
Introduction
Dedifferentiated liposarcoma (DDLPS) is a highly morbid mesenchymal tumor accounting for approximately 20% of all soft-tissue sarcomas [1]. A hallmark of DDLPS is the genomic amplification of Mouse Double Minute 2 Homolog (MDM2) [2–5]. MDM2 degrades p53, thus, an amplification in MDM2 results in reduced p53 activity and a shift towards pro-survival pathways. Our previous work has demonstrated that the amplification of MDM2 is directly tied to biological activity and clinical response to chemotherapy in this disease [6, 7]; furthermore, eliminating or reducing MDM2 activity may reduce of the oncogenicity of DDLPS tumors [7, 8]. A primary strategy to target MDM2 in DDLPS has been to sterically inhibit the ability of MDM2 to bind p53 [8–11]. These treatment modalities have shown promise pre-clinically but have yet to be proven clinically viable.
Recent in vitro studies suggest that pan-histone deacetylase (HDAC) inhibitors may modestly reduce MDM2 expression [12, 13] but have not been fully evaluated in DDLPS. While targeted HDAC inhibitors have been approved in the treatment of hematologic malignancies, they have shown limited activity in a number of solid tumor types with notable toxicities [14–18]. A benefit of targeted HDAC inhibition clinically would be the potential to avoid off-target toxicities associated with pan-HDAC inhibition. Whether targeted HDAC inhibition with defined specificity would be efficacious in DDLPS has yet to be explored.
The HDAC family of proteins is vast and is comprised of four groups and 18 members. To this end, our in silico analysis identified HDAC2 as a potential target for modulation of MDM2 in DDLPS. A recent study assessing gene expression in 1,332 mesenchymal tumors and normal tissues identified a specific increase in HDAC2 expression in DDLPS compared to other soft tissue sarcomas (STS) and normal tissue [19]. In lung cancer cell lines, MDM2 expression could be reduced by selectively knocking down HDAC2 [20]. In this study we demonstrate that selective inhibition of HDAC2 induced p53-dependent survivin downregulation through MDM2 proteasomal degradation [20]. Results presented here suggest that inhibition of HDAC2, specifically utilizing the HDAC1/2 inhibitor romidepsin, reduces MDM2 expression and promotes apoptosis in DDLPS. This may serve as a viable therapeutic modality for this highly morbid disease.
Results
Elevated HDAC2 levels are poorly prognostic in DDLPS
To assess the clinical significance of HDAC2 expression, we evaluated TCGA and MSKCC datasets, which included mRNA expression and DFS. As a continuous variable in the TCGA dataset, elevated HDAC2 expression was associated with a reduced disease-free survival (DFS) manifested as an increase in hazard of 5% per increment of HDAC2 reads (Hazard-ratio (HR) 1.7; 95% Confidence Interval (95%CI) 0.97–2.9; p = 0.06). To assess HDAC2 expression as a dichotomous predictor of DFS, subjects were stratified into HDAC2 High and HDAC2 Low groups utilizing a maximal inflection point test to identify the optimal cut off using both gene expression and DFS time to separate cohorts (methods, Supplementary Figure 2A), elevated HDAC2 was associated with poor DFS (median DFS: HDAC2 High 5.7 months, HDAC2 Low 31.1 months; HR 7.1, 95%CI 2.5–19.8, p < 0.001; Figure 1A). Elevated HDAC2 expression also correlated with poor overall survival (OS) as a continuous (HR 1.06; 95%CI 1.01–1.11; p = 0.02) and dichotomous predictor (median OS: HDAC2 High 18.5 months, HDAC2 Low 76.4 months; HR 4.9, 95%CI 1.1–21.9, p < 0.001; Figure 1B).
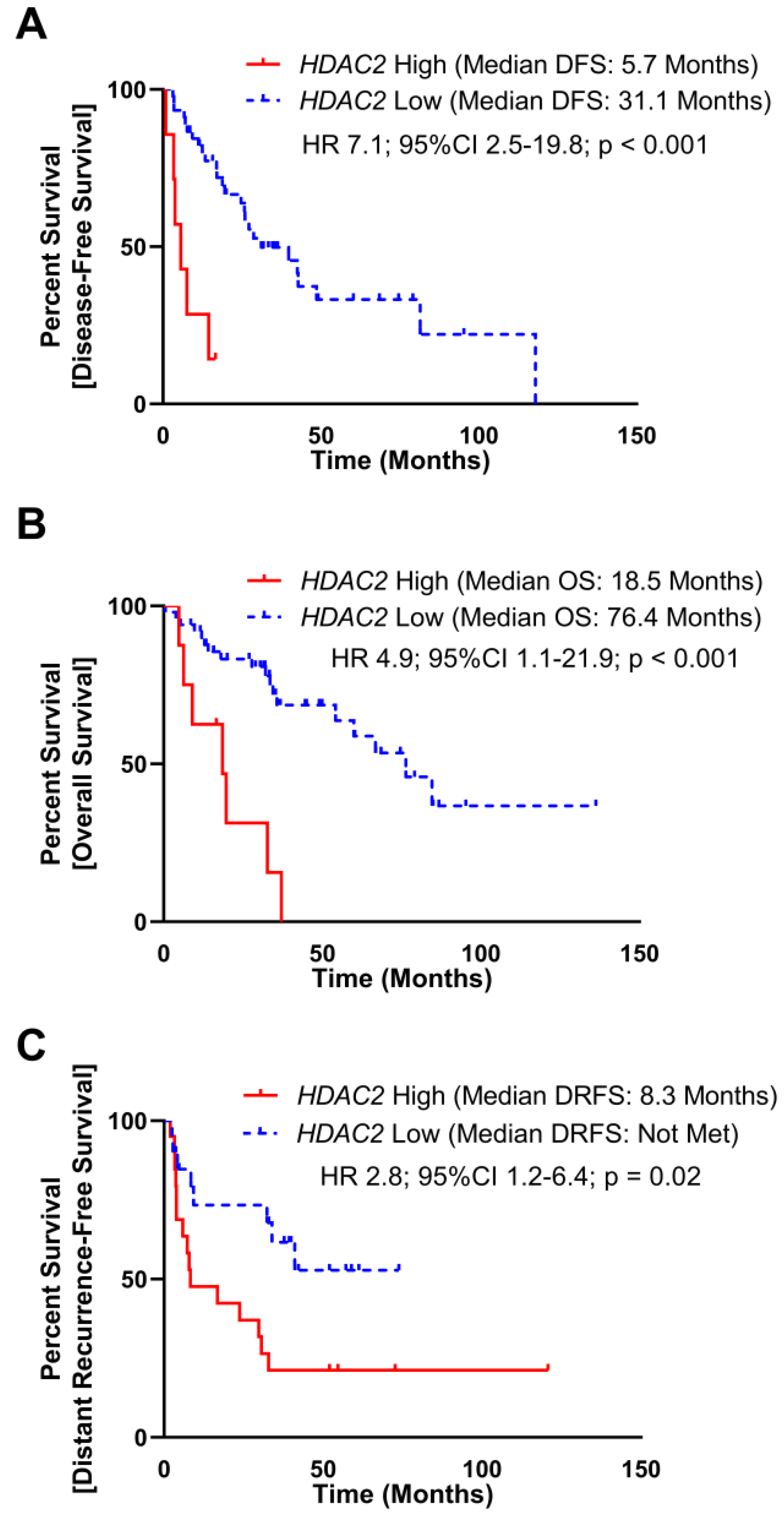
Figure 1: Elevated HDAC2 mRNA expression is poorly prognostic in DDLPS. (A) mRNA expression of HDAC2 and disease-free survival (DFS) data from 52 subjects with DDLPS from The Cancer Genome Atlas (TCGA) were split into two groups utilizing maximally selected rank statistic (Supplementary Figure 1A). Subjects with elevated HDAC2 expression experienced reduced DFS (median DFS: HDAC2 High 5.7 months, HDAC2 Low 31.1 months; HR 7.1, 95%CI 2.5–19.8, p < 0.001). (B) mRNA expression of HDAC2 and overall survival (OS) data from 58 subjects with DDLPS from the TCGA were split into two groups utilizing maximally selected rank statistics (Supplementary Figure 1A). Subjects with elevated HDCA2 expression experienced reduced OS (median OS: HDAC2 High 18.5 months, HDAC2 Low 76.4 months; HR 4.4, 95%CI 1.1–21.9, p = 0.04). (C) mRNA expression of HDAC2 and distant recurrence-free survival (DRFS) data from 40 subjects with DDLPS from the Memorial Sloan-Kettering Cancer Center Dataset (MSKCC) were split into two utilizing maximally selected rank statistic (Supplementary Figure 1B). Subjects with elevated HDAC2 expression experienced reduced DRFS (median DRFS: HDAC2 High 8.3 months, HDAC2 Low not met; HR 2.8, 95%CI 1.2–6.4, p = 0.02).
In the MSKCC dataset, elevated HDAC2 expression as a continuous variable was associated with a reduced distant recurrence-free survival (HR 2.2; 95%CI 1.1–4.8; p = 0.04). As a dichotomous predictor stratified at the maximal inflection point (Supplementary Figure 2B), elevated HDAC2 was associated with poor DRFS (median DRFS: HDAC2 High 8.3 months, HDAC2 Low not met; HR 2.8, 95%CI 1.2–6.4, p = 0.02; Figure 1C). The MSKCC dataset did not include OS data.
HDAC2 knockdown reduces MDM2 expression in DDLPS
To interrogate the effect of modulating HDAC2 on the expression of MDM2 we used shRNA targeted to HDAC2 in the DDLPS cell line LPS246. The addition of shRNA reduced HDAC2 protein expression compared to scrambled shRNA control. MDM2 expression was reduced in shRNA treated cells compared to scrambled shRNA control (Supplementary Figure 3).
Pharmacologic inhibition of HDAC2 reduces cell proliferation
To assess the effect of more targeted HDAC inhibition in DDLPS, we used a panel of four DDLPS cell lines whose MDM2 status was fully outlined previously [8]. Cells were treated with two HDAC inhibitors, either MI-192 (specific to HDAC2 and HDAC3) or romidepsin (specific to HDAC1 and HDAC2), for 72 hours [22, 23]. All cell lines treated with MI-192 or romidepsin showed similar sensitivity with IC50’s in the low micromolar (IC50 range: 2–34 μM) and nanomolar (IC50 range: 6–9 nM) range respectively and did not correlate with baseline MDM2 status (Figure 2A–2B).
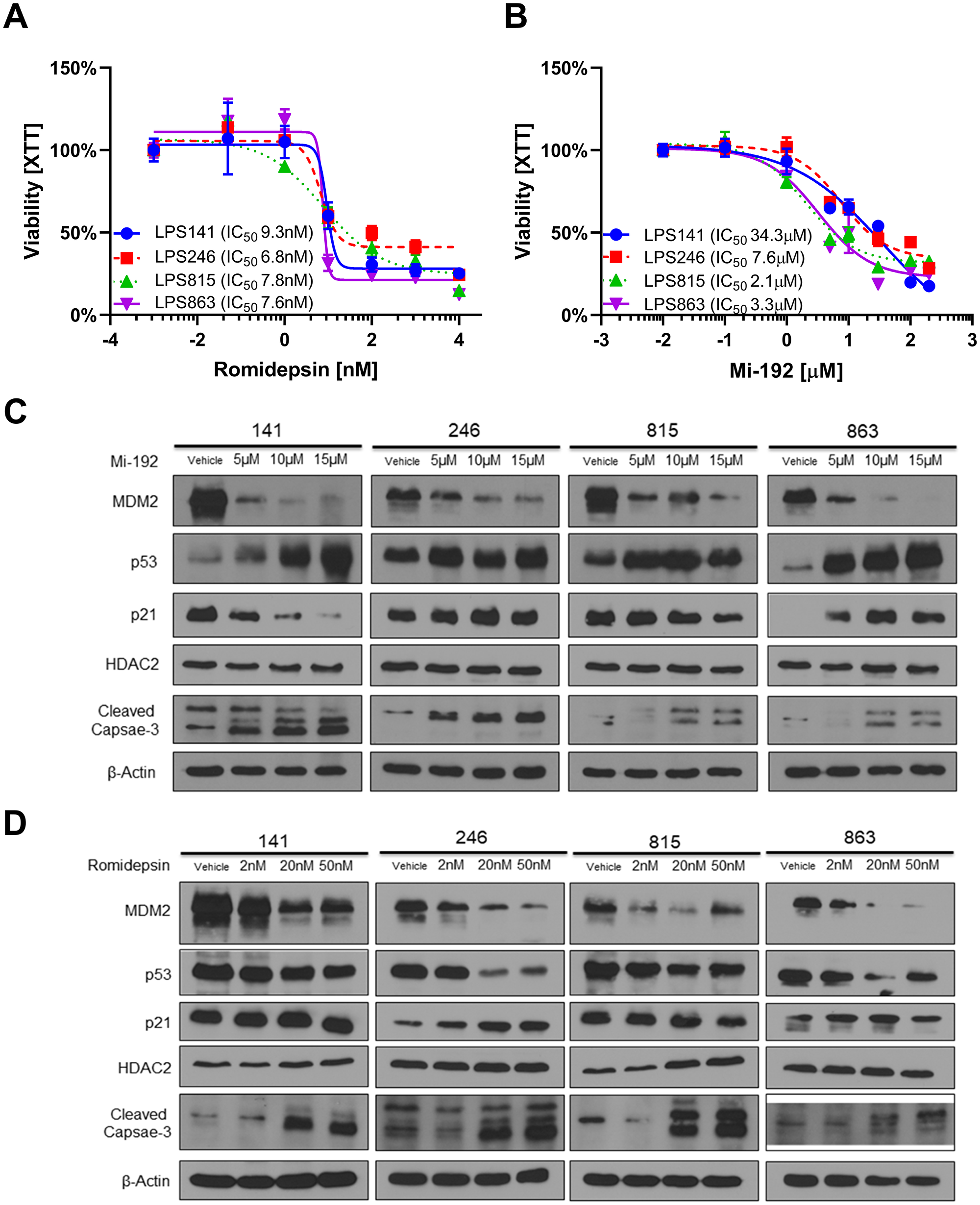
Figure 2: HDAC2 inhibitors reduce MDM2 expression in in vitro models of DDLPS. (A, B) Cellular viability in a panel of four DDLPS cell lines as measured by the XTT assay. Percent viability is relative to vehicle treated control. Cellular sensitivity to romidepsin (A) and MI-192 (B) did not correlate with baseline MDM2 status in these cells. (C) Protein expression for DDLPS cells treated for 48 hours with MI-192. MI-192 reduced MDM2 and p21 expression while increasing expression of p53 and cleaved capsase-3. (D) Protein expression for DDLPS cells treated for 48 hours with romidepsin. Romidepsin reduced MDM2 expression while increasing expression cleaved capsase-3. In the 246 and 863 cell lines, romidepsin reduced p53 expression.
HDAC inhibitors reduced MDM2 protein in vitro and activated p53-mediated cell death
To assess the molecular effect of HDAC2 inhibition in DDLPS, DDLPS cell lines were treated with either MI-192 or romidepsin at increasing doses for 48 hours before protein was collected to be measured by Western blot. Both MI-192 and romidepsin significantly reduced MDM2 protein expression compared to vehicle control with little effect on HDAC2 protein expression (Figure 2C–2D). MI-192 induced an increase in p53 expression; however, this effect was not seen consistently in romidepsin treated DDLPS cell line models. Finally, both MI-192 and romidepsin increased expression of cleaved-caspase 3, suggesting an induction of cellular apoptosis.
HDAC2 inhibition elicits anti-tumor effect and decreases MDM2 RNA levels in DDLPS xenografts
To test the effect of HDAC2 inhibition in vivo, we utilized a DDLPS mouse model subcutaneously xenografted with the LPS863 cell line. When tumors were palpable, mice were randomized to receive either vehicle control or romidepsin injected IP twice weekly. In DDLPS xenografts, romidepsin significantly delayed tumor growth as a single agent compared to vehicle control (Day 22: control 357.7 ± 231.7 mm3, romidepsin 233.8 ± 130.3 mm3; p = 0.001; Figure 3A). In excised tumors, romidepsin reduced tumor weight (fold change compared to control: control 1.0 ± 0.2, romidepsin 0.48 ± 0.14; p = 0.04; Figure 3B) and reduced MDM2 levels as measured by quantitative RT-PCR (fold change compared to control: control 1.0 ± 0.21, romidepsin 0.51 ± 0.07; p = 0.02; Figure 3C). No appreciable mouse toxicity in terms of body weight or fur changes were noted.
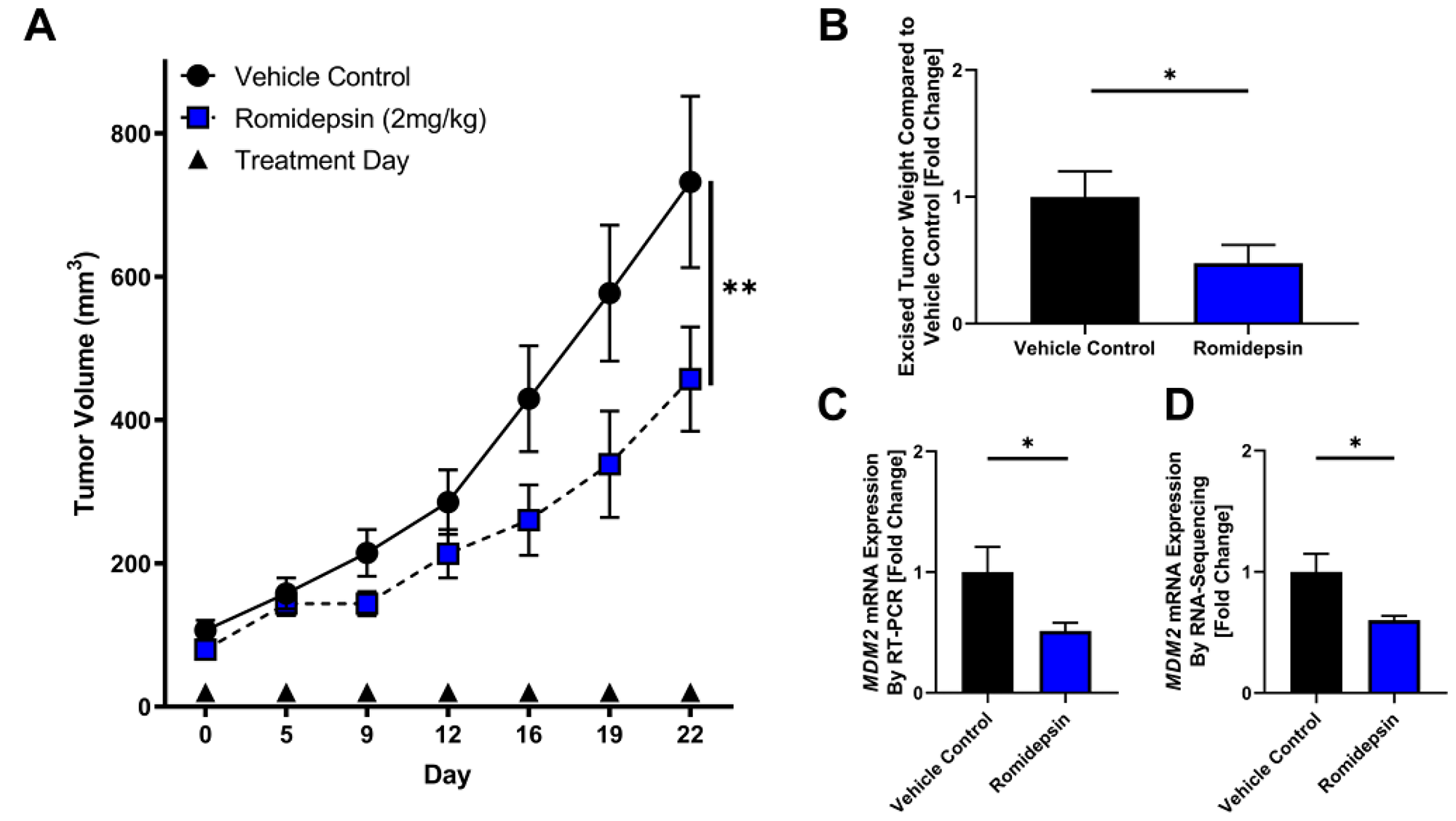
Figure 3: Romidepsin exhibits anti-tumor effect in xenograft model of DDLPS. (A–C) DDLPS xenograft models with bilateral flank injections of the LPS863 cell line were randomly divided into treatment with vehicle control or romidepsin. (A) Tumor growth was significantly reduced in mice treated with romidepsin compared to vehicle control (Day 22: control 357.7 ± 231.7 mm3, romidepsin 233.8 ± 130.3 mm3; p = 0.001). (B–D) Mice were sacrificed at day 22 and tumors excised for further analysis. (B) Excised tumors were weighed, demonstrating a lower mean tumor weight in mice treated with romidepsin compared to control (fold change compared to control: control 1.0 ± 0.2, romidepsin 0.48 ± 0.14; p = 0.04). (C) mRNA was collected from excised tumors and measured for MDM2 expression normalized to B2M. Romidepsin significantly lowered MDM2 expression compared to vehicle control (fold change compared to control: control 1.0 ± 0.21, romidepsin 0.51 ± 0.07; p = 0.02). (D) mRNA was collected from excised tumors and measured for MDM2 expression by RNA-sequencing. Romidepsin significantly lowered MDM2 expression compared to vehicle control (fold change compared to control: control 1.0 ± 0.15, romidepsin 0.60 ± 0.03; p = 0.04). *p < 0.05; **p < 0.01.
Four tumors from both vehicle control and romidepsin treated mice were selected at the time of tumor harvest for further analysis utilizing RNA sequencing. MDM2 was significantly reduced in tumors in the romidepsin arm compared to vehicle control (fold change compared to control: control 1.0 ± 0.15, romidepsin 0.60 ± 0.03; p = 0.04). To assess global regulatory changes induced by romidepsin treatment, Ingenuity Pathway Analysis was used to assess the most highly altered regulatory pathways by treatment group. Top altered regulators included that of the TP53 network (Listing of altered regulators, Supplementary Table 1).
DISCUSSION
Advanced DDLPS is associated with both poor prognosis and poor response to treatment. Pathognomonic to all DDLPS is the amplification of the MDM2 gene whose direct effect is to inhibit p53 activity [6, 24]. Pre-clinical studies have demonstrated that MDM2 action may be targetable; however, clinical development remains an on-going challenge with notable toxicity [8–11]. Here we demonstrate an alternate approach through the inhibition of HDAC2. This HDAC2 inhibition reduces MDM2 expression and abrogates tumor growth in vitro and in vivo.
We established HDAC2 as an epigenetic protein of interest in two independent datasets using baseline mRNA expression of DDLPS tumors. HDAC2 was the most highly co-expressed class I HDAC with MDM2. In these datasets, elevated HDAC2 was associated with poor DFS, DRFS, and OS. Clearly prospective studies will be necessary to identify the clinical and prognostic utility of HDAC2 expression in DDLPS. Our data does suggest that HDAC2 may play a role in the biology of DDLPS. We acknowledge that results from this study cannot be used to determine an appropriate HDAC2 mRNA clinical cutoff due to a lack of a reproducible sequencing platform between datasets and the need of a larger database of DDLPS patients from which to draw.
A prior study of HDAC inhibition demonstrated in vitro effect of pan-HDAC inhibition in DDLPS [12]. Our work has further evaluated HDAC2 specific medications MI-192 and romidepsin. Excitingly, romidepsin is an FDA approved agent in the treatment of other malignancies. In vitro, romidepsin showed strong effect in our panel of DDLPS cell lines, effectively reducing MDM2 expression while triggering apoptosis. In vivo, romidepsin demonstrated significant activity in a xenograft mouse model of DDLPS. Importantly, romidepsin treated tumors not only demonstrated significantly smaller tumors but also exhibited a reduction in MDM2 expression compared to vehicle control. Upstream regulatory analysis identified reactivation of p53 activity that may be a critical driver of romidepsin’s anti-tumor effects in DDLPS. Taken together, this pre-clinical analysis suggests that inhibition of HDAC2, specifically utilizing the HDAC1/2 inhibitor romidepsin, reduces MDM2 expression and may promote apoptosis in DDLPS. We have previously shown that MDM2 amplification and expression is associated with chemoresistance in DDLPS [7]. Further study of the HDAC2:MDM2 axis may provide further insight into mechanisms of chemoresistance in this disease. Additionally, HDAC2 modulation of MDM2 along with other DNA damaging agents and CDK4 inhibitors are under development.
In summary, to our knowledge, this is the first report of romidepsin's activity in MDM2-amplified tumors [16]. The data presented here suggests a potential role for HDAC2 inhibition in DDLPS as a modulator of the MDM2:p53 pathway. Further clinical trials will be needed to verify this hypothesis.
Materials and Methods
Clinical datasets
The Cancer Genome Atlas (TCGA) - Gene expression, as measured by RNA-seq, from 58 subjects with DDLPS from the TCGA and was collected and assessed utilizing cbioportal [25]. Disease-free survival (DFS) data was available for 52 subjects, while overall survival (OS) data was available for all 58 subjects. HDAC2 was considered as both a continuous and dichotomous predictor of clinical outcomes.
Memorial Sloan-Kettering Cancer Center Dataset (MSKCC) - Gene expression data from 40 subjects with DDLPS was downloaded from the GEO database as previously described [26]. Gene expression was measured by microarray. Distant recurrence-free survival (DRFS) and gene expression was collected for all 40 subjects. HDAC2 was considered as both a continuous and dichotomous predictor of clinical outcomes.
Cell culture
Patient-derived DDLPS cell lines (LPS246, LPS815, and LPS863) were brought directly into culture from patients from the SARC Sarcoma SPORE housed at The Ohio State University and maintained as previously described [8]. Dr. Jonathan Fletcher (Brigham and Women’s Hospital, Boston, MA, USA) generously provided us with LPS141 cells.
Chemical compounds and reagents
Romidepsin (Selleckchem) and MI-192 (Sigma) were dissolved in DMSO as recommended by the manufactures and stored at −80° C for in vitro experiments. Serial dilutions were made to ensure a final DMSO concentration was below 0.1%.
Proliferation assay
Exponentially growing DDLPS cell lines were seeded into 96-well plates and treated with the indicated compounds. Cell viability was determined by adding 20 μL of XTT reagent (Roche) as per manufacturer’s instructions. Romidepsin treatments ranged from 0.001–10 μM while MI-192 treatments ranged from 1–100 μM.
Western blotting
In 100 mm culture dishes, 750,000 cells were plated and allowed to attach overnight. Drug (MI-192, Romidepsin) concentrations were calculated based on the IC50 concentration of the DDLPS cell lines to each respective drug. Cells were lysed using SDS/laemmli lysis buffer. Lysates were clarified by sonication. Supernatant was used for protein analysis. Equal amount (25 ug) of cell extracts were resolved by SDS-polyacrylamide gel electrophoresis and transferred to a pretreated PVDF membrane for 90 minutes in a cold room. Membranes were incubated overnight at 4° C using Rabbit monoclonal antibodies of MDM2 (ThermoFisher), HDAC2 (Abcam), P53 (Santa Cruz), P21 (Santa Cruz), and Caspase 3 (Thermofisher), using optimized dilutions for protein detection. A mouse monoclonal antibody was used to detect beta-actin (Cell Signaling). Proteins were visualized using enhanced chemiluminescence (ECL) detection agents (Perkin Elmer).
mRNA expression
Xenografted tumors were lysed using a Precellys Evolution Tissue Homogenizer with Cryolys Cooling Reservoir. mRNA was extracted using the Qiagen RNeasy Mini Kit (Qiagen), and mRNA was quantified with a NanoDrop 2000 instrument. Bio-Rad’s iScript cDNA Synthesis Kit (Bio-Rad) was used to generate a cDNA library and iTaq™ Universal SYBR® Green Supermix was used for quantitative detection of transcripts via RT-PCR. Gene expression was evaluated by ΔΔct and normalized to B2M.
shRNA knockdown
293T packaging cells were seeded at 1.0 × 106 in 60 mm culture dishes and incubated for 24 hours. PSPAX2 lentiviral packaging plasmid (#12260), pMD2.g vsv-g envelope plasmid (#12259), and scramble shRNA control (#1864) were purchased from Addgene. HDAC2 shRNA were purchased from Sigma (SHCLNG-NM_001527). 2.5 ug PSPAX2, 2.5 ug pMD2.G, 5 ug shRNA, 250 uL serum free DMEM, and 20 uL Fugene 6 (Promega E2691) were added to a 1.7 mL tube and set for 30 minutes at room temperature. The reaction mixture was added to the 60mm dishes and incubated at 37 degrees for 18 hours. Media was replaced with DMEM containing 30% FBS and incubated for another 72 hours. The supernatant was collected and filtered through a 0.45 uM cellulose acetate membrane. To a 100 mm dish, LPS246 were seeded at 1.0 × 106. Thirty minutes prior to infection, polybrene was added to the plate for a final concentration of 8 ug/mL. 1mL of the filtered viral supernatant was added to each plate according to each intended treatment. After 72 hours, the media was replaced and puromycin was added to reach a final concentration of 3.0 ug/mL. Following another 72 hours, the cells were collected for analysis.
DDLPS xenograft model
Xenograft models were generated utilizing bilateral flank injections of the LPS863 cell line in 8-week-old, outbred, athymic nude (NCr-nu/nu) female mice. These mice were acquired from the athymic nude mouse colony maintained by the Target Validation Shared Resource (TVSR) at the Ohio State University; original breeders (strain #553 and 554) for the colony were received from the NCI Frederic facility. Bilateral flanks were injected with 1.5 × 106 cells mixed in PBS:Matrigel (2:1, v/v) with a final volume of 150 μL. Once palpable tumors were established, mice were randomly divided into two treatment arms: vehicle control or romidepsin treated. Romidepsin (2 mg/kg) was administered twice weekly by intraperitoneal (IP) injection. Tumor and body weight measurements were performed every 3–4 days until animals were harvested on day 22 as planned prior to study initiation. At harvest, tumors were weighed, measured and prepared for mRNA expression analysis as described. All animal experiments were carried out under protocols approved by the Ohio State University Institutional Animal Care and Use.
RNA sequencing
Four tumors from LPS863 xenografted athymic nude (NCr-nu/nu) female mice were randomly selected at the time of tumor harvest for both control and romidepsin treated mice as described above. Tumors were lysed using a Precellys Evolution Tissue Homogenizer with Cryolys Cooling Reservoir. mRNA was extracted using the Qiagen RNeasy Mini Kit (Qiagen) and treat with DNase I (Thermofisher). mRNA was quantified with a NanoDrop 2000 instrument. Using 200 ug RNA, a cDNA library was generated with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina. An Illumina Hiseq 4000 was used to generate 17–20 × 106 reads that passed filter cluster. Paired ends were 150 base pairs in length.
Statistical analysis
All data was analyzed in Graphpad Prism v.8.0.0. Spearman’s correlation was used to assess co-expression between genes. Survival curves were calculated using the log-rank test for significance and cox proportional hazard regression to calculate hazard ratios (HR) with 95% confidence intervals (95%CI). Assessment of HDAC2 as a continuous predictor of clinical outcomes was assessed individually for both datasets. In addition, two groups of patients with high (HDAC2 High) and low (HDAC2 Low) HDAC2 expression were delineated using maximally selected rank statistics as implemented in the maxstat R package (http://cran.r-project.org/web/packages/maxstat/index.html). Student’s t-test was used as appropriate. The IC50 dose response curves were calculated in utilizing a four-parameter, Hill-slope equation. Ingenuity Pathway Analysis (IPA, QIAGEN) was used to conduct upstream regulator analysis. All data are reported as mean ± SEM unless otherwise noted; p values <0.05 were considered significant. All in vitro data was replicated in ≥2 independent experiments.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the Ohio State University Comprehensive Cancer Center and the National Institutes of Health under grant number P30 CA016058. We thank the Target Validation Shared Resource (TVSR) at the Ohio State University Comprehensive Cancer Center for providing the athymic nude mice used in the preclinical studies as well as conducting the xenograft mouse studies described herein. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
FUNDING
This work was supported by an NCCN Young Investigator’s Award (to JLC), NIH K12 CA133250 award (to JLH).
References
1. Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014; 46:95–104. https://doi.org/10.1097/PAT.0000000000000050. [PubMed].
2. Beird HC, Wu CC, Ingram DR, Wang WL, Alimohamed A, Gumbs C, Little L, Song X, Feig BW, Roland CL, Zhang J, Benjamin RS, Hwu P, et al. Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin. Cold Spring Harb Mol Case Stud. 2018; 4:a002386. https://doi.org/10.1101/mcs.a002386. [PubMed].
3. Nilbert M, Rydholm A, Mitelman F, Meltzer PS, Mandahl N. Characterization of the 12q13-15 amplicon in soft tissue tumors. Cancer Genet Cytogenet. 1995; 83:32–36. https://doi.org/10.1016/S0165-4608(95)00016-X. [PubMed].
4. Binh MB, Sastre-Garau X, Guillou L, de Pinieux G, Terrier P, Lagacé R, Aurias A, Hostein I, Coindre JM. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol. 2005; 29:1340–47. https://doi.org/10.1097/01.pas.0000170343.09562.39. [PubMed].
5. Crago AM, Singer S. Clinical and molecular approaches to well differentiated and dedifferentiated liposarcoma. Curr Opin Oncol. 2011; 23:373–78. https://doi.org/10.1097/CCO.0b013e32834796e6. [PubMed].
6. Ricciotti RW, Baraff AJ, Jour G, Kyriss M, Wu Y, Liu Y, Li SC, Hoch B, Liu YJ. High amplification levels of MDM2 and CDK4 correlate with poor outcome in patients with dedifferentiated liposarcoma: A cytogenomic microarray analysis of 47 cases. Cancer Genet. 2017; 218–219:69–80. https://doi.org/10.1016/j.cancergen.2017.09.005. [PubMed].
7. Bill KLJ, Seligson ND, Hays JL, Awasthi A, Demoret B, Stets CW, Duggan MC, Bupathi M, Brock GN, Millis SZ, Shakya R, Timmers CD, Wakely PE Jr, et al. Degree of MDM2 Amplification Affects Clinical Outcomes in Dedifferentiated Liposarcoma. Oncologist. 2019; 24:989–96. https://doi.org/10.1634/theoncologist.2019-0047. [PubMed].
8. Bill KL, Garnett J, Meaux I, Ma X, Creighton CJ, Bolshakov S, Barriere C, Debussche L, Lazar AJ, Prudner BC, Casadei L, Braggio D, Lopez G, et al. SAR405838: A Novel and Potent Inhibitor of the MDM2:p53 Axis for the Treatment of Dedifferentiated Liposarcoma. Clin Cancer Res. 2016; 22:1150–60. https://doi.org/10.1158/1078-0432.CCR-15-1522. [PubMed].
9. Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. 2017; 10:123. https://doi.org/10.1186/s13045-017-0482-3. [PubMed].
10. de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, Hsu K, Zheng W, Macé S, Tuffal G, Thomas K, Schellens JH. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer. 2017; 76:144–51. https://doi.org/10.1016/j.ejca.2017.02.005. [PubMed].
11. Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, Johnson-Levonas AO, Lam R, Meister AK, Russo G, Knox CD, Rose S, Hong DS. Phase I Trial of the Human Double Minute 2 Inhibitor MK-8242 in Patients With Advanced Solid Tumors. J Clin Oncol. 2017; 35:1304–11. https://doi.org/10.1200/JCO.2016.70.7117. [PubMed].
12. Ou WB, Zhu J, Eilers G, Li X, Kuang Y, Liu L, Mariño-Enríquez A, Yan Z, Li H, Meng F, Zhou H, Sheng Q, Fletcher JA. HDACi inhibits liposarcoma via targeting of the MDM2-p53 signaling axis and PTEN, irrespective of p53 mutational status. Oncotarget. 2015; 6:10510–20. https://doi.org/10.18632/oncotarget.3230. [PubMed].
13. Peltonen K, Kiviharju TM, Järvinen PM, Ra R, Laiho M. Melanoma cell lines are susceptible to histone deacetylase inhibitor TSA provoked cell cycle arrest and apoptosis. Pigment Cell Res. 2005; 18:196–202. https://doi.org/10.1111/j.1600-0749.2005.00225.x. [PubMed].
14. Coiffier B, Pro B, Prince HM, Foss F, Sokol L, Greenwood M, Caballero D, Borchmann P, Morschhauser F, Wilhelm M, Pinter-Brown L, Padmanabhan S, Shustov A, et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012; 30:631–36. https://doi.org/10.1200/JCO.2011.37.4223. [PubMed].
15. Haigentz M Jr, Kim M, Sarta C, Lin J, Keresztes RS, Culliney B, Gaba AG, Smith RV, Shapiro GI, Chirieac LR, Mariadason JM, Belbin TJ, Greally JM, et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012; 48:1281–88. https://doi.org/10.1016/j.oraloncology.2012.05.024. [PubMed].
16. Amiri-Kordestani L, Luchenko V, Peer CJ, Ghafourian K, Reynolds J, Draper D, Frye R, Woo S, Venzon D, Wright J, Skarulis M, Figg WD, Fojo T, et al. Phase I trial of a new schedule of romidepsin in patients with advanced cancers. Clin Cancer Res. 2013; 19:4499–507. https://doi.org/10.1158/1078-0432.CCR-13-0095. [PubMed].
17. Stadler WM, Margolin K, Ferber S, McCulloch W, Thompson JA. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin Genitourin Cancer. 2006; 5:57–60. https://doi.org/10.3816/CGC.2006.n.018. [PubMed].
18. Noonan AM, Eisch RA, Liewehr DJ, Sissung TM, Venzon DJ, Flagg TP, Haigney MC, Steinberg SM, Figg WD, Piekarz RL, Bates SE. Electrocardiographic studies of romidepsin demonstrate its safety and identify a potential role for K(ATP) channel. Clin Cancer Res. 2013; 19:3095–104. https://doi.org/10.1158/1078-0432.CCR-13-0109. [PubMed].
19. Pacheco M, Nielsen TO. Histone deacetylase 1 and 2 in mesenchymal tumors. Mod Pathol. 2012; 25:222–30. https://doi.org/10.1038/modpathol.2011.157. [PubMed].
20. Seo SK, Hwang CS, Choe TB, Hong SI, Yi JY, Hwang SG, Lee HG, Oh ST, Lee YH, Park IC. Selective inhibition of histone deacetylase 2 induces p53-dependent survivin downregulation through MDM2 proteasomal degradation. Oncotarget. 2015; 6:26528–40. https://doi.org/10.18632/oncotarget.3100. [PubMed].
21. Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017; 171:950–65.e28. https://doi.org/10.1016/j.cell.2017.10.014. [PubMed].
22. Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002; 62:4916–21. [PubMed].
23. Boissinot M, Inman M, Hempshall A, James SR, Gill JH, Selby P, Bowen DT, Grigg R, Cockerill PN. Induction of differentiation and apoptosis in leukaemic cell lines by the novel benzamide family histone deacetylase 2 and 3 inhibitor MI-192. Leuk Res. 2012; 36:1304–10. https://doi.org/10.1016/j.leukres.2012.07.002. [PubMed].
24. Jour G, Gullet A, Liu M, Hoch BL. Prognostic relevance of Fédération Nationale des Centres de Lutte Contre le Cancer grade and MDM2 amplification levels in dedifferentiated liposarcoma: a study of 50 cases. Mod Pathol. 2015; 28:37–47. https://doi.org/10.1038/modpathol.2014.88. [PubMed].
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088. [PubMed].
26. Gobble RM, Qin LX, Brill ER, Angeles CV, Ugras S, O’Connor RB, Moraco NH, Decarolis PL, Antonescu C, Singer S. Expression profiling of liposarcoma yields a multigene predictor of patient outcome and identifies genes that contribute to liposarcomagenesis. Cancer Res. 2011; 71:2697–705. https://doi.org/10.1158/0008-5472.CAN-10-3588. [PubMed].