
Introduction
Triple negative breast cancer (TNBC) accounts for 15–20% of all breast cancers and is characterized by poor overall survival upon disease relapse. Unlike hormone receptor positive or HER2/neu positive breast cancer (BC), there is no effective targeted therapy for treatment of TNBC with the exception of those with germline BRCA1/2 mutations [1–5] or programmed death ligand-1 (PD-L1)-expressing TNBCs [6–8]. Despite these recent advances which led to moderate improvement in progression-free survival, the overall survival of patients with mTNBC has not changed. Therefore, there is still an unmet need to identify effective therapy targeting oncologic drivers.
In order to develop effective targeted therapy, it is critical to identify targetable genomic or transcriptomic drivers accountable for chemotherapy resistance. However, this is highly challenging for TNBC because these tumors are very heterogeneous, with at least four molecular sub-types proposed to date based on transcriptomic mRNA expression [9–11]. The molecular classifiers of Lehmann/Pietenpol [9, 10] defines six molecular subtypes of TNBC: basal-like 1, BL1; basal-like 2, BL2; mesenchymal, M; mesenchymal-stem-like, MSL; immune-modulatory, IM and luminal androgen receptor, LAR. The Burstein classifier defines 4 molecular subtypes of TNBC: basal-like immune-activated, BLIA; basal-like immunosuppressed, BLIS; luminal androgen receptor, LAR; and mesenchymal, MES [11]. Although these sub-types have deepened our understanding of the complexity of TNBC tumor biology, the associated molecular classifiers have not yet been incorporated in any routine clinical practice, nor have they changed the paradigm of treatment regimen selection. In addition to the challenges of tumor heterogeneity at the time of initial diagnosis, acquired chemotherapy resistance further complicates our understanding and treatment of TNBC. Such genomic evolution of TNBC through chemotherapy selection pressure is well recognized but poorly understood. Large-scale genomic databases such as The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) have provided valuable resources for understanding the biology of primary BC tumors [12, 13]. However, there is a lack of data obtained from tumor specimens collected through longitudinal studies.
Analyzing the genomic and transcriptomic changes between paired primary and recurrent TNBC can provide useful insights to improve our understanding of the underlying tumor heterogeneity and tumor evolution with chemotherapy therapy selection pressure, and potentially lead to identification of novel therapeutic targets. The phosphoinositide-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway is one of the most commonly altered oncogenic pathways identified in BC. Activation of the PI3K/AKT/mTOR pathway contributes to resistance to chemotherapy [14–16]. Despite the high frequency of alterations of the PIK3CA/AKT/mTOR pathway, the presence of these mutations does not warrant a significant response to single agent PI3K or mTOR inhibitors in early clinical trials [17, 18].
Previous genomic analysis comparing primary and metastatic breast cancers have provided inconsistent findings [19]. Some indicated concordant overall mRNA expression patterns between primary in-breast tumors and matched lymph nodes [20–22], while others identified discordant mRNA expression patterns [23] or somatic mutation profiles [24] of in-breast tumor and synchronous metastases. The current study is designed to characterize the genomic and transcriptomic alterations in paired longitudinal samples of primary and recurrent TNBC, with a focus on the PI3K/AKT/mTOR pathway. Our finding of AKT3 upregulation provides a rationale for ATK targeting in treatment of metastatic TNBCs. To our knowledge this is one of the few studies analyzing both genomic and transcriptomic changes between longitudinal paired primary and metastatic TNBC.
Results
Patient and disease characteristics (N = 10 patients)
The clinical characteristics, pathological features, treatment histories, and survival of a 10-pair TNBC cohort are described in Table 1. The majority of the tumors were infiltrating ductal carcinomas (IDC) (80%), stage I–III (90%), and the patients received standard-of-care chemotherapy with anthracycline and/or a taxane-containing regimen. Recurrence free survival (RFS) ranged from 2 to 39 months, and overall survival ranged from 9 to 92 months. Schematic paradigm of the treatment history and relapse pattern are shown in Figure 1, which illustrates the heterogeneity of treatment and duration of responses.
Table 1: TNBC patient characteristics, N = 10 patients
| Patient ID | Tumor location | Histology type | Age | Stage | Neo(adjuvant) chemotherapy | Radiation | RFS month | OS month |
|---|---|---|---|---|---|---|---|---|
| COH-1.1 | Breast primary | IDC | 64 | IIA | AC-T | Yes | 34 | 89 |
| COH-1.2 | Lung met | |||||||
| COH-2.1 | Breast primary | IDC | 51 | IIIA | AC | Declined | 17 | 60 |
| COH-2.2 | LN met | |||||||
| COH-2.3 | Bone met | |||||||
| COH-3.1 | Breast 2nd primary | IDC | 41 | IIIA | AC | Declined | 39 | 92 |
| COH-3.2 | LN met | |||||||
| COH-4.1 | LN primary | IDC | 44 | IIIB | Carbo/Taxol | Yes | 10 | 28 |
| COH-4.2 | Liver met | |||||||
| COH-5.1 | Breast primary | IDC | 39 | IIIC | AC | No | 2 | 9 |
| COH-5.2 | Skin met | |||||||
| COH-6.1 | Brain met | IDC | 50 | IIA | TAC | Declined | 31 | 67 |
| COH-6.2 | Soft tissue met | |||||||
| COH-7.1 | Breast primary | IDC | 58 | IA | Declined | Declined | 26 | 46 |
| COH-7.2 | LN met | |||||||
| COH-8.1 | Endometrium met | IDC | 38 | IV | N.A. | No | 11 | 58 |
| COH-8.2 | LN met | |||||||
| COH-9.1 | Breast primary | ILC | 45 | IIB | AC-T | Yes | 8 | 23 |
| COH-9.2 | Breast met | |||||||
| COH-10.1 | Breast primary | Metaplastic | 51 | IIB | AC-T | Yes | 16 | 58 |
| COH-10.2 | Lung met | |||||||
| COH-10.3 | Brain met |
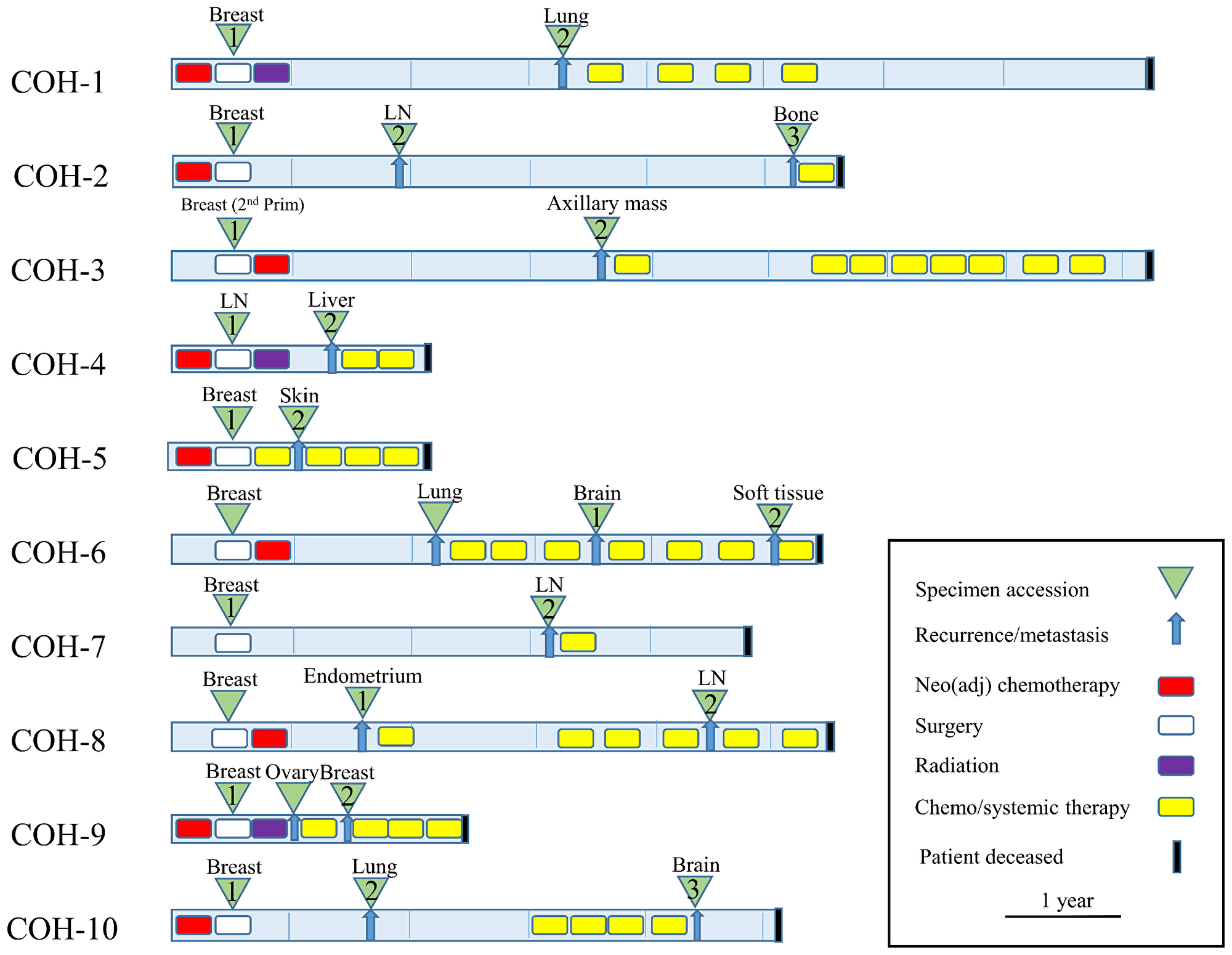
Figure 1: Clinical synopsis of treatment paradigm (N = 10 patients). TNBC specimens were collected at time of initial surgery and at first recurrence and/or metastasis (green triangles). Neo (adjuvant) chemotherapy, radiation, and lines of chemo/systemic therapy are also indicated.
Genomic profiling of paired TNBCs (N = 7 patients)
Genomic sequencing was successful in 7 paired specimens (N = 14 specimens; Figure 2). Due to lack of sufficient tumor tissue, sequencing could not be performed for 3 patients. A total of 324 genomic alterations including 67 known mutations/amplifications and 257 variants of unknown significance (VUS) were identified. Genomic alterations were identified in the following signaling pathways: cell cycle, p53, PI3K/mTOR, RAS/MAPK, and RTK/GF [25]. The most abundant genomic alterations were found in the PI3K/AKT/mTOR pathway (PI3K/AKT genes were altered in 5 out of 7 patients in this cohort). There was significant inter-patient genomic heterogeneity, but little intra-patient variability. These findings not only confirm the genomic heterogeneity of TNBCs, but also highlight the genomic stability of these tumors over time.
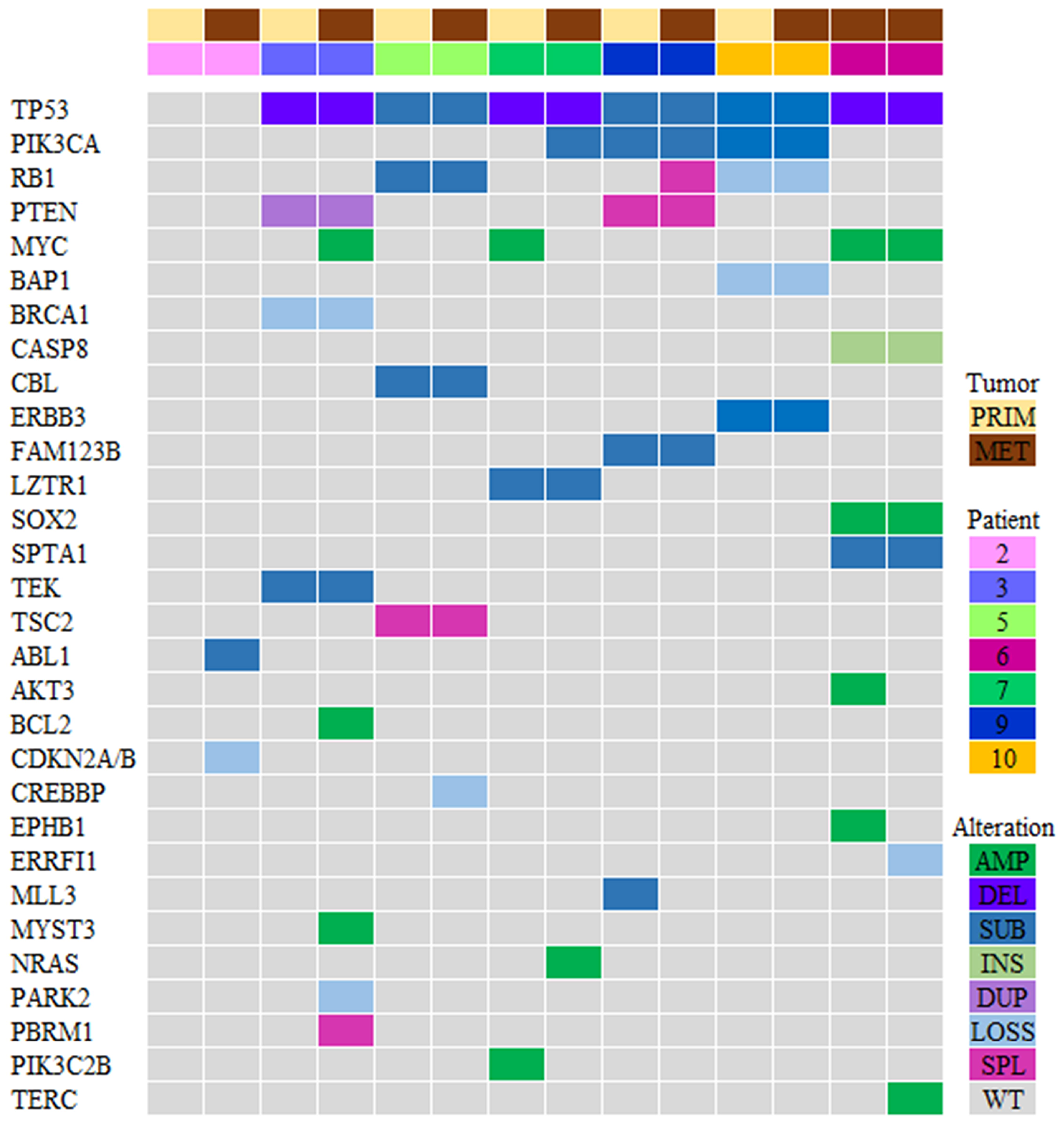
Figure 2: Genomic profiling of the PI3K pathway genes in TNBC (N = 7 patients). Tile plot illustrating patients ordered by primary/metastatic status (columns), and by mutation frequency (rows). Variants of unknown significance (VUS) were not included in this analysis. Prim, primary tumor; Met, metastatic tumor; Amp, amplification; Del, deletion; Sub, substitution; Ins, insertion; Dupl, duplication; Splice, splice site; WT, wild type.
Transcriptomic profiling of paired TNBCs (N = 10 patients; 22 specimens)
Principal coordinate analysis (PCoA) and hierarchical clustering were applied to assess the global expression pattern of the tumors, and showed clustering of samples for each patient. Only one distant metastasis COH-2.3 is dissimilar to COH-2.1 and COH-2.2, and more similar to COH-8 and COH-9 based on clustering (Figure 3A). The Euclidian distances analysis suggests that the mRNA expression is more concordant in intra-patient tumors than inter-patient tumors (Figure 3B).
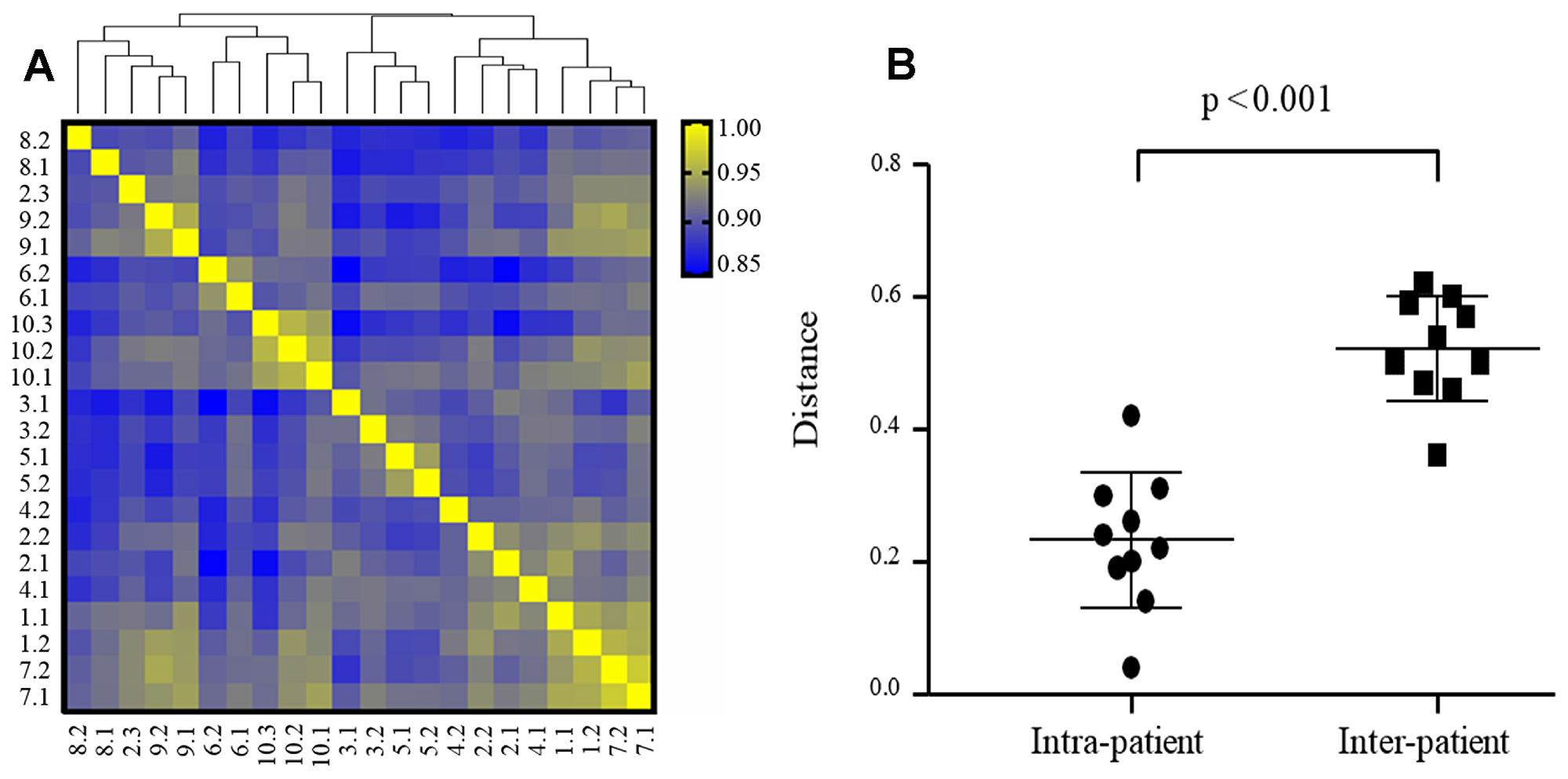
Figure 3: Intra- and inter-patient heterogeneity of mRNA expression in TNBC (N = 22 specimens). (A) Hierarchical cluster analysis of gene expression data (24,041 transcripts) from 22 TNBC specimens based on Pearson’s correlation. The color scale is based on the values of correlation coefficients. Distance between samples indicates the similarity of gene expression profiles. (B) mRNA expression is more concordant in intra-patient tumors than inter-patient tumors. Euclidian distances were calculated based on PC1, PC2, and PC3.
Hierarchical clustering of genes in the PI3 kinase (PI3K) pathway (N = 10 patients; 22 specimens)
We next used hierarchical clustering to study the transcriptomic variation of PI3K pathway among the paired TNBCs. Four groups were identified in our clustering analysis, with one unique sample COH-6.2 (distant metastasis) that could not be classified into any other group. The samples classified in each group had higher values in Pearson correlation coefficients (Figure 4A). Specifically, the 22 samples were classified into Group 1 (green rectangle), consisting of 4 samples from 3 patients, Group 2 (orange rectangle) consisting of 6 samples from 5 patients, Groups 3 (pink rectangle) with 5 samples from 4 patients, and Group 4 (blue) with 6 samples from 5 patients. Noticeably, whereas the metastatic samples of patient COH-3, COH-7 and COH-9 were classified into the same groups as their primary cancer samples, the metastatic samples of 7 other patients (COH-1, COH-2, COH-4, COH-5, COH-6, and COH-10) were clustered into different groups. The samples of COH-2 were classified into Group 1, 2 and 4, and samples of COH-10 were classified into Group 3 and 4. PCoA analysis revealed similar classification of the 22 samples based on the expression of genes involved in the PI3K pathway (Figure 4B). The molecular subtyping using Lehmann/Pietenpol classifier was performed and there was no association between the subtypes and the 4 clusters. In addition, there was no association between the 4 clusters and disease/treatment variables or survival. Taken together, these results suggest that the gene expression profile of the PI3K pathway reveals four distinctive patterns in these heterogeneous TNBC specimens.
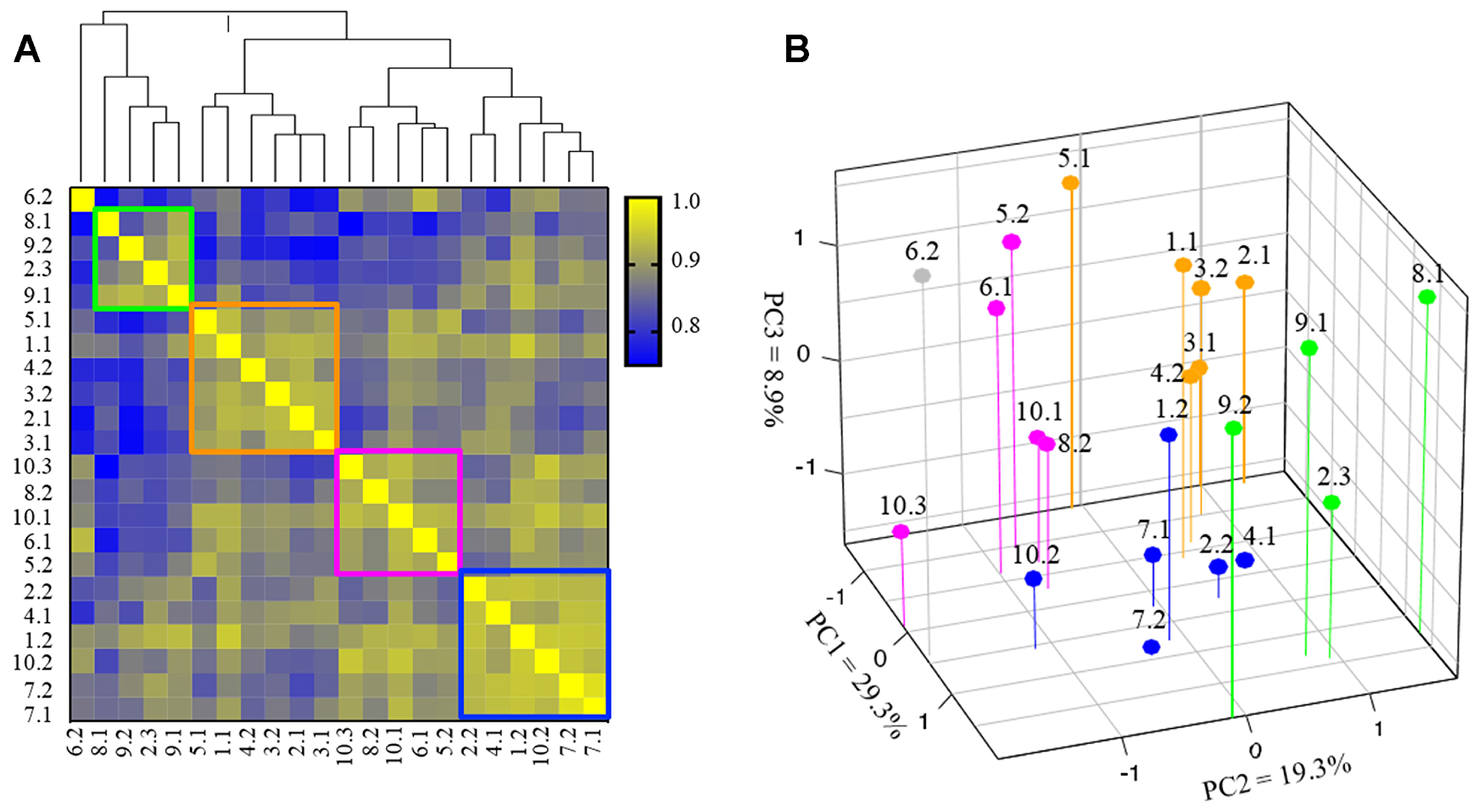
Figure 4: Subgroup clustering of PI3 kinase (PI3K) pathway genes (N = 22 specimens). (A) Hierarchical cluster analysis based on expression of 45 genes involved in PI3K pathway in 22 TNBC specimens. The color scale indicates the degree of correlation. Distance between samples indicate the similarity of gene expression profiles. Groups 1–4 are marked in green, orange, pink and blue boxes, respectively. COH-6.2 (gray) is an outlier and cannot be classified into any of the 4 groups. (B) PCoA based on gene expression involved in PI3K pathway (samples are color-coded into 4 groups as in 4A).
Distinctive gene expression patterns in paired TNBCs
Further analysis was conducted to identify key genes within the PI3K/AKT/mTOR pathway which attributed to the distinctive expression pattern. Among all the differentially expressed genes, AKT3, GSK3B (glycogen synthase kinase-3 beta), PI3KR1 (phosphoinositide-3-kinase regulatory subunit 1), GNAQ (G protein subunit alpha Q), and GNA11 (G protein subunit alpha 11) showed the strongest statistical significance (P < 0.0001; Figure 5A and 5B). The expression level of AKT3 was highest in Group 1, followed by Group 4, Group 2, and lastly Group 3. The highest level of AKT3 expression is similar to the highest level of four other genes, including GSK3B, GNAQ, PIK3R1, and GNA11. These data suggest that AKT3, together with other genes in the PI3K signaling cascade, may be key players driving the molecular discordance in TNBC.
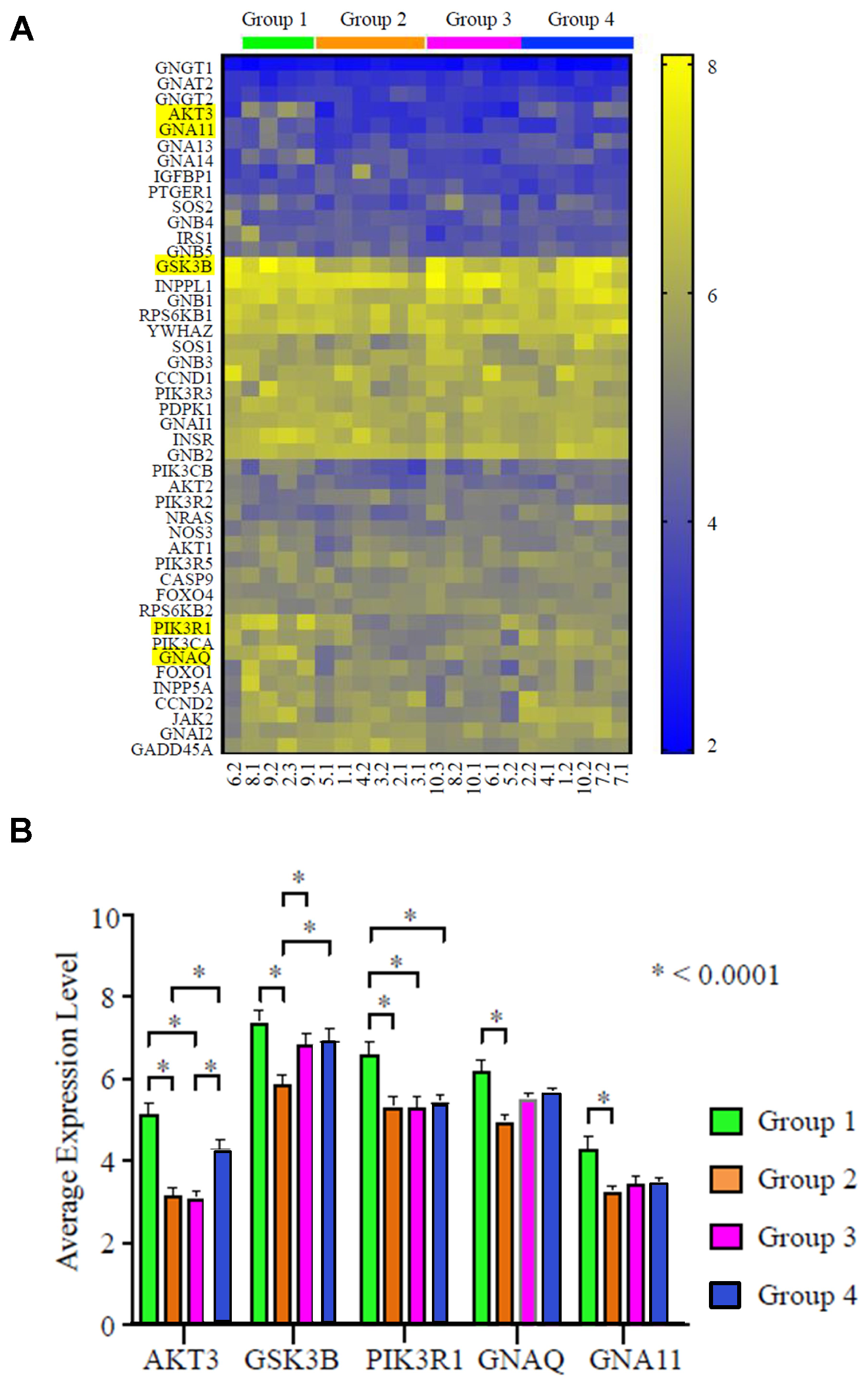
Figure 5: AKT3 is the most differentially expressed gene in the PI3K pathway in TNBC (N = 22 specimens). (A) Heat map of gene expression profile of the PI3K pathway. The four groups identified in Figure 3A are indicated with respective color bars labeled above the heat map. The top 5 genes with the largest expression variance are highlighted. (B) Relative mRNA expression level of the top 5 differentially expressed genes in the 4 respective groups; *P < 0.0001.
Differentially expressed genes and survival in public datasets
The association of these 5 differentially expressed genes with patient’s survival were tested using public datasets. AKT1 and AKT2 were also studied. Using the mRNA expression dataset in Rody et al. (N = 64) [26], increased expression of AKT3 (P = 0.0896), GNA11 (P = 0.0369) and GNAQ (P = 0.0714) were associated with a trend of shorter disease free survival (Supplementary Figure 1). In METABRIC (N = 299), increased expression of AKT1 (P = 0.0312), GNA11 (P = 0.0639), and GSK3B (P = 0.0351) were associated with shorter overall survival (Supplementary Figure 2). There was no association of AKT1, AKT2, GNAQ and PIK3R1 with overall survival.
AKT inhibitor ipatasertib is effective alone or in combination with chemotherapy in a PDX model of TNBC
The synergistic effect of ipatasertib and carboplatin was tested in vivo using a TNBC patient-derived xenograft (PDX) with the following alterations: PIK3CA E542K mutation, PTEN loss and TP53 H179R mutation. Tumor volume changes over time within each treatment group are shown in Figure 6A. Of note, mice in all treatment groups maintained their initial weight over the course of treatment, as shown in Figure 6B. Statistically significant tumor suppression was seen in both ipatasertib alone (P = 0.05), and in the combination ipatasertib and carboplatin group (P = 0.005) (Figure 6C).
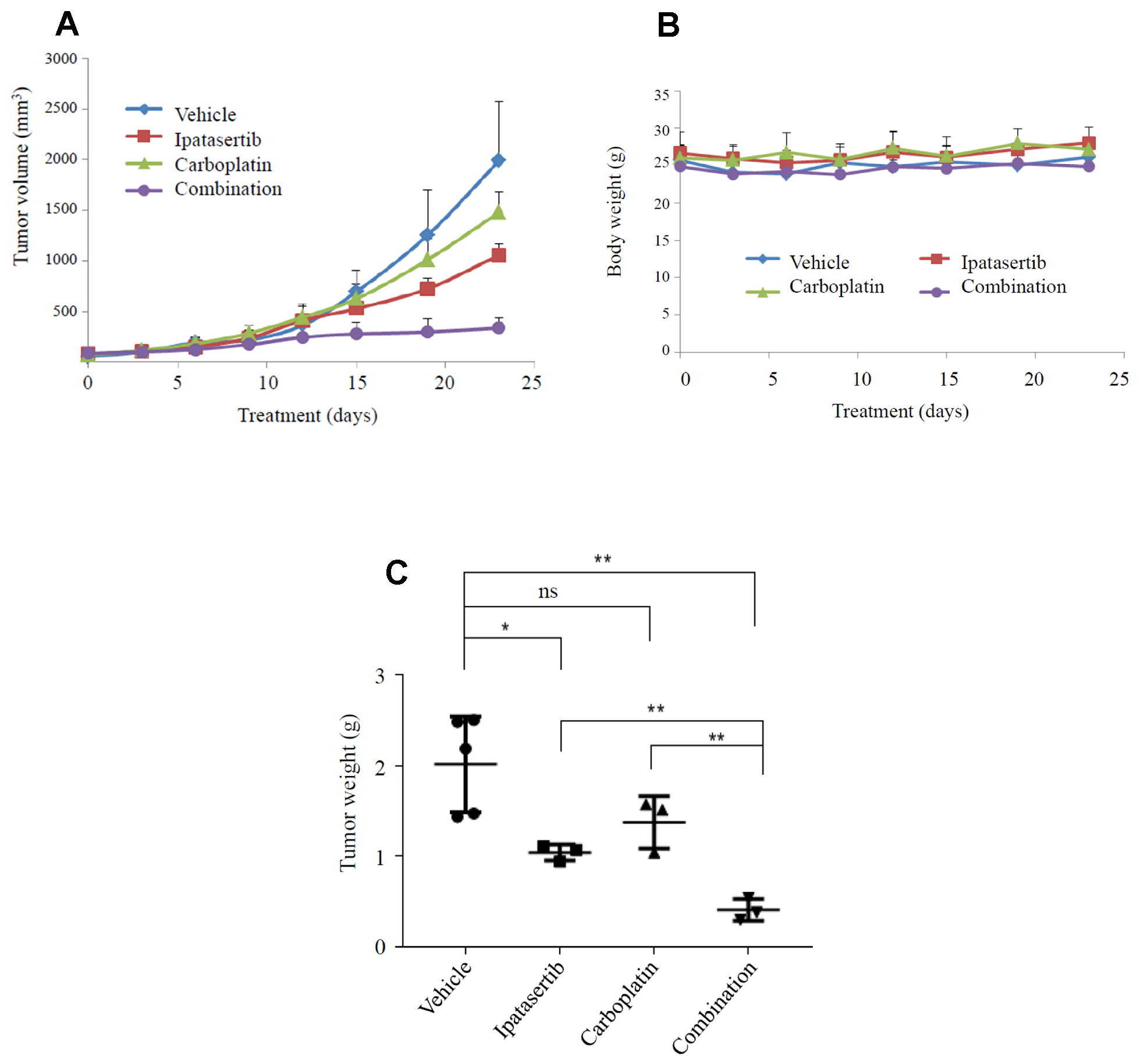
Figure 6: Synergistic effect of carboplatin and ipatasertib in a PDX model of TNBC. The dose of ipatasertib was 20 mg/kg for 9 days, followed by 30 mg/kg for 14 days; the dose of carboplatin was 10 mg/kg for two weeks, followed by 20 mg/kg for 1 week (total 3 doses). (A) PDX tumor volume changes over time; (B) Mice maintained their initial weight throughout the treatment; (C) Tumor volume among different groups: vehicle, ipatasertib, carboplatin, and combination ipatasertib plus carboplatin. *P = 0.05, **P = 0.005.
DISCUSSION
Analysis of genomic and transcriptomic changes between paired primary and recurrent TNBCs may provide insight into the underlying tumor heterogeneity and tumor evolution with chemotherapy/radiation therapy selection pressure. A better understanding of these changes may ultimately lead to identification of potential targets for overcoming resistance. In this study, we identified 4 unique groups of TNBCs based on the PI3K/AKT/mTOR pathway genes. The AKT inhibitor ipatasertib was tested in a PDX model of TNBC with PI3K/PTEN alterations and synergistic tumor suppression confirmed the efficacy of AKT targeting. Our study demonstrates the importance of genomic analysis in assisting targeted therapy development.
The serine-threonine protein kinase AKTs are central proteins in many cellular pathways such as cell survival, proliferation, glucose uptake, metabolism, angiogenesis, as well as radiation and drug response [27]. The three isoforms of AKT (AKT1, AKT2 and AKT3) have been proposed to have different physiological functions, properties, and expression patterns depending on the cell types [28]. Knockout mouse studies have shown AKT1 to be essential for cell survival, AKT2 to have a more prevalent role in glucose homeostasis, while AKT3 is involved in brain development [29]. Little is known about the influence of the different AKT isoforms in the genome and their effects in TNBC.
The five genes identified to display distinctive expression patterns in this cohort have been shown to be closely implicated in cancer biology. AKT3 is involved in various biological processes such as proliferation, differentiation, apoptosis, and tumorigenesis [30]. Several studies have suggested AKT3 to be an oncogene and a potential therapeutic target in TNBC [1–3]. One study has further correlated the expression of AKT3 with metastasis of TNBC [3]. The AKT3 copy number gain is frequently observed in TNBC (26%, copy number > 4) and associated shorter RFS [31]. In line with these studies, we found that AKT3 is significantly overexpressed in groups 1 and 4 compared with groups 2 and 3 (Figure 5), suggesting that AKT3 may be an important driver in TNBC progression and chemotherapy-resistance. Furthermore, GSK3B, a serine-threonine protein kinase downstream of AKT3 showed a similarly distinctive expression pattern. Given the reported role of GSK3B in mitosis, proliferation, motility and survival [32], it is likely involved in AKT3-regulated TNBC biology. PIK3R1 phosphorylates inositol and indirectly activates AKT3. The activating mutations of PI3KCA, and PI3K signaling pathway-related genes have also been frequently observed, and are known to activate AKT3 [9]. GNAQ and GNA11 can form a Gq alpha subunit, and both are subunits of guanine nucleotide–binding proteins. Gq alpha interacts with cell membrane receptors and plays a role in signaling multiple pathways, especially growth signaling [4]. Activating Q209L/P mutations in GNAQ or GNA11 (GNAQ/11) are present in approximately 5.6% of tumors and 80% of uveal melanomas. Combination of small molecules inhibiting MEK and PI3K enhances uveal melanoma cell death in a mutant GNAQ/GNA11-dependent manner [33, 34]. A phase II study with selumetinib (an ATP-independent inhibitor of mitogen-activated protein kinase) showed improved clinical activity compared with temozolomide in GNAQ/GNA11 mutant uveal melanoma [35]. Taken together, our findings suggest that the discordant expression of these genes may underlie the heterogeneity of TNBC and can serve as targets for novel combinations.
AKT targeting has made progress in recent clinical trials. Pan-AKT inhibitors have shown promising responses in patients with metastatic TNBC [36–38]. Ipatasertib is a highly selective ATP-competitive small-molecule AKT inhibitor, and showed activity in cell lines and xenograft models of a broad range of cancer types including breast cancer [39]. Sensitivity to ipatasertib was associated with high phosphorylated AKT levels, PTEN protein loss, or genetic mutations in PTEN and PIK3CA, whereas KRAS and BRAF mutations were typically associated with resistance to ipatasertib [39]. In a recent update of the Phase II LOTUS trial, paclitaxel in combination with ipatasertib showed a higher response rate and more durable responses in patients with TNBC tumors harboring PI3K/AKT/PTEN alterations [39].
DNA-damaging agents such as platinum drugs (cisplatin and carboplatin) are active in TNBC. In the TNT trial, patients with TNBC and germline BRCA1 and/or BRCA2 mutations were found to have a higher response rate and longer progression-free survival rate favoring carboplatin over docetaxel [40]. Our data from this PDX model showed promising synergistic effects combining ipatasertib and carboplatin. Hence, there is a strong rationale for testing this combination in the clinical setting. A phase II clinical trial combining ipatasertib and carboplatin is currently ongoing.
Our study is limited by its small sample size due to the challenge of obtaining longitudinal specimens over a long time course. Future studies elucidating the impact and biological significance of the 4 distinctive patterns of PI3K/AKT/mTOR pathway seen in this study are currently in progress.
Differential gene expression between paired primary and metastatic TNBCs was observed in this 10 patient cohort, with an increase in growth-promoting signals. Among the genes involved in PI3K pathway alterations, AKT3 appears to play a critical role and is a potential target for novel therapies for metastatic TNBC. Our mTNBC PDX model further confirms AKT as a potential target for treatment of TNBC. A phase I/II clinical is planned for further assessment of the clinical activity of this combination.
Materials and Methods
Patient selection
Longitudinal paired primary and metastatic TNBC specimens were identified through an Institutional Review Board (IRB)-approved protocol from patients with recurrence between 2002 and 2018. The eligibility criteria were: stage I–III breast cancer; ER negative, PR negative and HER-2neu negative defined by ASCO/CAP guideline; at least one tumor biospecimen available from initial surgery and metastatic biopsy. A total of 10 patients were studied in the current cohort. Of these, 2 patients had 3 samples each, resulting in a total of 22 specimens in the study. All pathology samples were formalin-fixed and paraffin-embedded (FFPE). Demographic data such as age, gender, date of birth, date of diagnosis, date of relapse, and date of death or last follow-up (if applicable) were obtained. Disease characteristics such as tumor grade, TNM stage, and ER/PR/HER2 status, as well as treatment variables including surgery, chemotherapy, and radiation therapy were also obtained.
mRNA expression of primary and recurrent TNBCs
Messenger RNA (mRNA) expression was profiled using GeneChip® Human Gene 2.0 ST array. Raw data were normalized and processed using Expression Console, and linear regression was performed using Limma to identify the differentially expressed genes between primary and recurrent/refractory TNBC. The expression of 24,041 transcripts was normalized by robust multi-array average (RMA) using R Bioconductor “oligo” package [41–44]. The annotation of the genes was constructed based on R Bioconductor “AnnotationDbi” package [45]. Gene expression was summarized by max approach. The similarity among samples was calculated based on Pearson correlation coefficients, which were used to construct hierarchical clustering and heat maps. Pathway-related heat maps were constructed, and hierarchical clustering based on the gene expression correlation of the genes defined in the pathways of Panther database (V3.4.1) were analyzed according to the methods described [46]. Pathways with differential gene conserved expression pattern were selected. The genes of the PI3 kinase pathway (P00048) defined in the Panther database (V3.4.1) [46] were selected for further analysis. The Principal Component Analysis (PCA) and Principal Coordinates Analysis (PCoA) were performed using the methods described in Past 3.14 [47]. The Euclidian distances between samples were calculated based on the coordinates of PC1, PC2 and PC3. The P-value of differential gene expression between the different groups of PI3K pathways were calculated by 2-way ANOVA multiple comparison using GraphPad Prism 7.
Gene expression and survival analyses using public databases
Gene expression and survival data of 64 patients with TNBC from Rody, et al. [26] and 299 patients with TNBC from METABRIC [12] were downloaded. The “high” and “low” groups were separated based on median mRNA expression values. Kaplan–Meier survival analyses, and were used to determine the survival differences between “high” and “low” mRNA expression groups. P-values were calculated by log-rank test using the Survival package in R [48]. The survival differences were considered to be statistically significant when P-values were < 0.05.
Akt inhibitor ipatasertib and carboplatin in patient derived xenograft (PDX) model
After obtaining informed written patient consent, TNBC tumor samples were obtained from patients at the time of surgery or biopsy at COH under protocol approved by COH Institutional Review Board (IRB). Fresh primary tumor tissues (2–3 mm in diameter) were surgically implanted into the mammary fat pad of 6- to 8-week-old female NOD/SCID/IL2Rgamma-null (NSG) mice. Once the xenograft was established, the tumor was removed, cut into small fragments, and subsequently passaged from mouse to mouse to expand the xenograft number. These mice were then used for the experiment. When the xenografts were palpable, animals were randomized into 4 groups and treated daily by oral gavage with vehicle, carboplatin, ipatasertib or a combination of both agents. Tumor volumes were assessed using calipers one to two times a week. The dose of ipatasertib was 20 mg/kg for 9 days, followed by 30 mg/kg for 14 days; the dose of carboplatin was 10 mg/kg for two weeks, followed by 20 mg/kg for 1 week (total 3 doses). The mice were sacrificed at day 23. Tumor volumes were calculated using the formula (width) 2 × length × 0.52. Body weight was monitored weekly as an indicator of drug-induced toxicity and overall health of the mice. All animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at COH in accordance with all applicable federal, state, and local requirements and institutional guidelines.
Ethics
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee, and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all participants included in the study. All tumor specimens were identified through a City of Hope IRB-approved retrospective protocol from patients consented to City of Hope Biorepository Protocol.
Abbreviations
AC: Adriamycin/cyclophosphamide; AC-T: Adriamycin/cyclophosphamide followed by paclitaxel; Amp: Amplification; BC: Breast cancer; Carbo/paclitaxel: Carboplatin/paclitaxel; Del: Deletion; Dupl: Duplication; FFPE: Formalin-fixed and paraffin-embedded; IDC: Infiltrating ductal carcinomas; Ins: Insertion; IRB: Institutional Review Board; ILC: Invasive lobular carcinoma; LN: Lymph node; mTOR: Mammalian target of rapamycin; mRNA: Messenger RNA; Met: Metastatic tumor; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; NSG: NOD/SCID/IL2Rgamma-null; OS: Overall survival; PDX: Patient-derived xenograft; PI3K: Phosphoinositide-3-kinase; Prim: Primary tumor; PCA: Principal Component Analysis; PCoA: Principle coordinate analysis; PD-L1: Programmed death ligand-1; RFS: Relapse-free survival; RMA: Robust multi-array average; Splice: Splice site; Sub: Substitution; TCGA: The Cancer Genome Atlas; TNBC: Triple negative breast cancer; VUS: Variants of unknown significance; WT: Wild type.
ACKNOWLEDGMENTS
The authors thank the STOP Cancer Foundation for their support. Research reported in this publication included work performed in the Pathology Research Services Core, Biostatistics and Mathematical Modeling Core, Integrative Genomics Core, and Bioinformatics Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICTS OF INTEREST
YY has contracted research sponsored by Merck, Eisai, Novartis, Puma, Genentech, and Pfizer; is a consultant for Puma, and is on the Speakers Bureau for Eisai. The other authors have no competing interests.
FUNDING
Yuan Yuan, MD, PhD was supported by STOP Cancer Foundation (PI Yuan Yuan MD PhD) and NCI K-12 Career Development Award (K12CA001727, PI Joanne Mortimer). Zhen Chen PhD was supported by NIH R00HL122368. Research reported in this publication included work performed in the Pathology Research Services Core, Biostatistics and Mathematical Modeling Core, Integrative Genomics Core, and Bioinformatics Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA033572.
References
1. Hong S, Funchain P, Haddad A, Crowe J, Dalpiaz N, Abraham J. Complete Durable Response From Carboplatin and Olaparib in a Heavily Pretreated Triple-Negative Metastatic Breast Cancer With Germline BRCA2 and “BRCAness” Mutations. J Oncol Pract. 2016; 12:270–72. https://doi.org/10.1200/JOP.2016.010710. [PubMed].
2. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017; 377:523–33. https://doi.org/10.1056/NEJMoa1706450. [PubMed].
3. Robson ME, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung NM, Armstrong A, Wu W, Goessl CD, Runswick S, et al. OlympiAD: Phase III trial of olaparib monotherapy versus chemotherapy for patients (pts) with HER2-negative metastatic breast cancer (mBC) and a germline BRCA mutation (gBRCAm). American Society of Clinical Oncology. 2017; 35:Supplemental 18. https://doi.org/10.1200/JCO.2017.35.18_suppl.LBA4.
4. Litton JK, Rugo HS, Ettl J, Hurvitz S, Gonçalves A, Lee K, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roché H, Im YH, Quek RGW, et al. EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician's choice of therapy in patients with advanced breast cancer and a germline BRCA mutation. Cancer Res. 2018; 78:Abstract GS6-07. https://doi.org/10.1158/1538-7445.SABCS17-GS6-07.
5. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roché H, Im YH, Quek RG, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018; 379:753–63. https://doi.org/10.1056/NEJMoa1802905. [PubMed].
6. Adams S, Diamond J, Hamilton E, Pohlmann P, Tolaney S, Molinero L, Zou W, Liu B, Waterkamp D, Funke R, Powderly J. Safety and clinical activity of atezolizumab (anti-PDL1) in combination with nab-paclitaxel in patients with metastatic triple-negative breast cancer. Cancer Res. 2016; 76: Abstract P2-11-06. https://doi.org/10.1158/1538-7445.SABCS15-P2-11-06.
7. Emens LA, Cruz C, Eder JP, Braiteh F, Chung C, Tolaney SM, Kuter I, Nanda R, Cassier PA, Delord JP, Gordon MS, ElGabry E, Chang CW, et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019; 5:74–82. https://doi.org/10.1001/jamaoncol.2018.4224. [PubMed].
8. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, et al, and IMpassion130 Trial Investigators. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018; 379:2108–21. https://doi.org/10.1056/NEJMoa1809615. [PubMed].
9. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011; 121:2750–67. https://doi.org/10.1172/JCI45014. [PubMed].
10. Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME, Pietenpol JA. Refinement of triple-negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS One. 2016; 11:e0157368. https://doi.org/10.1371/journal.pone.0157368. [PubMed].
11. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015; 21:1688–98. https://doi.org/10.1158/1078-0432.CCR-14-0432. [PubMed].
12. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, et al, and METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–52. https://doi.org/10.1038/nature10983. [PubMed].
13. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. https://doi.org/10.1038/nature11412. [PubMed].
14. LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat. 2008; 11:32–50. https://doi.org/10.1016/j.drup.2007.11.003. [PubMed].
15. McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, Malaponte G, Nicoletti F, Libra M, Bäsecke J, Maksimovic-Ivanic D, Mijatovic S, Montalto G, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011; 226:2762–81. https://doi.org/10.1002/jcp.22647. [PubMed].
16. Steelman LS, Navolanic PM, Sokolosky ML, Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Wong EW, Stadelman KM, Terrian DM, Leslie NR, Martelli AM, Stivala F, et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene. 2008; 27:4086–95. https://doi.org/10.1038/onc.2008.49. [PubMed].
17. Arsenic R, Lehmann A, Budczies J, Koch I, Prinzler J, Kleine-Tebbe A, Schewe C, Loibl S, Dietel M, Denkert C. Analysis of PIK3CA mutations in breast cancer subtypes. Appl Immunohistochem Mol Morphol. 2014; 22:50–56. https://doi.org/10.1097/PDM.0b013e318297afea. [PubMed].
18. LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016; 34:3803–15. https://doi.org/10.1200/JCO.2014.59.0018. [PubMed].
19. Szekely B, Bossuyt V, Li X, Wali VB, Patwardhan GA, Frederick C, Silber A, Park T, Harigopal M, Pelekanou V, Zhang M, Yan Q, Rimm DL, et al. Immunological differences between primary and metastatic breast cancer. Ann Oncol. 2018; 29:2232–39. https://doi.org/10.1093/annonc/mdy399. [PubMed].
20. Ellsworth RE, Seebach J, Field LA, Heckman C, Kane J, Hooke JA, Love B, Shriver CD. A gene expression signature that defines breast cancer metastases. Clin Exp Metastasis. 2009; 26:205–13. https://doi.org/10.1007/s10585-008-9232-9. [PubMed].
21. Krøigård AB, Larsen MJ, Thomassen M, Kruse TA. Molecular concordance between primary breast cancer and matched metastases. Breast J. 2016; 22:420–30. https://doi.org/10.1111/tbj.12596. [PubMed].
22. Suzuki M, Tarin D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: clinical implications. Mol Oncol. 2007; 1:172–80. https://doi.org/10.1016/j.molonc.2007.03.005. [PubMed].
23. Vecchi M, Confalonieri S, Nuciforo P, Viganò MA, Capra M, Bianchi M, Nicosia D, Bianchi F, Galimberti V, Viale G, Palermo G, Riccardi A, Campanini R, et al. Breast cancer metastases are molecularly distinct from their primary tumors. Oncogene. 2008; 27:2148–58. https://doi.org/10.1038/sj.onc.1210858. [PubMed].
24. Schrijver WA, Selenica P, Lee JY, Ng CK, Burke KA, Piscuoglio S, Berman SH, Reis-Filho JS, Weigelt B, van Diest PJ, Moelans CB. Mutation profiling of key cancer genes in primary breast cancers and their distant metastases. Cancer Res. 2018; 78:3112–21. https://doi.org/10.1158/0008-5472.CAN-17-2310. [PubMed].
25. Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sánchez V, Kurupi R, Moore PD, Pinto JA, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014; 4:232–45. https://doi.org/10.1158/2159-8290.CD-13-0286. [PubMed].
26. Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D, Schmidt M, Müller V, Holtrich U, Kaufmann M. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011; 13:R97. https://doi.org/10.1186/bcr3035. [PubMed].
27. Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013; 13:234–44. https://doi.org/10.2174/1568009611313030002. [PubMed].
28. Häggblad Sahlberg S, Mortensen AC, Haglöf J, Engskog MK, Arvidsson T, Pettersson C, Glimelius B, Stenerlöw B, Nestor M. Different functions of AKT1 and AKT2 in molecular pathways, cell migration and metabolism in colon cancer cells. Int J Oncol. 2017; 50:5–14. https://doi.org/10.3892/ijo.2016.3771. [PubMed].
29. Hollander MC, Maier CR, Hobbs EA, Ashmore AR, Linnoila RI, Dennis PA. Akt1 deletion prevents lung tumorigenesis by mutant K-ras. Oncogene. 2011; 30:1812–21. https://doi.org/10.1038/onc.2010.556. [PubMed].
30. Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014; 74:964–73. https://doi.org/10.1158/0008-5472.CAN-13-2175. [PubMed].
31. O’Hurley G, Daly E, O’Grady A, Cummins R, Quinn C, Flanagan L, Pierce A, Fan Y, Lynn MA, Rafferty M, Fitzgerald D, Pontén F, Duffy MJ, et al. Investigation of molecular alterations of AKT-3 in triple-negative breast cancer. Histopathology. 2014; 64:660–70. https://doi.org/10.1111/his.12313. [PubMed].
32. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003; 4:257–62. https://doi.org/10.1016/S1535-6108(03)00248-4. [PubMed].
33. O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer. 2013; 13:412–24. https://doi.org/10.1038/nrc3521. [PubMed].
34. Khalili JS, Yu X, Wang J, Hayes BC, Davies MA, Lizee G, Esmaeli B, Woodman SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ-and GNA11-dependent manner. Clin Cancer Res. 2012; 18:4345–55. https://doi.org/10.1158/1078-0432.CCR-11-3227. [PubMed].
35. Carvajal RD, Sosman JA, Quevedo F, Milhem MM, Joshua AM, Kudchadkar RR, Linette GP, Gajewski T, Lutzky J, Lawson DH, Lao CD, Flynn PJ, Albertini MR, et al. Phase II study of selumetinib (sel) versus temozolomide (TMZ) in gnaq/Gna11 (Gq/11) mutant (mut) uveal melanoma (UM). J Clin Oncol. 2013; 31:Supplemental 18. https://doi.org/10.1200/jco.2013.31.18_suppl.cra9003.
36. Kim SB, Dent R, Im SA, Espié M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko MJ, Kapp AV, Chan WY, Singel SM, et al, and LOTUS investigators. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017; 18:1360–72. https://doi.org/10.1016/S1470-2045(17)30450-3. [PubMed].
37. Dent R, Im SA, Espie M, Blau S, Tan AR, Isakoff SJ, Oliveira M, Saura C, Wongchenko M, Kapp AV, Chan WY, Singel SM, Maslyar DJ, et al. Overall survival (OS) update of the double-blind placebo (PBO)-controlled randomized phase 2 LOTUS trial of first-line ipatasertib (IPAT)+ paclitaxel (PAC) for locally advanced/metastatic triple-negative breast cancer (mTNBC). J Clin Oncol. 2018; 36:Supplemental 15. https://doi.org/10.1200/JCO.2018.36.15_suppl.1008.
38. Schmid P, Wheatley D, Baird R, Chan S, Abraham J, Tutt A, Kristeleit H, Patel G, Bathakur U, Bishop J, Harper-Wynne C, Perren T, Stein R, et al. A phase II, double blind, rand omised, placebo-controlled study of the AKT Inhibitor AZD5363 in combination with paclitaxel in triple-negative advanced or metastatic breast cancer (TNBC)(NCT02423603). Cancer Res. 2016; 76:Abstract OT1-03-13. https://doi.org/10.1158/1538-7445.SABCS15-OT1-03-13.
39. Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, Lee LB, Risom T, Gross S, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013; 19:1760–72. https://doi.org/10.1158/1078-0432.CCR-12-3072. [PubMed].
40. Tutt A, Tovey H, Cheang MC, Kernaghan S, Kilburn L, Gazinska P, Owen J, Abraham J, Barrett S, Barrett-Lee P, Brown R, Chan S, Dowsett M, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat Med. 2018; 24:628–637. https://doi.org/10.1038/s41591-018-0009-7. [PubMed].
41. Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010; 26:2363–67. https://doi.org/10.1093/bioinformatics/btq431. [PubMed].
42. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003; 19:185–93. https://doi.org/10.1093/bioinformatics/19.2.185. [PubMed].
43. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003; 31:e15. https://doi.org/10.1093/nar/gng015. [PubMed].
44. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–64. https://doi.org/10.1093/biostatistics/4.2.249. [PubMed].
45. Pagès H, Carlson M, Falcon S, Li N. AnnotationDbi: Annotation Database Interface. R package version 1.38.2. Bioconductor version: Release (35). 2017.
46. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017; 45:D183–89. https://doi.org/10.1093/nar/gkw1138. [PubMed].
47. Hammer Ø, Harper DAT, Ryan PD. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol Electronica. 2001; 4:9.
48. Bair E, Tibshirani R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2004; 2:E108. https://doi.org/10.1371/journal.pbio.0020108. [PubMed].