
Introduction
With a prevalence of 170 000 adults in the US alone, meningioma is now recognized as the most common primary intracranial tumor [1, 2]. Typically, meningiomas arise from the outermost layer of arachnoid mater cap cells [3]. Although most are benign, approximately 20% show aggressive behavior with 5-year recurrence rates of 50-80% in grade II and III cases [4]. Complete surgical resection is the primary goal during disease management; however, this is only achieved in ~50% of cases due the anatomical complexity and proximity to crucial nearby structures, particularly in tumors occupying the skull base [5, 6]. As approximately 25% of intracranial meningiomas involve the petroclival region, cavernous sinus, temporal fossa, and foramen magnum, complete surgical resection by neurosurgeons is challenging [3]. The identification of predictors of meningioma recurrence is critical to advance our understanding of tumorigenesis and improve the possibility of targeted therapy for these patients [7–15]. This is important since high-grade meningiomas frequently recur and are associated with high rates of morbidity and mortality [16].
Since the introduction of next generation sequencing (NGS), brain tumors are classified based on their molecular parameters as reported in the 2016 World Health Organization (WHO) classification [17]. However, for meningiomas, histological features remain the main differentiating factor, as our understanding of the genomic aberrations that drive these tumors remains incomplete. The 2016 World Health Organization (WHO) classification classes meningiomas into 3 grades and ~15 histopathological subtypes. Tumors are classed as WHO grade I–III based on mitotic activity, neuronal invasion and other aggressive features (e.g. macronuclei, hypercellularity and necrosis). Specific histologies are used to specify tumor grade such as those with clear-cell or chordoid histological morphologies are defined as grade II, whilst papillary meningiomas are classified as grade III. The controversy in these grading systems necessitates the need for more detailed investigations to identify mutational markers, which can be further integrated into combined histological-molecular classifications.
In this regard, Neurofibromin 2 (merlin, NF2) is recognized as the main tumor suppressor gene in meningioma, as it is observed in 40 to 60% of early-stage tumors [18, 19]. NF2 non-mutated tumors are reported to express other isolated chromosomal alterations and gene mutations at relatively high frequencies [20]. Most commonly, TNF receptor-associated factor 7 (TRAF7), Smoothened, frizzled family receptor (SMO), Krupple-like factor 4 (KLF4), and v-akt murine thymoma viral oncogene homolog 1 (AKT1) have been identified. More recently, a mutation in Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform (PIK3CA) was detected by targeted sequencing of 150 meningiomas [21]. When investigating the correlation between clinicopathological features and genotype, 80% of NF2-mutated meningiomas were found in the calvarium. To date, skull base lesions have not been shown to express other non-NF2 mutations [22].
In Saudi Arabia, the prevalence of brain tumors is approximately 0.3%; the second highest in the Middle East [23]. In studies assessing the epidemiology of primary brain tumors in Saudi Arabia, grade I meningiomas are the most commonly diagnosed pathological type, with overall recurrence rates of between 10.5 to 22.0% [24]. In this study, we reveal key genetic hotspots that contribute to the tumorigenesis and progression of meningioma in Saudi Arabia with special focus on skull based cases. We identified novel mutations in non-NF2 skull base tumors that may be related to tumor prognosis including Fibroblast growth receptor-3 (FGFR3). Mutations in this gene have not been demonstrated in human CNS tumors, despite its implications in several other forms of neoplasia.
Results
Cohort demographics
The study cohort comprised 71 patients with histologically proven meningioma (grades I, II, and III) reviewed by a board-certified neuropathologist (MA). The median patient age was 54.8 years (range 27–96 years). Most of the patients were female (51/71, 71.83%), while 20/71 (28.17%) were male. According to the WHO grade analysis, 57/71 patients had grade I (80.28%), 13/71 (18.30%) had grade II and 1/71 (1.420%) had grade III tumors (Figure 1A). A total of 12/71 patients (17.14%) received radiation therapy. A total of 51/71 of the tumors were found in the skull base (71.83%). Of these, 21 were anterior, 17 were middle and 13 were posterior tumors (Figure 1B). A total of 60/71 (84.51%) were primary tumors, and 10/71 were recurrent tumors (14.08%). According to WHO criteria, 22/71 (31.42%) of the tumors were meningothelial, 6/71 (8.57%) were atypical, 6/71 (8.57%) were transitional, 6 were grade f (8.57%), 3/71 (4.28) were secretory, and 2/71 (2.86%) were chordoid (Figure 1C). The patient characteristics are summarized in Figure 1 and Supplementary Table 1.
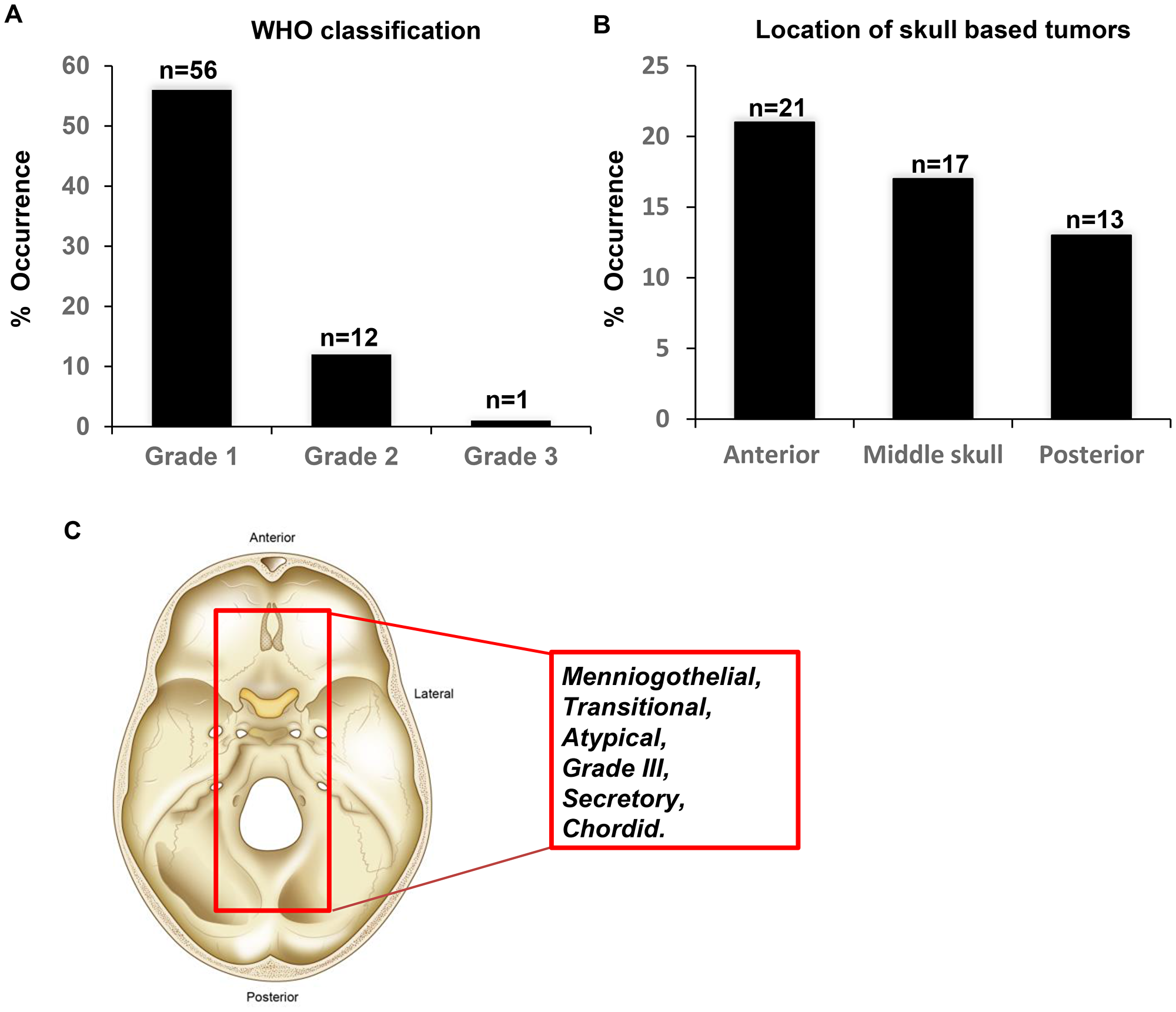
Figure 1: Characteristics of the meningioma’s investigated in the study. (A) Tumor grade according to the most recent WHO classification. (B) Location of the skull based tumors. (C) Schematic demonstrating the histology of the identified tumors from the study cohort.
NGS analysis
Mutations were detected in 68/71 patients (95.77%) with an average of 1.56 ± 1.07 genomic alterations (GAs) per patient (Figure 2B, Supplementary Table 2). Novel mutations found in three genes NF2, FGFR3 and PIK3CA. For NF2, 35 cases carried the missense mutation c.1060G>A (p.Ala354Thr), However, two of the 35 cases carried known pathogenic stop-gained mutation: c.784C>T (p.Arg262Ter). Another two novel missense mutations: c.925G>A (p.Glu309Lys) and c.1376T>C (p.Leu459Pro) found in two cases. Both mutations were predicted to have a damaging phenotype with PolyPhen-2 HVAR scores equaling 0.909 for c.925G>A (p.Glu309Lys) mutation and 1 for c.1376T>C (p.Leu459Pro) mutation. MutationTaster predictions showed a disease-causing result with a rank score equal to 0.81 for both mutations, while PROVEAN results suggested that both mutations are deleterious with scores equal to -3,32 and -6.32, respectively. For frameshift deletions, one mutation was found in one case.
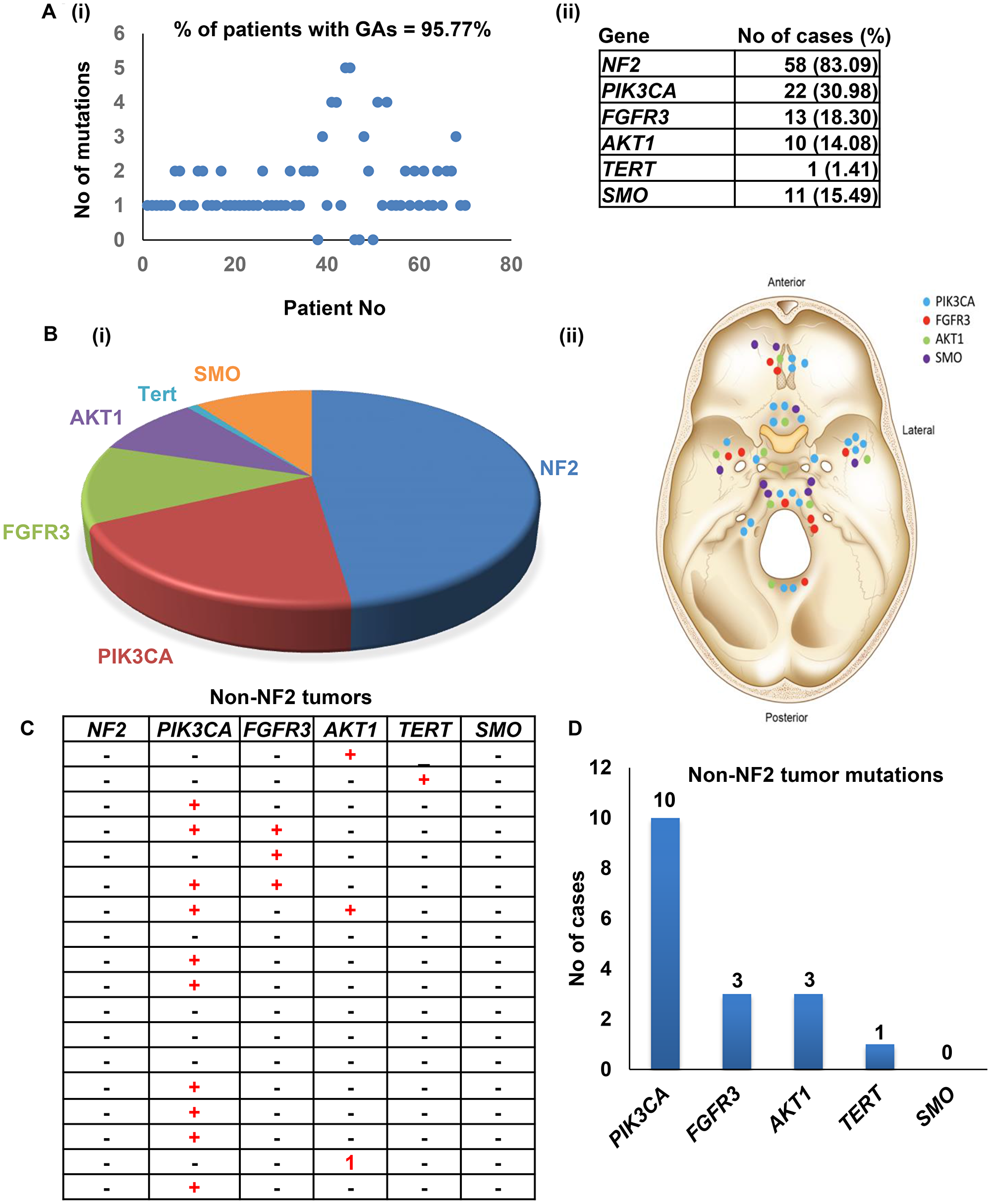
Figure 2: Meningioma mutations identified in the study. (A) Frequencies of genomic alterations identified in the study cohort. (B) Number of patients harboring the most commonly identified mutations. (C–D) Mutational frequencies in non-NF2 tumors.
Two novel missense mutations (c.932T>C (p.Val311Ala) and c.1376G>C (p.Arg459Pro)) in the FGFR3 gene were found in two cases, and both mutations were predicted to be disease-causing with rank scores equal to 0.81. PolyPhen-2 data suggested that c.1376G>C (p.Arg459Pro) mutation harbored a damaging PolyPhen-2 HVAR score equal to 0.996, with PROVEAN results suggesting a deleterious mutation with a score of -5.80. The other mutation was predicated to be benign and neutral. Both patients were WHO Grade 1, received radiotherapy and showed no tumor recurrence.
For the PIK3CA gene, 4 of the cases carried different novel exonic mutations. One case had a missense mutation that was predicted to be a disease-causing by MutationTaster but benign and neutral by PolyPhen-2 and PROVEAN. In addition, six cases were found harbor two reported pathogenic and likely pathogenic mutations; c.3140A>T (p.His1047Leu)/ c.3140A>G (p.His1047Arg) and c.112C>T (p.Arg38Cys). Pathogenic missense mutations in AKT1(c.49G>A (p.Glu17Lys)) and SMO(c.1234C>T (p.Leu412Phe)) genes were found in 5 and 4 cases, respectively. For the SMARCB1 gene, one reported synonymous mutation (c.897G>A (p.Ser299=)) that was likely benign was found in 5 cases (Figure 2A and 2B).
NF2 mutations were identified in (58/71) tumors and different patterns of genetic alterations were observed according to the NF2 status. Tumors with NF2 mutations harbored 1.83 GAs, and tumors with non-NF2 mutations harbored 1.24 GAs (Figure 2C). The NF2-positive tumors were predominantly of grade I (43/52, 82.69%), which was comparable to tumors lacking NF2 mutations (14/18, 77.77%). PI3KCA was the most frequently mutated gene in the non-NF2 mutated tumors (Figure 2C and 2D). Importantly, we identified 13 skull base tumors lacking NF2 mutations.
Hotspot TERTp mutational analysis
Screening of hotspot TERTp mutations, such as c.−146: G > A and c.−124: G > A, identified only a single patient (1/71) affected by the c.−124: G > A mutation. The patient did not possess a c.−146: G > A mutation (clinical timeline in Figure 3A). The patient was a male, aged 68 years, who had recurrent atypical WHO grade II biparietal meningioma. The patient underwent debulking resection. The patient was negative for mutations in NF2, PIK3CA, FGFR3, AKT1, and SMO. Pre-operative MRI of axial, sagittal and coronal views of the patient demonstrated a biparietal large fungating Meningioma (Figure 3B).
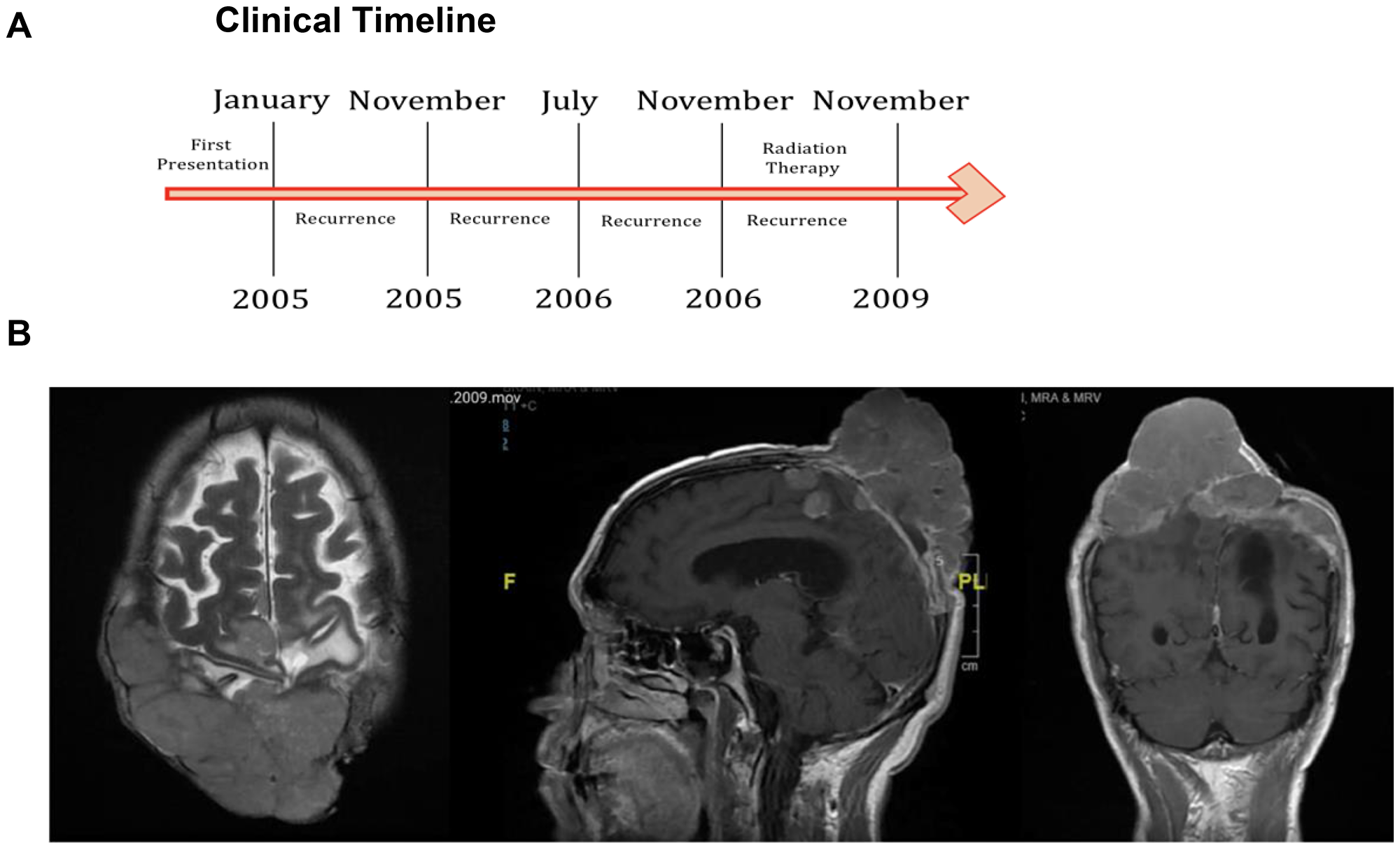
Figure 3: TERTp mutation identified in a single case. (A) Clinical timeline of the identified patient. (B) Pre-operative MRI of axial, sagittal and coronal views of the patient.
DISCUSSION
An array of genetic mutations has been proposed as causative for meningioma, including NF2, MN1, ARID1B, SEMA4D, and MUC2. NF2 and MN1 are classified as driver mutations that play a role in meningioma progression [25]. In this study, 71 tumors from histologically proven meningioma (grades I, II, & III) in the skull base were genetically analyzed. Based on histological analysis, the majority of patients were diagnosed with grade I meningothelial meningioma, and genomic alterations were identified in almost all cases (69/71). Among the meningiomas, the most common mutations were in NF2 (59/71), PIK3CA (22/71), FGFR3 (13/71), SMO (11/71) and AKT1 (10/71), with Tertp (1/71) mutations the least frequent. This was comparable to previous findings in which NF2 was mutated in ~79% of cases [26]. Of note, we identified 12/71 patients who did not harbor NF2 alterations [26].
NF2-mutated meningiomas show higher chromosomal instability during progression than non-NF2 mutated meningiomas [27–32]. In this study, tumors with NF2 mutations harbored 1.83 GAs per patient compared to the average of 1.24 GAs in non-NF2 mutations, consistent with this instability. The NF2-positive tumors were predominantly of grade I 43/52 (82.69%), which was comparable to tumors lacking NF2 mutations (77.77%). Thus, we did not observe an association between the tumor status and NF2 alterations reported in other cohorts [29]. Tumors in the skull base are traditionally amenable to dissection and debulking with resection. We observed significantly different rates (p ≤ 0,05) of recurrence (7/52, 13.41%) in tumors harboring NF2 mutations compared to tumor harboring non-NF2 mutations (8/18, 44.44%), suggesting that NF2 drivers are associated with improved outcomes. This information is useful for appropriate resection and accurate evaluation of meningioma prognosis, highlighting that non-NF2 tumors may benefit from targeted treatments due to their lower mutation rates but higher likelihood of recurrence. It is therefore important that diagnostic tests are not restricted to frequent NF2 mutational profiles, and diverse genetic profiling is readily available in clinical settings.
The least frequent mutation was in TERTp (1/71 tumors). Telomerases are unique reverse transcriptases that maintain telomere length during cell division [33]. Telomerase activity is robust in embryonic cells but suppressed in mature somatic cells during adulthood. Telomerases are expressed in up to ~90% of solid tumors [34, 35]. Several studies have reported the frequent occurrence of TERTp mutations in gliomas that result in altered telomere lengthening and increased tumor survival due to their ability to escape cell senescence [36]. Indeed, the presence of TERTp mutations in meningioma is associated with a poorer prognosis and shorter overall survival [1]. We found that TERTp mutations were much less frequent in skull base meningiomas, with grade I tumors showing no incidence (0/56). The TERTp mutation was observed in a male patient aged 68 years who had recurrent atypical WHO grade II meningioma and underwent debulking resection. Further genetic profiling of a larger number of higher grade meningiomas that were encountered in this study (only ≥ 12 tumors higher than grade I) will be required to confirm whether TERTp indeed directly influences prognosis. If proven to be the case, this subgroup of meningioma patients may require more frequent follow-up and aggressive management to optimize their survival outcomes [1].
We observed alterations in PI3KCA (22/71 cases) and AKT1 (10/71 cases). PI3KCA was the most mutated gene in non-NF2 tumors. Mutations in these survival genes are typically associated with therapeutic resistance and chemotherapy-induced mutagenesis [37–40]. This highlights the importance of genetic assessment following surgical resection; if PI3KCA- and AKT1-mutated tumors recur and undergo malignant progression to a higher histological grade, postoperative adjuvant treatment using known and well-characterized inhibitors should be employed, as opposed to radiotherapy or chemotherapy interventions. In this regard, taselisib and fulvestrant (Faslodex) have shown antitumor activity and may be recommended [41–45].
With regard to the potential of our datasets to guide precision therapy, smoothened (encoded by the SMO gene) is a Frizzled G protein-coupled receptor and a critically important component of the hedgehog signaling pathway [46]. SMO mutations were identified in 11/71 (15.74%) cases in our cohort, leading to a characterized predisposition and vulnerability to meningioma formation [47, 48]. SMO is a molecular target of vismodegib, the first small-molecule hedgehog inhibitor to be approved by the FDA [49] that has shown promise in the treatment of basal cell carcinomas [50]. Of note, vismodegib has been included in current clinical trials on progressive meningioma (NCT02523014) in patients harboring SMO mutations. In lung cancer studies, SMO amplifications and subsequent activation of the hedgehog pathway confer resistance to anti-EGFR drugs [46]. Phase II trials of erlotinib or gefitinib, two targeted EGFR inhibitors, demonstrated limited efficacy in progressive meningioma patients, thought to be due to the expression of EGFR in the trial participants [51]. We anticipate that the subset of SMO mutation-harboring tumors we identified will show a response to vismodegib therapy, but the efficacy of anti-EGFR therapeutics is unlikely due to drug resistance. Genetically profiling erlotinib- and gefitinib-resistant meningioma would further support these findings.
An interesting occurrence was the identification of single FGFR3 mutations in three patients. Two novel missense mutations in the FGFR3 gene were found in two cases, and PolyPhen-2 data suggested that one mutation harbored a damaging PolyPhen-2 score (HVAR score equal to 0.996). Previous studies have highlighted FGFR3 mutations in an array of malignancies, including breast cancer, bladder cancer, prostate cancer, and squamous non-small cell lung carcinoma (sqNSCLC) [52–57]. Interesting FGFR3 mutations are typically associated with low-grade cancers and favorable prognoses [55, 56, 58], and patients harboring these mutations had WHO grade I tumors, with no recurrence in our cohort. Clinical trials for sqNSCLC with the selective FGFR1-4 inhibitor erdafitinib are currently ongoing (NCT03827850), and a favorable therapeutic outcome is predicted in FGFR3-mutated meningiomas (Supplementary Figure 1). Thus, FGFR3 mutations may be a prognostic biomarker for a favorable outcome in meningioma patients, who may further respond to erdafitinib treatment.
Materials and Methods
Patients and samples
Tissue samples were collected from 71 patients diagnosed with meningioma according to clinical and pathological criteria from 2008 to 2016. Tissues were treated and archived as formalin-fixed paraffin-embedded (FFPE) samples in the Pathology Department, KFMC, Riyadh, KSA. This study was performed with IRB#16-310 following the relevant ethical guidelines and regulations from the King Fahad Medical City, Riyadh, KSA.
DNA extraction and quantification
FFPE blocks were retrieved from the Pathology Department, KFMC, Riyadh, KSA. Genomic DNA was extracted from FFPE samples using GeneRead DNA FFPE Kits (Qiagen, USA) according to the manufacturer’s protocol. DNA samples were quantified by Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, USA) using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, USA).
TERT promoter (TERTp) mutational screening
All 71 samples were screened for the presence of hotspot TERTp mutations (c.−146: G > A and c.−124: G > A). PCR was performed using the primer pairs Fw: 5′-CAGCGCTGCCTGAAACTC-3′ and Rev: 5′-GTCCTGCCCCTTCACCTT-3′ [7]. HotStarTaq DNA Polymerase PCR kits were used for PCR (Qiagen, USA) following a touchdown PCR program with annealing temperatures of 64 to 55° C, resulting in PCR products of ~235 bp. Products were purified using the Agencourt AMPure PCR purification system (Agencourt Bioscience, USA) and sequenced using the BigDye Terminator Cycle Sequencing kit (PE Applied Biosystems, USA). Runs were performed on ABI 3730xl capillary sequencers as previously described [59].
Targeted sequencing
All samples underwent targeted sequencing using a customized panel designed by Thermo Fisher Scientific that has been verified for both sensitivity and specificity. The panel includes the following 66 known cancer genes: HRAS STK11, TERT, FGFR3, CARD11, GNA11, JAK2, TP53, VHL, MYCN, JAK3, CDKN2A, SMARCB1, KRAS, FLT3, ALK, NF1, NF2, SRC, MLH1, ERBB2, FGFR1, DDX3X, CTNNB1, CIC, RET, MPL, MSH2, MSH6, SMAD4, RB1, CYLD, EGFR, PDGFRA, KIT, KDR, GNAS, BCL2, MEN1, CDH1, BCL1, ATRX, GNAQ, PTEN, IDH2, CDK6, PTCH1, AKT1, ATM, APC, PTPN11, NRAS, MET, HNF1A, FGFR2, MYC, SMO, ABL1, NOTCH1, BRAF, FBXW7, NPM1, PIK3CA, BCL6, IDH1, and ERBB4.
Libraries were manually prepared using the Ion AmpliSeq™ Library Kit 2.0 (Thermo Fisher Scientific, USA). Templates were prepared using the KingFisher System (Thermo Fisher Scientific, USA) and Ion AmpliSeq™ Kit for Chef DL8 (Thermo Fisher Scientific, USA) and Ion 530 Chip Kits (Thermo Fisher Scientific, USA). Templates were sequenced using the Ion S5XL system and Ion S5 sequencing solution (Thermo Fisher Scientific, USA). All sequencing experiments were performed in the Saudi Human Genome Project Laboratory, KFMC, Riyadh, KSA.
Sequence analysis and bioinformatics
Sanger sequencing for TERTp mutations was performed using SeqMan NGen® (version 5.07 DNASTAR, USA). The NGS data pipeline was implemented by the bioinformatics teams of the Saudi Human Genome Projects, KACST and KFMC, Riyadh, KSA. Briefly, the quality of the NGS reads were measured, and low-quality reads were excluded. Reads were aligned to the reference genome using Torrent Aligner Software (Torrent Mapping Alignment Programs (TMAP)) and the human genome reference (GRCh37/hg19). Variants were detected for each sequenced sample using the Torrent Variant Caller (TVC) program. Resultant files were saved in the variant call format (VCF). The VCF file was used for annotation against dbSNP, the 1000 Genomes Project, the Human Gene Mutation Database, and the Catalog of Somatic Mutations in Cancer. A local genome database (SGP777) that includes specific Arab variants was compared.
Previously identified exonic mutations with pathogenic effects were reported, whilst mutations with benign effects were excluded. Novel exonic mutations were assessed for the prediction of damage effects using the in silico prediction tools MutationTaster (http://www.mutationtaster.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), and PROVEAN (http://provean.jcvi.org/index.php).
Statistical analysis
P-values < 0.05 were considered statistically significant. Secure electronic databases were created for data storage and analysis. Data were entered and analyzed using SPSS version 23.
Conclusions
While our cohort was focused on skull base meningioma’s, investigation of sequencing data in our cohort has identified a number of non-NF2 mutations in skull base meningioma’s, This highlights a range of mutations outside the known cancer driver NF2 that may be linked to meningioma prognosis. Taken together, these findings highlight how genetic profiling can guide optimal treatment strategies, prognostic prediction, and patient management for skull based meningioma. Further studies in a range of meningioma grades are now required to confirm the prognostic significance of the identified mutants.
ACKNOWLEDGMENTS
The authors gratefully acknowledge King Abdulaziz City for Science and Technology and the Saudi Human Genome Project for the technical support.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare
FUNDING
This study was supported by King Fahad Medical City IRF 17-65 (MA).
References
1. Lu VM, Goyal A, Lee A, Jentoft M, Quinones-Hinojosa A, Chaichana KL. The prognostic significance of TERT promoter mutations in meningioma: a systematic review and meta-analysis. J Neurooncol. 2019; 142:1–10. https://doi.org/10.1007/s11060-018-03067-x. [PubMed].
2. Yao A, Pain M, Balchandani P, Shrivastava RK. Can MRI predict meningioma consistency?: a correlation with tumor pathology and systematic review. Neurosurg Rev. 2018; 41:745–753. https://doi.org/10.1007/s10143-016-0801-0. [PubMed].
3. Dijkstra BM, Jeltema HJ, Kruijff S, Groen RJ. The application of fluorescence techniques in meningioma surgery-a review. Neurosurg Rev. 2018. [Epub ahead of print]. https://doi.org/10.1007/s10143-018-01062-4. [PubMed].
4. Adeberg S, Hartmann C, Welzel T, Rieken S, Habermehl D, von Deimling A, Debus J, Combs SE. Long-term outcome after radiotherapy in patients with atypical and malignant meningiomas--clinical results in 85 patients treated in a single institution leading to optimized guidelines for early radiation therapy. Int J Radiat Oncol Biol Phys. 2012; 83:859–864. https://doi.org/10.1016/j.ijrobp.2011.08.010. [PubMed].
5. De La Garza-Ramos R, Flores-Rodriguez JV, Martinez-Gutierrez JC, Ruiz-Valls A, Caro-Osorio E. Current standing and frontiers of gene therapy for meningiomas. Neurosurg Focus. 2013; 35:E4. https://doi.org/10.3171/2013.8.FOCUS13305. [PubMed].
6. Fathi AR, Roelcke U. Meningioma. Curr Neurol Neurosci Rep. 2013; 13:337. https://doi.org/10.1007/s11910-013-0337-4. [PubMed].
7. Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, Hovland P, Davidorf FH. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011; 48:856–859. https://doi.org/10.1136/jmedgenet-2011-100156. [PubMed].
8. Forest F, Yvorel V, Vassal F, Trombert B, Dumollard JM, Nuti C, Péoc’h M. BRAF V600 point mutation is not present in relapsing meningioma. Clin Neuropathol. 2015; 34:164–165. https://doi.org/10.5414/NP300836. [PubMed].
9. Gils C, Frøslev-Friis C. Hypercoagulability in a patient with meningioma and Factor VLeiden mutation. [Article in Danish] Ugeskr Laeger. 2014; 176:V06140351. [PubMed].
10. Guaraldi F, Corazzini V, Gallia GL, Grottoli S, Stals K, Dalantaeva N, Frohman LA, Korbonits M, Salvatori R. Genetic analysis in a patient presenting with meningioma and familial isolated pituitary adenoma (FIPA) reveals selective involvement of the R81X mutation of the AIP gene in the pathogenesis of the pituitary tumor. Pituitary. 2012; 15:S61–S67. https://doi.org/10.1007/s11102-012-0391-y. [PubMed].
11. Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer. 2012; 11:565–570. https://doi.org/10.1007/s10689-012-9548-0. [PubMed].
12. Mordechai O, Postovsky S, Vlodavsky E, Eran A, Constantini S, Dotan E, Cagnano E, Weyl-Ben-Arush M. Metastatic rhabdoid meningioma with BRAF V600E mutation and good response to personalized therapy: case report and review of the literature. Pediatr Hematol Oncol. 2015; 32:207–211. https://doi.org/10.3109/08880018.2014.936058. [PubMed].
13. Raffalli-Ebezant H, Rutherford SA, Stivaros S, Kelsey A, Smith M, Evans DG, Kilday JP. Pediatric intracranial clear cell meningioma associated with a germline mutation of SMARCE1: a novel case. Childs Nerv Syst. 2015; 31:441–447. https://doi.org/10.1007/s00381-014-2558-5. [PubMed].
14. Ruiz J, Capilla E, Diaz JF, Ruiz JA, Andrade J, Hernandez T, Mollejo M, Melendez B. Secretory meningioma with KLF4 K409Q mutation in collision with glioma. Clin Neuropathol. 2015; 34:322–329. https://doi.org/10.5414/NP300860. [PubMed].
15. Strom RG, Shvartsbeyn M, Rosenblum MK, Hameed MR, Nafa K, Mikolaenko I, Babu RP. Melanocytic tumor with GNA11 p.Q209L mutation mimicking a foramen magnum meningioma. Clin Neurol Neurosurg. 2012; 114:1197–1200. https://doi.org/10.1016/j.clineuro.2012.02.030. [PubMed].
16. Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK, Horowitz P, Schumacher SE, Esaulova E, Mei Y, Chevalier A, Ducar M, Thorner AR, et al. Genomic landscape of high-grade meningiomas. NPJ Genom Med. 2017; 2:15. https://doi.org/10.1038/s41525-017-0014-7. [PubMed].
17. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016; 33:237–247. https://doi.org/10.1007/s10014-016-0271-7. [PubMed].
18. Choy W, Kim W, Nagasawa D, Stramotas S, Yew A, Gopen Q, Parsa AT, Yang I. The molecular genetics and tumor pathogenesis of meningiomas and the future directions of meningioma treatments. Neurosurg Focus. 2011; 30:E6. https://doi.org/10.3171/2011.2.FOCUS1116. [PubMed].
19. Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006; 5:1045–1054. https://doi.org/10.1016/S1474-4422(06)70625-1. [PubMed].
20. Domingues P, Gonzalez-Tablas M, Otero A, Pascual D, Ruiz L, Miranda D, Sousa P, Goncalves JM, Lopes MC, Orfao A, Tabernero MD. Genetic/molecular alterations of meningiomas and the signaling pathways targeted. Oncotarget. 2015; 6:10671–10688. https://doi.org/10.18632/oncotarget.3870. [PubMed].
21. Zadeh G, Karimi S, Aldape KD. PIK3CA mutations in meningioma. Neuro Oncol. 2016; 18:603–04. https://doi.org/10.1093/neuonc/now029. [PubMed].
22. Yuzawa S, Nishihara H, Yamaguchi S, Mohri H, Wang L, Kimura T, Tsuda M, Tanino M, Kobayashi H, Terasaka S, Houkin K, Sato N, Tanaka S. Clinical impact of targeted amplicon sequencing for meningioma as a practical clinical-sequencing system. Mod Pathol. 2016; 29:708–716. https://doi.org/10.1038/modpathol.2016.81. [PubMed].
23. Ibrahim AW. C.N.S. tumors in eastern Saudi Arabia. Neurosurg Rev. 1992; 15:295–302. [PubMed].
24. Alnaami I, Sarhan L, Alqahtani A, Alghamdi A, Alkhashrami S, Mostafa O. Does brain tumor epidemiology differ from place to another? Saudi single tertiary care center experience. Biomed Res. 2018; 29:2982–2987. https://doi.org/10.4066/biomedicalresearch.29-18-816.
25. Huang G, Feng J, Hao S, Li D, Wang K, Wang L, Wu Z, Wan H, Zhang L, Zhang J. CASP8, XRCC1, WRN, NF2, and BRIP1 polymorphisms analysis shows their genetic susceptibility for meningioma risk and the association with tumor-related phenotype in a Chinese population. World Neurosurg. 2018; 114:e883–e891. https://doi.org/10.1016/j.wneu.2018.03.108. [PubMed].
26. Goutagny S, Yang HW, Zucman-Rossi J, Chan J, Dreyfuss JM, Park PJ, Black PM, Giovannini M, Carroll RS, Kalamarides M. Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clin Cancer Res. 2010; 16:4155–4164. https://doi.org/10.1158/1078-0432.CCR-10-0891. [PubMed].
27. Abdelmontalab FY, Fadl EI, Abushama H, Kreskowski K, Liehr T. Molecular cytogenetic study of the NF2 gene deletion in meningioma in sudanese patients. Balkan J Med Genet. 2013; 16:29–32. https://doi.org/10.2478/bjmg-2013-0028. [PubMed].
28. Angus SP, Oblinger JL, Stuhlmiller TJ, DeSouza PA, Beauchamp RL, Witt L, Chen X, Jordan JT, Gilbert TS, Stemmer-Rachamimov A, Gusella JF, Plotkin SR, Haggarty SJ, et al, and Children’s Tumor Foundation Synodos for NF2 Consortium. EPH receptor signaling as a novel therapeutic target in NF2-deficient meningioma. Neuro Oncol. 2018; 20:1185–1196. https://doi.org/10.1093/neuonc/noy046. [PubMed].
29. Benmaamar R. Non-NF2 mutations in meningioma. Lancet Oncol. 2013; 14:e91. https://doi.org/10.1016/S1470-2045(13)70005-6. [PubMed].
30. Burns SS, Akhmametyeva EM, Oblinger JL, Bush ML, Huang J, Senner V, Chen CS, Jacob A, Welling DB, Chang LS. Histone deacetylase inhibitor AR-42 differentially affects cell-cycle transit in meningeal and meningioma cells, potently inhibiting NF2-deficient meningioma growth. Cancer Res. 2013; 73:792–803. https://doi.org/10.1158/0008-5472.CAN-12-1888. [PubMed].
31. Burns SS, Chang LS. Generation of noninvasive, quantifiable, orthotopic animal models for NF2-associated schwannoma and meningioma. Methods Mol Biol. 2016; 1427:59–72. https://doi.org/10.1007/978-1-4939-3615-1_4. [PubMed].
32. Chow HY, Dong B, Duron SG, Campbell DA, Ong CC, Hoeflich KP, Chang LS, Welling DB, Yang ZJ, Chernoff J. Group I paks as therapeutic targets in NF2-deficient meningioma. Oncotarget. 2015; 6:1981–1994. https://doi.org/10.18632/oncotarget.2810. [PubMed].
33. Penna GC, Pestana A, Cameselle JM, Momesso D, de Andrade FA, Vidal AP, Araujo Junior ML, Melo M, Fernandes PV, Corbo R, Vaisman M, Sobrinho-Simões M, Soares P, Vaisman F. TERTp mutation is associated with a shorter progression free survival in patients with aggressive histology subtypes of follicular-cell derived thyroid carcinoma. Endocrine. 2018; 61:489–498. https://doi.org/10.1007/s12020-018-1642-0. [PubMed].
34. Duan WC, Wang L, Li K, Wang WW, Zhan YB, Zhang FJ, Yu B, Bai YH, Wang YM, Ji YC, Zhou JQ, Liu XZ, Zhang ZY. IDH mutations but not TERTp mutations are associated with seizures in lower-grade gliomas. Medicine (Baltimore). 2018; 97:e13675. https://doi.org/10.1097/MD.0000000000013675. [PubMed].
35. Li X, Wei J, Liu Y, Li P, Fan L, Wang Y, Li M, Zhao D, Yu Z, Ye J, Guo Y, Yan Q, Guo S, Wang Z. Primary astrocytic tumours and paired recurrences have similar biological features in IDH1, TP53 and TERTp mutation and MGMT, ATRX loss. Sci Rep. 2017; 7:13038. https://doi.org/10.1038/s41598-017-13272-9. [PubMed].
36. Lee Y, Koh J, Kim SI, Won JK, Park CK, Choi SH, Park SH. The frequency and prognostic effect of TERT promoter mutation in diffuse gliomas. Acta Neuropathol Commun. 2017; 5:62. https://doi.org/10.1186/s40478-017-0465-1. [PubMed].
37. Bredemeier M, Kasimir-Bauer S, Kolberg HC, Herold T, Synoracki S, Hauch S, Edimiris P, Bankfalvi A, Tewes M, Kimmig R, Aktas B. Comparison of the PI3KCA pathway in circulating tumor cells and corresponding tumor tissue of patients with metastatic breast cancer. Mol Med Rep. 2017; 15:2957–2968. https://doi.org/10.3892/mmr.2017.6415. [PubMed].
38. Ruiz A, Sebagh M, Saffroy R, Allard MA, Bosselut N, Hardoin G, Vasseur J, Hamelin J, Adam R, Morere JF, Lemoine A. Chronological occurrence of PI3KCA mutations in breast cancer liver metastases after repeat partial liver resection. BMC Cancer. 2019; 19:169. https://doi.org/10.1186/s12885-019-5365-2. [PubMed].
39. Tessitore A, Bruera G, Mastroiaco V, Cannita K, Cortellini A, Cocciolone V, Dal Mas A, Calvisi G, Zazzeroni F, Ficorella C, Ricevuto E, Alesse E. KRAS and 2 rare PI3KCA mutations coexisting in a metastatic colorectal cancer patient with aggressive and resistant disease. Hum Pathol. 2018; 74:178–182. https://doi.org/10.1016/j.humpath.2018.01.021. [PubMed].
40. Wang Y, Zhang H, Lin M, Wang Y. Association of FGFR2 and PI3KCA genetic variants with the risk of breast cancer in a Chinese population. Cancer Manag Res. 2018; 10:1305–1311. https://doi.org/10.2147/CMAR.S164084. [PubMed].
41. Dickler MN, Saura C, Richards DA, Krop IE, Cervantes A, Bedard PL, Patel MR, Pusztai L, Oliveira M, Cardenas AK, Cui N, Wilson TR, Stout TJ, et al. Phase II study of taselisib (GDC-0032) in combination with fulvestrant in patients with HER2-negative, hormone receptor-positive advanced breast cancer. Clin Cancer Res. 2018; 24:4380–4387. https://doi.org/10.1158/1078-0432.CCR-18-0613. [PubMed].
42. Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, Savage HM, Sampath D, Salphati L, Lin RS, Jin H, Parmar H, Hsu JY, et al. Phase I dose-escalation study of Taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017; 7:704–715. https://doi.org/10.1158/2159-8290.CD-16-1080. [PubMed].
43. Morgillo F, Della Corte CM, Diana A, Mauro CD, Ciaramella V, Barra G, Belli V, Franzese E, Bianco R, Maiello E, de Vita F, Ciardiello F, Orditura M. Phosphatidylinositol 3-kinase (PI3Kalpha)/AKT axis blockade with taselisib or ipatasertib enhances the efficacy of anti-microtubule drugs in human breast cancer cells. Oncotarget. 2017; 8:76479–76491. https://doi.org/10.18632/oncotarget.20385. [PubMed].
44. Orditura M, Della Corte CM, Diana A, Ciaramella V, Franzese E, Famiglietti V, Panarese I, Franco R, Grimaldi A, Lombardi A, Caraglia M, Santoriello A, Procaccini E, et al. Three dimensional primary cultures for selecting human breast cancers that are sensitive to the anti-tumor activity of ipatasertib or taselisib in combination with anti-microtubule cytotoxic drugs. Breast. 2018; 41:165–171. https://doi.org/10.1016/j.breast.2018.08.002. [PubMed].
45. Tamura K, Kodaira M, Shimizu C, Yonemori K, Yunokawa M, Shimomura A, Kobayashi T, Nakano K, Tomomatsu J, Ito Y, Tanaka J, Kuriki H, Gu Z, Takahashi S. Phase I study of taselisib in Japanese patients with advanced solid tumors or hormone receptor-positive advanced breast cancer. Cancer Sci. 2018; 109:1592–1601. https://doi.org/10.1111/cas.13561. [PubMed].
46. Arnhold V, Boos J, Lanvers-Kaminsky C. Targeting hedgehog signaling pathway in pediatric tumors: in vitro evaluation of SMO and GLI inhibitors. Cancer Chemother Pharmacol. 2016; 77:495–505. https://doi.org/10.1007/s00280-016-2962-5. [PubMed].
47. Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, Listewnik ML, Dias-Santagata D, Thorner AR, Van Hummelen P, Brastianos PK, Reardon DA, Wen PY, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol. 2016; 18:649–55. https://doi.org/10.1093/neuonc/nov316. [PubMed].
48. Boetto J, Apra C, Bielle F, Peyre M, Kalamarides M. Selective vulnerability of the primitive meningeal layer to prenatal Smo activation for skull base meningothelial meningioma formation. Oncogene. 2018; 37:4955–4963. https://doi.org/10.1038/s41388-018-0328-7. [PubMed].
49. Dlugosz A, Agrawal S, Kirkpatrick P. Vismodegib. Nat Rev Drug Discov. 2012; 11:437–438. https://doi.org/10.1038/nrd3753. [PubMed].
50. Dirix L, Rutten A. Vismodegib: a promising drug in the treatment of basal cell carcinomas. Future Oncol. 2012; 8:915–928. https://doi.org/10.2217/fon.12.82. [PubMed].
51. Norden AD, Raizer JJ, Abrey LE, Lamborn KR, Lassman AB, Chang SM, Yung WK, Gilbert MR, Fine HA, Mehta M, Deangelis LM, Cloughesy TF, Robins HI, et al. Phase II trials of erlotinib or gefitinib in patients with recurrent meningioma. J Neurooncol. 2010; 96:211–217. https://doi.org/10.1007/s11060-009-9948-7. [PubMed].
52. Ach T, Schwarz-Furlan S, Ach S, Agaimy A, Gerken M, Rohrmeier C, Zenk J, Iro H, Brockhoff G, Ettl T. Genomic aberrations of MDM2, MDM4, FGFR1 and FGFR3 are associated with poor outcome in patients with salivary gland cancer. J Oral Pathol Med. 2016; 45:500–509. https://doi.org/10.1111/jop.12394. [PubMed].
53. Atala A. Re: mutations in TERT promoter and FGFR3 and telomere length in bladder cancer. J Urol. 2016; 195:1168–1169. https://doi.org/10.1016/j.juro.2016.01.020. [PubMed].
54. Bersani C, Sivars L, Haeggblom L, DiLorenzo S, Mints M, Ahrlund-Richter A, Tertipis N, Munck-Wikland E, Nasman A, Ramqvist T, Dalianis T. Targeted sequencing of tonsillar and base of tongue cancer and human papillomavirus positive unknown primary of the head and neck reveals prognostic effects of mutated FGFR3. Oncotarget. 2017; 8:35339–35350. https://doi.org/10.18632/oncotarget.15240. [PubMed].
55. Beukers W, van der Keur KA, Kandimalla R, Vergouwe Y, Steyerberg EW, Boormans JL, Jensen JB, Lorente JA, Real FX, Segersten U, Orntoft TF, Malats N, Malmstrom PU, et al. FGFR3, TERT and OTX1 as a urinary biomarker combination for surveillance of patients with bladder cancer in a large prospective multicenter study. J Urol. 2017; 197:1410–1418. https://doi.org/10.1016/j.juro.2016.12.096. [PubMed].
56. Blanca A, Requena MJ, Alvarez J, Cheng L, Montironi R, Raspollini MR, Reymundo C, Lopez-Beltran A. FGFR3 and cyclin D3 as urine biomarkers of bladder cancer recurrence. Biomark Med. 2016; 10:243–53. https://doi.org/10.2217/bmm.15.120. [PubMed].
57. Cao W, Ma E, Zhou L, Yuan T, Zhang C. Exploring the FGFR3-related oncogenic mechanism in bladder cancer using bioinformatics strategy. World J Surg Oncol. 2017; 15:66. https://doi.org/10.1186/s12957-017-1125-4. [PubMed].
58. Bersani C, Haeggblom L, Ursu RG, Giusca SE, Marklund L, Ramqvist T, Nasman A, Dalianis T. Overexpression of FGFR3 in HPV-positive tonsillar and base of tongue cancer is correlated to outcome. Anticancer Res. 2018; 38:4683–4690. https://doi.org/10.21873/anticanres.12774. [PubMed].
59. Linsler S, Kraemer D, Driess C, Oertel J, Kammers K, Rahnenfuhrer J, Ketter R, Urbschat S. Molecular biological determinations of meningioma progression and recurrence. PLoS One. 2014; 9:e94987. https://doi.org/10.1371/journal.pone.0094987. [PubMed].