
Introduction
Pheochromocytoma and paraganglioma (PPGL) are chromaffin cell-derived neuroendocrine tumors [1]. While having been considered mostly sporadic for long time, it has become clear over the last 2 decades that 30–40% of these tumors are due to underlying germline mutations in one of several susceptibility genes [2–4]. Nearly 30 genes have been identified with germline or somatic mutations in PPGL [3]. Apart from the well-known genes such as RET, NF1, VHL, SDHx, TMEM127, and MAX, many of the new genes have been identified in the last 7 years that include HIF2A-EPAS1, FH, H-RAS, PHD1, MDH2, ATRX, H3F3A, CSDE1, MAML3 and IRP1 (summarized in reference [4]). These genes involve at least 3 signal transduction pathways, the pseudohypoxemia, the tyrosine kinase, and the WNT pathways [3, 5]. Much more recently discovered predisposing genes for PPGL include DLST [6], SLC25A11 [7] and DNMT3A [8]. The discovery of new genes has been propagated by major advances in technology. Next Generation Sequencing (NGS) technology has made sequencing of the whole exome fairly routinely available. Many studies have been published on the molecular genetics of PPGL using this technology [3–5, 9–12]. These studies have shown some ethnic differences in the rates of mutations and the underlying genetic landscape [12–15]. There have not been comprehensive studies from the Middle East region and none from the Arabic population. These populations are homogeneous with high rates of consanguinity making them ideal for studying hereditary diseases [16]. In this paper, we report the underlying genetic mutations and the genotype/phenotype correlation in a large series of patients with PPGL from the highly consanguineous population of Saudi Arabia.
Results
Patients' characterstics
A total of 101 patients were included in this study and their clinical and pathological characteristics are summarized in Table 1.
Table 1: Age, sex and pathological features of 101 cases of PPGL
| Characteristic | Number or Frequency |
|---|---|
| Age (yrs) median (Range) | 38 (8–81) |
| Sex F: M | 61:40 |
| Tumor size (cm), Median (Range) | 5 (1–24) |
| Vascular Invasion | 10 (9.9%) |
| Capsular invasion | 19 (18.8%) |
| Distant Metastasis | 10 (9.9%) |
| Sites | |
| PCC (4 Bilateral) | 32 (31.7%) |
| Abdominal PGL | 26 (25.7%) |
| Head/Neck PGL (2 bilateral) | 39 (38.6%) |
| Other sites | 2 (1.99%) |
| Multiple sites (including 4 bilateral PCC and 2 bilateral head/neck PGL) | 8 (7.9%) |
Mutational analysis and genotype/phenotype correlation
Germline mutations were detected in 30 cases by PCR and direct Sanger sequencing. The remaining cases were subjected to whole exome sequencing using NGS as direct Sanger sequencing either did not reveal mutations in the known genes that were tested or was unsuccessful. NGS identified additional 7 cases with pathogenic variants. These 7 cases with NGS-detected mutations were subsequently confirmed by Sanger sequencing. Overall, of 101 sporadic cases of PPGL, 37 (36.6%) had germline mutations while 64 patients (63.4%) had no mutations in any of the genes known to be involved in the pathogenesis of PPGL (Table 2 and Figure 1). The most commonly mutated gene was SDHB (21/101 cases, 20.8%) followed by SDHC (4/101, 3.9%). SDHD, VHL and MAX genes were mutated in 3 (3%), 2 (2%) and 2 (2%) cases, respectively. The following genes were mutated in 1 patient each (1%), RET, SDHA, SDHAF2, TMEM127 and NF1.
Table 2: Overall results of genomic profiling of 101 cases of apparently sporadic PPGL
| Diagnosis | Total | Mutation-positive | SDHB | SDHD | SDHC | SDHA | SDHAF2 | RET | VHL | NF1 | MAX | TMEM127 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unilateral PCC (1 metastatic) | 28 | 4 | 1 | 1 | 1 | 1 | ||||||
| Bilateral PCC | 4 | 3 | 1 | 2 | ||||||||
| Abd. PGL (6 metastatic) | 26 | 16 | 12 | 2 | 1 | 1 | ||||||
| H/N PGL (2 metastatic) | 37 | 10 | 5 | 2 | 2 | 1 | ||||||
| Bilateral H/N PGL | 2 | 2 | 1 | 1 | ||||||||
| Abd. and H/N PGL | 1 | 1 | 1 | |||||||||
| PCC, abd. and H/N PGL | 1 | 1 | 1 | |||||||||
| Bladder PGL | 1 | 0 | ||||||||||
| Intradural PGL | 1 | 0 | ||||||||||
| Total | 101 | 37 (36.6%) | 21 (20.8%) | 3 (3%) | 4 (3.9%) | 1 (1%) | 1 (1%) | 1 (1%) | 2 (2%) | 1 (1%) | 2 (2%) | 1 (1%) |
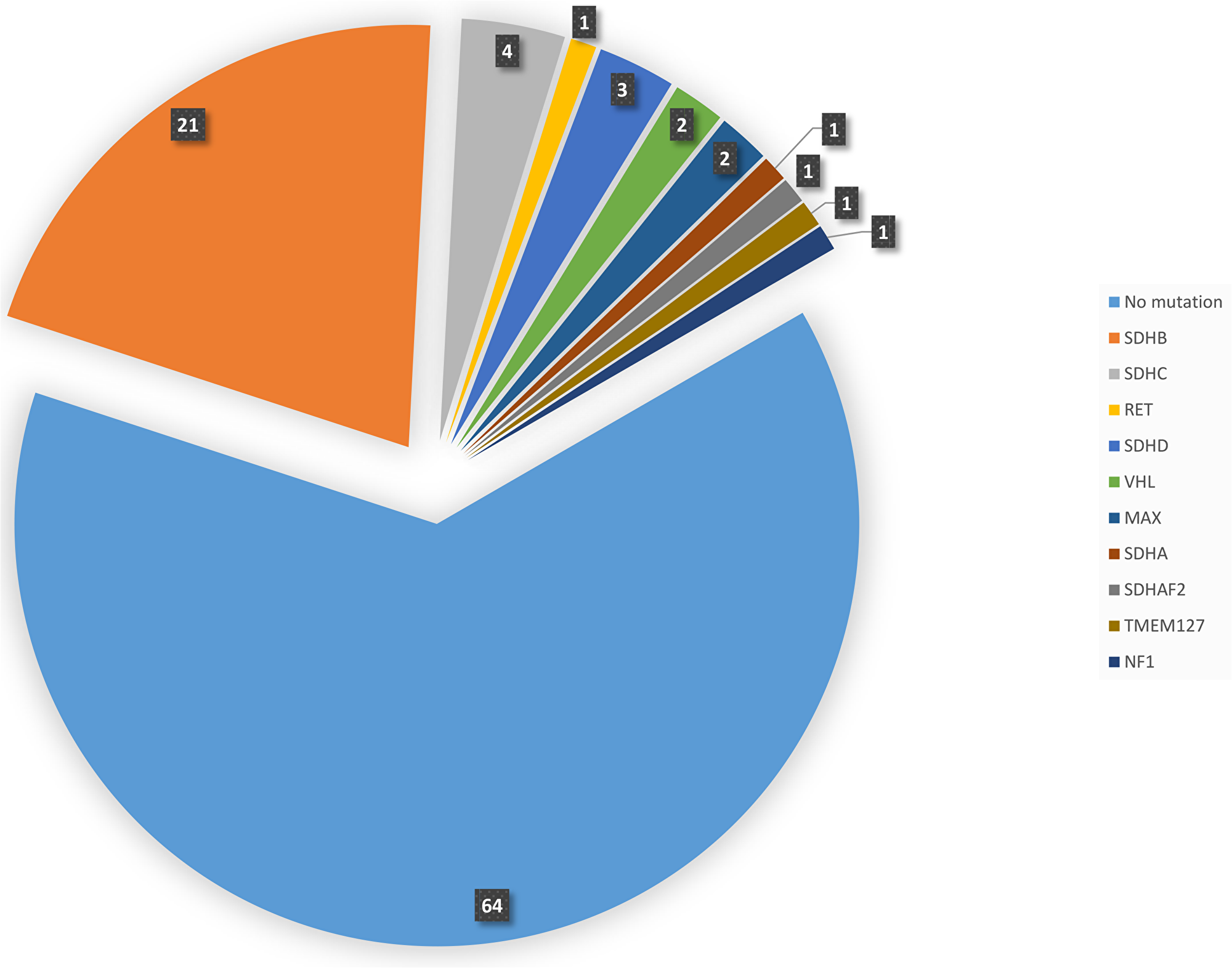
Figure 1: Pie diagram showing the distribution and number of cases with germline mutations in different gene.
SDHB mutations
The majority of cases with SDHB mutations presented with abdominal PGL (14/21 cases, 66.7%) (Table 3) including one patient with combined abdominal and head and neck PGL. Five cases presented with head/neck PGL of whom one case had bilateral carotid body tumors associated with the c.689G>A, p.R230H mutation. Two cases presented with PCC (one isolated and one with mediastinal and head and neck PGL). Six cases (28.6%) developed distant metastasis (Table 3). SDHB is the most commonly mutated gene in our series (21/37 mutations, 56.8%). The non-sense mutation c.268C>T, p.R90X was the most frequent mutation occurring in 12 out of 21 cases (57%) with SDHB mutations. In 4 cases (33%) with this mutation, the disease was metastatic (Table 3).
Table 3: SDHB mutations (NM_003000.2) in 21 cases of PPGL
| Diagnosis | Metastatic | Tissue | Sequencing | Mutation Nucleotide change | Mutation Amino acid change | Variant status |
|---|---|---|---|---|---|---|
| Abdominal PGL | Yes | Blood | Sanger | c.412G>A | p.D138N | Known Pathogenic [51] |
| Abdominal PGL | Yes | Blood | Sanger | c.689G>A | p.R230H | Known Pathogenic [52] |
| Abdominal PGL | Yes | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | Yes | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | Yes | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | Sanger | c.343C>T | p.R115X | Known Pathogenic [54] |
| Abdominal PGL | No | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Abdominal PGL | No | FFPE | NGS | c.637dupA | p.M213fs | Novel, likely pathogenic |
| H/N PGL | No | Blood | Sanger | c.689G>A | p.R230H | Known Pathogenic [55] |
| H/N PGL | No | Blood | Sanger | c.409A>G | p.K137E | Novel, likely pathogenic |
| H/N PGL | No | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| H/N PGL | No | FFPE | Sanger | c.79C>T | p.R27X | Known Pathogenic [2] |
| Bilateral H/N PGL | No | Blood | Sanger | c.689G>A | p.R230H | Known pathogenic [55] |
| Abdominal and H/N PGL | Yes | FFPE | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| Unilateral PCC | No | Blood | Sanger | c.268C>T | p.R90X | Known Pathogenic [53] |
| PCC, mediastinal PGL, H/N PGL | No | Blood | Sanger | c.689G>A | p.R230H | Known Pathogenic [54] |
SDHC mutations
Four patients had SDHC mutations. These mutations were missense in 3 cases and a splice site mutation in one case (Tables 2 and 4). Two of these 4 cases had abdominal PGL while the other two had head and neck PGL. All of these were benign tumors.
SDHD mutations
The most common variant found was c.34G>A, p. G12S occurring in 7/10 cases with SDHD variants (70%). However, the role of this variant in the pathogenesis of PPGL has been controversial. Excluding this variant, another 3 cases had germline SDHD mutations (Table 4). They all presented with head and neck PGL (bilateral in one case). Interestingly, a case with the nonsense mutation c.15G>A, p.W5X presented with multiple recurrent head and neck PGLs (Table 4).
Table 4: Mutations in RET, VHL, NF1, SDHA, SDHC, SDHD, SDHAF2, TEMEM127 and MAX
| Diagnosis | Gene | Tissue | Sequencing | Nucleotide change | Amino Acid Change | Variant status |
|---|---|---|---|---|---|---|
| Abdominal PGL | RET | FFPE | Sanger | c.1900T>C | p.C634R | Known Pathogenic [56] |
| Unilateral PCC | VHL | Blood | Sanger | c.482G>A | p.R161Q | Known Pathogenic [2, 57] |
| Bilateral PCC | VHL | Blood | Sanger | c.355T>C | p.F119L | Known Pathogenic [58] |
| Unilateral PCC | NF1 | Blood | NGS | c.4150G>A | p.E1384K | Novel, VUS |
| Unilateral H/N PGL | SDHA | Blood | Sanger | c.994C>T | p.P332S | Novel, likely pathogenic |
| Abdominal PGL | SDHC | FFPE | Sanger | c.305T>C | p.L102P | Novel, likely pathogenic |
| Abdominal PGL | SDHC | FFPE | Sanger | c.329C>T | p.P110L | Novel, likely pathogenic |
| H/N PGL | SDHC | Blood | NGS | c.78-2A>T | Splice site mutation | Known Pathogenic [59, 60] |
| H/N PGL | SDHC | Blood | Sanger | c.164A>G | p.H55R | Novel, likely pathogenic |
| H/N PGL | SDHD | FFPE | Sanger | c.184G>A | p.A62T | Novel, likely pathogenic |
| H/N PGL | SDHD | FFPE | Sanger | c.335C>T | p.T112I | Novel, likely pathogenic |
| Bilateral H/N PGL | SDHD | Blood | Sanger | c.15G>A | p.W5X | Likely pathogenic [2, 61] |
| Metastatic PCC | SDHAF2 | Blood | NGS | c.438C>A | p.N146K | Novel, VUS |
| Abdominal PGL | TMEM127 | Blood | NGS | c.281G>A | p.R94Q | Novel, likely pathogenic |
| Bilateral PCC | MAX | Blood | NGS | c.196C>T | p.R66X | Novel, likely pathogenic |
| Bilateral PCC | MAX | Blood | NGS | c.161T>A | p.I54N | Novel, likely pathogenic |
RET mutations
Only one patient without family or personal history of multiple endocrine neoplasia type 2 presented with abdominal PGL and his genetic testing revealed the well-known c.1900T>C, p.C634R (Table 4). In addition, the c.2071G>A, p.G691S variant is a single nucleotide polymorphism (SNP) that was quite common occurring in 6 cases (5 PCC and 1 abdominal PGL).
Other genes
A number of other known genes were mutated and presented with interesting manifestations (Table 4). VHL mutations were detected in 2 cases (Table 4). These cases presented with bilateral PCC in one case and unilateral PCC in another case (Table 4). MAX gene was mutated in 2 cases both of whom presented with bilateral PCC. A novel SDHAF2 variant was found in one case of metastatic PCC. By in Silico analysis (Mutation taster and Polyphen2), this variant is disease causing and probably damaging with a score of 0.999. Other genes included RET, TMEM127, SDHA and NF1. Each was mutated once. The tumor type, location and the mutations in these genes are detailed in Table 4. The patient with NF1 variant does not have features of neurofibromatosis. However, the variant was confirmed on Sanger sequencing and the in Silico analysis predicted it to be highly damaging.
Mutations by tumor type
Mutations in PCC
Our study included a total of 32 cases of PCC. These include 26 cases with unilateral, 4 with bilateral and 2 metastatic PCC. Twenty-four cases (75%) had no detectable germline mutations. The other 8 (8/32, 25%) cases harbored underlying germline mutations. The underlying genes and mutations are detailed in Table 5. Bilateral PCC occurred in 4 cases and the underlying genes were MAX in 2 cases, VHL in 1 case and no identifiable mutation in another case. In one unusual case with metastatic PCC, the underlying mutation was a novel SDHAF2 variant that is likely pathogenic (Tables 4 and 5).
Table 5: Germline mutations in patients with PCC, abdominal PGL and head and neck PGL
| Category | Diagnosis | Gene | Mutation (Nucleotide change) | Mutation (Amino Acid Change) |
|---|---|---|---|---|
| Pheochromocytoma | Unilateral PCC | SDHB | c.268C>T | p.R90X |
| Unilateral PCC | NF1 | c.4150G>A | p.E1384K | |
| Metastatic PCC | SDHAF2 | c.438C>A | p.N146K | |
| Unilateral PCC | VHL | c.482G>A | p.R161Q | |
| Bilateral PCC | VHL | c.355T>C | p.F119L | |
| Bilateral PCC | MAX | c.196C>T | p.R66X | |
| Bilateral PCC | MAX | c.161T>A | p.I54N | |
| Unilateral PCC, Mediastinal PGL, H/N PGL | SDHB | c.689G>A | p.R230H | |
| Abdominal Paraganglioma | Abdominal PGL | RET | c.1900T>C | p.C634R |
| Abdominal PGL | SDHB | c.637dupA | p.M213fs | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHB | c.343C>T | p.R115X | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Abdominal PGL | SDHC | c.305T>C | p.L102P | |
| Abdominal PGL | SDHC | c.329C>T | p.P110L | |
| Abdominal PGL | TMEM127 | c.281G>A | p.R94Q | |
| Metastatic Abdominal PGL | SDHB | c.412G>A | p.D138N | |
| Metastatic Abdominal PGL | SDHB | c.689 G>A | p.R230H | |
| Metastatic abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Metastatic Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Metastatic Abdominal PGL | SDHB | c.268C>T | p.R90X | |
| Metastatic abdominal and H/N PGL | SDHB | c.268C>T | p.R90X | |
| Head and neck paraganglioma | Unilateral H/N PGL | SDHA | c.994C>T | p.P332S |
| Unilateral H/N PGL | SDHB | c.409A>G | p.K137E | |
| Unilateral H/N PGL | SDHB | c.268C>T | p.R90X | |
| Unilateral H/N PGL | SDHB | c.79C>T | p.R27X | |
| Unilateral H/N PGL | SDHB | c.689G>A | p.R230H | |
| Unilateral H/N PGL | SDHC | c.78-2A>T | Splice site | |
| Unilateral H/N PGL | SDHC | c.164A>G | p.H55R | |
| Unilateral H/N PGL | SDHD | c.184G>A | p.A62T | |
| Unilateral H/N PGL | SDHD | c.335C>T | p.T112I | |
| Bilateral H/N PGL | SDHB | c.689G>A | p.R230H | |
| Bilateral H/N PGL | SDHD | c.15G>A | p.W5X |
Mutations in PGL
Overall, 69 cases of PGL were included, of which 26 abdominal PGL (6 metastatic), 35 head and neck PGL, 1 combined abdominal and head/neck PGL, 1 combined PCC, abdominal and head/neck PGLs, 1 bladder PGL, 2 bilateral head/neck PGL, 2 metastatic head and neck PGL and 1 intradural spinal PGL (Table 2). The spinal PGL occurred in a 28-year old man who presented with a 14-month history of lower back pain radiating to both hips and difficulty in walking. He had no hyperadrenergic symptoms. MRI of the spine showed an intradural tumor of 1.4 cm size at lumbar spine 3 (L3) compressing the cauda equina. Surgical resection and histolpathological examination confirmed a diagnosis of PGL with typical microscopic picture and positive Chromogranin A stain. Germline mutations were found in 29 of 69 (42%) of these PGLs while 40 cases were negative for any underlying mutation. These cases with positive mutations include 17 cases of abdominal PGL (5 metastatic), 11 cases of head and neck PGL (two bilateral) and one case of abdominal and head and neck PGLs (Table 5). The details of these mutations are summarized in Table 5.
Multiple PPGL
Four patients had bilateral PCC (2 with MAX, 1 VHL and 1 without identifiable mutation) (Table 5), one patient had synchronous abdominal and head/neck PGL (SDHB R90X mutation) (Table 5), two had bilateral head and neck PGLs (one SDHB and 1 SDHD mutation) (Table 5), and 1 patient had metachronous PCC, abdominal PGL and head and neck PGL (p.R230H SDHB mutation) (Table 5).
Metastatic PCC and PGL
Overall, 10/101 patients (10%) had metastatic PPGL and 7 of them (70%) had underlying germline mutations. Two patients had metastatic PCC (one had no mutation and one had an SDHAF2 mutation) (Tables 4 and 5). Six patients had abdominal PGL and SDHB mutations; one of them had both abdominal and head and neck PGLs (Table 3) and 2 patients had metastatic head and neck PGL (both negative for mutations). The first patient with head and neck PGL was a 38-year old lady who presented with a 7-cm left carotid body tumor that could not be completely resected as it was invading the surrounding structures and the carotid artery. Histopathologcal examination of the resected tumor confirmed the diagnosis of PGL with positive chromogranin A and synaptophysin stains and a Ki67 proliferation index of 3%. There was clear capsular and vascular invasion and positive margin. CT scan of the chest, abdomen and pelvis and MIBG whole body scan showed MIBG-avid bilateral lung metastases up to 1 cm in size. The patient was treated with surgery twice, chemotherapy and 3 doses of MIBG (cumulative dose 364 mCi) but continued to have progression of the lung metastases. She is currently stable with persistent bilateral lung metastases. The second patient was a 48-year old man who presented with a 4-year history of gradually enlarging right mid and upper neck mass with dizziness, headache and pain. CT scan of the neck, chest abdomen and pelvis showed a 6 × 4 cm right carotid body tumor and innumerable bilateral 1-1.5 cm lung, liver and skeletal metastases. These lesions were only faintly positive on MIBG but vey avid on octreotide whole body scan. Histopathological examination of the resected right carotid body tumor and biopsy from a liver lesion confirmed the diagnosis of metastatic carotid body tumor. The patient was supposed to start chemotherapy but was lost for follow up and is likely to have died secondary to the metastatic PGL.
DISCUSSION
In this study, we have comprehensively defined the genomic profile of PPGL from a previously unstudied highly consanguineous Arab population. Overall, we found a high rate (36.6%) of germline mutations in this series of patients with PPGL without family history of these tumors. In our study, SDHB mutations are the most common mutations in PPGL (20.8%) in general and in PGL in particular (30.4%). SDHC and SDHD mutations are much less common occurring in 3.9% and 3% respectively. Other genetic mutations are rare (Figure 1). None of the more recently described genes (DLST, SLC25A11 and DNMT3A) was found mutated in this study. Metastatic PPGL occurred in about 28.6% of patients (6/21) with SDHB mutations and in a patient with SDHAF2 mutation.
Mutations involved in the pathogenesis of PPGL have been recently classified into 4 categories; 1. pseudohypoxemia group involving mainly the succinate dehydrogenase subgroup (SDHA, SDHB, SDHC, SDHD and SDHAF2), fumarate hydratase (FH) and the VHL-EPAS1 subgroup; 2. The Tyrosine kinase group (RET, NF1, MAX, TMEM127 and HRAS); 3. WNT-related pathway (somatic mutations in MAML3 and CSDE1); and 4. adrenocortical admixture group [3]. The last group is less clear than others and its existence is controversial [17, 18].
Our study showed that most mutations involve the pseudohypoxemia group with the vast majority occurring in SDHB and to a lesser extent in SDHC and SDHD. These mutations occurred commonly in PGL but very rarely in PCC. SDHB was the most commonly mutated gene (56.8% of patients with positive mutations and 20.8% of all cases studied) and c.268C>T, p.R90X was the most common mutation (Tables 2 and 3). The high frequency of this mutation suggests that it might be a founder mutation in the studied population. By contrast, in the recently published TCGA data from an international consortium population, SDHB germline mutations occurred only in 9.8% (17/173 cases) and the p.R90X mutation occurred only in 1/17 cases [3]. The most frequent SDHB mutation in TCGA data was p.I127S (5/17 cases), which was not found in our patients [3]. This suggests significant ethnic differences in the molecular genetics of PPGL. By contrast, in the original report of germline mutations in non-syndromic PCC from Europe, SDHB mutations were found in 12 of 271 (4.4%) apparently sporadic PPGL (mostly PCC) [2] which is not much different from our study in which 2 out of 32 (6.3%) PCC carry SDHB mutations. A more recent report from Spain in which 329 sporadic single non-familial PPGL were tested for mutations showed an overall prevalence of germline mutations of 14%. Similar to our study, PGL were more commonly mutated (28.7% vs. 42% in our study) and most mutations occurred in SDHB (63% vs. 56.8% in our study) followed by SDHD (13% vs. 8% in our study) and SDHC (4.3% vs. 10.8% in our study) with other genes being rarely mutated [19]. In another study from USA, a review of 129 cases of PPGL who underwent surgery was undertaken and of 42 patients that were tested, 21 (50%) were positive for germline mutations. However, some of those cases were syndromic PPGL [20]. Another study from India reviewed 150 cases of PPGL of whom 30 cases were syndromic PPGL. These cases were tested only for 5 genes (RET, VHL, SDHB, SDHD and SDHC). All 30 syndromic PPGL were positive for germline mutations while in 120 cases of this series with sporadic PPGL, 19 (15.8%) had germline mutations with VHL, SDHB and SDHD being the most commonly mutated genes [21].
In the current study, SDHC mutations were the second most common mutations in PGL occurring in 4 out of 37 cases (10.8%) with positive mutations. This is unusual as the literature cited SDHC-associated PGL to be much less common than SDHB and SDHD-associated PGL [22–25]. In a review of a number of series with more than 3000 patients with PPGL, SDHC mutations occurred only in 1% of cases (31/3193 cases) [1]. In the TCGA data, SDHC mutations were not found [3]. Similarly, in a recent report from Europe, SDHC mutations were not found in 87 cases of PCC [24]. An SDHC founder mutation (c.397C>T, p. Arg133Ter) was detected in about 70% of a cohort of 29 French Canadian patients presenting mostly (70%) with head and neck PGL and with distant metastasis in 30% of them [26]. In our study, two of the SDHC-associated PGL occurred in the head and neck and two were abdominal PGL. All of these 4 cases were benign PGL. Other studies have shown that SDHC-related PGL commonly occur in the head and neck region but also in the mediastinum and abdominal regions [22, 23].
SDHD mutations occurred in three cases with head and neck PGL, two unilateral and one bilateral. Apart from these 3 cases, the most common SDHD variant found in this study was the c.34G>A, p.G12S occurring in 7 cases. The pathogenicity of this variant has been controversial with some studies suggesting that it is pathogenic while others suggested that is a non-pathogenic SNP [27–29]. This variant was not reported in the TCGA data [3]. If considered pathogenic, SDHD mutations would be the second most common mutations after SDHB (10/48, 20.8%). However, we considered it to be non-pathogenic as it also occurred in the normal Saudi Genome Project database (MIF 0.046%). In fact, all SDHD mutations were also rare in the TCGA data occurring only in 3 out of 173 cases (2%) [3]. However, TCGA study excluded head and neck PGL in which SDHD mutation most commonly occur [30].
Other pseudohypoxia genes were much less frequently mutated. SDHA mutations occurred only once in a patient with carotid body tumor (Table 4). Mutations in this gene are rare but can be associated with aggressive metastatic PGL similar to those of SDHB [31, 32]. One patient had an SDHAF2 variant of unknown significance and presented with metastatic PCC. SDHAF2 mutations are extremely rare and their association with metastatic PPGL has not been reported. However, this variant was assessed to be disease causing by mutation taster and probably damaging with a score of 0.999 on Polyphen2 analysis. In a large study from Spain and Netherland with more than 430 patients with SDHB- and SDHD- negative sporadic and familial PPGL tested specifically for SDHAF2, none was found to have SDHAF2 mutation [33]. SDHAF2 mutations were not also reported in TCGA data [3]. VHL mutations were detected in two patients; one with unilateral and one with bilateral PCC. VHL mutations have been commonly reported in hereditary PPGL and can be associated with unilateral or bilateral PCC or less frequently with PGL [2, 34].
Of the tyrosine kinase group, RET mutation occurred in only 1 case of abdominal PGL. Interestingly, MAX mutations occurred in 2 cases of PCC, both of whom presented with bilateral PCC. MAX mutations-associated PCC tend to be bilateral and may metastasize to distant sites [35]. In a study of 972 cases from the European-American-Asian Pheochromocytoma and Paraganglioma Registry without mutations in the common PPGL genes, 58 had mutations in less commonly mutated genes (SDHA, TMEM127, SDHAF2) including 8 cases of PCC with MAX mutations. Two of these 8 cases were bilateral PCC [32]. We also found one case with a TMEM127 mutation in an abdominal PGL. TMEM127 mutations are generally more frequent than MAX mutations and may present with PCC, abdominal or head and neck PGL [32, 36].
Distant metastases are associated with unfavorable prognosis and are essentially incurable [37, 38]. Genetic markers that may predict development of metastatic PPGL can be of significant value for early and more effective intervention and closer follow up [39–44]. SDHB mutations have been associated with increased risk of distant metastasis [45–49]. The association between other gene mutations and distant metastasis are less clear [40, 43]. In this study, SDHB mutations remain the most frequently associated mutations with distant metastasis occurring in 6 out of 21 cases (28.6%). Only one additional case with an SDHAF2 variant developed distant metastasis. However, 3 cases with distant metastasis had no detectable mutations in any of known or potential candidate gene.
Our study is the first to evaluate the rates, types of germline mutations and the phenotype-genotype correlation in a large series of PPGL from an Arab population. However, it has some shortcomings including limitation of our study to germline mutations. Some previously reported mutations are somatic, particularly the WNT-related MAML4 fusion and HRAS mutations [3]. We have not included patients with positive family history of PPGL since our aim was to assess the rates of undiagnosed hereditary cases and to discover cases that seem sporadic. However, data from this study from an Arab population strongly supports genetic screening for patients with PPGL in this population since the frequency of germline mutations is high. It also suggests that for targeted sequencing, SDHB should be the first gene to be tested, especially in patients with abdominal or metastatic PGL. Other genes, especially SDHC and SDHD should be tested when mutations are not found in SDHB or the clinical presentation suggests a likely genetic mutation (e. g. VHL and MAX in bilateral PCC).
In conclusion, approximately 37% of our patients with non-familial PPGL harbor germline mutations in different susceptibility genes. The most commonly mutated gene is SDHB presenting mostly with abdominal PGL and less frequently with head and neck PGL. It is also associated with the highest risk of distant metastasis. SDHC and SDHD mutations are the second most common genetic alterations in PGL. VHL and MAX mutations occur mainly in PCC and tend to present with bilateral disease. Other genetic alterations are rare but have unique presentations.
Patients and methods
Patients
We studied all patients of PPGL who have no definite family history of such tumors and were seen at the King Faisal Specialist Hospital and Research Center (KFSHRC), Riyadh, Saudi Arabia during the period of January 2003-January 2019. KFSHRC is the main tertiary care center in Saudi Arabia where most cases of PPGL are referred to. Over this period, we managed 101 patients with non-familial PPGL. To avoid selection bias, we excluded patients with known familial PPGL syndromes. All cases underwent surgery and the histopathological examination confirmed the diagnosis of PPGL. The patients’ characteristics are summarized in Table 1.
Samples
We obtained an Institutional Review Board approval from the Office of Research Affairs of the KFSHRC. Informed consents for all prospective blood sample collection were obtained from the patients or their guardians. We collected blood samples from 53 cases. However, in cases where blood samples were not available for testing, we used formalin fixed paraffin embedded (FFPE) non-tumorous tissue (48 patients). To ensure that testing is for germline mutations, the tissue was carefully selected by an experienced pathologist (H. A) from previously surgically removed normal tissues avoiding any tumor tissue. Genomic DNA was isolated from peripheral blood leucocytes using the Gentra Blood Kit (Qiagen Corp, Valencia, CA, USA) according to the manufacturer’s instructions. For isolation of DNA from FFPE, DNA was extracted using a commercial DNA extraction kit (QIAamp DNA FFPE Tissue Kit, QIAGEN, Catalog No. 56404) according to the manufacturer’s instructions. DNA was quantified using a nanodrop2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and its purity was assured by the A260/280 ratio of ≥ 1.8 indicating good purity DNA. We performed polymerase chain reaction (PCR) and direct Sanger sequencing using Big Dye terminator v3.1 cycle sequencing reaction kit and an ABI PRISM 3730XL genetic analyzer (Applied Biosystems) to detect mutations in RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, VHL, MAX and TMEM127. The primers and PCR conditions have been published previously [2, 11, 50]. When a pathogenic mutation was found in one of these genes, no further testing was performed in the remaining genes. This approach led to identification of 30 pathogenic or likely pathogenic variants in any one of these genes (Table 2). We subjected the remaining cases in whom no mutation was found by PCR and Sanger sequencing or when Sanger sequencing was unsuccessful or unclear to NGS-based whole exome analysis. Variants reported by NGS were subsequently confirmed by direct Sanger Sequencing.
Whole exome sequencing
Whole exome sequencing was achieved using the Ion Proton platform (AmpliSeq kit). Briefly, 100 ng DNA from each sample were collected and the extracted DNA is then amplified using AmpliSeq HiFi mix (Life Technologies, Carlsbad, CA, USA) for 10 cycles. The resultant PCR products were then pooled followed by primer digestion using FuPa reagent (Life Technologies, Carlsbad, CA, USA). A ligation step was then conducted using Ion P1 and Ion Xpress Barcode adapters. After that the libraries were purified and quantified using qPCR and the Ion Library Quantification Kit (Life Technologies, Carlsbad, CA, USA). The next step included emulsion of the libraries using Ion OneTouch System to attach the DNA fragments to the Ion Sphere particles. The final step in the library preparation included enrichment of the Ion Sphere particles using Ion OneTouch ES (Life Technologies, Carlsbad, CA, USA). Once the library became ready, they are loaded on the sequencing chip which is then inserted into the Ion Proton instrument (Life Technologies, Carlsbad, CA, USA) for sequencing.
Bioinformatics analysis
For bioinformatics analysis, we used the Torrent Suite (https://github.com/iontorrent/TS) analysis kit, using the manufacturer’s recommended parameters for base calling and alignment. The first step after base calling is to check the reads for quality and trim the low-quality parts. Then the reads are aligned to the reference human genome (version hg19) using the manufacturer’s recommended parameters. After the alignment, we used the variant calling pipeline of the Torrent suite both of which are based on the BWA-GATK pipeline, but they were more tuned to the Ion Torrent technology, by including flow signal information and library of common sequencing error motifs to improve the accuracy.
After variant calling, we ran the in-house developed annotation pipeline. This pipeline is based on the Annovar package (http://annovar.openbioinformatics.org), which includes about 40 databases. We also added more information tracks including variant frequencies from the database of the Saudi Human Genome Program.
To speed up the analysis, the list of annotated variants per each sample was filtered to remove intronic and synonymous variants. The remaining variants were then prioritized based on the following criteria: 1) Existence in the set of genes which are known to be related to the disease, 2) the effect score (whether the variant is truncating/damaging or not), 3) frequency in public databases and Saudi population.
Quality of sequencing
The sequencing quality was assessed using different metrics. These showed that the target regions are well covered by the NGS reads (99.6% total average coverage at 1× and 96.88% average coverage at 20×) with an average depth of 224 (i. e. each base in the target region is covered by 224 reads on average). Genetic variants detected by NGS were subsequently confirmed by PCR and targeted Sanger sequencing of the exons/introns in which these variants were found.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work is supported by intramural financial support of the Research Centre of the King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia.
References
1. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, and Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014; 99:1915–42. https://doi.org/10.1210/jc.2014-1498. [PubMed].
2. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, et al, and Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002; 346:1459–66. https://doi.org/10.1056/NEJMoa020152. [PubMed].
3. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, Ling S, Jefferys SR, de Cubas AA, et al, and Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell. 2017; 31:181–93. https://doi.org/10.1016/j.ccell.2017.01.001. [PubMed].
4. Alrezk R, Suarez A, Tena I, Pacak K. Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging. Front Endocrinol (Lausanne). 2018; 9:515. https://doi.org/10.3389/fendo.2018.00515. [PubMed].
5. Buffet A, Ben Aim L, Leboulleux S, Drui D, Vezzosi D, Libé R, Ajzenberg C, Bernardeschi D, Cariou B, Chabolle F, Chabre O, Darrouzet V, Delemer B, et al, and French Group of Endocrine Tumors (GTE) and COMETE Network. Positive Impact of Genetic Test on the Management and Outcome of Patients With Paraganglioma and/or Pheochromocytoma. J Clin Endocrinol Metab. 2019; 104:1109–18. https://doi.org/10.1210/jc.2018-02411. [PubMed].
6. Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Currás-Freixes M, Letón R, Torres-Pérez R, Richter S, Pita G, Herráez B, Cianchetta G, Honrado E, et al. Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. Am J Hum Genet. 2019; 104:651–664. https://doi.org/10.1016/j.ajhg.2019.02.017. [PubMed] Erratum in: Recurrent Germline DLST Mutations in Individuals with Multiple Pheochromocytomas and Paragangliomas. [Am J Hum Genet. 2019]. https://doi.org/10.1016/j.ajhg.2019.04.010. [PubMed].
7. Buffet A, Morin A, Castro-Vega LJ, Habarou F, Lussey-Lepoutre C, Letouzé E, Lefebvre H, Guilhem I, Haissaguerre M, Raingeard I, Padilla-Girola M, Tran T, Tchara L, et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018; 78:1914–22. https://doi.org/10.1158/0008-5472.CAN-17-2463. [PubMed].
8. Remacha L, Currás-Freixes M, Torres-Ruiz R, Schiavi F, Torres-Pérez R, Calsina B, Letón R, Comino-Méndez I, Roldán-Romero JM, Montero-Conde C, Santos M, Pérez LI, Pita G, et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet Med. 2018; 20:1644–51. https://doi.org/10.1038/s41436-018-0003-y. [PubMed].
9. Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, Sanna M, Yao M, Robledo M, Neumann HP, Woodward ER, Latif F, Abbs S, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol. 2015; 2015:138573. https://doi.org/10.1155/2015/138573. [PubMed].
10. Crona J, Delgado Verdugo A, Maharjan R, Stålberg P, Granberg D, Hellman P, Björklund P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab. 2013; 98:E1266–71. https://doi.org/10.1210/jc.2012-4257. [PubMed].
11. Burnichon N, Vescovo L, Amar L, Libé R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin PF, Jeunemaitre X, Favier J, Gimenez-Roqueplo AP. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011; 20:3974–85. https://doi.org/10.1093/hmg/ddr324. [PubMed].
12. Ben Aim L, Pigny P, Castro-Vega LJ, Buffet A, Amar L, Bertherat J, Drui D, Guilhem I, Baudin E, Lussey-Lepoutre C, Corsini C, Chabrier G, Briet C, et al. Targeted next-generation sequencing detects rare genetic events in pheochromocytoma and paraganglioma. J Med Genet. 2019; 56:513–20. https://doi.org/10.1136/jmedgenet-2018-105714. [PubMed].
13. Goncalves J, Lussey-Lepoutre C, Favier J, Gimenez-Roqueplo AP, Castro-Vega LJ. Emerging molecular markers of metastatic pheochromocytomas and paragangliomas. Ann Endocrinol (Paris). 2019; 80:159–62. https://doi.org/10.1016/j.ando.2019.04.003. [PubMed].
14. Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers (Basel). 2019; 11. https://doi.org/10.3390/cancers11040436. [PubMed].
15. Brito JP, Asi N, Bancos I, Gionfriddo MR, Zeballos-Palacios CL, Leppin AL, Undavalli C, Wang Z, Domecq JP, Prustsky G, Elraiyah TA, Prokop LJ, Montori VM, Murad MH. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: a systematic review. Clin Endocrinol (Oxf). 2015; 82:338–45. https://doi.org/10.1111/cen.12530. [PubMed].
16. el-Hazmi MA, al-Swailem AR, Warsy AS, al-Swailem AM, Sulaimani R, al-Meshari AA. Consanguinity among the Saudi Arabian population. J Med Genet. 1995; 32:623–26. https://doi.org/10.1136/jmg.32.8.623. [PubMed].
17. Crona J, Backman S, Welin S, Taïeb D, Hellman P, Stålberg P, Skogseid B, Pacak K. RNA-Sequencing Analysis of Adrenocortical Carcinoma, Pheochromocytoma and Paraganglioma from a Pan-Cancer Perspective. Cancers (Basel). 2018; 10. https://doi.org/10.3390/cancers10120518. [PubMed].
18. Flynn A, Dwight T, Harris J, Benn D, Zhou L, Hogg A, Catchpoole D, James P, Duncan EL, Trainer A, Gill AJ, Clifton-Bligh R, Hicks RJ, Tothill RW. Pheo-Type: A Diagnostic Gene-expression Assay for the Classification of Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab. 2016; 101:1034–43. https://doi.org/10.1210/jc.2015-3889. [PubMed].
19. Currás-Freixes M, Inglada-Pérez L, Mancikova V, Montero-Conde C, Letón R, Comino-Méndez I, Apellániz-Ruiz M, Sánchez-Barroso L, Aguirre Sánchez-Covisa M, Alcázar V, Aller J, Álvarez-Escolá C, Andía-Melero VM, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015; 52:647–56. https://doi.org/10.1136/jmedgenet-2015-103218. [PubMed].
20. Asban A, Kluijfhout WP, Drake FT, Beninato T, Wang E, Chomsky-Higgins K, Shen WT, Gosnell JE, Suh I, Duh QY. Trends of genetic screening in patients with pheochromocytoma and paraganglioma: 15-year experience in a high-volume tertiary referral center. J Surg Oncol. 2018; 117:1217–22. https://doi.org/10.1002/jso.24961. [PubMed].
21. Pandit R, Khadilkar K, Sarathi V, Kasaliwal R, Goroshi M, Khare S, Nair S, Raghavan V, Dalvi A, Hira P, Fernandes G, Sathe P, Rojekar A, et al. Germline mutations and genotype-phenotype correlation in Asian Indian patients with pheochromocytoma and paraganglioma. Eur J Endocrinol. 2016; 175:311–23. https://doi.org/10.1530/EJE-16-0126. [PubMed].
22. Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F, Coupier I, Thieblot P, Lecomte P, et al, and PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009; 94:2817–27. https://doi.org/10.1210/jc.2008-2504. [PubMed].
23. Else T, Marvin ML, Everett JN, Gruber SB, Arts HA, Stoffel EM, Auchus RJ, Raymond VM. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J Clin Endocrinol Metab. 2014; 99:E1482–86. https://doi.org/10.1210/jc.2013-3853. [PubMed].
24. Sbardella E, Cranston T, Isidori AM, Shine B, Pal A, Jafar-Mohammadi B, Sadler G, Mihai R, Grossman AB. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine. 2018; 59:175–82. https://doi.org/10.1007/s12020-017-1310-9. [PubMed].
25. Khadilkar K, Sarathi V, Kasaliwal R, Pandit R, Goroshi M, Shivane V, Lila A, Bandgar T, Shah NS. Genotype-phenotype correlation in paediatric pheochromocytoma and paraganglioma: a single centre experience from India. J Pediatr Endocrinol Metab. 2017; 30:575–81. https://doi.org/10.1515/jpem-2016-0375. [PubMed].
26. Bourdeau I, Grunenwald S, Burnichon N, Khalifa E, Dumas N, Binet MC, Nolet S, Gimenez-Roqueplo AP. A SDHC Founder Mutation Causes Paragangliomas (PGLs) in the French Canadians: New Insights on the SDHC-Related PGL. J Clin Endocrinol Metab. 2016; 101:4710–18. https://doi.org/10.1210/jc.2016-1665. [PubMed].
27. Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000; 60:6822–25. [PubMed].
28. Cascón A, Ruiz-Llorente S, Cebrián A, Letón R, Tellería D, Benítez J, Robledo M. G12S and H50R variations are polymorphisms in the SDHD gene. Genes Chromosomes Cancer. 2003; 37:220–21. https://doi.org/10.1002/gcc.10212. [PubMed].
29. Lendvai N, Tóth M, Valkusz Z, Bekő G, Szücs N, Csajbók E, Igaz P, Kriszt B, Kovács B, Rácz K, Patócs A. Over-representation of the G12S polymorphism of the SDHD gene in patients with MEN2A syndrome. Clinics (São Paulo). 2012; 67:85–89. https://doi.org/10.6061/clinics/2012(Sup01)15. [PubMed].
30. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000; 287:848–51. https://doi.org/10.1126/science.287.5454.848. [PubMed].
31. Jha A, de Luna K, Balili CA, Millo C, Paraiso CA, Ling A, Gonzales MK, Viana B, Alrezk R, Adams KT, Tena I, Chen A, Neuzil J, et al. Clinical, Diagnostic, and Treatment Characteristics of SDHA-Related Metastatic Pheochromocytoma and Paraganglioma. Front Oncol. 2019; 9:53. https://doi.org/10.3389/fonc.2019.00053. [PubMed].
32. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, Wellner U, Malinoc A, Taschin E, Barbon G, Lanza V, Söderkvist P, Stenman A, et al, and European-American-Asian Pheochromocytoma-Paraganglioma Registry Study Group. Clinical Characterization of the Pheochromocytoma and Paraganglioma Susceptibility Genes SDHA, TMEM127, MAX, and SDHAF2 for Gene-Informed Prevention. JAMA Oncol. 2017; 3:1204–12. https://doi.org/10.1001/jamaoncol.2017.0223. [PubMed].
33. Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suárez C, Hussain AK, Vriends AH, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010; 11:366–72. https://doi.org/10.1016/S1470-2045(10)70007-3. [PubMed].
34. Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, Nathanson KL, States LJ, Tomlinson GE, Villani A, Voss SD, Schiffman JD, Wasserman JD. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res. 2017; 23:e68–75. https://doi.org/10.1158/1078-0432.CCR-17-0547. [PubMed].
35. Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G, Gómez-Graña A, de Cubas AA, Inglada-Pérez L, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011; 43:663–67. https://doi.org/10.1038/ng.861. [PubMed].
36. Abermil N, Guillaud-Bataille M, Burnichon N, Venisse A, Manivet P, Guignat L, Drui D, Chupin M, Josseaume C, Affres H, Plouin PF, Bertherat J, Jeunemaître X, Gimenez-Roqueplo AP. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J Clin Endocrinol Metab. 2012; 97:E805–09. https://doi.org/10.1210/jc.2011-3360. [PubMed].
37. Toledo R, Jimenez C. Recent advances in the management of malignant pheochromocytoma and paraganglioma: focus on tyrosine kinase and hypoxia-inducible factor inhibitors. F1000Res. 2018; 7. https://doi.org/10.12688/f1000research.13995.1. [PubMed].
38. Hamidi O. Metastatic pheochromocytoma and paraganglioma: recent advances in prognosis and management. Curr Opin Endocrinol Diabetes Obes. 2019; 26:146–54. https://doi.org/10.1097/MED.0000000000000476. [PubMed].
39. Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, Bertagna X, Schlumberger M, Jeunemaitre X, Gimenez-Roqueplo AP, Plouin PF. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007; 92:3822–28. https://doi.org/10.1210/jc.2007-0709. [PubMed].
40. Bickmann JK, Sollfrank S, Schad A, Musholt TJ, Springer E, Miederer M, Bartsch O, Papaspyrou K, Koutsimpelas D, Mann WJ, Weber MM, Lackner KJ, Rossmann H, Fottner C. Phenotypic variability and risk of malignancy in SDHC-linked paragangliomas: lessons from three unrelated cases with an identical germline mutation (p.Arg133*). J Clin Endocrinol Metab. 2014; 99:E489–96. https://doi.org/10.1210/jc.2013-3486. [PubMed].
41. Burnichon N, Buffet A, Gimenez-Roqueplo AP. Pheochromocytoma and paraganglioma: molecular testing and personalized medicine. Curr Opin Oncol. 2016; 28:5–10. https://doi.org/10.1097/CCO.0000000000000249. [PubMed].
42. Fishbein L, Ben-Maimon S, Keefe S, Cengel K, Pryma DA, Loaiza-Bonilla A, Fraker DL, Nathanson KL, Cohen DL. SDHB mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr Relat Cancer. 2017; 24:L51–55. https://doi.org/10.1530/ERC-17-0086. [PubMed].
43. van Hulsteijn LT, Dekkers OM, Hes FJ, Smit JW, Corssmit EP. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet. 2012; 49:768–76. https://doi.org/10.1136/jmedgenet-2012-101192. [PubMed].
44. Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, de la Fouchardière C, Valdes N, Riccardi F, Do Cao C, Bertherat J, Goichot B, Beuschlein F, et al, and European Network for the Study of Adrenal Tumors (ENS@T). Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J Clin Endocrinol Metab. 2019; 104:2367–74. https://doi.org/10.1210/jc.2018-01968. [PubMed].
45. Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X, and COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003; 63:5615–21. [PubMed].
46. King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, Wesley R, Lodish M, Raygada M, Gimenez-Roqueplo AP, McCormack S, Eisenhofer G, Milosevic D, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011; 29:4137–42. https://doi.org/10.1200/JCO.2011.34.6353. [PubMed].
47. Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006; 91:4505–09. https://doi.org/10.1210/jc.2006-0423. [PubMed].
48. Turkova H, Prodanov T, Maly M, Martucci V, Adams K, Widimsky J Jr, Chen CC, Ling A, Kebebew E, Stratakis CA, Fojo T, Pacak K. Characteristics and outcomes of metastatic sdhb and sporadic pheochromocytoma/paraganglioma: an national institutes of health study. Endocr Pract. 2016; 22:302–14. https://doi.org/10.4158/EP15725.OR. [PubMed].
49. Hamidi O, Young WF Jr, Gruber L, Smestad J, Yan Q, Ponce OJ, Prokop L, Murad MH, Bancos I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin Endocrinol (Oxf). 2017; 87:440–50. https://doi.org/10.1111/cen.13434. [PubMed].
50. Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005; 23:8812–18. https://doi.org/10.1200/JCO.2005.03.1484. [PubMed].
51. Lefebvre S, Borson-Chazot F, Boutry-Kryza N, Wion N, Schillo F, Peix JL, Brunaud L, Finat A, Calender A, Giraud S. Screening of mutations in genes that predispose to hereditary paragangliomas and pheochromocytomas. Horm Metab Res. 2012; 44:334–38. https://doi.org/10.1055/s-0032-1306308. [PubMed].
52. Koenig W, Hehr R, Ditschuneit HH, Kuhn K, Ernst E, Rosenthal J, Hombach V. Lovastatin alters blood rheology in primary hyperlipoproteinemia: dependence on lipoprotein(a)? J Clin Pharmacol. 1992; 32:539–45. https://doi.org/10.1177/009127009203200609. [PubMed].
53. Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001; 69:49–54. https://doi.org/10.1086/321282. [PubMed].
54. Bayley JP, van Minderhout I, Weiss MM, Jansen JC, Oomen PH, Menko FH, Pasini B, Ferrando B, Wong N, Alpert LC, Williams R, Blair E, Devilee P, Taschner PE. Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma. BMC Med Genet. 2006; 7:1. https://doi.org/10.1186/1471-2350-7-1. [PubMed].
55. Klein RD, Jin L, Rumilla K, Young WF Jr, Lloyd RV. Germline SDHB mutations are common in patients with apparently sporadic sympathetic paragangliomas. Diagn Mol Pathol. 2008; 17:94–100. https://doi.org/10.1097/PDM.0b013e318150d67c. [PubMed].
56. Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, Di Fiore PP. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995; 267:381–83. https://doi.org/10.1126/science.7824936. [PubMed].
57. Couvé S, Ladroue C, Laine E, Mahtouk K, Guégan J, Gad S, Le Jeune H, Le Gentil M, Nuel G, Kim WY, Lecomte B, Pagès JC, Collin C, et al. Genetic evidence of a precisely tuned dysregulation in the hypoxia signaling pathway during oncogenesis. Cancer Res. 2014; 74:6554–64. https://doi.org/10.1158/0008-5472.CAN-14-1161. [PubMed].
58. Kang HC, Kim IJ, Park JH, Shin Y, Jang SG, Ahn SA, Park HW, Lim SK, Oh SK, Kim DJ, Lee KW, Choi YS, Park YJ, et al. Three novel VHL germline mutations in Korean patients with von Hippel-Lindau disease and pheochromocytomas. Oncol Rep. 2005; 14:879–83. https://doi.org/10.3892/or.14.4.879. [PubMed].
59. Peczkowska M, Cascon A, Prejbisz A, Kubaszek A, Cwikła BJ, Furmanek M, Erlic Z, Eng C, Januszewicz A, Neumann HP. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008; 4:111–15. https://doi.org/10.1038/ncpendmet0726. [PubMed].
60. Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, Morris Q, Barash Y, Krainer AR, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015; 347:1254806. https://doi.org/10.1126/science.1254806. [PubMed].
61. Beristain E, Vicente MA, Guerra I, Gutiérrez-Corres FB, Garin I, Perez de Nanclares G. Disomy as the genetic underlying mechanisms of loss of heterozigosity in SDHD-paragangliomas. J Clin Endocrinol Metab. 2013; 98:E1012–16. https://doi.org/10.1210/jc.2012-4083. [PubMed].