
Introduction
Ovarian cancer is a lethal gynecological malignancy that accounts for 5%–6% of all cancer-related deaths. In 2012 alone, a total of 238,700 new cases had been identified with 151,900 cases succumbing to mortality [1]. Epithelial ovarian cancer (EOC), the most common type of ovarian cancer, has been classified into the following subtypes according to histopathological findings: high-grade serous (HGSC), low-grade serous, mucinous, endometrioid, and clear cell carcinoma. Given that EOC generally develops with few specific symptoms, majority of the patients are diagnosed at advanced stages [2]. Therefore, diagnosing EOC at an early stage is a need that remains clearly unmet [3]. The standard treatment for EOC has been primary debulking surgery aiming for no macroscopically identifiable residual tumor followed by adjuvant platinum–taxane-based combination chemotherapy. Recently, molecular targeted therapies, including bevacizumab, poly ADP-ribose polymerase (PARP) inhibitors, and anti-programmed cell death (PD)-1 antibodies, have been helping to overcome the poor prognosis of EOC [4, 5]. Given that the clinical benefits obtained through these therapies are believed to be associated with specific molecular aberrations, identification of reliable biomarkers through molecular testing is warranted for clinical application. In the SOLO2/ENGOT-Ov21 trial, which aimed to investigate the efficacy of PARP inhibitor, clinical benefits were found in patients bearing germline BRCA1/2 mutations, indicating that mutation testing for BRCA1/2 should be considered in part of clinical practice [4, 6]. Similarly, microsatellite instability testing has been offered to individuals with EOC considering that it has been shown to reflect the extent of tumor mutational burden and is expected to be a predictive biomarker for superior response to anti-PD-1/PD-L1 antibodies [7]. Although the development of these molecular targeted therapies has shed light on novel treatment options for EOC, their efficacy is still limited and mortality in EOC cases with advanced stages remains substantially problematic.
Currently, accumulating evidence has indicated that microRNA (miRNA) plays a critical role in the pathogenesis of human cancers, including EOC [8–10]. Thus far, miRNA-34a (miR-34a) has been shown to be a key regulator of tumor suppression by controlling factors related to cell biology, such as proliferation, invasion, apoptosis, and chemoresistance [11, 12]. Corney et al. revealed that the miR-34 family, which was downregulated in EOC harboring p53 mutations, could promote tumor progression by regulating MET expression in EOC [13]. Previously, we identified unique miRNA expression pro-files that could discriminate ovarian cancer histotypes with frequently reduced miR-34a expression in HGSC [14]. Recently, Rokavec et al. showed that interleukin-6 receptor (IL-6R), which was a direct target of miR-34a, was repressed via IL-6R/signal transducer and activator of transcription (STAT) 3/miR-34a feedback loop in colorectal cancer [15]. Considering that the IL-6R/STAT3 signaling pathway is upregulated and believed to be a potential therapeutic target in EOC [16, 17], we hypothesized that the reduced miR-34a expression may play an important role in the pathogenesis of HGSC through derepression of the IL-6R/STAT3 signaling pathway.
This study therefore aimed to investigate the biological significance of reduced miR-34a expression in HGSC and evaluate the viability of miR-34a as a therapeutic application for HGSC via a series of in vitro and in vivo experiments. In addition, we evaluated the relationship between miR-34a expression and clinicopathological characteristics, including IL-6R expression status, in patients with HGSC.
Results
Involvement of miR-34a in the pathogenesis of HGSC
To examine the biological significance of reduced miR-34a expression in HGSC, we conducted miRNA mimic-based replacement experiments using two HGSC cell lines, KF28 (p53 dominant-negative mutant) and A2780 (p53 wild type), in which miR-34a expression was significantly lower compared with noncancerous ovarian surface epithelium (OSE) (Figure 1A). We first measured in vitro proliferation and invasion using cells with stable miR-34a overexpression (Figure 1B-1C). Accordingly, HGSC cell lines with miR-34a overexpression had reduced cell proliferation and invasion compared with either cells expressing a nontargeting control (mock) or parental cells (null) (Figure 2A-2B, Supplementary Figure 1). We further examined the responsiveness of these cell lines to cytotoxic agents, including cisplatin and paclitaxel. Both miR-34a-transfected HGSC cells showed significantly greater cell death in response to either cisplatin or paclitaxel compared with mock and null controls (Figure 2C).
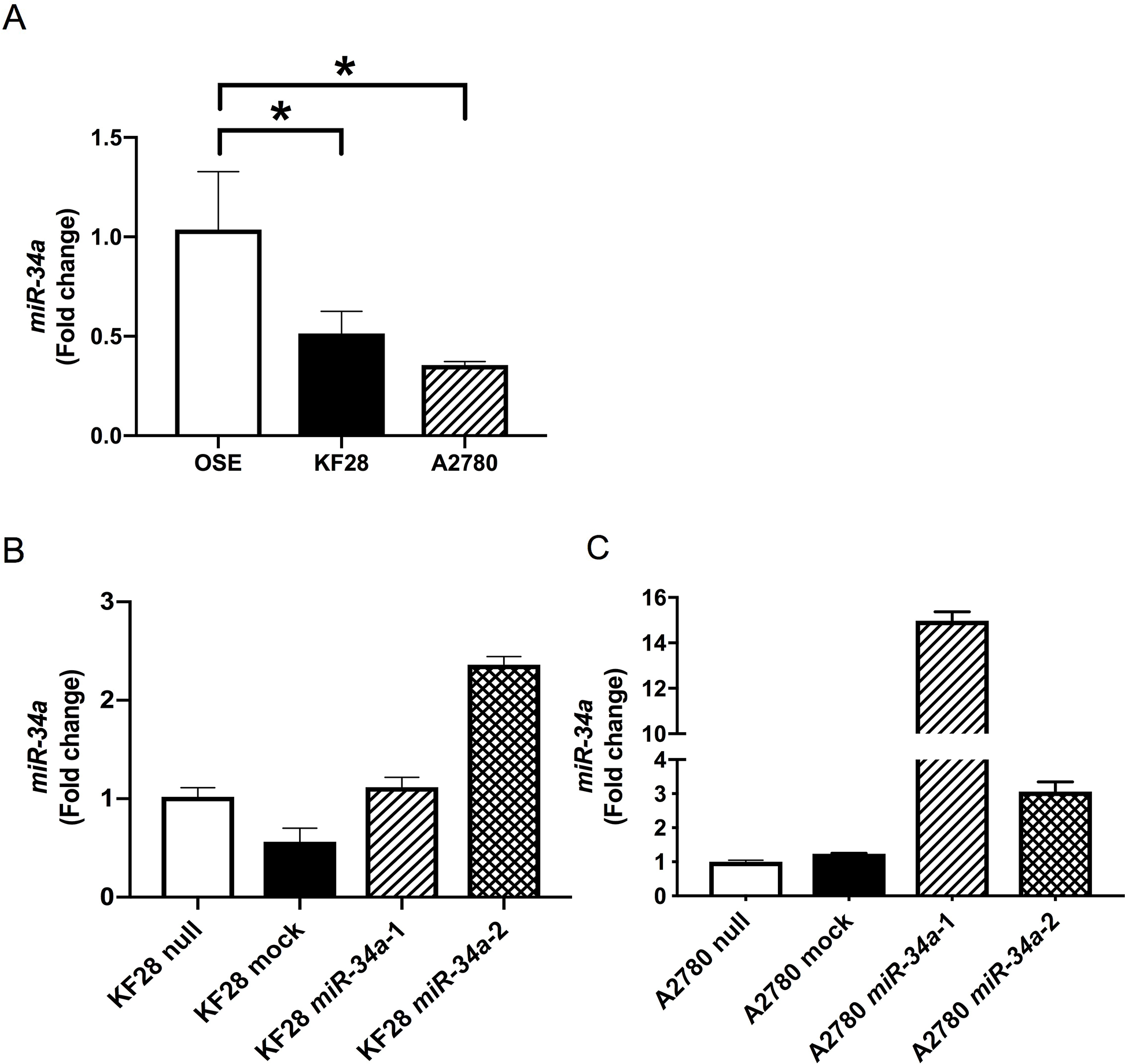
Figure 1: Establishment of HGSC cell lines with stable miR-34a overexpression miR-34a expression was quantified using the comparative method in real-time RT-PCR analysis. (A) miR-34a expressions were significantly lower in HGSC cell lines (KF28 and A2780) than in OSE cell line (B, C) HGSC cell lines with stable miR-34a overexpression were established in KF28 (B) and A2780 (C) cells. * means P < 0.05.
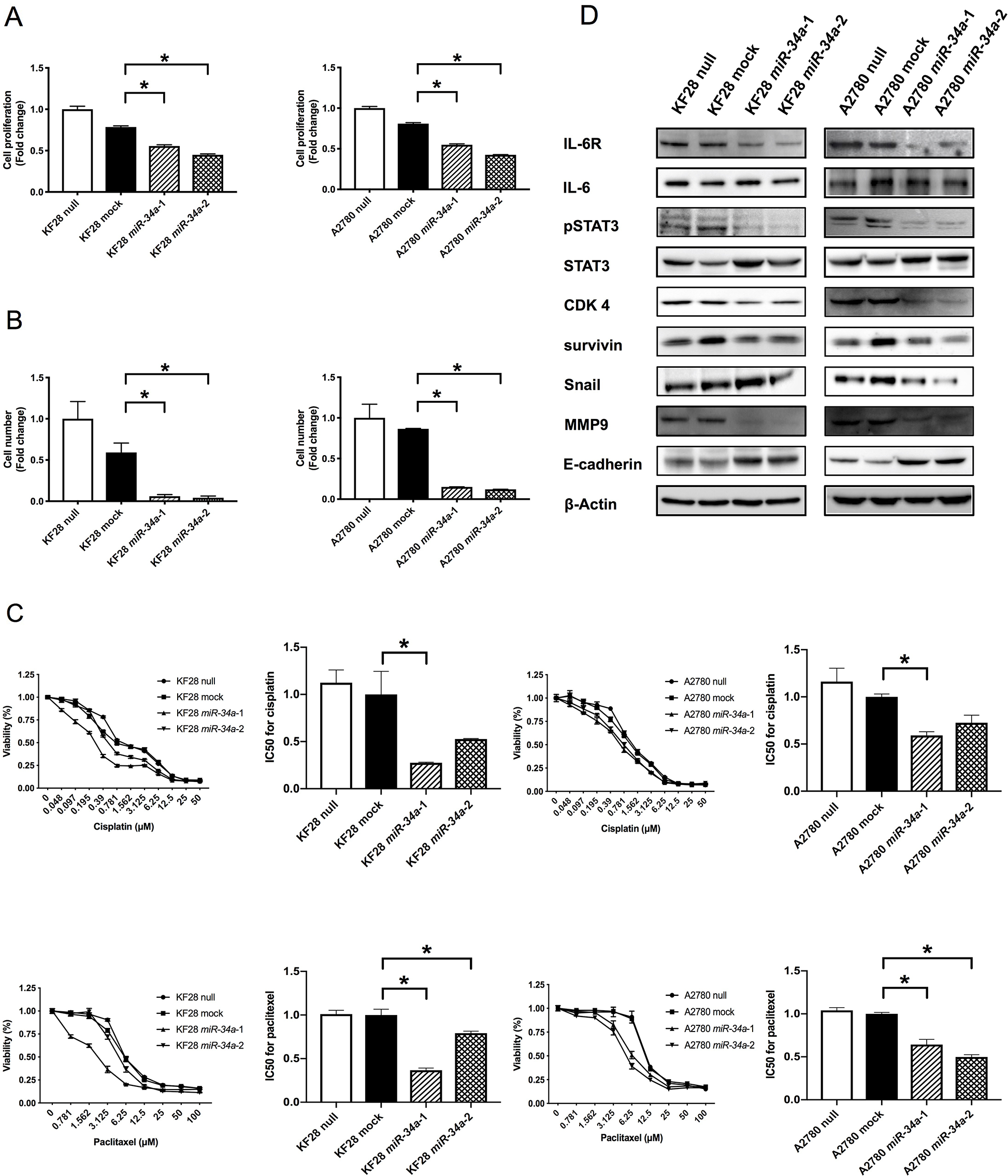
Figure 2: . Involvement of miR-34a in HGSC pathogenesis in vitro. (A) Cell proliferation ability was analyzed using the MTS assay in KF28 and A2780 cells. HGSC cell lines with miR-34a overexpression had lower cell proliferation ability compared with negative control cells. (B) Cell invasion ability was analyzed using the invasion assay in KF28 and A2780 cells. HGSC cell lines with miR-34a overexpression had lower cell invasion ability compared with negative control cells. (C) Responsiveness of HGSC cell lines to cytotoxic agents was analyzed using the cytotoxicity assay in KF28 and A2780 cells for cisplatin and paclitaxel. miR-34a-transfected HGSC cells showed significantly greater cell death due to cytotoxic agents compared with negative control cells. (D) Protein expression of HGSC cell lines was analyzed using Western blot analysis. Forced expression of miR-34a reduced IL-6R expression affecting downstream of the IL-6R/STAT3 signaling pathway. β-Actin was used as loading control. * means P < 0.05.
Next, we addressed whether miR-34a in HGSC could target IL-6R, which has been shown to be highly expressed in EOC [16, 18]. Accordingly, miR-34a overexpression reduced IL-6R expression and subsequently decreased STAT3 phosphorylation at tyrosine 705 without affecting overall levels of IL-6 or STAT3 (Figure 2D, Supplementary Figure 3). In addition, CDK4 and survivin, mediators of cell proliferation, decreased in HGSC cell lines with stable miR-34a overexpression (Figure 2D, Supplementary Figure 3). Furthermore, miR-34a overexpression increased E-cadherin expression and was associated with reduced Snail and MMP9 expression, indicating that epithelial–mesenchymal transition (EMT) was inhibited in these cell lines.
To clarify the functional interaction between miR-34a and IL-6R in HGSC, we investigated whether restoration of IL-6R expression could reverse the miR-34a-induced inhibitory effect on cell proliferation in HGSC cell lines. Accordingly, IL-6R overexpression in A2780 cells co-transfected with a miR-34a mimic partially rescued suppression of cell proliferation (Supplementary Figure 2A).
Involvement of miR-34a in HGSC tumorigenicity in vivo
To further evaluate the role of miR-34a in HGSC tumorigenicity, KF28 cells with stable miR-34a overexpression were injected subcutaneously into BALB/c nude mice, after which tumor growth was monitored for up to 6 weeks (Figure 3A). In the KF28 null, mock, and miR-34a-1 groups, 5 mice were used and in the KF28 miR-34a-2 group, 3 mice were used because tumor formation was not observed in 2 out of 5 mice. Mice injected with KF28 cells stably expressing miR-34a showed an overall reduction in tumor burden compared with those in the null or mock groups (Figure 3B). Consistent with increased miR-34a levels, IL-6R expression was significantly reduced in KF28 cells overexpressing miR-34a (Figure 3C). Representative images of subcutaneous tumor xenographs are shown in Figure 3D.
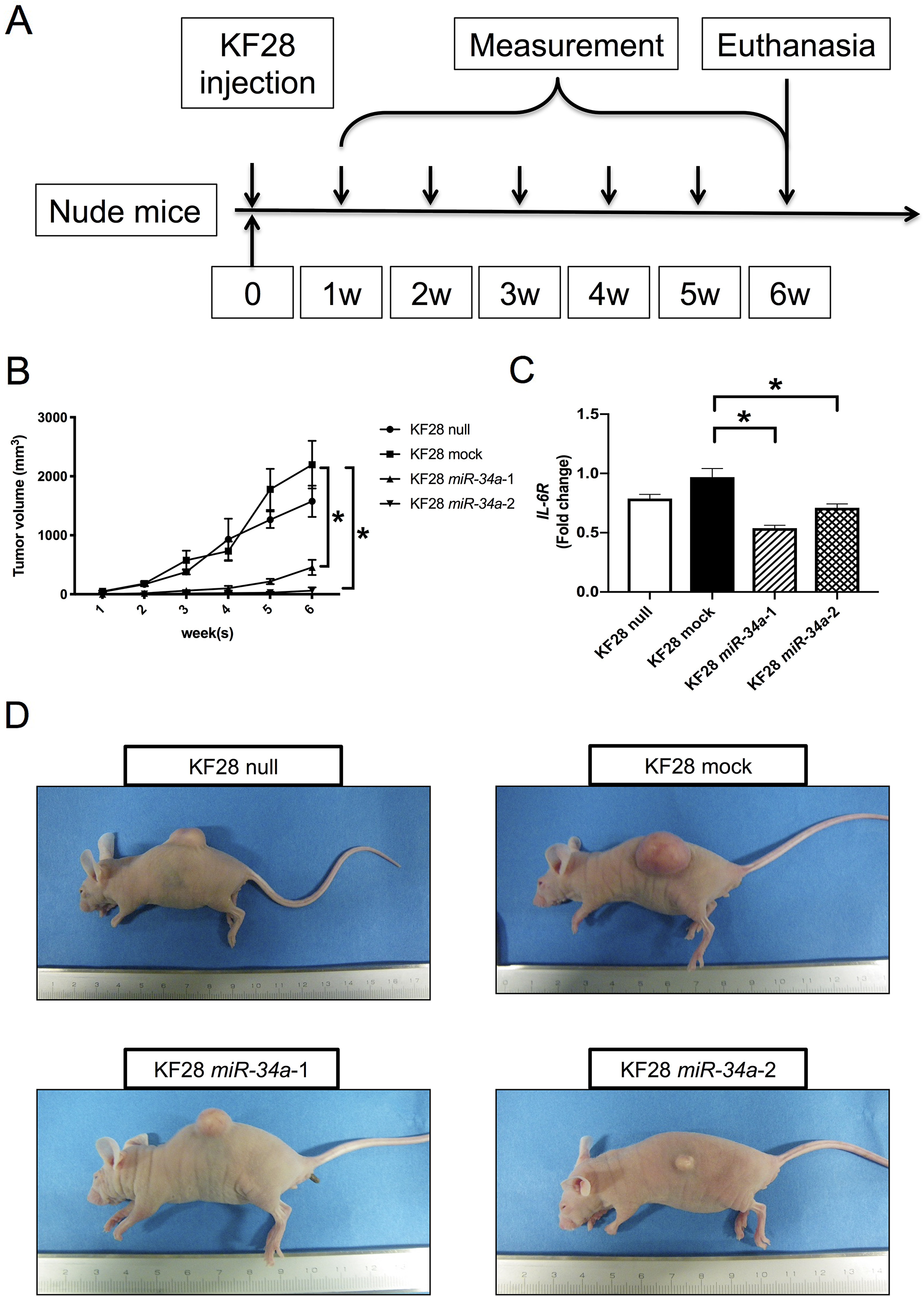
Figure 3: . Replacement of miR-34a inhibiting HGSC tumorigenicity in vivo. (A) KF28 cells were injected subcutaneously into nude mice. Tumor growths were monitored up to 6 weeks. (B) An overall reduction in the tumor volume was observed in mice inoculated with KF28 cells having stable miR-34a overexpression. (C) IL-6R mRNA expression of the tumors were quantified using the comparative method in real-time RT-PCR analysis. (D) Representative images of the gross tumor morphology showing tumor formation in mice. * means P < 0.05.
miR-34a expression in patients with HGSC
Considering our findings in vitro and in vivo, we were motivated to investigate the clinical relevance of miR-34a in patients with HGSC. We examined primary specimens derived from 33 patients with HGSC and assessed miR-34a expression using real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis. Clinical characteristics of patients are presented in Table 1. Accordingly, patients with advanced-stage disease (stages III and IV) had significantly lower miR-34a expression than those with early-stage disease (stages I and II) (Figure 4A). Additionally, patients without residual tumor after initial surgery showed a significant reduction in miR-34a expression (Figure 4B). We subsequently measured IL-6R expression in the same cohort using immunohistochemical analysis. Although no statistical correlation was found, there was a trend of inverse correlation between miR-34a expression and IL-6R immunohistochemistry (IHC) score (Figure 4C). Next, we explored the underlying mechanisms for reduced miR-34a expression in the clinical specimens. Both the deletion of the miR-34a genomic locus (1p36.22) and hypermethylation of the promoter region have been reported as possible mechanisms for miR-34a downregulation in EOC [19]. Using a TaqMan PCR-based method for copy number analysis, loss of heterozygosity for the miR-34a genomic region was observed in 9 patients (29.0 %) within our cohort (Figure 4D, Supplementary Table 1). To examine the CpG methylation status of the miR-34a promoter lesion, the MethyLight analysis was performed. Accordingly, miR-34a promoter methylation was observed in 25 patients (75.8 %) with percentage of methylated reference values ranging from 0.58 to 44.26 (Figure 4D, Supplementary Table 1).
Table 1: Clinicopathological characteristics of the 33 patients with HGSC
| Parameters | n = 33 |
|---|---|
| Patient age (years, mean ± SD) | 55.2 ± 7.9 |
| BMI (kg/m2, mean ± SD) | 19.3 ± 2.9 |
| FIGO stage | |
| I (%) | 4 (12.1) |
| II (%) | 4 (12.1) |
| III (%) | 23 (69.7) |
| IV (%) | 2 (6.1) |
| Follow-up period [months, median (IQR)] | 27 (11-72) |
| Recurrence or progression (%) | 9 (27.3) |
| Platinum sensitive (%) | 7 (77.8) |
| Platinum resistant (%) | 2 (22.2) |
| Death at the observation time point (%) | 5 (15.2) |
| Residual tumor after initial surgery | |
| R = 0 (%) | 19 (57.6) |
| R ≠ 0 (%) | 14 (42.4) |
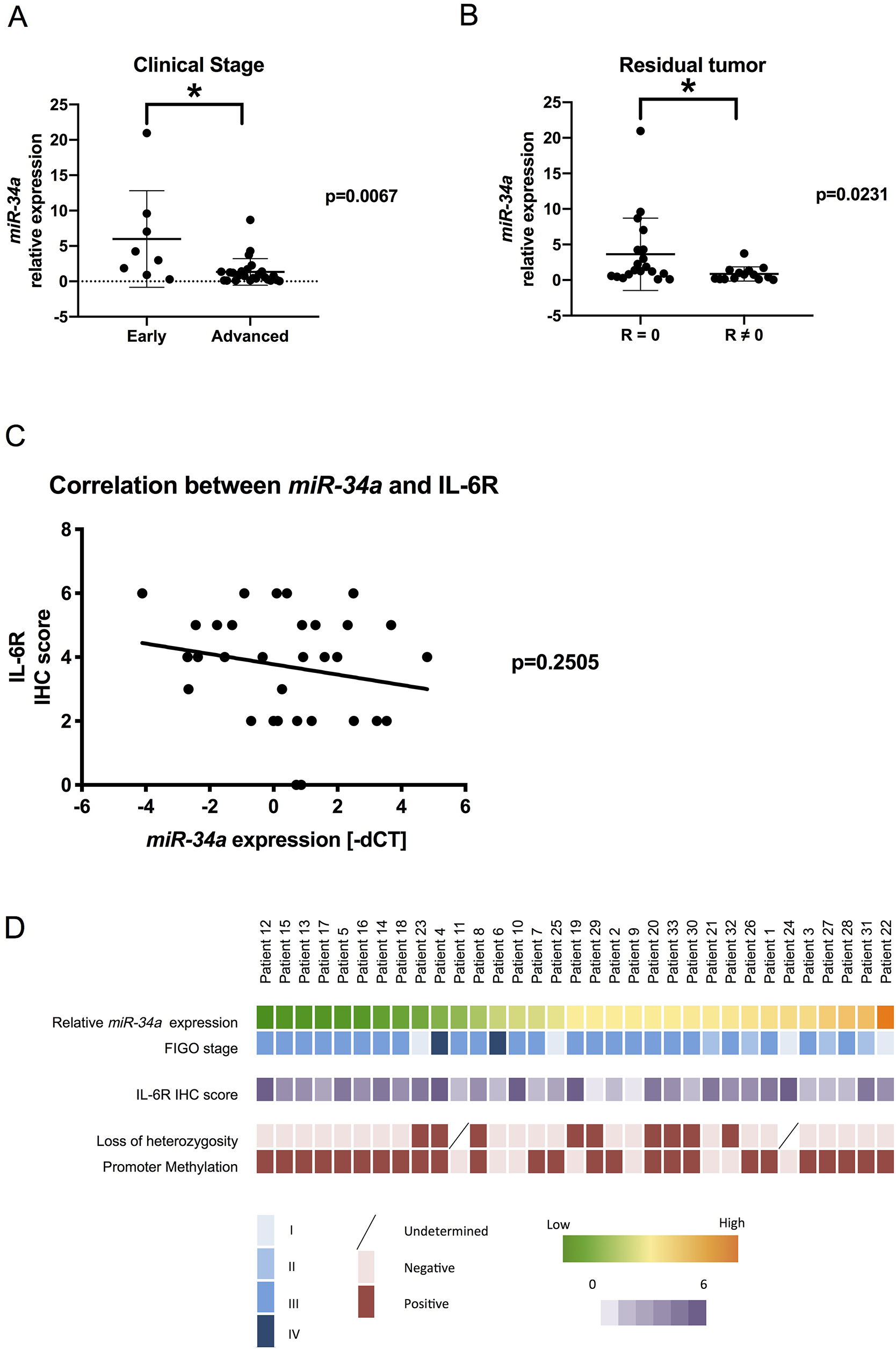
Figure 4: Clinical relevance of miR-34a in patients with HGSC miR-34a expression was quantified using the comparative method in real-time RT-PCR analysis. (A) Patients with advanced-stage disease had significantly lower miR-34a expression levels than those with early-stage disease. (B) Patients with residual tumors had significantly lower miR-34a expressions than those without residual tumors after surgery. R: residual tumors. (C) Linear regression analysis of the correlation between miR-34a expression (−ΔCT) and IL-6R IHC score showed an inverse trend. (D) Clinicopathological characteristics of patients were described using a heat map. * means P < 0.05
DISCUSSION
Accumulating evidence from miRNA profiling has demonstrated that miRNAs play essential roles in ovarian carcinogenesis and impact different clinical aspects [20, 21]. Upregulated miRNAs that are present in cancer cells and contribute to cancer development by inhibiting tumor suppressor genes are considered oncogenic miRNAs, whereas downregulated miRNAs that suppress cancer development by inhibiting proto-oncogenes are known as tumor suppressor miRNAs. In the HGSC cell lines used herein, ectopic expression of miR-34a significantly reduced proliferation and invasion abilities through the downregulation of IL-6R expression and downstream STAT3 signaling, suggesting that redu-ced miR-34a expression might play an important role in the malignant potential of HGSC cells. This phenotype was observed regardless of the original p53 status, thus establishing miR-34a as an independent tumor suppressor miRNA. Since STAT3 activation is induced by phosphorylation at a critical tyrosine residue (tyrosine 705), and this phosphorylation is mainly regulated in the STAT3 signaling pathway [22] we have investigated STAT3 phosphorylation at tyrosine 705 in the present study. In addition, as cell confluence might affect STAT3 phosphorylation [23] we uniformed the cell confluence at approximately 80% in null, mock, and miR-34a overexpression cells for Western blot analysis. Based on this setting, downregulation of pSTAT3 expression may not be affected by cell confluence but triggered by miR-34a replacement. It is also known that STAT3 activates its expression with binding to own promoter [24], and therefore downregulation of the STAT3 signaling pathway may lead to downregulation of total STAT3 expression. However, in the present study, total STAT3 expressions were not substantially changed in miR-34a overexpression cells. We previously reported that downregulation of the IL-6R/STAT3 signaling pathway did not affect total STAT3 expression in three ovarian cancer cell lines, as consistent with this study [17, 25]. These findings suggest that positive feedback regulation of STAT3 may be tissue and/or condition dependent. An earlier finding showing that the IL-6R/STAT3/ miR-34a feedback loop promoted EMT in colorectal cancer prompted us to investigate the existence of the same feedback loop in HGSC [15]. However, downregulation of IL-6R by siRNA did not induce miR-34a expression in the HGSC cell line, A2780 (Supplementary Figure 2B), suggesting that IL-6R/STAT3/miR-34a feedback is tumor specific. Corney et al. demonstrated that frequent miR-34 family downregulation was correlated with EOC metastasis [13]. In addition, a recent report showed a significant association between decreased miR-34a expression and worse prognosis in patients with EOC [26, 27]. Consistent with the aforementioned reports, we also identified that reduced miR-34a expression in HGSC was associated with advanced clinical stage. Taken together, these results indicated that miR-34a might be involved in HGSC pathogenesis through its effects on tumor progression [21]. The underlying mechanisms for reduced miR-34a expression in HGSC can be largely explained by p53 loss-of-function mutations, which were detected in 96% of the patients with this EOC subtype [13, 27, 28]. However, consistent with previous reports [13, 27], we also observed a wide range of promoter methylation and/or copy number alterations at the miR-34a locus in our cohort, although no clear association between either methylation or copy number change and miR-34a expression was observed.
Bioinformatic approaches and in vitro experiments have validated various molecules, including MET, Snail, E2F3a, and histone deacetylase 1 (HDAC1), as direct targets of miR-34a in EOC [13, 26, 29, 30]. Reduced miR-34a expression may affect the invasive behavior of EOC by inducing EMT via alleviating the repression of MET or Snail [13, 26]. Similarly, miR-34a has been shown to suppress ovarian cancer cell proliferation and chemoresistance by controlling HDAC1 expression [30]. Here we demonstrate that miR-34a exerted tumor-suppressive effects in HGSC by regulating the IL-6R/STAT3 signaling pathway and the associated expression of Snail, MMP9, CDK4, and survivin. These IL-6R/STAT3-mediated regulatory proteins could perhaps also be direct targets of miR-34a as shown in earlier studies [11, 31]. The ability of miR-34a to directly bind to the 3′-UTR of IL-6R has been demonstrated in the context of colorectal cancer [15]. The present study provides independent evidence that miR-34a regulates the IL-6R/STAT3 pathway in ovarian cancer by demonstrating that IL-6R reexpression is able to mitigate the effects of miR-34a mimetic oligos (Supplementary Figure 2A). Although the involvement of the IL-6R/STAT3 signaling pathway in EOC has been explored with regard to tumor progression and chemoresistance [16, 17, 25, 32–34], the use of antibodies (siltuximab and tocilizumab) targeting this signaling pathway has not generated clear clinical relevance [32, 33, 35]. Alternative approaches for repressing IL-6R signaling in the context of HGSC are thus needed.
MiRNA-based therapy has great potential for becoming a more powerful tool in cancer treatment by simultaneous modulation of multiple genes involved in cancer-related signaling pathways. Based on our findings from in vitro analyses, we further investigated the viability of miR-34a as a therapeutic application for HGSC in vivo. The results obtained from mice transplanted with stable miR-34a overexpression xenographs showed that replacement of miR-34a could effectively reduce tumorigenicity. Furthermore, IL-6R mRNA expression, which is directly regulated by miR-34a, was significantly reduced in KF28 cells overexpressing miR-34a (Figure 3C). We also demonstrated that pSTAT3 expression level in KF28 cells overexpressing miR-34a was downregulated compared with that in control by Western blot analysis (Supplementary Figure 4). However, the effect of miR-34a replacement on STAT3 activation was not significant compared with that on IL-6R mRNA expression. We hypothesized the etiologies accounting for this difference owing to the following reasons. First, there might be survival advantages in this in vivo experiment. Namely, the STAT3 signaling pathway is essential to survive so that its activation in tumors obtained from survived mice may be maintained to some extent in miR-34a replacement group. This explanation can be supported by the fact that tumors were harvested after a long duration since the transplantation of the cancer cells. Second, the STAT3 signaling pathway is regulated by many factors other than IL-6R; therefore, miR-34a cannot strongly affect STAT3 activation compared with IL-6R mRNA expression that is directly regulated by miR-34a. These etiologies may produce the difference in the effect of miR-34a on its correlating targets. A previous study has shown that the soluble form of IL-6R not only potentiates the effects of secreted IL-6 but also widens the range of cells affected by IL-6 [36]; in this regard, miR-34a may affect the soluble form of IL-6R and can be a potential rationale for reducing the tumorigenicity.
In our animal model, miR-34a was stably integrated into the genome of the tumor cells. The main obstacle has been the low efficiency of exogenous miRNA delivery into cancer cells using currently available methods [11]. Further research is required to develop more efficient strategies to reexpress miR-34a. In other types of tumors, miR-34a replacement therapeutic strategies have also been investigated in preclinical setting. For multiple myeloma, the combination of miR-34a with other anticancer agents strongly inhibits tumor growth and appears as a promising strategy [37]. Combined with radiotherapy, miR-34a can improve the efficacy of lung cancer radiotherapy by inducing senescence via targeting c-Myc [38]. Furthermore, the development of bioengineered RNAs as novel therapeutic agents has been paid attention, and bioengineered miR-34a prodrug has been demonstrated to be effective in controlling osteosarcoma tumor growth [39]. To date, only one phase I clinical trial has demonstrated the oncosuppressive effects of miR-34a in patients with solid tumors [40]. However, adverse effects, such as myelosuppression and liver damage, were also observed. Although unmet requirements still have to be resolved before miR-34a can be used in clinical practice, improvements in drug delivery systems and management of adverse effects may make it applicable for clinical use in the near future.
In conclusion, we show that reduced miR-34a expression is strongly correlated with HGSC pathogenesis through its regulation of the pro-inflammatory IL-6R/STAT3 pathway and its subsequent targets. Accordingly, the present study provides the rationale for miR-34a replacement as a promising therapeutic target strategy for HGSC. Nonetheless, further work is required to establish a safe and efficient delivery system for miR-34a and similar tumor-suppressive miRNAs.
Materials and Methods
Clinical samples and cell lines
This study involved surgically obtaining tumor specimens of primary ovarian cancer from patients receiving treatment at Department of Obstetrics and Gynecology, The Jikei University School of Medicine. The Ethics Review Committee of The Jikei University School of Medicine approved of the study protocol [approval number: 27-076(7961), 28-063(8326)], and all patients provided written informed consent prior study participation. Tumors were staged according to the International Federation of Gynecology and Obstetrics staging system (2014). This study used two ovarian serous carcinoma cell lines, KF28 and A2780. KF28, a single-cell clone of the human ovarian serous carcinoma cell line [41], was obtained as a kind gift from Dr. Kikuchi Y (Ohki Memorial Kikuchi Cancer Clinic for Women, Saitama, Japan), whereas A2780 cells were purchased from KAC Inc. (Tokyo, Japan). KF28 was negative for mycoplasma contamination tested using Cycleave™ PCR Mycoplasma Detection Kit (Takara Bio Inc., Shiga, Japan). KF28 and A2780 cells were cultured in RPMI-1640 (Sigma-Aldrich, Tokyo, Japan) containing 10% fetal bovine serum (FBS). As a normal control, we used human OSE cell lines established as described previously [42]. OSE cells were maintained in RPMI-1640 (Sigma-Aldrich, Tokyo, Japan) containing 10% FBS.
Establishment of HGSC cell lines with stable miR-34a overexpression
Stable miR-34a overexpression clones were established in KF28 and A2780 cells through plasmid vector transfection and Lipofectamine™ 2000 Transfection Reagent (Invitrogen). As a selectable marker, neomycin resistance gene was used and cultured with G-418-containing medium, after which the surviving colonies expressing the neomycin resistance gene were collected.
RNA and DNA extraction
Freshly excised surgical specimens were stored in RNAlater™ Solution (Thermo Fisher Scientific, MA, USA) at 4°C for 24 h and subsequently frozen at −80°C prior to RNA extraction. We used either the manual method using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction or the automated method using the gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, Gladbach, Germany) for homogenization and the Maxwell™ RSC simplyRNA Tissue Kit (Promega Corporation, Madison, WI, USA) for total RNA extraction. For DNA extraction, freshly excised surgical specimens were stored at −80°C. Two extraction methods were utilized, including the manual method using the Gentra Puregene Tissue Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s instruction and the automated method using the gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, Gladbach, Germany) for homogenization and the Maxwell™ RSC blood DNA Kit (Promega Corporation, Madison, WI, USA).
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Extracted RNAs for IL-6R mRNA assessment were subjected to reverse transcription using qScript cDNA SuperMix™ (Quantabio, Beverly, MA). Complementary DNAs (cDNAs) were subjected to quantitative real-time RT-PCR using PerfeCTa SYBR™ Green FastMix (Quantabio, Beverly, MA). Meanwhile, miRNAs were assessed by subjecting them to reverse transcription using the TaqMan™ MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). cDNAs were subjected to quantitative real-time RT-PCR analysis using TaqMan™ Fast Advanced Master Mix (Applied Biosystems, Foster City, CA, USA). All PCR reactions were performed in 96-well plates using the StepOnePlus™ real-time PCR System (Applied Biosystems). Glyceraldehyde 3-phosphate dehydrogenase was used as an endogenous control during mRNA PCR, whereas SNORD38B labeled with FAM reporter dye (Applied Biosystems) was used as an endogenous control during miRNA PCR. In the experiments, parental cells or negative control were set as the reference. miR-34a expression was quantified using the comparative method (2−ΔΔCT), where CT = threshold cycle, ΔΔ CT = (CTmiR-34a − CT SNORD38B) − (CT reference − CT SNORD38B).
Transfection assay
For miRNA transfection, A2780 cells were seeded into 6-cm dishes at a density that would yield 70% confluency after 24 h and were subsequently transfected. This cell line was transfected using the mirVana™ miRNA mimic (Thermo Fisher Scientific) specific for miR-34a or mirVana™ miRNA mimic Negative Control (Thermo Fisher Scientific) at a final concentration of 50 nM. For siRNA transfection, Stealth RNAi™ siRNAs for IL-6R (Invitrogen) and negative control (Invitrogen) were used at a final concentration of 5 nM. For transfection of the IL-6R gene, plasmid vector pEZ-M61-IL-6R (EX-A0457-M61, GeneCopoeia™) or empty pReciever-M61 (EX-NEG-M61, GeneCopoeia™) were used at a final concentration of 1.5 nM. RNA and DNA transfection were performed using Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen) and Lipofectamine™ 2000 Transfection Reagent (Invitrogen), respectively, according to the manufacturer’s instruction.
Western blot analysis
Cells at approximately 80% confluence were collected and subjected to Western blot analysis. Total cell lysates were prepared in 1× radioimmunoprecipitation assay lysis buffer, after which protein concentration was analyzed using the DC™ Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). Total protein was resolved on gradient NuPage 4%–12% Bis-Tris gels (Thermo Fisher Scientific). Proteins were then transferred onto membranes using an iBlot1 Gel Transfer Device (Thermo Fisher Scientific). The membranes were incubated with primary antibodies at 4°C with gentle agitation. All antibodies were diluted in Tris-buffered saline containing 0.1% Tween 20 and 5% bovine serum albumin. Horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse antibody (Cell Signaling Technology; 1:10000) was diluted in Tris-buffered saline with 0.1% Tween 20 and 5% nonfat milk for 1 h at room temperature with gentle agitation. Positive immunoreactions were detected using the ImmunoStar LD chemiluminescence system (Wako, Tokyo, Japan). Antibodies against CDK4 (clone D9G3E; 1:1000), survivin (clone 91G4B7; 1:1000), Snail (clone L70G2; 1:1000), STAT3 (clone 79D7; 1:2000), phosphorylated STAT3 (Tyr705) (cloneD3A7; 1:500), and β-Actin (clone 13E5; 1:1000) were obtained from Cell Signaling Technology (Beverly, MA), whereas those against IL-6Ra (clone C-20; 1:1000) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal antibodies against MMP-9 (clone 56-2A4; 1:500), MDM4 (clone GR238615-19, 1:1000), and E-cadherin (clone EP700Y; 1:50000) were purchased from Abcam (Cambridge, UK). Antibodies against IL-6 (clone 21865-1-Ap; 1:1000) were obtained from Proteintech (Rosemont, IL)
MTS assay
MTS assay was performed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, WI) following the manufacturer’s protocol. Briefly, MTS solution was added to each of the 96-well plates and incubated for 1 h, after which absorbance was measured at 490 nm using a microplate reader.
Cell proliferation assay
KF28 and A2780 cells were seeded into 96-well plates at a density of 8 × 103 cells per well in triplicate at the least. MTS solution was added 1 h before each of the desired time points and cells were incubated at 37°C. Data were collected as the average absorbance of the wells, and each experiment was repeated three times with values being presented as mean ± standard error of the mean (SEM).
Invasion assay
KF28 and A2780 cells were seeded onto the top chamber of 24-well matrigel-coated polyethylene terephthalate membrane inserts with 8-mm pores (Corning, Tewksbury, MA). The bottom chamber was filled with 0.75 ml of medium with 10% FBS as a chemoattractant. Insert cups with 0.5 ml of medium but without FBS were then prepared. After incubation for 48 h, the filter membrane was fixed with 100% methanol and stained using Wright–Giemsa staining. The degree of invasiveness was quantified by counting the number of cells in at least three random fields of view per filter using ×100 magnification. All experiments were repeated three times with values being presented as mean ± SEM.
Cytotoxicity assay
KF28 and A2780 cells were seeded into 96-well plates at a density of 1 × 104 and 1 × 103 cells per well, respectively, with varying concentrations of paclitaxel and cisplatin (see Figure 2). In vitro chemosensitivity was measured after 96 h using the MTS assay as described. Data were collected as the average absorbance of three wells in each of the three independent experiments with values being presented as mean ± SEM. The IC50 that inhibited the absorbance was defined as the paclitaxel and cisplatin concentration at 490nm to 50%.
Immunohistochemical analysis
Immunohistochemical analysis of IL-6R expression was performed on 3-mm paraffin sections of formalin-fixed, paraffin-embedded tissues using the Ventana Discovery XT automated stainer (Ventana 20 Medical Systems, Tucson, AZ). After deparaffinization, antigen retrieval was carried out using CC1buffer (Cell Conditioning 1; citrate buffer pH 6.0, Ventana Medical Systems). Two investigators (R. Y. and J. S.S.) scored the expression levels based on the stain intensity and extent. IHC score was conducted entirely independent of all clinical variables. Similar to our previous study [30], positively stained tumor cells were graded using a semiquantitative five-category system: 0, <5% positive cells; 1, 6%–25%; 2, 26%–50%; 3, 51%–75%; and 4, 76%–100%. The intensity of positively stained tumor cells was determined using scores of 0 to 2 as follows: 0 (none), 1 (weak; intensity was less than that in the positive control), and 2 (strong; intensity was equal to or greater than that in the positive control). The addition of these two systems provided the overall score.
Copy number assay
Chromosome 1p36.22, in which the miR-34a gene is located, was targeted for this analysis. RNaseP was used as the endogenous control. For TaqMan™ Copy Number Assay, 1p36.22 labeled with FAM reporter dye (Applied Biosystems) and RNaseP labeled with TAMRA (Applied Biosystems) were used. PCR was performed as described previously, after which data were analyzed using CopyCaller™ Software v2.1 (Thermo Fisher Scientific).
MethyLight analysis
Bisulfite modification was performed using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s instructions. Two sets of primers designed specifically for bisulfite-converted DNA were used: a methylated set for the miR-34a promoter and a reference set, COL2A1, to normalize for input DNA. The specificity of the reactions for methylated DNA was confirmed separately using SssI (New England Biolabs, Frankfurt, Germany)-treated human white blood cell DNA (heavily methylated). Methyl-specific primers failed to amplify genomic DNA not subjected to bisulfite treatment. The percentage of fully methylated molecules at a specific locus was calculated by dividing the miR-34a promoter/COL2A1 ratio of a sample by that of SssI-treated controls and multiplying by 100. The threshold for detectable methylation was set using a CT value < 35, whereas a CT value ≥ 35 was defined as methylation negative. Identification of CpG islands and primer sequences were obtained from Reimer et al.’s report [29], and all primer sequences are presented in Supplementary Table 2.
Animal experiment
BALB/cSlc-nu/nu female mice aged 5 weeks were obtained from Sankyo Laboratory Co. Ltd. (Tokyo, Japan) and housed under a 12-h darkness/light cycle in an animal facility at The Jikei University School of Medicine with a controlled temperature (20°C–25°C) and humidity (40%–70%). Food and water were provided ad libitum throughout the study. Mice were allowed to acclimatize for 1 week, after experiments were performed. KF28 cells were injected subcutaneously into 20 mice to construct human cancer models and assess the potential availability of miR-34a. These 20 mice were divided into four groups and subcutaneously injected with the following cells (5 × 106/in 50 μl PBS): (1) null KF28 cells (n = 5), (2) mock KF28 cells (n = 5), (3) KF28 miR-34a-1 cells (n = 5), and (4) KF28 miR-34a-2 cells (n = 5). Seven days after the subcutaneous injection of KF28 cells, tumor formations were confirmed in 18 mice (no tumor formation in two mice injected with KF28 miR-34a-2). Observation was maintained for 42 days, and tumor sizes were measured by diameter in three directions (diameters 1, 2, and 3) weekly. Tumor volumes were calculated using the following equation: Tumor Volume (mm3) = Diameter 1 (mm) × Diameter 2 (mm) × Diameter 3 (mm). On the last day, all mice were euthanized by cervical dislocation while under general anesthesia (induced using an isoflurane inhalation). The tumors were immediately excised with some portions thereof being stored at −80°C for biochemical analysis and the others being fixed in 4% paraformaldehyde for histological study. The protocol for these animal experiments was reviewed and approved by the Institutional Animal Care and Use Committee of The Jikei University (No. 2015-114) and conformed to the Guidelines for the Proper Conduct of Animal Experiments of the Science Council of Japan (2006).
Statistical analysis
Statistical analyses were performed using Prism 8.01 software (GraphPad, Inc.). The Mann–Whitney U test was used to compare the means between two independent groups of in vitro assays, in vivo assays, and clinical samples. For the assessment of tumor growth in the animal model, we calculated the area under the curve corresponding to the tumor growth curve and analyzed using ordinary one-way ANOVA for multiple comparisons. Associations between clinicopathological parameters and miRNA expressions were analyzed using the chi-square test, Mann–Whitney U test, and linear regression analysis. Statistical significance was set at P < 0.05.
Abbreviations
miRNA: microRNA; HGSC: high-grade serous carcinoma; PARP: poly ADP-ribose polymerase; IL-6R: interleukin-6 receptor; STAT3: Signal Transducers and Activator of Transcription 3; EOC: epithelial ovarian cancer; EMT: epithelial–mesenchymal transition; RT-PCR: reverse transcription-polymerase chain reaction; FBS: fetal bovine serum; cDNAs: complementary DNAs; SEM: standard error of the mean; CDK4: cyclin-dependent kinase 4; MMP, 9: matrix metalloproteinase, 9; IC50: concentration producing 50% inhibition; OSE: ovarian surface epithelium; HDAC1: histone deacetylase 1; FIGO: International Federation of Gynecology and Obstetrics; IHC: immunohistochemistry.
ACKNOWLEDGMENTS
We thank all members of the Obstetrics and Gynecology Department of The Jikei University School of Medicine for their enthusiastic clinical practice.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This study was supported by Japan Society for the Promotion of Science KAKENHI Grant Number JP16K11159 (NY) (https://www.jsps.go.jp/j-grantsinaid/).
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65:87–108. https://doi.org/10.3322/caac.21262. [PubMed].
2. Howladers N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. SEER Cancer Statistics Review, 1975-2014, National Cancer Institute. 2018. https://seer.cancer.gov/csr/1975_2014/.
3. Heintz A, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan H, Pecorelli S, Beller U. Carcinoma of the ovary. Int J Gynaecol Obstet. 2006; 95:S161–S192. https://doi.org/10.1016/S0020-7292(06)60033-7. [PubMed].
4. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, Korach J, Huzarski T, Poveda A, Pignata S, Friedlander M, Colombo N, Harter P, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017; 18:1274–84. https://doi.org/10.1016/s1470-2045(17)30469-2. [PubMed].
5. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, Kanai M, Mori Y, Matsumoto S, Chikuma S, Matsumura N, Abiko K, Baba T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2015; 33:4015–22. https://doi.org/10.1200/jco.2015.62.3397. [PubMed].
6. Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016; 27:1449–55. https://doi.org/10.1093/annonc/mdw142. [PubMed].
7. Gadducci A, Guerrieri ME. Immune Checkpoint Inhibitors in Gynecological Cancers: Update of literature and perspectives of clinical research. Anticancer Res. 2017; 37:5955–65. https://doi.org/10.21873/anticanres.12042. [PubMed].
8. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017; 16:203–22. https://doi.org/10.1038/nrd.2016.246. [PubMed].
9. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006; 9:189–98. https://doi.org/10.1016/j.ccr.2006.01.025. [PubMed].
10. Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006; 66:7390–4. https://doi.org/10.1158/0008-5472.can-06-0800. [PubMed].
11. Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014; 6:214–30. https://doi.org/10.1093/jmcb/mju003. [PubMed].
12. Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer. 2012; 12:613–26. https://doi.org/10.1038/nrc3318. [PubMed].
13. Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, Nikitin AY. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 2010; 16:1119–28. https://doi.org/10.1158/1078-0432.ccr-09-2642. [PubMed].
14. Yanaihara N, Noguchi Y, Saito M, Takenaka M, Takakura S, Yamada K, Okamoto A. MicroRNA gene expression signature driven by miR-9 Overexpression in ovarian clear cell carcinoma. PLoS One. 2016; 11:e0162584. https://doi.org/10.1371/journal.pone.0162584. [PubMed].
15. Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, Slotta-Huspenina J, Bader FG, Greten FR, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014; 124:1853–67. https://doi.org/10.1172/jci73531. [PubMed].
16. Isobe A, Sawada K, Kinose Y, Ohyagi-Hara C, Nakatsuka E, Makino H, Ogura T, Mizuno T, Suzuki N, Morii E, Nakamura K, Sawada I, Toda A, et al. Interleukin 6 receptor is an independent prognostic factor and a potential therapeutic target of ovarian cancer. PLoS One. 2015; 10:e0118080. https://doi.org/10.1371/journal.pone.0118080. [PubMed].
17. Yanaihara N, Anglesio MS, Ochiai K, Hirata Y, Saito M, Nagata C, Iida Y, Takakura S, Yamada K, Tanaka T, Okamoto A. Cytokine gene expression signature in ovarian clear cell carcinoma. Int J Oncol. 2012; 41:1094–100. https://doi.org/10.3892/ijo.2012.1533. [PubMed].
18. Rath KS, Funk HM, Bowling MC, Richards WE, Drew AF. Expression of soluble interleukin-6 receptor in malignant ovarian tissue. Am J Obstet Gynecol. 2010; 203:230.e1–8. https://doi.org/10.1016/j.ajog.2010.03.034. [PubMed].
19. Slabakova E, Culig Z, Remsik J, Soucek K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017; 8:e3100. https://doi.org/10.1038/cddis.2017.495. [PubMed].
20. Davidson B, Trope CG, Reich R. The clinical and diagnostic role of microRNAs in ovarian carcinoma. Gynecol Oncol. 2014; 133:640–6. https://doi.org/10.1016/j.ygyno.2014.03.575. [PubMed].
21. Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot CV, Zhao Y, Reynolds S, Cheng H, Rupaimoole R, Cogdell D, Nykter M, Broaddus R, et al. Integrated analyses identify a master microRNA regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell. 2013; 23:186–99. https://doi.org/10.1016/j.ccr.2012.12.020. [PubMed].
22. Rébé C, Végran F, Berger H, Ghiringhelli F. STAT3 activation: A key factor in tumor immunoescape. JAK-STAT. 2013; 2:e23010. https://doi.org/10.4161/jkst.23010. [PubMed].
23. Su HW, Yeh HH, Wang SW, Shen MR, Chen TL, Kiela PR, Ghishan FK, Tang MJ. Cell confluence-induced activation of signal transducer and activator of transcription-3 (Stat3) triggers epithelial dome formation via augmentation of sodium hydrogen exchanger-3 (NHE3) expression. J Biol Chem. 2007; 282:9883–94. https://doi.org/10.1074/jbc.M606754200. [PubMed].
24. Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012; 119:e110–9. https://doi.org/10.1182/blood-2011-09-381483. [PubMed].
25. Yanaihara N, Hirata Y, Yamaguchi N, Noguchi Y, Saito M, Nagata C, Takakura S, Yamada K, Okamoto A. Antitumor effects of interleukin-6 (IL-6)/interleukin-6 receptor (IL-6R) signaling pathway inhibition in clear cell carcinoma of the ovary. Mol Carcinog. 2016; 55:832–41. https://doi.org/10.1002/mc.22325. [PubMed].
26. Dong P, Xiong Y, Watari H, Hanley SJ, Konno Y, Ihira K, Yamada T, Kudo M, Yue J, Sakuragi N. MiR-137 and miR-34a directly target Snail and inhibit EMT, invasion and sphere-forming ability of ovarian cancer cells. J Exp Clin Cancer Res. 2016; 35:132. https://doi.org/10.1186/s13046-016-0415-y. [PubMed].
27. Schmid G, Notaro S, Reimer D, Abdel-Azim S, Duggan-Peer M, Holly J, Fiegl H, Rossler J, Wiedemair A, Concin N, Altevogt P, Marth C, Zeimet AG. Expression and promotor hypermethylation of miR-34a in the various histological subtypes of ovarian cancer. BMC Cancer. 2016; 16:102. https://doi.org/10.1186/s12885-016-2135-2. [PubMed].
28. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15. https://doi.org/10.1038/nature10166. [PubMed].
29. Reimer D, Hubalek M, Kiefel H, Riedle S, Skvortsov S, Erdel M, Hofstetter G, Concin N, Fiegl H, Muller-Holzner E, Marth C, Altevogt P, Zeimet AG. Regulation of transcription factor E2F3a and its clinical relevance in ovarian cancer. Oncogene. 2011; 30:4038–49. https://doi.org/10.1038/onc.2011.119. [PubMed].
30. Lv T, Song K, Zhang L, Li W, Chen Y, Diao Y, Yao Q, Liu P. miRNA-34a decreases ovarian cancer cell proliferation and chemoresistance by targeting HDAC1. Biochem Cell Biol. 2018; 96:663–71. https://doi.org/10.1139/bcb-2018-0031. [PubMed].
31. Misso G, Di Martino MT, De Rosa G, Farooqi AA, Lombardi A, Campani V, Zarone MR, Gulla A, Tagliaferri P, Tassone P, Caraglia M. Mir-34: a new weapon against cancer? Mol Ther Nucleic Acids. 2014; 3:e194. https://doi.org/10.1038/mtna.2014.47. [PubMed].
32. Guo Y, Nemeth J, O’Brien C, Susa M, Liu X, Zhang Z, Choy E, Mankin H, Hornicek F, Duan Z. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin Cancer Res. 2010; 16:5759–69. https://doi.org/10.1158/1078-0432.ccr-10-1095. [PubMed].
33. Coward J, Kulbe H, Chakravarty P, Leader D, Vassileva V, Leinster DA, Thompson R, Schioppa T, Nemeth J, Vermeulen J, Singh N, Avril N, Cummings J, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011; 17:6083–96. https://doi.org/10.1158/1078-0432.ccr-11-0945. [PubMed].
34. Wang Y, Niu XL, Qu Y, Wu J, Zhu YQ, Sun WJ, Li LZ. Autocrine production of interleukin-6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett. 2010; 295:110–23. https://doi.org/10.1016/j.canlet.2010.02.019. [PubMed].
35. Dijkgraaf EM, Santegoets SJ, Reyners AK, Goedemans R, Wouters MC, Kenter GG, van Erkel AR, van Poelgeest MI, Nijman HW, van der Hoeven JJ, Welters MJ, van der Burg SH, Kroep JR. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-alpha2b in patients with recurrent epithelial ovarian cancer. Ann Oncol. 2015; 26:2141–9. https://doi.org/10.1093/annonc/mdv309. [PubMed].
36. Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005; 65:10794–800. https://doi.org/10.1158/0008-5472.can-05-0623. [PubMed].
37. Zarone MR, Misso G, Grimaldi A, Zappavigna S, Russo M, Amler E, Di Martino MT, Amodio N, Tagliaferri P, Tassone P, Caraglia M. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci Rep. 2017; 7:17949. https://doi.org/10.1038/s41598-017-18186-0. [PubMed].
38. He X, Yang A, McDonald DG, Riemer EC, Vanek KN, Schulte BA, Wang GY. MiR-34a modulates ionizing radiation-induced senescence in lung cancer cells. Oncotarget. 2017; 8:69797–807. https://doi.org/10.18632/oncotarget.19267. [PubMed].
39. Zhao Y, Tu MJ, Wang WP, Qiu JX, Yu AX, Yu AM. Genetically engineered pre-microRNA-34a prodrug suppresses orthotopic osteosarcoma xenograft tumor growth via the induction of apoptosis and cell cycle arrest. Sci Rep. 2016; 6:26611. https://doi.org/10.1038/srep26611. [PubMed].
40. Beg MS, Brenner AJ, Sachdev J, Borad M, Kang YK, Stoudemire J, Smith S, Bader AG, Kim S, Hong DS. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs. 2017; 35:180–8. https://doi.org/10.1007/s10637-016-0407-y. [PubMed].
41. Yamamoto K, Kikuchi Y, Kudoh K, Nagata I. Modulation of cisplatin sensitivity by taxol in cisplatin-sensitive and -resistant human ovarian carcinoma cell lines. J Cancer Res Clin Oncol. 2000; 126:168–72. https://doi.org/10.1007/s004320050027. [PubMed].
42. Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, Ishii N, Yanaihara N, Yamada K, Takikawa O, Kawaguchi R, Isonishi S, Tanaka T, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res. 2005; 11:6030–9. https://doi.org/10.1158/1078-0432.ccr-04-2671. [PubMed].