
Introduction
The proteostasis network ensures intracellular homeostasis in spite of endogenous and exogenous perturbations that lead to changes in protein conformation and abundance [1]. Autophagy is a major player in the proteostasis network, allowing for the regulated turnover of large protein aggregates and even entire organelles. Moreover, components of the autophagy machinery dynamically interact with multiple signaling pathways to optimize cellular adaptation to cell-autonomous or environmental stress signals [2, 3].
Cystic fibrosis (CF) transmembrane conductance regulator (CFTR) is a unique member of the ATP-binding cassette (ABC) transporters family that acts as a cyclic adenosine monophosphate (cAMP)-regulated anion channel mediating chloride/bicarbonate transport across epithelia [4, 5]. Emerging evidence indicates that CFTR does not merely function as an anion channel but that it also orchestrates the proteostasis network. Indeed, CFTR operates in a context-specific dynamic system of interactor proteins that is connected to, and influenced by, the proteostasis network [6–8]. Functional perturbation of CFTR can result from inherited loss-of-function mutations that cause CF [5] or pharmacological inhibition of CFTR channel activity [8]. In either case, CFTR inhibition leads to a major derangement of cellular proteostasis. CFTR malfunction increases the generation of reactive oxygen species (ROS) that induce post-translational modifications of transglutaminase 2 (TGM2), a versatile multifunctional enzyme that catalyzes several post-translational modifications of target proteins [9, 10]. TGM2 undergoes small ubiquitin like-modifier (SUMOylation), a post-translational modification that inhibits TGM2 ubiquitination leading to persistent high TGM2 protein levels and TGM2 activation as the result of permissive elevated Ca2+ levels [11]. Activated TGM2 targets a plethora of substrates, among which the essential autophagy protein Beclin 1 (BECN1) [6–8], that is essential for autophagosome formation [2]. BECN1 targeting by TGM2 causes its dislodgement, as well as that of several BECN1 interactors, away from the endoplasmic reticulum, leading to its functional sequestration in intracellular aggregates [6–8]. Inactivation of the protein complex organized around BECN1 has two major negative effects on cellular proteostasis. First, the functional sequestration of phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) and of UV radiation resistance-associated-gene (UVRAG), two major components of the BECN1 complex, negatively impacts on intracellular trafficking in CF epithelial cells, as it reduces the availability of phosphatidyl-inositol-3-phosphate (PtdIns3P) at early endosomes and perturbs endosomal fusion/maturation and trafficking [6, 12, 13]. Second, BECN1 inactivation disables autophagy, leading to defective autophagosome formation and accumulation of the autophagic substrate sequestosome 1 (SQSTM1) at endosomal level with subsequent reduced availability of the small GTPase RAB5 and RAB7, which are essential for endosomal maturation [14]. In addition, the ubiquitin-binding-protein SQSTM1 accumulates at the plasma membrane (PM) and favors the disposal of several surface proteins, including epidermal growth factor receptor (EGFR) and CFTR itself [7, 8]. Importantly, defective CFTR results in the activation of the innate immune system at the mucosal surface, as it leads to TGM2-mediated sequestration of the anti-inflammatory peroxisome proliferator-activated receptor-γ (PPARγ), and of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inhibitor alpha (NFKBIA) within histone-deacetylase 6 (HDAC6)+/vimentin+ intracellular aggresomes. The neutralization of NFKBIA results in nuclear translocation of the pro-inflammatory transcription factor of NF-κB, thus increasing the transactivation of genes coding for pro-inflammatory cytokines [7, 11]. Thus, downstream of the CFTR defect and ROS generation, TGM2 activation can function as a rheostat of the post- translational network.
CFTR, TGM2 and autophagy
Notably, CFTR, TGM2 and autophagy are engaged in a feed-forward loop, meaning that CFTR dysfunction activates TGM2 and disables autophagy, while the inhibition of TGM2 and the restoration of autophagy re-establishes CFTR function at the cell surface. This intimate connection between CFTR, TGM2 and autophagy, can be conceived as a common platform for the surveillance of cellular homeostasis. In this perspective, CFTR can be viewed as a major sensor of stress, that alerts the autophagy machinery when a stressful event risks to perturb cellular physiology. Accordingly, CFTR malfunction in macrophages of CF patients compromises the autophagy-mediated clearance of Pseudomonas aeruginosa and Burkholderia cepacia [15, 16]. Defective bacterial clearance can be reverted by restoring CFTR function at the cell surface. In addition, the interaction of functional CFTR with caveolin-1 (CAV1) in airways is required to avoid excessive Toll-like receptor 4 (TLR4) signaling upon exposure to bacterial products [17].
The correction of deficient proteostasis by means of so-called proteostasis regulators constitutes an emerging strategy for palliating CFTR malfunction arising from loss-of-function mutations in the CFTR gene [18–20]. The approximately 2000 CFTR mutations have been categorized in 6 classes according to their impact on the synthesis (class I), processing (class II), gating (class III), conductance (class IV), quantity (amount) (class V) or recycling (class VI) of the CFTR protein [5]. Among these mutants, the most frequent one is the class II F508del-CFTR mutant that occurs in 70 to 90% of CF patients either in a heterozygous or homozygous form. Mutation-specific drugs have been approved by regulatory instances (such as the Food and Drug Administration, FDA, and the European Medicine Agency, EMA) and directly target the mutated CFTR protein to increase its PM expression (correctors) or improve its ion transport function (potentiators) [5]. In contrast, proteostasis regulators aim at targeting the cellular environment in which mutant CFTR traffics and functions [18–20]. Proteostasis regulators interrupt the feed-forward loop between CFTR, TGM2 and autophagy to reestablish autophagy flux that is deranged by, but can also impact on, the CFTR defect. In line with this evidence, the proteostasis regulator cysteamine, a TGM2 inhibitor that prevents TGM2-mediated BECN1 sequestration, can reestablish autophagic flux and restore the function of the F508del-CFTR mutant at the epithelial surface, both in patients and in mice models bearing a similar CFTR mutation [18–20]. Interestingly, the beneficial effects of cysteamine on both CFTR function and autophagy persist for several weeks after cysteamine withdrawal. Thus, restoration of proteostasis results in transient homeostasis before the system again loses its balance. Of note, in CF patients bearing class II CFTR mutations, treatment with epigallocatechin-gallate (EGCG, an inhibitor of the autophagy-inhibitory acetyl transferase EP300) [21] can prolong the beneficial effects of cysteamine with respect to autophagy induction and restore CFTR function in nasal respiratory epithelial cells [18, 19]. Preclinical studies involving F508del-CFTR mice indicate that the aforementioned combination treatment (cysteamine plus EGCG) loses its capacity to restore CFTR function in a Becn1 haploinsufficient (Becn1+/-) background [19]. Thus, autophagy is required for sustaining a functional CFTR at the cell surface.
CF AND CD: NEW MECHANISM OF CONNECTION
CF features
CF is best known for its respiratory phenotype, which results from increased viscosity of the mucus in the lung, increased pulmonary infections, and chronic inflammation [5, 22]. Thus, defective CFTR function ultimately drives inflammation, persistent and untreatable bacterial colonization and recurrent chest infections, mostly by Pseudomonas aeruginosa, Staphylococcus aureus and Burkholderia cepacia, causing chronic progressive lung disease with bronchiectasis and alveolar destruction culminating in respiratory insufficiency [23]. Beyond its respiratory manifestations, CF is a systemic disease because CFTR is expressed in, and is relevant to the function of, many tissues, including the small and large intestines, pancreas, the biliary tree, the male reproductive tract and sweat glands [5, 24]. Gastrointestinal symptoms of CF are not only attributable to thick and sticky mucus in the intestine and in pancreatic ducts that lead to exocrine pancreatic insufficiency, but are also due to a constitutive intestinal inflammation owing to CFTR malfunction [5, 24]. Preclinical evidence indicates that defective autophagy downstream to CFTR malfunction is pivotal for driving the disease phenotype at the intestinal level. Thus, CF mice feed with a standard diet usually succumb to intestinal obstruction after weaning unless they are orally treated with cysteamine to restore autophagy and to consequently rescue CFTR function [18].
CD pathogenesis
The intestine of CF patients is exposed to a particularly high antigenic load due to the exocrine pancreatic insufficiency. Indeed, patients suffering from CF may exhibit increased levels of antibodies against dietary antigens, increased fecal calprotectin levels, alteration of the intestinal microbiota, as well as increased intestinal permeability [24]. In particular, a ~4% prevalence of positive anti-TGM2-IgA autoantibodies, a serological marker of celiac disease (CD), has been reported in several cohorts of CF patients [25–28]. Moreover, the prevalence of CD is three times higher in CF patients than in the general population [28]. Moreover, mice that are CFTR deficient due to the CFTR knock-out mice or the F508del-CFTR knock-in mutation differed from wild type mice in thus far that they developed signs of ileal inflammation when they were fed with the gluten component gliadin. Thus, inherited CFTR malfunction favors gliadin responsiveness [28]. Based on these results, the question arises as to whether CFTR might be conceived as a protective mucosal shield that usually prevents CD. CD manifests as a permanent intolerance to dietary proteins from wheat, rye and barley, occurring in ~1 % of individuals worldwide. In the CD intestine, the ingestion of gluten proteins including gliadin results into an adaptive immune response against gliadin-derived peptides with an autoimmune component [29, 30]. It is known that some peptide fractions from gliadin are particularly pathogenic. Thus, the peptide P31-43 must ignite an innate immune response in epithelial cells, while the peptide P57-68 can induce cognate immune responses by T cells and antibodies in a subset of genetically susceptible individuals bearing the human leukocyte antigen (HLA) DQ2/DQ8 [31–33]. In intestinal epithelia from celiac patients, P31-43 enters the endosomal compartment, triggers TGM2 activation, perturbs endosomal maturation and trafficking, and activates the NF-κB pathway [34], which are all features reminiscent of those occurring in respiratory epithelia from CF patients.
CFTR and P31-43 peptide relationship
Recently, we have demonstrated that CFTR and autophagy are major players in the pathogenesis of CD. In intestinal epithelial, P31-43 encounters CFTR in clathrin+ vesicles and binds to, and reduces the ATPase activity of the nuclear-binding-domain-1 (NBD1) of CFTR, thus impairing CFTR function [28]. Similarly, to the inherited CFTR defect associated with CF, the gliadin-induced CFTR malfunction occurring in CD results in the activation of TGM2 and perturbs the autophagy machinery. Indeed, P31-43-mediated inhibition of CFTR drives TGM2 activation that covalently crosslinks P31-43, CFTR and TGM2 in a trimolecular complex, thus amplifying the detrimental effects of gliadin. This results in reduced activity of the BECN1 complex with reduced availability of PtdIns3P and UVRAG at the endosomal level together with SQSTM1 accumulation. In addition, CFTR malfunction suffices to increase the production of interleukin-15 (IL15), a pro-inflammatory cytokine critical for CD pathogenesis [29, 32, 33, 35–37], as the result of TGM2-mediated NF-κB activation, exactly as this occurs in CF epithelia [28].
In sum, important alterations of cellular activity such as TGM2 activation and autophagy inhibition related to CFTR malfunction (due to mutations in CF or gliadin-derived peptide in CD), represent an "infernal trio" [38] (characterized by three alterations: inhibition of CFTR, activation of TGM2, disablement of autophagy) that eliminate a loops of cellular stress.
Drug repurposing in CD?
Of note the CFTR chloride channel function constitutes a potential therapeutic target in CD. Thus, maintaining CFTR in an open conformational state by means of pharmacological potentiators (such as the FDA/EMA-approved drug Ivacaftor, formerly called VX-770), can avoid P31-43 binding to NBD1, thus preventing its inhibitory effect on CFTR. As a consequence, CFTR potentiators can protect intestinal epithelial cells from P31-43 induced stress response as they control TGM2 activation, restore the function of the BECN1 complex, prevent the accumulation of SQSTM1 and restore endosomal trafficking [28]. Importantly, CFTR potentiators prevent IL15 upregulation and control P31-43 driven immune activation [28]. These effects were initially observed in cultured human epithelial cells and then reproduced in vivo, in gliadin-sensitive BALB/C mice, as well as in non-obese diabetic (NOD) mice which are particularly susceptible to oral challenge with gliadin [39–42]. In these models of gliadin sensitivity, pretreatment with the CFTR potentiator Ivacaftor, a drug approved for the treatment of CF patients bearing particular CFTR mutations [5, 43], prevents the gliadin-induced suppression of the CFTR-mediated chloride current (as observable in the small intestine mounted in Ussing chambers responding to the CFTR stimulator forskolin) and controls the gliadin-induced intestinal inflammation. Importantly, Ivacaftor also controls the adaptive immune response, and instead promotes a tolerogenic response to gliadin in peripheral blood mononuclear cells (PBMC) from celiac patients that are co-cultured with gliadin-challenged intestinal epithelial cells [28].
Based on these findings, other additional strategies might intercept central mechanisms of both CD and CF, targeting the aforementioned ‘infernal trio’ [38]. First, it may be possible to use other CFTR potentiators including Vrx-532 [28] or genistein [44] that would be expected to act similarly to Ivacaftor [28]. Second, the inhibition of TGM2 with cysteamine or other, more specific agents that are currently in development [6, 11, 28, 45–49], should restore BECN1 and autophagy, thereby protecting CftrF508del mice from the increased responsiveness to gliadin [50]. Finally, autophagy stimulation could be achieved by inhibition of the acetyltransferase EP300 (examples: aspirin, epigallocatechin gallate, EGCG, and spermidine) [21, 51, 52], neutralizing BECN1 inhibitory proteins from the BCL2 family (examples: ABT737, navitoclax, venetoclax) [53] or inhibitors of the mechanistic target of rapamycin complex-1 (mTORC1; examples: rapamycin, tacrolimus) [54].
It is important to note that each of the aforementioned strategies for the treatment of CD (for which one of the primary causes apparently is the gluten-mediated CFTR inhibition) has been successfully applied to CF (which is due to inherited loss-of-function CFTR mutations): i) Ivacaftor has initially been designed for improving the function of specific CFTR mutants in CF and is right now FDA and EMA-approved for the treatment of CF [43, 55–58]; ii) cysteamine can be used to inhibit TGM2 and is able to restore CFTR protein and function at the PM of cultured cells from patients with the CFTRF508del/F508del mutation [18, 19, 47]; iii) the autophagy activator EGCG, combined with cysteamine, improves and prolongs its rescuing effect [19, 58]. Altogether, these findings illustrate the clinical feasibility of tackling the ‘infernal trio’.
Conclusions
In conclusion, CFTR can be inhibited in two apparently different diseases, in CF, where CFTR is mutated, and in CD, where CFTR is inhibited by gluten/gliadin-derived peptides [59] (Figure 1). In both conditions, CFTR inhibition ultimately compromises autophagy, thus reducing the capacity of cells to withstand stress and maintain tissue homeostasis. Downstream of CFTR, TGM2 plays a major role to connect CFTR malfunction and autophagy inhibition in a vicious cycle, as TG2 inhibition can interrupt this feed-forward loop [9, 10, 49]. As a perspective, stimulation of CFTR function by pharmacological potentiators, inhibition of TGM2, as well as reactivation of autophagy by suitable drugs, may be used for the treatment of both CF and CD.
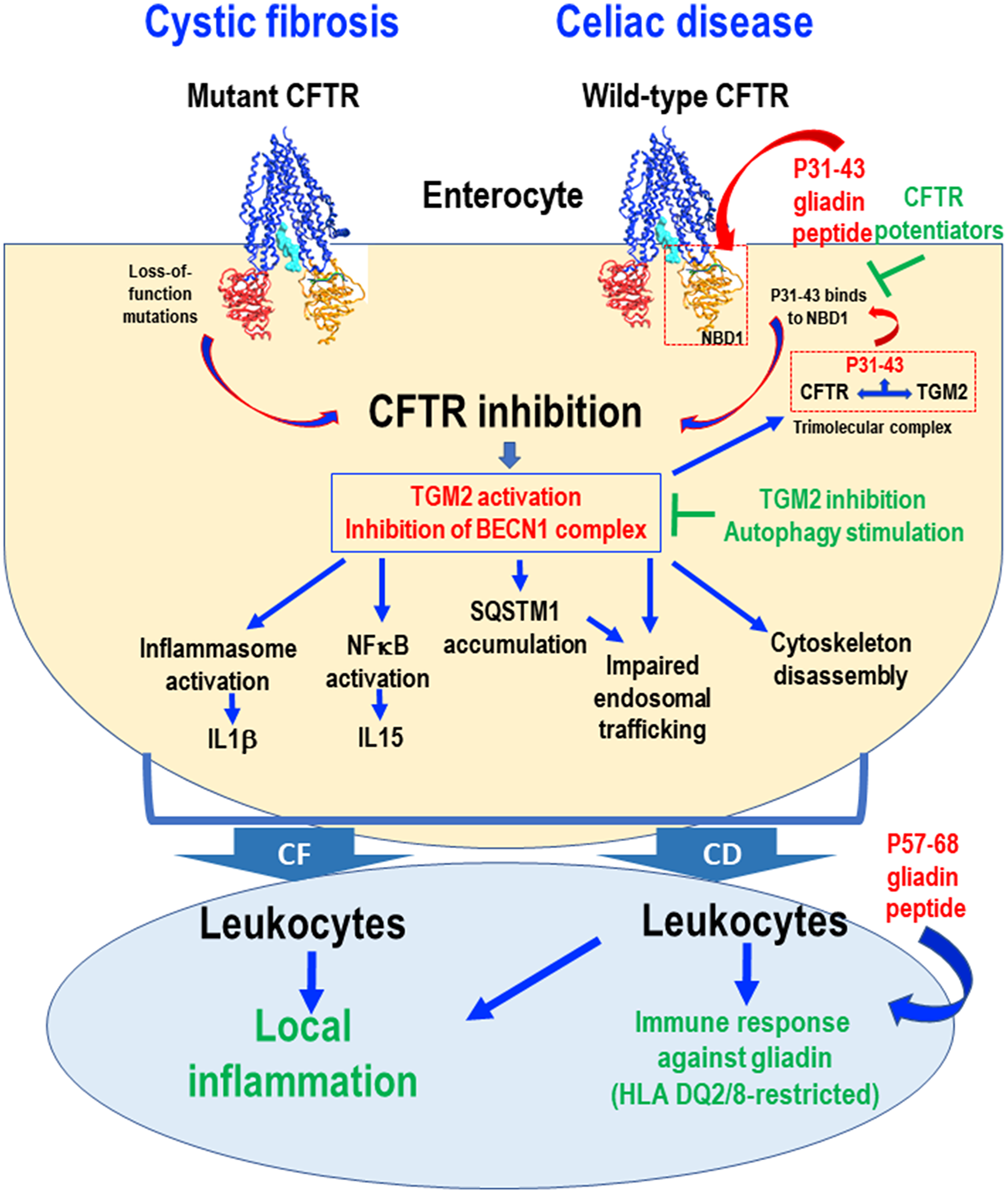
Figure 1: Schematic view of common pathogenic events downstream of CFTR inhibition in cystic fibrosis (CF) and celiac disease (CD). Loss-of-function mutations in the CFTR gene cause CFTR inhibition in CF. In the intestine from CD patients, the gliadin-derived P31-43 peptide interacts with, and binds to, specific residues of the NBD1 domain of CFTR, if the domain is in its inactive conformation, thus competing with ATP binding and blocking CFTR function. In both CF and CD epithelial cells, CFTR inhibition disrupts cellular proteostasis through two effects (i) transglutaminase-2 (TGM2) activation and (ii) BECN1 complex inhibition. In CD, TGM2 accessorily is recruited to a tripartite complex that stabilizes P31-43 binding to CFTR, thus worsening CFTR inhibition. In both conditions, CFTR inhibition leads to impaired endosomal trafficking, cytoskeleton disassembly, inflammasome activation resulting in interleukin-1β (IL1β) secretion, NF-κB activation and consequent interleukin-15 (IL15) production. Stressed enterocytes stimulate local inflammation in both CF and CD. In the gut from CD patients, this ignites the immune responses against gliadin, in particular P57-68, in a context of HLA-DQ2/DQ8. This pathogenic cascade can be interrupted by CFTR potentiators that prevent P31-43 binding to CFTR or by reconstitution of cellular proteostasis by TGM2 inhibition or BECN1 complex activation.
It might be important to explore the possibility to combine such close-to-etiological treatments with suitable life style interventions to avoid excessive gluten/gliadin uptake, as well as with non-specific measures designed to dampen inflammation and to improve the gut barrier function.
As a final note, it should be mentioned that new findings suggest that CFTR dysfunction contributes to the pathogenesis of other inflammatory state affecting epithelia [60, 61]. Thus, it is tempting to speculate, yet remains to be demonstrated, that impaired autophagy and CFTR malfunction are often associated among each other to drive the phenotype of a variety of human diseases.
Abbreviations
CFTR: cystic fibrosis transmembrane conductance regulator; CF: cystic fibrosis; CD: celiac disease; HLA: human leukocyte antigen; ROS: reactive oxygen species; TGM2: tissue transglutaminase 2; BECN1: Beclin 1; NBD1: nuclear binding domain 1; FDA: Food and Drug Administration; VX-770: Ivacaftor; NF-κB: nuclear factor kappa B; IL-15: interleukin-15; NOD mice: non-obese diabetic mice.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
A patent application by LM is pending (filing date, July 26, 2017. N° 102017000085714).
FUNDING
This project was supported by the Italian Institute of Health, under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases "RescueCFTRpreclinic” (to LM and GK), by the Specific Targeted Research Project FunMeta (ERC-2011-AdG-293714 to LR). GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). This work was supported in part by grants from AIRC (to MP) the Italian Ministry of Health (Ricerca Corrente and Ricerca Finalizzata), Fondazione Fibrosi Cistica (FFC# to MP), Regione Lazio (Gruppi di ricerca to MP). The authors also acknowledge the support of the grant from the Russian Government Programme for the Recruitment of the Leading Scientists into the Russian Institutions of Higher Education 14.W03.31.0029 (to MP).
References
1. Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015; 84:435–64. https://doi.org/10.1146/annurev-biochem-060614-033955. [PubMed].
2. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010; 40:280–93. https://doi.org/10.1016/j.molcel.2010.09.023. [PubMed].
3. Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019; 176:11–42. https://doi.org/10.1016/j.cell.2018.09.048. [PubMed].
4. Gadsby DC, Vergani P, Csanády L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006; 440:477–83. https://doi.org/10.1038/nature04712. [PubMed].
5. Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015; 16:45–56. https://doi.org/10.1038/nrg3849. [PubMed].
6. Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D’Apolito M, Guido S, Masliah E, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol. 2010; 12:863–75. https://doi.org/10.1038/ncb2090. [PubMed].
7. Villella VR, Esposito S, Bruscia EM, Maiuri MC, Raia V, Kroemer G, Maiuri L. Targeting the Intracellular Environment in Cystic Fibrosis: Restoring Autophagy as a Novel Strategy to Circumvent the CFTR Defect. Front Pharmacol. 2013; 4:1. https://doi.org/10.3389/fphar.2013.00001. [PubMed].
8. Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S, Pettoello-Mantovani M, Carnuccio R, De Matteis MA, Luini A, Maiuri MC, Raia V, Kroemer G, Maiuri L. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 2013; 20:1101–15. https://doi.org/10.1038/cdd.2013.46. [PubMed].
9. Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002; 27:534–39. https://doi.org/10.1016/S0968-0004(02)02182-5. [PubMed].
10. Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol. 2012; 294:1–97. https://doi.org/10.1016/B978-0-12-394305-7.00001-X. [PubMed].
11. Luciani A, Villella VR, Vasaturo A, Giardino I, Raia V, Pettoello-Mantovani M, D’Apolito M, Guido S, Leal T, Quaratino S, Maiuri L. SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J Immunol. 2009; 183:2775–84. https://doi.org/10.4049/jimmunol.0900993. [PubMed].
12. Marat AL, Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J. 2016; 35:561–79. https://doi.org/10.15252/embj.201593564. [PubMed].
13. Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008; 10:776–87. https://doi.org/10.1038/ncb1740. [PubMed].
14. Szatmári Z, Sass M. The autophagic roles of Rab small GTPases and their upstream regulators: a review. Autophagy. 2014; 10:1154–66. https://doi.org/10.4161/auto.29395. [PubMed].
15. Ferrari E, Monzani R, Villella VR, Esposito S, Saluzzo F, Rossin F, D’Eletto M, Tosco A, De Gregorio F, Izzo V, Maiuri MC, Kroemer G, Raia V, Maiuri L. Cysteamine re-establishes the clearance of Pseudomonas aeruginosa by macrophages bearing the cystic fibrosis-relevant F508del-CFTR mutation. Cell Death Dis. 2017; 8:e2544. https://doi.org/10.1038/cddis.2016.476. [PubMed].
16. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F, Davis IC, Valvano MA, Amer AO. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr ΔF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. 2013; 288:2049–58. https://doi.org/10.1074/jbc.M112.411728. [PubMed].
17. Zhang PX, Murray TS, Villella VR, Ferrari E, Esposito S, D’Souza A, Raia V, Maiuri L, Krause DS, Egan ME, Bruscia EM. Reduced caveolin-1 promotes hyperinflammation due to abnormal heme oxygenase-1 localization in lipopolysaccharide-challenged macrophages with dysfunctional cystic fibrosis transmembrane conductance regulator. J Immunol. 2013; 190:5196–206. https://doi.org/10.4049/jimmunol.1201607. [PubMed].
18. De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, Maiuri MC, Pettoello-Mantovani M, Guido S, et al. Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy. 2014; 10:2053–74. https://doi.org/10.4161/15548627.2014.973737. [PubMed].
19. Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A, Grassia R, Leone CA, Guido S, et al. A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2016; 23:1380–93. https://doi.org/10.1038/cdd.2016.22. [PubMed].
20. Romani L, Oikonomou V, Moretti S, Iannitti RG, D’Adamo MC, Villella VR, Pariano M, Sforna L, Borghi M, Bellet MM, Fallarino F, Pallotta MT, Servillo G, et al. Thymosin α1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat Med. 2017; 23:590–600. https://doi.org/10.1038/nm.4305. [PubMed].
21. Izzo V, Pietrocola F, Sica V, Durand S, Lachkar S, Enot D, Bravo-San Pedro JM, Chery A, Esposito S, Raia V, Maiuri L, Maiuri MC, Kroemer G. Metabolic interactions between cysteamine and epigallocatechin gallate. Cell Cycle. 2017; 16:271–79. https://doi.org/10.1080/15384101.2016.1249550. [PubMed].
22. Gavina M, Luciani A, Villella VR, Esposito S, Ferrari E, Bressani I, Casale A, Bruscia EM, Maiuri L, Raia V. Nebulized hyaluronan ameliorates lung inflammation in cystic fibrosis mice. Pediatr Pulmonol. 2013; 48:761–71. https://doi.org/10.1002/ppul.22637. [PubMed].
23. Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015; 372:351–62. https://doi.org/10.1056/NEJMra1300109. [PubMed].
24. Ooi CY, Durie PR. Cystic fibrosis from the gastroenterologist’s perspective. Nat Rev Gastroenterol Hepatol. 2016; 13:175–85. https://doi.org/10.1038/nrgastro.2015.226. [PubMed].
25. Walkowiak J, Blask-Osipa A, Lisowska A, Oralewska B, Pogorzelski A, Cichy W, Sapiejka E, Kowalska M, Korzon M, Szaflarska-Popławska A. Cystic fibrosis is a risk factor for celiac disease. Acta Biochim Pol. 2010; 57:115–18. https://doi.org/10.18388/abp.2010_2382. [PubMed].
26. Fluge G, Olesen HV, Gilljam M, Meyer P, Pressler T, Storrösten OT, Karpati F, Hjelte L. Co-morbidity of cystic fibrosis and celiac disease in Scandinavian cystic fibrosis patients. J Cyst Fibros. 2009; 8:198–202. https://doi.org/10.1016/j.jcf.2009.02.002. [PubMed].
27. De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013; 3:a009753. https://doi.org/10.1101/cshperspect.a009753. [PubMed].
28. Villella VR, Venerando A, Cozza G, Esposito S, Ferrari E, Monzani R, Spinella MC, Oikonomou V, Renga G, Tosco A, Rossin F, Guido S, Silano M, et al. A pathogenic role for cystic fibrosis transmembrane conductance regulator in celiac disease. EMBO J. 2019; 38:e100101. https://doi.org/10.15252/embj.2018100101. [PubMed].
29. Meresse B, Malamut G, Cerf-Bensussan N. Celiac disease: an immunological jigsaw. Immunity. 2012; 36:907–19. https://doi.org/10.1016/j.immuni.2012.06.006. [PubMed].
30. Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol. 2013; 13:294–302. https://doi.org/10.1038/nri3407. [PubMed].
31. Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003; 362:30–37. https://doi.org/10.1016/S0140-6736(03)13803-2. [PubMed].
32. Cerf-Bensussan N, Meresse B. Coeliac disease &gluten sensitivity: epithelial stress enters the dance in coeliac disease. Nat Rev Gastroenterol Hepatol. 2015; 12:491–92. https://doi.org/10.1038/nrgastro.2015.120. [PubMed].
33. Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol. 2009; 2:8–23. https://doi.org/10.1038/mi.2008.75. [PubMed].
34. Barone MV, Troncone R, Auricchio S. Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int J Mol Sci. 2014; 15:20518–37. https://doi.org/10.3390/ijms151120518. [PubMed].
35. Setty M, Discepolo V, Abadie V, Kamhawi S, Mayassi T, Kent A, Ciszewski C, Maglio M, Kistner E, Bhagat G, Semrad C, Kupfer SS, Green PH, et al. Distinct and Synergistic Contributions of Epithelial Stress and Adaptive Immunity to Functions of Intraepithelial Killer Cells and Active Celiac Disease. Gastroenterology. 2015; 149:681–91.e10. https://doi.org/10.1053/j.gastro.2015.05.013. [PubMed].
36. Jabri B, Abadie V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat Rev Immunol. 2015; 15:771–83. https://doi.org/10.1038/nri3919. [PubMed].
37. DePaolo RW, Abadie V, Tang F, Fehlner-Peach H, Hall JA, Wang W, Marietta EV, Kasarda DD, Waldmann TA, Murray JA, Semrad C, Kupfer SS, Belkaid Y, et al. Co-adjuvant effects of retinoic acid and IL-15 induce inflammatory immunity to dietary antigens. Nature. 2011; 471:220–24. https://doi.org/10.1038/nature09849. [PubMed].
38. Maiuri L, Villella VR, Piacentini M, Raia V, Kroemer G. Defective proteostasis in celiac disease as a new therapeutic target. Cell Death Dis. 2019; 10:114. https://doi.org/10.1038/s41419-019-1392-9. [PubMed].
39. Moon SH, Kim J, Kim MY, Park H, Song TJ, Kim SA, Lee SS, Seo DW, Lee SK, Kim MH. Sensitization to and Challenge with Gliadin Induce Pancreatitis and Extrapancreatic Inflammation in HLA-DQ8 Mice: An Animal Model of Type 1 Autoimmune Pancreatitis. Gut Liver. 2016; 10:842–50. https://doi.org/10.5009/gnl15484. [PubMed].
40. Papista C, Gerakopoulos V, Kourelis A, Sounidaki M, Kontana A, Berthelot L, Moura IC, Monteiro RC, Yiangou M. Gluten induces coeliac-like disease in sensitised mice involving IgA, CD71 and transglutaminase 2 interactions that are prevented by probiotics. Lab Invest. 2012; 92:625–35. https://doi.org/10.1038/labinvest.2012.13. [PubMed].
41. Larsen J, Weile C, Antvorskov JC, Engkilde K, Nielsen SM, Josefsen K, Buschard K. Effect of dietary gluten on dendritic cells and innate immune subsets in BALB/c and NOD mice. PLoS One. 2015; 10:e0118618. https://doi.org/10.1371/journal.pone.0118618. [PubMed].
42. Galipeau HJ, Rulli NE, Jury J, Huang X, Araya R, Murray JA, David CS, Chirdo FG, McCoy KD, Verdu EF. Sensitization to gliadin induces moderate enteropathy and insulitis in nonobese diabetic-DQ8 mice. J Immunol. 2011; 187:4338–46. https://doi.org/10.4049/jimmunol.1100854. [PubMed].
43. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, et al, and VX08-770-102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011; 365:1663–72. https://doi.org/10.1056/NEJMoa1105185. [PubMed].
44. Esposito S, Villella VR, Ferrari E, Monzani R, Tosco A, Rossin F, D’Eletto M, Castaldo A, Luciani A, Silano M, Bona G, Marseglia GL, Romani L, et al. Genistein antagonizes gliadin-induced CFTR malfunction in models of celiac disease. Aging (Albany NY). 2019; 11:2003–19. https://doi.org/10.18632/aging.101888. [PubMed].
45. Maiuri L, Luciani A, Giardino I, Raia V, Villella VR, D’Apolito M, Pettoello-Mantovani M, Guido S, Ciacci C, Cimmino M, Cexus ON, Londei M, Quaratino S. Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down-regulation. J Immunol. 2008; 180:7697–705. https://doi.org/10.4049/jimmunol.180.11.7697. [PubMed].
46. Lionetti E, Gatti S, Pulvirenti A, Catassi C. Celiac disease from a global perspective. Best Pract Res Clin Gastroenterol. 2015; 29:365–79. https://doi.org/10.1016/j.bpg.2015.05.004. [PubMed].
47. Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, Guido S, Pettoello-Mantovani M, Carnuccio R, Scholte B, De Matteis A, Maiuri MC, Raia V, et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy. 2012; 8:1657–72. https://doi.org/10.4161/auto.21483. [PubMed].
48. Luciani A, Villella VR, Vasaturo A, Giardino I, Pettoello-Mantovani M, Guido S, Cexus ON, Peake N, Londei M, Quaratino S, Maiuri L. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut. 2010; 59:311–19. https://doi.org/10.1136/gut.2009.183608. [PubMed].
49. Rossin F, Villella VR, D’Eletto M, Farrace MG, Esposito S, Ferrari E, Monzani R, Occhigrossi L, Pagliarini V, Sette C, Cozza G, Barlev NA, Falasca L, et al. TG2 regulates the heat-shock response by the post-translational modification of HSF1. EMBO Rep. 2018; 19:e45067. https://doi.org/10.15252/embr.201745067. [PubMed].
50. Villella VR, Esposito S, Ferrari E, Monzani R, Tosco A, Rossin F, Castaldo A, Silano M, Marseglia GL, Romani L, Barlev NA, Piacentini M, Raia V, et al. Autophagy suppresses the pathogenic immune response to dietary antigens in cystic fibrosis. Cell Death Dis. 2019; 10:258. https://doi.org/10.1038/s41419-019-1500-x. [PubMed].
51. Pietrocola F, Lachkar S, Enot DP, Niso-Santano M, Bravo-San Pedro JM, Sica V, Izzo V, Maiuri MC, Madeo F, Mariño G, Kroemer G. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015; 22:509–16. https://doi.org/10.1038/cdd.2014.215. [PubMed].
52. Pietrocola F, Castoldi F, Markaki M, Lachkar S, Chen G, Enot DP, Durand S, Bossut N, Tong M, Malik SA, Loos F, Dupont N, Mariño G, et al. Aspirin Recapitulates Features of Caloric Restriction. Cell Rep. 2018; 22:2395–407. https://doi.org/10.1016/j.celrep.2018.02.024. [PubMed].
53. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007; 26:2527–39. https://doi.org/10.1038/sj.emboj.7601689. [PubMed].
54. Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017; 16:487–511. https://doi.org/10.1038/nrd.2017.22. [PubMed].
55. Tosco A, Villella VR, Castaldo A, Kroemer G, Maiuri L, Raia V. Repurposing therapies for the personalised treatment of cystic fibrosis. Expert Opin Orphan Drugs. 2018; 6:361–73. https://doi.org/10.1080/21678707.2018.1483231.
56. Maiuri L, Raia V, Kroemer G. Strategies for the etiological therapy of cystic fibrosis. Cell Death Differ. 2017; 24:1825–44. https://doi.org/10.1038/cdd.2017.126. [PubMed].
57. Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014; 13:29–36. https://doi.org/10.1016/j.jcf.2013.06.008. [PubMed].
58. De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014; 13:674–80. https://doi.org/10.1016/j.jcf.2014.09.005. [PubMed].
59. Maiuri L, Villella VR, Raia V, Kroemer G. The gliadin-CFTR connection: new perspectives for the treatment of celiac disease. Ital J Pediatr. 2019; 45:40. https://doi.org/10.1186/s13052-019-0627-9. [PubMed].
60. Raju SV, Solomon GM, Dransfield MT, Rowe SM. Acquired Cystic Fibrosis Transmembrane Conductance Regulator Dysfunction in Chronic Bronchitis and Other Diseases of Mucus Clearance. Clin Chest Med. 2016; 37:147–58. https://doi.org/10.1016/j.ccm.2015.11.003. [PubMed].
61. Thiagarajah JR, Ko EA, Tradtrantip L, Donowitz M, Verkman AS. Discovery and development of antisecretory drugs for treating diarrheal diseases. Clin Gastroenterol Hepatol. 2014; 12:204–09. https://doi.org/10.1016/j.cgh.2013.12.001. [PubMed].