance of T22 in Hsp90 modulation of the heat shock response, we measured Hsf1 activity in yHsp90 T22A (normal ATPase activity), T22E (reduced ATPase activity) and T22I (elevated ATPase activity)-expressing yeast cells by transforming them with a heat shock element HSE-lacZ reporter plasmid. We confirmed that yHsp90-T22I displayed a higher basal and heat-induced (39˚C for 40 min) Hsf1 activity compared to WT cells [39] (Figure 3B), while Hsf1 activity was significantly diminished in the phospho-mimetic yHsp90-T22E mutant. At the same time, Hsf1 activity in yHsp90-T22A mutant yeast was equivalent to that of yeast expressing WT yHsp90. These data suggest that the ability of yHsp90 to regulate Hsf1 closely parallels its inherent ATPase activity, but that temperature sensitivity itself is not an accurate predictor of the impact of these Hsp90 mutations on the heat shock response.
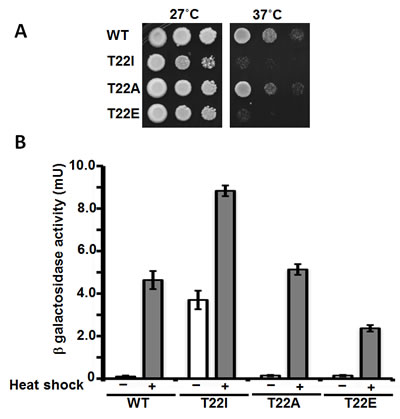
Figure 3. A) Yeast expressing WT yHsp90-His6 or the phospho mutants T22A and T22E, as well as T22I, were grown to mid-log phase (107 cells/ml) and then a 1:10 dilution series was spotted on YPDA agar. The plates were incubated either at 27˚C or 37˚C for 3 days to test for temperature sensitivity. B) Yeast cells expressing WT yHsp90-His6 or phospho mutants T22A and T22E, or T22I, were transformed with a Heat Shock Element (HSE)-lacZ reporter. HSE activity was measured in unstressed (light bars) and heat shocked (40 min at 39˚C, dark bars) cells. The data are expressed as mean +/- standard deviation derived from four independent experiments.
PHOSPHORYLATION OF THREONINE 22 CONFERS INCREASED SENSITIVITY TO HSP90 INHIBITORS
There are currently 18 Hsp90 inhibitors in various stages of clinical evaluation as targeted anti-cancer agents. Uncovering modifications to Hsp90 that might enhance sensitivity to these inhibitors at the cellular level would certainly aid their continued clinical development. To that end, we have examined the sensitivity of T22 mutants to Hsp90 inhibitors. We expressed non-phospho (T22A) and phospho-mimetic (T22E) Hsp90 mutants, as well as yHsp90-T22I, as the sole Hsp90 species in the PP30 yeast strain lacking the pleiotropic drug resistance pump, Pdr5 [40, 41]. Although the S. cerevisiae genome contains 30 plasma membrane ATP-binding cassette (ABC) proteins [42], Pdr5 has been identified as the major mediator of Hsp90 inhibitor efflux [40, 41].
Yeast cells were grown to exponential phase and were spotted at 107, 106 and 105 cells/ml on YPDA plates containing 40 or 60 µM GA or radicicol (RD). The synthetic Hsp90 inhibitors, ganetespib (formerly STA-9090, Synta Pharmaceuticals) and SNX-2112 (Serenex/Pfizer) were also included in these assays (at 60 or 80 µM). Our data show that the drug sensitivity of y-Hsp90-T22A-expressing yeast was equivalent to or less than that of yeast expressing WT yHsp90. In contrast, yeast expressing yHsp90-T22I were uniformly hypersensitive to all of the Hsp90 inhibitors examined (Figure 4A, B). Interestingly, yeast expressing the phospho-mimetic yHsp90-T22E were also more sensitive than either WT or yHsp90-T22A-expressing yeast to the four Hsp90 inhibitors (Figure 4A, B). However, this increased sensitivity was evident only at higher drug concentrations. These data identify T22 as an important determinant of Hsp90 inhibitor sensitivity in yeast and suggest that T22 phosphorylation status may contribute to drug sensitivity in vivo.
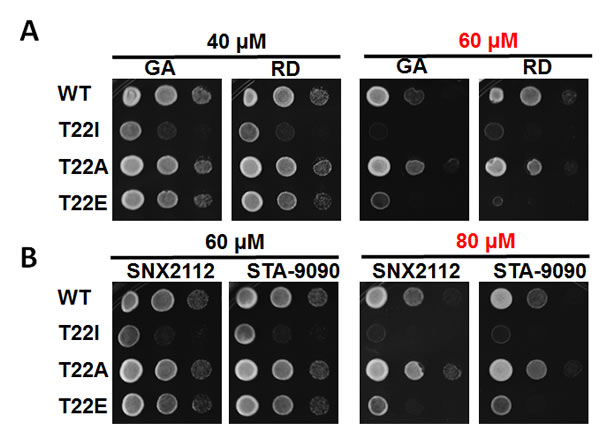
Figure 4: PP30 pdr5∆ yeast strain expressing only WT y Hsp90, yHsp90-T22I, yHsp90-T22A, or yHsp90-T22E were grown to mid-log phase (107 cells/ml) and then a 1:10 dilution series was spotted on YPDA agar containing indicated concentrations of the Hsp90 inhibitors geldanamycin (GA), radicicol (RD), SNX2112, or STA-9090. Plates were incubated at 25˚C for 4 days.
CONCLUDING REMARKS
Hsp90 phosphorylation has been known for nearly 30 years [25], however only recently are we beginning to appreciate its significance in fine-tuning Hsp90 chaperone activity [4]. Our recent work has demonstrated that the Hsp90 client kinase CK2 phosphorylates a conserved threonine residue (T22) in the N-domain of yHsp90. This residue participates in a hydrophobic interaction with lysine 378 of the catalytic loop in the Hsp90 middle domain, helping to stabilize the ATPase-competent state induced by ATP binding to the N-domain. Phospho-mimetic mutation of this residue in both yeast and human Hsp90 alters chaperone function (see Figure 5 and [33]). Here, we have provided additional data showing that mutation of this residue impacts both the heat shock response and sensitivity to Hsp90 inhibitors.
It is unlikely that the regulation of Hsp90 function, in all its complexity, is universal for all client proteins of this chaperone. Indeed, a recent report [2] has suggested that different clients and co-chaperone complexes can be accommodated by subtly different Hsp90 conformational states. Perhaps phosphorylation of Hsp90, at T22 and additional amino acid residues, participates in modulating the equilibrium among these client-dependent conformational states.
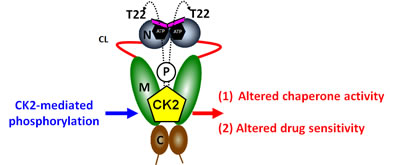
Figure 5: Casein kinase 2 (CK2) phosphorylates a conserved threonine residue (T22) in the N-domain of yHsp90. This residue is an important determinant of Hsp90 ATPase activity and drug sensitivity; T22 phosphorylation affects Hsp90 chaperone function and alters its sensitivity to Hsp90 inhibitors. For further details, please see [33].
ACKNOWLEDGMENTS
We gratefully acknowledge our colleagues and collaborators, Laurence H. Pearl, Peter W. Piper, Chris Prodromou, Barry Panaretou, Wanping Xu, Kristin Beebe, Cara Vaughan and Andrew Truman, for their scientific contributions and stimulating discussions. We thank Weiwen Ying of Synta Pharmaceuticals, Inc. for supplying ganetespib and Timothy Haystead (Duke University) for supplying SNX-2112. This work was supported by the Intramural Research Program of the National Cancer Institute.
REFERENCES
1. Mayer MP. Gymnastics of molecular chaperones. Mol Cell. 2010; 39:321-331.
2. Street TO, Lavery LA, Agard DA. Substrate binding drives large-scale conformational changes in the hsp90 molecular chaperone. Mol Cell. 2011; 42:96-105.
3. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010; 11:515-528.
4. Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010; 10:537-549.
5. Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem. 2008; 283:18473-18477.
6. Rudiger S, Freund SM, Veprintsev DB, Fersht AR. CRINEPT-TROSY NMR reveals p53 core domain bound in an unfolded form to the chaperone Hsp90. Proc Natl Acad Sci U S A. 2002; 99:11085-11090.
7. Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007; 32:517-530.
8. Sharp S, Workman P. Inhibitors of the HSP90 molecular chaperone: current status. Adv Cancer Res. 2006; 95:323-348.
9. Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem. 2009; 9:1479-1492.
10. Pearl LH, Prodromou C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem. 2006; 75:271-294.
11. Picard D. Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci. 2002; 59:1640-1648.
12. Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006; 440:1013-1017.
13. Prodromou C, Pearl LH. Structure and functional relationships of Hsp90. Curr Cancer Drug Targets. 2003; 3:301-323.
14. Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem. 2008; 15:2702-2717.
15. Tsutsumi S, Mollapour M, Graf C, Lee CT, Scroggins BT, Xu W, Haslerova L, Hessling M, Konstantinova AA, Trepel JB, Panaretou B, Buchner J, Mayer MP, Prodromou C, Neckers L. Hsp90 charged-linker truncation reverses the functional consequences of weakened hydrophobic contacts in the N domain. Nat Struct Mol Biol. 2009; 16:1141-1147.
16. Hainzl O, Lapina MC, Buchner J, Richter K. The charged linker region is an important regulator of Hsp90 function. J Biol Chem. 2009; 284:22559-22567.
17. Pearl LH, Prodromou C. Structure and in vivo function of Hsp90. Curr Opin Struct Biol. 2000; 10:46-51.
18. Cunningham CN, Krukenberg KA, Agard DA. Intra- and intermonomer interactions are required to synergistically facilitate ATP hydrolysis in Hsp90. J Biol Chem. 2008; 283:21170-21178.
19. Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997; 89:239-250.
20. Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999; 42:260-266.
21. Chang HC, Nathan DF, Lindquist S. In vivo analysis of the Hsp90 cochaperone Sti1 (p60). Mol Cell Biol. 1997; 17:318-325.
22. Panaretou B, Siligardi G, Meyer P, Maloney A, Sullivan JK, Singh S, Millson SH, Clarke PA, Naaby-Hansen S, Stein R, Cramer R, Mollapour M, Workman P, Piper PW, Pearl LH, Prodromou C. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol Cell. 2002; 10:1307-1318.
23. Scroggins BT, Neckers L. Post-translational modification of heat shock protein 90: impact on chaperone function. Expert Opin Drug Discov. 2007; 2:1403-1414.
24. Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. Hsp90 is regulated by a switch point in the C-terminal domain. EMBO Rep. 2009; 10:1147-1153.
25. Dougherty JJ, Puri RK, Toft DO. Phosphorylation in vivo of chicken oviduct progesterone receptor. J Biol Chem. 1982; 257:14226-14230.
26. Mollapour M, Tsutsumi S, Neckers L. Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle. 2010; 9:1-7.
27. McLaughlin SH, Smith HW, Jackson SE. Stimulation of the weak ATPase activity of human hsp90 by a client protein. J Mol Biol. 2002; 315:787-798.
28. Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J Biol Chem. 1995; 270:28654-28659.
29. Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, Lee MJ, Lee S, Morra G, Bourboulia D, Scroggins BT, Colombo G, Blagg BS, Panaretou B, Stetler-Stevenson WG, Trepel JB, Piper PW et al. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function. Mol Cell. 2010; 37:333-343.
30. Miyata Y, Yahara I. The 90-kDa heat shock protein, HSP90, binds and protects casein kinase II from self-aggregation and enhances its kinase activity. J Biol Chem. 1992; 267:7042-7047.
31. Lees-Miller SP, Anderson CW. Two human 90-kDa heat shock proteins are phosphorylated in vivo at conserved serines that are phosphorylated in vitro by casein kinase II. J Biol Chem. 1989; 264:2431-2437.
32. Wandinger SK, Suhre MH, Wegele H, Buchner J. The phosphatase Ppt1 is a dedicated regulator of the molecular chaperone Hsp90. EMBO J. 2006; 25:367-376.
33. Mollapour M, Tsutsumi S, Truman AW, Xu W, Vaughan CK, Beebe K, Konstantinova A, Vourganti S, Panaretou B, Piper PW, Trepel JB, Prodromou C, Pearl LH, Neckers L. Threonine 22 phosphorylation attenuates hsp90 interaction with cochaperones and affects its chaperone activity. Mol Cell. 2011; 41:672-681.
34. Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995; 15:3917-3925.
35. Prodromou C, Panaretou B, Chohan S, Siligardi G, O’Brien R, Ladbury JE, Roe SM, Piper PW, Pearl LH. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000; 19:4383-4392.
36. Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009; 28:602-613.
37. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell. 1998; 94:471-480.
38. Hjorth-Sorensen B, Hoffmann ER, Lissin NM, Sewell AK, Jakobsen BK. Activation of heat shock transcription factor in yeast is not influenced by the levels of expression of heat shock proteins. Mol Microbiol. 2001; 39:914-923.
39. Harris N, MacLean M, Hatzianthis K, Panaretou B, Piper PW. Increasing Saccharomyces cerevisiae stress resistance, through the overactivation of the heat shock response resulting from defects in the Hsp90 chaperone, does not extend replicative life span but can be associated with slower chronological ageing of nondividing cells. Mol Genet Genomics. 2001; 265:258-263.
40. Millson SH, Prodromou C, Piper PW. A simple yeast-based system for analyzing inhibitor resistance in the human cancer drug targets Hsp90alpha/beta. Biochem Pharmacol. 2010; 79:1581-1588.
41. Piper PW, Millson SH, Mollapour M, Panaretou B, Siligardi G, Pearl LH, Prodromou C. Sensitivity to Hsp90-targeting drugs can arise with mutation to the Hsp90 chaperone, cochaperones and plasma membrane ATP binding cassette transporters of yeast. Eur J Biochem. 2003; 270:4689-4695.
42. Jungwirth H, Kuchler K. Yeast ABC transporters-- a tale of sex, stress, drugs and aging. FEBS Lett. 2006; 580:1131-1138.
43. Lees-Miller SP, Anderson CW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. 1989; 264:17275-17280.
44. Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics. 2010; 9:1314-1323.
45. Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A. 2008; 105:13793-13798.
46. Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, Gerke V, Schmidt-Arras DE, Berdel WE, Muller-Tidow C, Mann M, Serve H. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009; 36:326-339.
47. Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, Mitchell J, Gygi SP, Rush J, Polakiewicz RD et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008; 105:692-697.
48. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, Sullivan L, Mitchell J, Wetzel R et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131:1190-1203.
49. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010; 143:1174-1189.
50. Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, Cantley LC, Gygi SP, Rush J, Comb MJ. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010; 3:ra64.
51. Rose DW, Wettenhall RE, Kudlicki W, Kramer G, Hardesty B. The 90-kilodalton peptide of the heme-regulated eIF-2 alpha kinase has sequence similarity with the 90-kilodalton heat shock protein. Biochemistry. 1987; 26:6583-6587.
52. Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007; 282:9364-9371.
53. Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2007; 104:2199-2204.
54. Huang SY, Tsai ML, Chen GY, Wu CJ, Chen SH. A systematic MS-based approach for identifying in vitro substrates of PKA and PKG in rat uteri. J Proteome Res. 2007; 6:2674-2684.
55. Ogiso H, Kagi N, Matsumoto E, Nishimoto M, Arai R, Shirouzu M, Mimura J, Fujii-Kuriyama Y, Yokoyama S. Phosphorylation analysis of 90 kDa heat shock protein within the cytosolic arylhydrocarbon receptor complex. Biochemistry. 2004; 43:15510-15519.
56. Kurokawa M, Zhao C, Reya T, Kornbluth S. Inhibition of apoptosome formation by suppression of Hsp90beta phosphorylation in tyrosine kinase-induced leukemias. Mol Cell Biol. 2008; 28:5494-5506.
57. Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006; 127:635-648.
58. Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, Brown RA, Witze ES, Schweppe RE, Resing KA, Ahn NG. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009; 34:115-131.
59. Wisniewski JR, Nagaraj N, Zougman A, Gnad F, Mann M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010; 9:3280-3289.
60. Han G, Ye M, Liu H, Song C, Sun D, Wu Y, Jiang X, Chen R, Wang C, Wang L, Zou H. Phosphoproteome analysis of human liver tissue by long-gradient nanoflow LC coupled with multiple stage MS analysis. Electrophoresis. 2010; 31:1080-1089.
61. Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007; 104:1488-1493.
62. Hoffert JD, Wang G, Pisitkun T, Shen RF, Knepper MA. An automated platform for analysis of phosphoproteomic datasets: application to kidney collecting duct phosphoproteins. J Proteome Res. 2007; 6:3501-3508.
63. Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008; 105:10762-10767.
64. Xia Q, Cheng D, Duong DM, Gearing M, Lah JJ, Levey AI, Peng J. Phosphoproteomic analysis of human brain by calcium phosphate precipitation and mass spectrometry. J Proteome Res. 2008; 7:2845-2851.
65. Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci Signal. 2009; 2:ra46.
66. Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010; 3:ra3.
67. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007; 316:1160-1166.
68. Duval M, Le Boeuf F, Huot J, Gratton JP. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell. 2007; 18:4659-4668.
69. Ballif BA, Carey GR, Sunyaev SR, Gygi SP. Large-scale identification and evolution indexing of tyrosine phosphorylation sites from murine brain. J Proteome Res. 2008; 7:311-318.
70. Jorgensen C, Sherman A, Chen GI, Pasculescu A, Poliakov A, Hsiung M, Larsen B, Wilkinson DG, Linding R, Pawson T. Cell-specific information processing in segregating populations of Eph receptor ephrin-expressing cells. Science. 2009; 326:1502-1509.
71. Bonnette PC, Robinson BS, Silva JC, Stokes MP, Brosius AD, Baumann A, Buckbinder L. Phosphoproteomic characterization of PYK2 signaling pathways involved in osteogenesis. J Proteomics. 2010; 73:1306-1320.
72. Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics. 2008; 7:1389-1396.