
Introduction
Adenomatous Polyposis Coli (APC) is a 312 kDa protein that functions as a tumor suppressor by acting as a requisite scaffolding protein that stabilizes the β-catenin destruction complex. Somatic mutations in the APC gene are the most prevalent initiating event in colorectal carcinogenesis [1, 2]. These deleterious APC mutations mostly occur in a specific region, the APC mutation cluster region (MCR), and singularly define Familial Adenomatous Polyposis (FAP), a genetic condition predisposing to the development of colorectal adenomatous polyps and early onset colorectal adenocarcinoma [3, 4]. Mechanistically, these APC mutations are thought to disrupt the role of APC in promoting β-catenin degradation to temper proliferative canonical Wnt signaling [1, 5], and in modulating retinoic acid-dependent intestinal development [6–9]. Over time, additional genetic insults to cancer driver genes KRAS and TP53 together with the underlying APC defect eventually progress FAP adenomas to colorectal cancer [10, 11].
APC is widely assumed as the only relevant gene mutated in early adenomas, but it is only recently that next-generation sequencing of colon adenomas have resulted in data to confirm this long-standing hypothesis [12, 13]. In our recent work to define a comprehensive genomic landscape of adenomas and at-risk mucosa, we found that patient-derived FAP adenomas do not just have the expected somatic APC mutations, but also recurrent mutations in Wnt pathway genes and in novel genes previously not linked to progressing colon adenomas to adenocarcinomas. Surprisingly, when the APC gene is excluded, one gene with frequent accumulating genomic alterations CCR4-NOT Transcription Complex Subunit 3 (CNOT3), which was observed in 5 out of 25 sequenced FAP adenomas [13].
Transcription complexes are comprised of multiple proteins that perform combinatorial regulatory functions critical to proper transcription. The CCR4-NOT complex is a highly conserved, mRNA transcription regulator [14, 15] that functions mainly through the deadenylation and ubiquitination activity of its CCR4 (CNOT6) and NOT4 (CNOT4) subunits, respectively [16, 17]. CNOT3 is thought to primarily serve as a scaffolding protein in CCR4-NOT but there is growing evidence linking CNOT3 mutations to disease [18, 19]. In the COSMIC database, the two most common CNOT3 mutations in cancers are E → K mutations at amino acid positions 20 and 70, which are the CNOT3 mutations we found in our adenoma samples [13, 20]. Unfortunately, there are no published work that details a unique mechanistic role for the NOT3 domain where the two E → K mutations are located to help guide us to the possible effects of these two mutations. A partial mechanistic role was reported by Suzuki et al that described a cooperative function for the N-terminal and C-terminal ends of CNOT3 in mRNA decay [21]. Overall, more work needs to be done to fully understand the mechanistic role of the NOT3 domain. Additionally, because the CCR4-NOT complex has numerous biological activities, the functional consequences of the CNOT3 E20K and E70K mutations will be very difficult to ascertain using traditional in vitro methods. To circumvent this, we turn to zebrafish and employ in vivo functional genomics analyses.
From our experiments, cnot3 knockdown and apc deficiency are rescued by human CNOT3 and the E20K variant but not by E70K, suggesting that E70K is an inactivating mutation. We also provide mechanistic evidence that CNOT3 is genetically linked to APC through CtBP1. Taken together with our findings that CNOT3 mutations are present in approximately 20% of FAP adenomas, we conclude that proper CNOT3 function is important for intestinal development and that CNOT3 inactivation might work in concert with APC deficiency to prevent intestinal differentiation and potentially advance colon adenomas to a more transformed state.
Results
CNOT3 E20K and E70K are common mutations in cancer tissues
We previously reported that CNOT3 mutations occur in FAP adenoma [19]. To determine how prevalent these mutations are in cancer, we mined the publicly available cBIOPORTAL for Cancer Genomics database [19]. From our analyses, CNOT3 alterations (mutations, deletions, and amplifications) are common across multiple types of cancers in sequenced patient and cell line tumor samples. Remarkably, the CNOT3 E20K and E70K mutations we found in adenoma tissues are the two most common mutations found in numerous cancer types (Figure 1A). Additionally, of all the CCR4-NOT supercomplex subunits, the scaffold protein CNOT3 has the most number of E → K mutations (Figure 1B). These results suggest that the CNOT3 E → K variants identified in FAP adenomas are clinically relevant, thus justifying the need for their functional characterization.
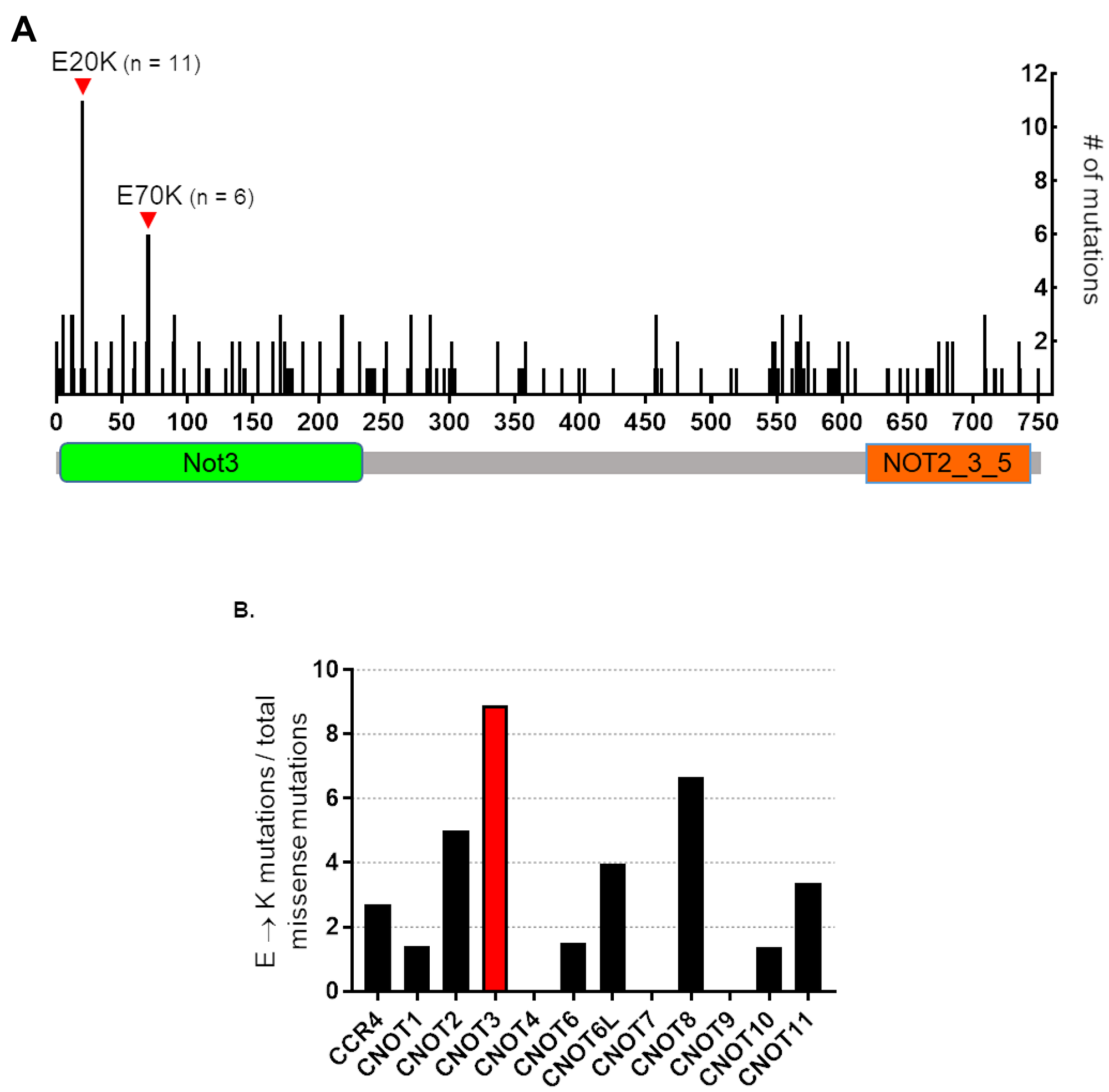
Figure 1: Human CNOT3 E to K mutations in cancer. (A) Schematic representation of CNOT3 protein showing the numerous mutations throughout the CNOT3 amino acid sequence in cancers. The two most common missense mutations are E20K and E70K, which occur in the Not3 domain (green box) and are the two mutations that we discovered during FAP adenoma sequencing experiments [13]. (B) Percentage of E to K mutations compared to total missense mutations in the twelve CCR4-NOT supercomplex subunits over all cancer samples in the cBIOPORTAL database. CNOT3 is presented in red.
CNOT3 is required for intestinal differentiation
We used zebrafish embryonic development as an unbiased, whole organism readout for identifying the in vivo activity of CNOT3 and the functional consequences of the E20K and E70K mutations. We first characterized the relevant CNOT3 orthologue in zebrafish to be cnot3a (See Supplementary Figures 1 and 2 and Supplementary Table 1). We then interrogated the involvement of cnot3 in embryonic intestinal formation by staining cnot3a morphants for primordial intestine marker gata6 and intestinal differentiation marker fabp2. At 48 hpf, when intestinal premordium (pg) is already present, gata6 staining is comparable in cnot3a morphants and control group (Figure 2A). From 72 hpf to 96 hpf, cnot3a morphants exhibited minimal increase in fabp2 (g) staining while control embryos have significantly increased staining from one timepoint to the next (Figure 2B). Our results show that cnot3a knockdown hinders intestinal differentiation and suggest that proper cnot3a expression plays a role in this important developmental process.
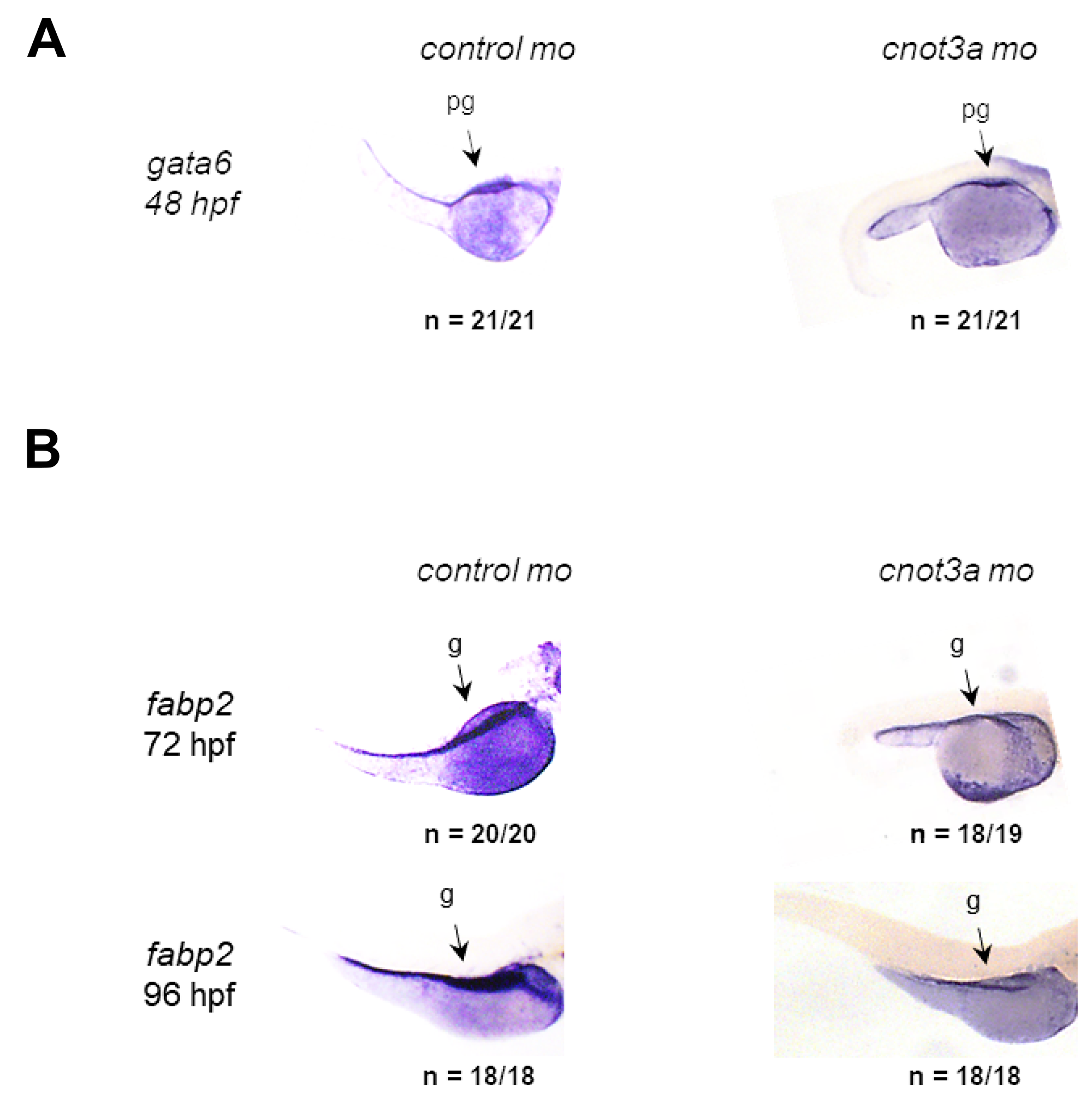
Figure 2: CNOT3 is required for intestinal differentiation. WISH staining for (A) the gut precursor marker gata6 (pg) at 48 hpf and (B) the differentiated gut-specific marker fabp2 (g) in 72 and 96 hpf control and cnot3a morphants.
CNOT3 and E20K rescue cnot3a depletion but E70K variant does not
To validate that human CNOT3 function translates into the zebrafish, we co-injected CNOT3 mRNA with cnot3a morpholino in 1-2 cell stage embryos and used the highly penetrant lordosis phenotype caused by cnot3a knockdown (Supplementary Figure 2) as a readout for complementation. Our results show that the co-injected group had significantly fewer embryos with the lordosis phenotype compared to the cnot3a morpholino-only group, thus confirming that wildtype CNOT3 compensates for cnot3 knockdown (Figure 3A and 3B).
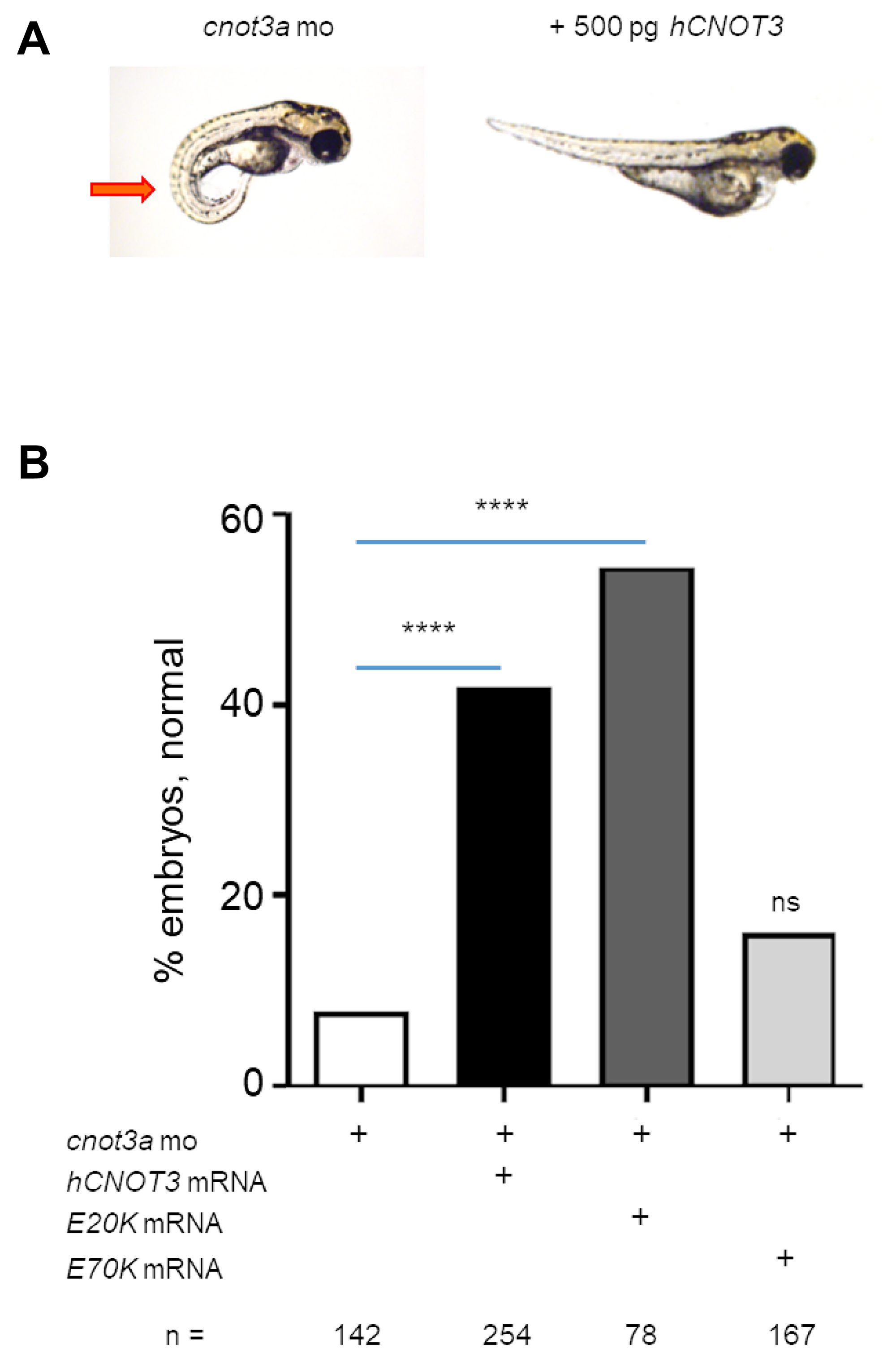
Figure 3: Functional characterization of CNOT3 variants. (A) The lordosis phenotype (red arrow) caused by cnot3a knockdown was rescued by co-injection of 250 pg wildtype human CNOT3 (hCNOT3) with 4 ng cnot3a morpholino (mo). (B) Percentage of rescue after co-injection of hCNOT3, E20K, or E70K variants with cnot3a mo (n > 75 per group). Significance of co-injection rescue was determined using Fisher’s exact test compared to cnot3a mo injection alone. ns = not significant. ****= p-value < 0.0001.
Next, we determine whether the two CNOT3 mutations identified in FAP adenomas [13] affect wildtype CNOT3 function in vivo by co-injecting each one with cnot3a mo into 1-2 cell stage zebrafish embryos. Our results show that CNOT3 E20K variant rescues the observed cnot3a morphant lordosis phenotype significantly while E70K does not (Figure 3B). Our data suggests that CNOT3 E70K is an inactivating mutation.
Human CNOT3 E70K variant cannot rescue intestinal differentiation in the apc-deficient zebrafish model (apcmcr)
To determine the genetic relationship of apc and cnot3a, we measure cnot3a expression by qRT-PCR in apc mutant (apcmcr) and wildtype sibling zebrafish embryos. We find that apcmcr fish have decreased cnot3a expression compared to control (Figure 4A) suggesting that apc regulates cnot3a.
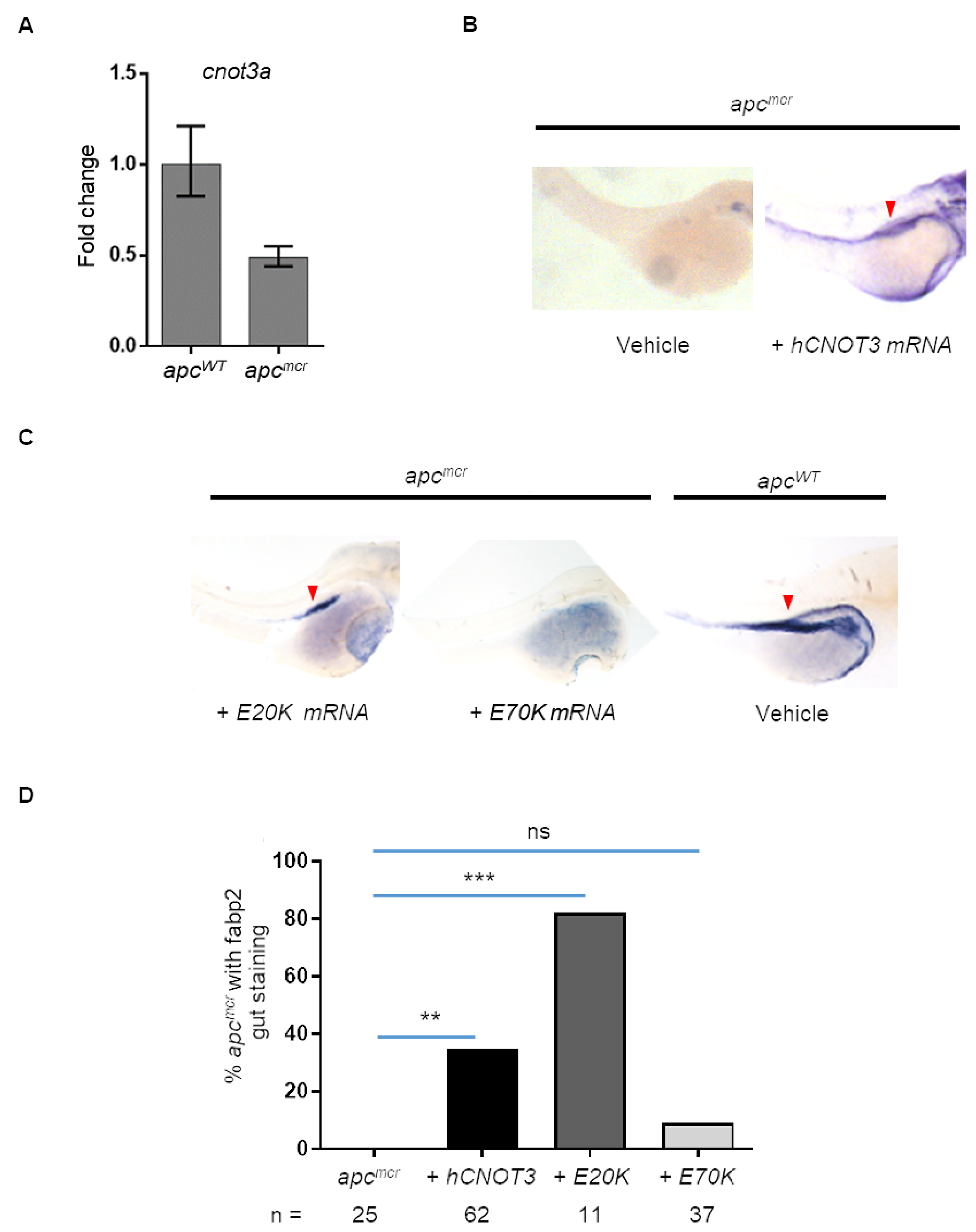
Figure 4: Human CNOT3 rescues intestinal defects in zebrafish apcmcr embryos. (A) Graph shows cnot3a mRNA expression relative to 18s in 72 hpf embryos. (B) apcmcr and wildtype embryos, WISH staining for gut marker fabp2 for vehicle and hCNOT3 injected apcmcr embryos. (C) WISH staining for hCNOT3 E20K and E70K-injected apcmcr embryos and vehicle-injected apcWT. Note that the red arrowhead = fabp2 staining. (D) Percentage of apcmcr fish with gut marker staining from (A and B) at 72 hpf (n > 10 per group). Statistical significance of CNOT3 mRNA injection rescue was determined using Fisher’s exact test compared to uninjected apcmcr embryos alone. ns = not significant; *** = p-value = 0.0003; ** = p-value = 0.0043.
Based on this result, we wondered whether the intestinal differentiation defect of apcmcr zebrafish could be rescued by human CNOT3 mRNA injection. The apcmcr zebrafish is an established tool for understanding the genetics of colon cancer progression, and using the restoration of intestinal differentiation in homozygous apcmcr embryos as a readout has helped previously to uncover novel genes downstream of APC [6–8, 22–24]. Thus, to find out if cnot3a functions downstream of apc, we injected CNOT3 mRNA to 1-2 cell apcmcr embryos and screened for intestinal differentiation at 72 hpf. Using the intestinal development marker fabp2, we observe that introduction of CNOT3 mRNA to apcmcr embryos does initiate intestinal development (Figure 4B).
We exploit the finding that CNOT3 rescues apcmcr intestinal differentiation to determine the functional consequences of CNOT3 E20K and E70K mutations by injecting CNOT3 E20K and E70K mRNA into 1 - 2 cell stage apcmcr zebrafish embryos. Similar to CNOT3, the E20K variant rescues intestinal differentiation but E70K injection does not (Figure 4C–4D). The apcmcr intestinal differentiation rescue assay results provide additional evidence that E70K is an inactivating mutation. CNOT3 mRNA injections were confirmed by PCR (Supplementary Figure 3).
CNOT3 is regulated by transcriptional repressor CTBP1
Our data from the previous section suggests that CNOT3 is downstream of APC during intestinal development (Figure 4A–4D). We have also shown elsewhere that APC regulates CtBP1 that in turn controls retinoic acid (RA) biosynthesis [6–8]. This led us to perform a series of qRT-PCR analyses to clarify how CNOT3 fits in this model. We first explored how ctbp1 knockdown, which we have previously shown to restore intestinal differentiation in apcmcr zebrafish [8], affects cnot3a levels. We observe that knockdown of the transcriptional corepressor ctbp1 in both apc-deficient (Figure 5A) and apc wildtype (WT) (Figure 5B) embryos leads to a 2-3 fold increase in cnot3a expression suggesting that cnot3a is downstream of ctbp1. We have also previously reported that RA biosynthesis, which is regulated by CTBP1, partially rescues intestinal development [6–8]. To determine if CNOT3 rescue of intestinal differentiation depends on RA biosynthesis, we exposed apc mutant fish to retinoic acid. Our qRT-PCR results show that cnot3a expression does not change with RA supplementation (Figure 5C). Similarly, exposure to the RA inhibitor, DEAB, also does not affect cnot3a expression (Figure 5D). These series of qRT-PCR experiments imply that CtBP1 regulates CNOT3 independent of retinoic acid status.
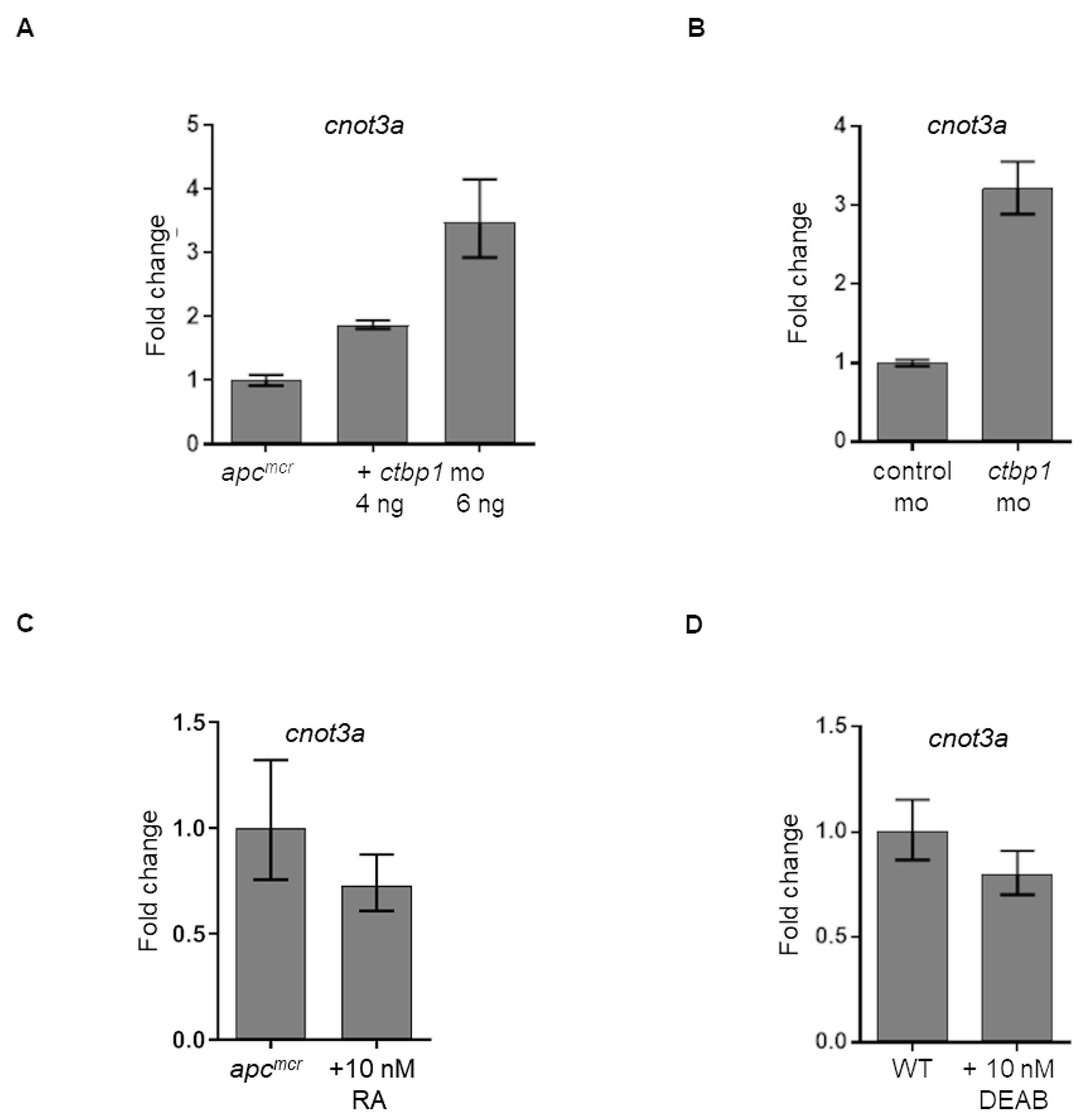
Figure 5: Regulation of cnot3a expression by ctbp1. (A–D) Graphs showing cnot3a mRNA expression relative to 18s using qRT-PCR assay. mRNA expression of cnot3a in (A, C) apcmcr and (B, D) wildtype embryos after injection of ctbp1 morpholino (mo) (A–B), and after exposure to (C) 10 nM retinoic acid (RA) or (D) RA inhibitor DEAB. Values represent mean ± SD. Graphs shown above is representative of 2 independent pooled embryo samples (3 technical replicates each).
A possible mechanism by which CTBP1 protein regulates CNOT3 is by working as a transcriptional repressor of CNOT3. By mining a previously published CtBP1 CHiP-Seq dataset [25], we found evidence that CtBP1 binds approximately (-) 4,000 bp (chr19:54,637,229-54,637,258) of the CNOT3 transcription start site (chr19:54,641,436) in chromosome 19 (Figure 6). Viewing ENCODE data using the UCSC Genome browser, this presumptive CtBP1 binding site possess characteristics of a transcription factor (TF) binding site including (i) DNase I hypersensitivity (31 / 125 cell types), (ii) RNA Polymerase II, EGR1, and ZNF143 CHiP-Seq pulldown, and (iii) H3K4Me1 and H3K27Ac signal [26–33]. Thus, we propose that CtBP1 binding to this site could help localize repression elements to suppress CNOT3 expression. Taken together, our results suggest a novel mechanism wherein APC regulates CNOT3 via CTBP1 independent of retinoic acid (Figure 7).
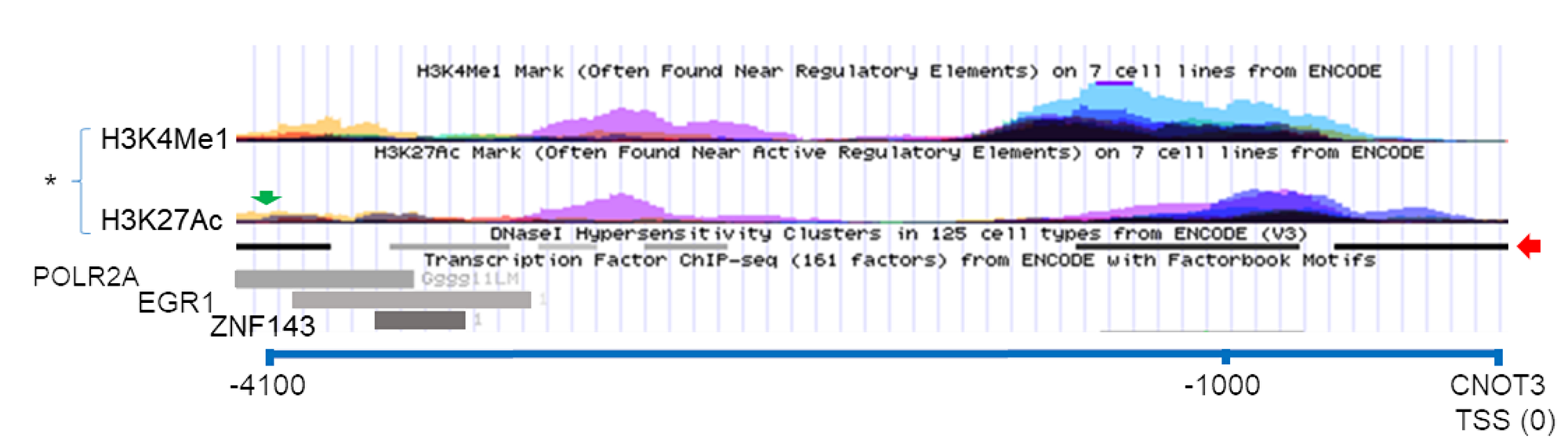
Figure 6: Possible CTBP1 binding site near the CNOT3 gene. USCS Genome Browser screen capture showing the CTBP1 binding site (green arrow) in chromosome 19 (-) 4100 bp of the CNOT3 Translational Start Site (TSS) from CTBP1-CHiP Seq dataset (33). (*) = H3K4Me1 and H3K27Ac signal from seven different high-throughput ENCODE cell line datasets. Red arrow show DNA regions with hypersensitivity to DNAse I treatment. Transcription Factor POLR2A, EGR1, and ZNF143 ChIP-Seq pulldown are represented as gray rectangles.
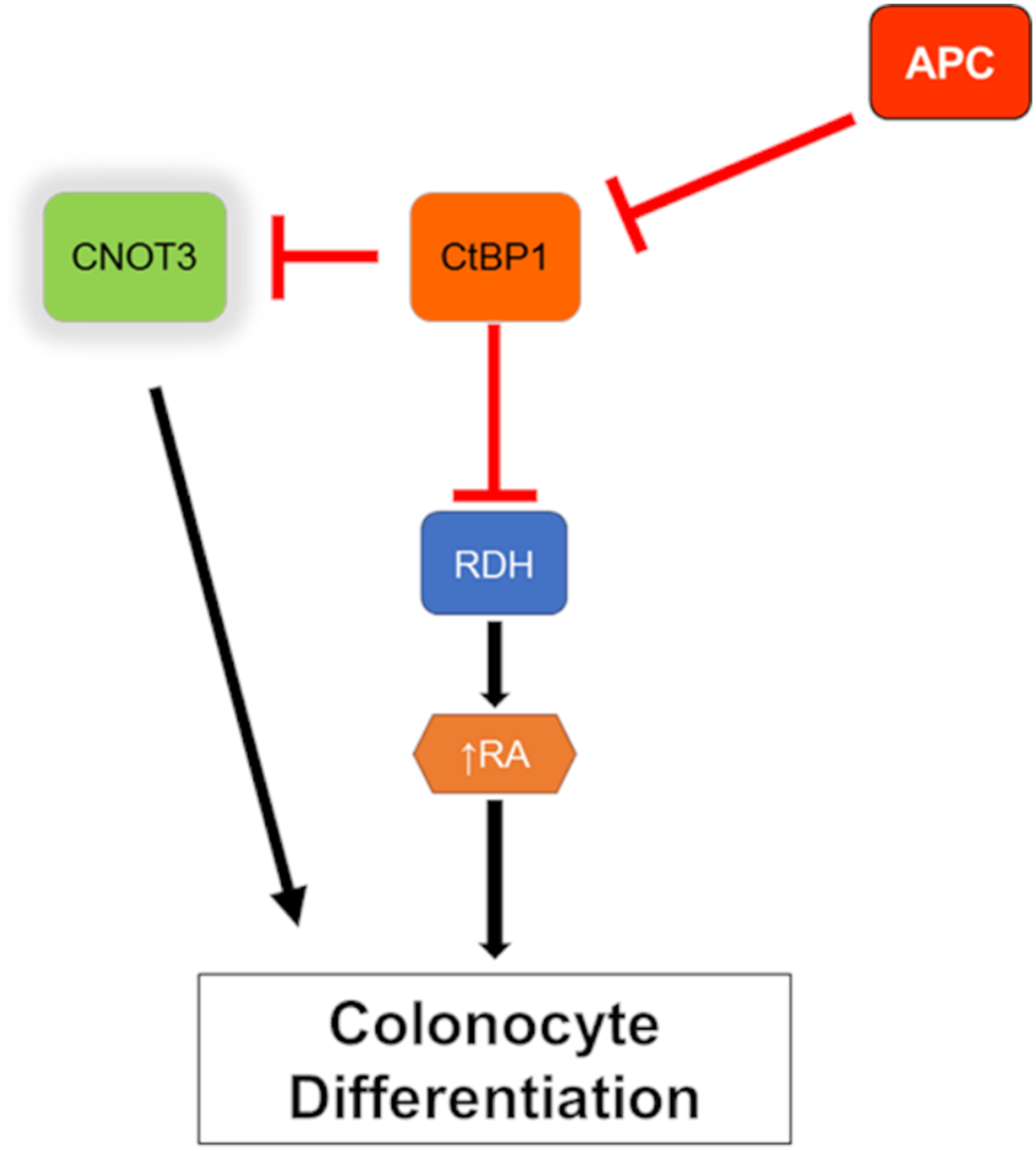
Figure 7: Model of CNOT3 involvement in intestinal differentiation. Red line indicates inhibition and black arrow indicates activation.
CNOT3 mutations are again present in an expanded cohort of fap adenoma samples
To confirm our previously published observation that five of twenty-five (20%) FAP adenoma samples carry CNOT3 mutations [13], we further interrogated the mutational status of CNOT3 in colorectal premalignancy by performing deep sequencing in all the exons using Ion Torrent (IT) in a new cohort of 37 adenomas of 14 FAP patients with paired germline samples (Supplementary Tables 2 and 3). The mean depth obtained for CNOT3 from the IT was 5,047x. Seven adenomas (19%) harbored somatic CNOT3 mutations, with the K286E mutation found in four of the samples (Table 1 and Supplementary Table 4). Comparing these CNOT3 mutations against the sequenced tumor sample data in the COSMIC and cBIOPORTAL databases, we discovered that they are all novel mutations, thus expanding the spectrum of CNOT3 alterations reported in tumor studies and confirming that approximately 20% of FAP adenomas have CNOT3 mutations.
Table 1: CNOT3 mutations found in an extended cohort of FAP adenomas
| Sample ID | Mutation |
|---|---|
| MDAC14_P05 | E120A |
| MDAC34_P04 | D285E |
| MDAC24_P02 | K286E |
| MDAC29_P01 | K286E |
| MDAC32_P01 | K286E |
| MDAC34_P02 | K286E |
| MDAC24_P01 | Frameshift at G488 |
DISCUSSION
The implementation of high throughput genomic sequencing has created a growing clinical need to streamline the evaluation of novel mutations in disease-causing genes for functional relevance and, therefore, their possible contribution to disease progress. Previous work on the APC tumor suppressor gene has shown it as being involved in numerous processes for normal development and homeostasis [34]. Central among these roles is APC control of intestinal differentiation and its ability to suppress turnover and proliferation of intestinal epithelia [35]. Although these roles are clearly the basis of APC function as the major driver of both sporadic and hereditary colon tumorigenesis [1–4], APC dysregulation alone though does not lead to colon cancer. Additional hits in other genes like KRAS and TP53 are needed together with APC dysfunction to cause colon cancer [10, 11]. It is unclear, however, whether additional genes also contributed to tumorigenesis. Following up our previous work defining the genetic profile of adenomas and at-risk mucosa from FAP adenomas [13], here we use the zebrafish to rapidly functionally characterize two clinically-relevant CNOT3 mutations and also offer evidence that CNOT3 plays a critical role in intestinal differentiation downstream of APC.
Our work is the first to provide evidence that the CNOT3 E70K mutation, a common but previously uncharacterized mutation in cancers, is an inactivating mutation. Our work details two functional assays pointing to CNOT3 E70K being an inactivated variant (Figures 3 and 4). One plausible scenario based on the intestinal differentiation assay, is that the CNOT3 E70K inactivating mutation could be working with APC mutation to better prevent differentiation while waiting for other tumor-promoting mutations in KRAS and TP53 to occur [10, 11]. Our data also hints of CNOT3 E20K being an activating mutation as it rescues both complementation and intestinal differentiation assays better than wildtype CNOT3. This was unexpected though not unprecedented as both gain-of-function and inactivating mutations in TP53 have been reported to be tumor-promoting [36, 37]. Note that a limitation of our study is that we do not directly demonstrate that either CNOT3 E20K or E70K mutations are oncogenic. However, our study does create a rationale for studying CNOT3 mutations in the context of adenoma progression to colon carcinoma.
We have previously shown that CtBP1 is downstream of APC and regulates RDHs during intestinal differentiation [6–8]. Based on our current findings that (i) cnot3a depletion leads to stalled intestinal differentiation (Figure 2), (ii) apc mutation in zebrafish results in decreased cnot3a expression (Figure 4A), (iii) CNOT3 mRNA is able to restore differentiation of apcmcr intestine (Figure 4B), and (iv) ctbp1 regulates cnot3a expression (Figure 5A, 5B) independent of RA biosynthesis (Figure 5C, 5D), we conclude that CNOT3 is connected to APC through CtBP1 and that CNOT3 works in parallel with retinoic acid to effect intestinal differentiation.
Finally, the prevalence of CNOT3 mutations in the TCGA PanCancer dataset that contains 10,967 samples from 32 studies showed a Somatic Mutation Frequency (SMF) for APC of 7.3%, while CNOT3 was 1.3% [19]. In this dataset, the Somatic Mutation Frequency (SMF) of APC is 7.3%, while CNOT3 is 1.3%. In the colon cancer subset (594 samples), APC and CNOT3 were altered in 66.67% and 1.18% of cancers, respectively. The percentage of CNOT3 mutations (~20%) in FAP adenoma samples that we previously reported [13] and corroborated in this manuscript is higher compared to the TCGA data that contains only sporadic and inherited colon cancer samples. Since CNOT3 mutations occur more favorably in FAP patients, our observations are very relevant in the context of personalized medicine and justifies the need to further characterize the novel “CNOT3-mutant FAP adenoma” subset in more detail.
Materials and Methods
Zebrafish maintenance
Wild-type TU and apcWT/mcr Danio rerio (zebrafish) were maintained as previously described [38]. Fertilized embryos were collected following natural spawnings in 1× E3 medium (286 mg/L NaCl, 13 mg/L KCl, 48 mg/L CaCl2·2H2O, 40 mg/L MgSO4, 0.01% methylene blue) and allowed to develop at 28.5° C.
Morpholino and RNA microinjections
For RNA rescue experiments, full length human wildtype CNOT3, E20K, and E70K variant RNA transcripts were transcribed from linearized plasmid DNA using mMESSAGE mMACHINE transcription kit (ThermoFisher Scientific, Waltham, MA). For microinjections, 1-2 nl of RNA was injected with our without 4 ng cnot3a morpholino into embryos at the one-to-two cell stages. Overexpression of mRNA transcript was assessed by PCR. Statistical analyses were performed using Fisher’s exact test (GraphPad Prism v 7.02). Parental CNOT3 plasmid was obtained from Origene (Rockville, MD).
A complete list of morpholinos and PCR primers used are provided in Supplementary Table 5.
Zebrafish cnot3a and cnot3b in situ hybridization probes. 400-bp cnot3a and cnot3b ORF gene fragments were chemically synthesized and attached to pUC57-Kan by Genewiz (South Plainfield, NJ). Sense and antisense probes were made using T3 and T7 DIG RNA Labeling kit (Roche, Basel, Switzerland), respectively, from linearized cnot3a and cnot3b pUC57 plasmids.
In situ hybridization
Whole organism in situ hybridization (WISH) was performed as previously described using digoxigenin-labeled riboprobes for gata6 and fabp2 (fatty acid binding protein 2, intestinal) [39]. Embryos were cleared in 2:1 benzyl benzoate / benzyl alcohol solution and documented using an Olympus SZX12/DP71 imaging system (Olympus Corporation, Shinjuku, Tokyo, Japan).
Quantitative RT-PCR (zebrafish)
RNA from zebrafish embryo lysates was isolated using the RNeasy kit (Qiagen, Hilden, Germany). cDNA was synthesized from 1 μg of total RNA using iScript (Bio-Rad, Hercules, CA). Intron-spanning primers, when possible, were designed using the Universal ProbeLibrary Assay Design Center (Roche). A complete list of primer sets is provided in Supplementary Table 5.
PCR master mix was prepared with the FastStart Essential DNA Probe Master kit and Universal Probe Library probes according to the manufacturer’s protocols (Roche). PCR was performed in triplicate using the LightCycler 96 System (Roche) with 45 cycles of amplification and annealing temperature of 60°C for two to three biological replicates. Relative change in gene expression was determined by normalizing against 18S rRNA and comparing test group with control.
Quantitative RT-PCR (human samples)
RNA was extracted from 23 colorectal adenoma and 10 matched normal mucosa samples from 10 different patients diagnosed with FAP using Trizol (Invitrogen). cDNA was synthesized using cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was performed using SYBR Green Mastermix (Applied Biosystems). CNOT3 specific primers have been previously reported [40] and β-actin was used as endogenous control. Experiments were performed in triplicates and the relative expression was calculated by the ΔCt method using RNA from HCT116 as a reference.
Bioinformatics analyses
The publicly available cancer database COSMIC was used to identify CNOT3 point mutations in tumor samples [20]. cBIOPORTAL was used to identify CNOT3 mRNA mutations from publicly available databases [19]. The USCS Genome Browser was used to visualize the transcription and regulatory elements in the human CNOT3 chromosome 19 region data from the ENCODE Project (GrCh37 Assembly) [26, 27]. Pairwise global and local protein sequence alignment was performed using programs available from The European Bioinformatics Institute (EBI) [41]. Mutassessor and Polyphen-2 were used to predict possible functional effect of CNOT3 amino acid changes [42, 43].
Subjects and samples
Targeted sequencing (AmpliSeq) was performed in 37 colorectal adenomas and matched normal mucosa samples from 14 patients with FAP collected at MD Anderson (Supplementary Tables 2 and 3). Informed consent was obtained from all individuals and the Institutional Review Board approved this study. Tissues were retrieved from the endoscopy suite and immediately flash-frozen or preserved in RNAlater (Life Technologies) and then stored at −80° C according to internal protocols. Blood was collected in EDTA tubes and stored appropriately for subsequent extraction of germline DNA. Genomic DNA was extracted from whole blood using the Blood & Cell Culture DNA Mini Kit (Qiagen) and from tissues using the QIAmp DNA Mini Kit (Qiagen). Confirmation of the diagnosis of adenomatous polyps was performed by an expert gastrointestinal pathologist (M.W.T.) in all of those biopsied that rendered enough tissue for both nucleic acid extraction and pathology confirmation. Evidence of high-grade dysplasia could not be verified in all of the samples due to the limited availability of representative tissue material.
Ampliseq sequencing
Ampliseq sequencing was performed using the next-generation sequencing platform IT Personal Genome Machine (PGM; Life Technologies) by the Sequencing and Non-Coding RNA Program at MD Anderson, using the Ion PGM 200 Sequencing Kit on an Ion 318 Chip Kit (Life Technologies). A multiplex custom targeted gene panel was designed with the Ion AmpliSeq Designer and included the entire coding sequence of CNOT3 (2,432 base pair, 95% coverage of the exonic sequence). IT Variant Caller v4.2 was run in the somatic low stringency proton mode to detect variants against hg19 on each adenoma and normal BAM file. Then, normal variants were subtracted from matched adenoma variants to create a list of somatic candidates for each adenoma and normal pair. Events located within the first and last 15% of the bases of the read were excluded. Then, a list of somatic candidates went through the following quality control steps: 1) Mutation allele frequencies were re-evaluated after removing variant reads where the mutation lies within the first 15% or last 15% of the bases of the reads; 2) Mutations with more than 2 variant alleles were excluded; 3) Mutations must be covered by a minimum of 100 reads. If a mutation allele frequency is 2-5%, at least 10 reads must show the variant allele. If a mutation allele frequency >5%, at least 25 reads must show the variant allele. Finally, the candidates were imported into a database by vtools [44], which included 5 different functional in silico prediction analysis by Polyphen2, SIFT, Mutation Taster, Mutation Assessor and Condel, and annotated with ANNOVAR [45].
Study approval
The use of zebrafish in these studies was in accordance with an approved IACUC protocol (#17-03) and within institutional guidelines.
Statistics
Statistical tests used are reported in the figure legends, where all data presented indicate mean ± SEM, unless otherwise specified. A p-value less than 0.05 was considered significant. All representative experiments were conducted on 2–3 separate occasions with a minimum of 3 individual samples. All statistical analyses were conducted using Graphpad Prism v 7.02 (GraphPad Software, Inc.).
Abbreviations
FAP: Familial Adenomatous Polyposis; CNOT3: CCR4-NOT Subunit 3; APC: Adenomatous Polyposis Coli; RDH: Retinol Dehydrogenase; CRC: Colorectal Cancer; CtBP1: C-Terminal Binding Protein.
ACKNOWLEDGMENTS
We wish to thank the OMRF Core Labs. We thank the patients and their families for their participation. We thank the staff of the ncRNA Program at MD Anderson Cancer Center for the generation of the IonTorrent sequencing data. We thank the Clinical Cancer Prevention Research Core for their assistance obtaining the samples for this study.
CONFLICTS OF INTEREST
EV reports a consulting role for Janssen Research and Development.
FUNDING
This work was supported by NCI/NIH (R01 CA116468NIH, (DAJ)), Samuel Waxman Cancer Research Foundation, Oklahoma Center for Adult Stem Cell Research (OCASCR) and Oklahoma Medical Research Foundation (OMRF); R03CA176788 (US National Institutes of Health/National Cancer Institute), The University of Texas MD Anderson Cancer Center Institutional Research Grant (IRG) Program and a gift from the Feinberg Family to E.V., the Janice Davis Gordon Memorial Postdoctoral Fellowship in Colorectal Cancer Prevention (Division of Cancer Prevention/MD Anderson Cancer Center) to E.B.. Cancer Prevention Educational Award (R25T CA057730, U.S. National Institutes of Health/National Cancer Institute) to K.C, and P30 CA016672 (US National Institutes of Health/National Cancer Institute) to the University of Texas Anderson Cancer Center Core Support Grant.
References
1. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998; 58:1130–34. [PubMed].
2. Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006; 101:385–98. [PubMed].
3. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001; 10:721–33. https://doi.org/10.1093/hmg/10.7.721. [PubMed].
4. Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat. 1993; 2:425–34. https://doi.org/10.1002/humu.1380020602. [PubMed].
5. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007; 56:417–25. https://doi.org/10.1136/gut.2006.093310. [PubMed].
6. Nadauld LD, Sandoval IT, Chidester S, Yost HJ, Jones DA. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J Biol Chem. 2004; 279:51581–89. https://doi.org/10.1074/jbc.M408830200. [PubMed].
7. Nadauld LD, Shelton DN, Chidester S, Yost HJ, Jones DA. The zebrafish retinol dehydrogenase, rdh1l, is essential for intestinal development and is regulated by the tumor suppressor adenomatous polyposis coli. J Biol Chem. 2005; 280:30490–95. https://doi.org/10.1074/jbc.M504973200. [PubMed].
8. Nadauld LD, Phelps R, Moore BC, Eisinger A, Sandoval IT, Chidester S, Peterson PW, Manos EJ, Sklow B, Burt RW, Jones DA. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J Biol Chem. 2006; 281:37828–35. https://doi.org/10.1074/jbc.M602119200. [PubMed].
9. Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, Jones DA. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J Biol Chem. 2004; 279:34397–405. https://doi.org/10.1074/jbc.M314021200. [PubMed].
10. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011; 6:479–507. https://doi.org/10.1146/annurev-pathol-011110-130235. [PubMed].
11. Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009; 137:623–34. https://doi.org/10.1016/j.cell.2009.02.037. [PubMed].
12. Lin SH, Raju GS, Huff C, Ye Y, Gu J, Chen JS, Hildebrandt MAT, Liang H, Menter DG, Morris J, Hawk E, Stroehlein JR, Futreal A, et al. The somatic mutation landscape of premalignant colorectal adenoma. Gut. 2018; 67:1299–1305. https://doi.org/10.1136/gutjnl-2016-313573. [PubMed].
13. Borras E, San Lucas FA, Chang K, Zhou R, Masand G, Fowler J, Mork ME, You YN, Taggart MW, McAllister F, Jones DA, Davies GE, Edelmann W, et al. Genomic Landscape of Colorectal Mucosa and Adenomas. Cancer Prev Res (Phila). 2016; 9:417–27. https://doi.org/10.1158/1940-6207.CAPR-16-0081. [PubMed].
14. Bartlam M, Yamamoto T. The structural basis for deadenylation by the CCR4-NOT complex. Protein Cell. 2010; 1:443–52. https://doi.org/10.1007/s13238-010-0060-8. [PubMed].
15. Collart MA, Panasenko OO. The Ccr4—not complex. Gene. 2012; 492:42–53. https://doi.org/10.1016/j.gene.2011.09.033. [PubMed].
16. Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R. The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell. 2001; 104:377–86. https://doi.org/10.1016/S0092-8674(01)00225-2. [PubMed].
17. Albert TK, Hanzawa H, Legtenberg YI, de Ruwe MJ, van den Heuvel FA, Collart MA, Boelens R, Timmers HT. Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 2002; 21:355–64. https://doi.org/10.1093/emboj/21.3.355. [PubMed].
18. De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M, Lahortiga I, Lucà R, Yan J, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013; 45:186–90. https://doi.org/10.1038/ng.2508. [PubMed].
19. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088. [PubMed].
20. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–11. https://doi.org/10.1093/nar/gku1075. [PubMed].
21. Suzuki T, Kikuguchi C, Sharma S, Sasaki T, Tokumasu M, Adachi S, Natsume T, Kanegae Y, Yamamoto T. CNOT3 suppression promotes necroptosis by stabilizing mRNAs for cell death-inducing proteins. Sci Rep. 2015; 5:14779. https://doi.org/10.1038/srep14779. [PubMed].
22. Haramis AP, Hurlstone A, van der Velden Y, Begthel H, van den Born M, Offerhaus GJ, Clevers HC. Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 2006; 7:444–49. https://doi.org/10.1038/sj.embor.7400638. [PubMed].
23. Hurlstone AF, Haramis AP, Wienholds E, Begthel H, Korving J, Van Eeden F, Cuppen E, Zivkovic D, Plasterk RH, Clevers H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature. 2003; 425:633–37. https://doi.org/10.1038/nature02028. [PubMed].
24. Valvezan AJ, Huang J, Lengner CJ, Pack M, Klein PS. Oncogenic mutations in adenomatous polyposis coli (Apc) activate mechanistic target of rapamycin complex 1 (mTORC1) in mice and zebrafish. Dis Model Mech. 2014; 7:63–71. https://doi.org/10.1242/dmm.012625. [PubMed].
25. Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M, Torchia J. TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012; 46:636–49. https://doi.org/10.1016/j.molcel.2012.03.027. [PubMed].
26. Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012; 489:91–100. https://doi.org/10.1038/nature11245. [PubMed].
27. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. https://doi.org/10.1101/gr.229102. [PubMed].
28. Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, Nguyen N, Paten B, Zweig AS, Karolchik D, Kent WJ. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014; 30:1003–05. https://doi.org/10.1093/bioinformatics/btt637. [PubMed].
29. Simone D, Calabrese FM, Lang M, Gasparre G, Attimonelli M. The reference human nuclear mitochondrial sequences compilation validated and implemented on the UCSC genome browser. BMC Genomics. 2011; 12:517. https://doi.org/10.1186/1471-2164-12-517. [PubMed].
30. Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013; 49:825–37. https://doi.org/10.1016/j.molcel.2013.01.038. [PubMed].
31. Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, Moore J, Pierce BG, Dong X, Virgil D, Birney E, Hung JH, Weng Z. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013; 41:D171–76. https://doi.org/10.1093/nar/gks1221. [PubMed].
32. ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. https://doi.org/10.1038/nature11247. [PubMed].
33. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, et al. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. https://doi.org/10.1038/nature11232. [PubMed].
34. Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007; 120:3327–35. https://doi.org/10.1242/jcs.03485. [PubMed].
35. Creamer B, Shorter RG, Bamforth J. The turnover and shedding of epithelial cells. I. The turnover in the gastro-intestinal tract. Gut. 1961; 2:110–18. https://doi.org/10.1136/gut.2.2.110. [PubMed].
36. van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000; 6:2138–45. [PubMed].
37. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010; 2:a001107. https://doi.org/10.1101/cshperspect.a001107. [PubMed].
38. Westerfield M. The zebrafish book : a guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene (OR): M. Westerfield; 1993.
39. Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008; 3:59–69. https://doi.org/10.1038/nprot.2007.514. [PubMed].
40. Venturini G, Rose AM, Shah AZ, Bhattacharya SS, Rivolta C. CNOT3 is a modifier of PRPF31 mutations in retinitis pigmentosa with incomplete penetrance. PLoS Genet. 2012; 8:e1003040. https://doi.org/10.1371/journal.pgen.1003040. [PubMed].
41. Brooksbank C, Bergman MT, Apweiler R, Birney E, Thornton J. The European Bioinformatics Institute’s data resources 2014. Nucleic Acids Res. 2014; 42:D18–25. https://doi.org/10.1093/nar/gkt1206. [PubMed].
42. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013; Chapter 7: Unit7.20. https://doi.org/10.1002/0471142905.hg0720s76. [PubMed].
43. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118. https://doi.org/10.1093/nar/gkr407. [PubMed].
44. San Lucas FA, Wang G, Scheet P, Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics. 2012; 28:421–22. https://doi.org/10.1093/bioinformatics/btr667. [PubMed].
45. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. https://doi.org/10.1093/nar/gkq603. [PubMed].