
Introduction
The definition and nature of driver genes have been discussed intensely in the last decade. Here, for the sake of simplicity, we highlight a manageable number of cancer driver genes as therapeutic targets based on the 138 driver genes identified by B. Vogelstein and his group and treat these genes as common feature points of multiple cancer genome mutational landscapes, a kind of focus that is critical for targeted drug development [1–5].
Since the majority of targeted therapies address only one oncogene, resistance develops eventually even if one of the actual drivers was targeted. The rapid development of drug resistance is due to the fact that more than a single driver may exist in a given tumor [6–8]. In addition, all tumor types are heterogeneous and certain cancer cell subpopulations or subclones tend to persist [9–11]. After the initial shock caused by the inhibition of a single key driver other non-targeted drivers come to the forefront. They are activated via feedback loops and/or with increased expression at the protein level and sometimes newly acquired mutations within the already mutated genes or in others [12–15]. All tumors with multiple driver mutations in their genome have the potency to show primary resistance to monotherapies therefore we need to target multiple drivers with combination therapies to overcome resistance [16–23]. The driver mutation profile of a tumor of an individual patient would be a valuable primary basis for a more precise and cost-effective approach of combination therapy design than combinatorial methods [26–29].
Here we present sequencing-based, targeted combination therapy studies on colon, lung and multiple myeloma cell lines and also on patient-derived surviving cultures in order to underpin the potency of the individual driver gene pattern based combinational method. We also would like to propose that the pursuit for simplicity may offer highly efficient approaches.
Results
Targeting drivers in multiple myeloma cell line models
Multiple myeloma is a currently incurable hematological tumor with several well characterized cell line models [30–32]. In the case of RPMI8226, U266 and LP1 myeloma cell lines our targeted drug design was based on data from the Multiple Myeloma Cell Line Characterization Project [24] and the COSMIC database [25]. As a subsequent step, we eliminated the mutations that did not manifest in amino acid changes. Novel unknown mutations that lead to actual amino acid changes were considered structural changes that may result in functional alterations, therefore they are potential molecular targets that can be inhibited by specific pharmaceuticals. Frameshift mutations were automatically regarded as total function loss. By using this simple approach we identified the potential drivers and their associated targets for each myeloma cell line (Table 1).
Table 1: Driver genes and the associated driver targets of multiple myeloma cell lines
| RPMI8226 | U266 | LP1 | |||
|---|---|---|---|---|---|
| Drivers | Target(s) | Drivers | Target(s) | Drivers | Target(s) |
| KDM6A | KDM6A, SETD2 | KDM6A | KDM6A, SETD2 | KDM6A | KDM6A, SETD2 |
| ARID1A | AKT, PI3K, mTOR, AR | ARID1A | AKT, PI3K, mTOR, AR | CASP8 | DNMT1, DNMT3A, HDAC |
| MAP3K1 | MEK, AR | MAP3K1 | MEK, AR | MAP3K1 | MEK, AR |
| ARID1B | AKT, PI3K, mTOR, AR | ARID1B | AKT, PI3K, mTOR, AR | ARID1B | AKT, MTOR, AR |
| MLL3 | HDAC, proteasome | MLL3 | HDAC, proteasome | MLL3 | HDAC, proteasome |
| NOTCH1 | NOTCH | NOTCH1 | NOTCH | CDKN2A | CDKs |
| KRAS | KRAS, Ftase, MEK | KRAS | KRAS, Ftase, MEK | TP53 | HDAC, AURKA, MYC, CDKs, BCL2 |
| TP53 | CDKs, MYC, HDAC, AURKA, BCL2, PLK1 | TP53 | CDKs, MYC, HDAC, AURKA, BCL2, Topoisomerase II | ||
| JAK3 | JAKs, AR | JAK3 | JAKs, AR | ||
The three cell lines share many common tumor suppressor genes (TSGs) with at least one loss-of-function mutation: KDM6A, ARID1A (in LP1, ARID1A’s partner, ARID2B is mutated), MAP3K1, TP53 and MLL. RPMI8226 and U266 have some other mutated drivers in common (e.g., KRAS, JAK3, NOTCH and CDH1).
Although IC50 values have been typically utilized for the characterization of the efficacy of a given compound, here we introduce the IC95 values and the corresponding Combination Indices (CI95s), because we found that these values are more informative when the research objective is to decrease the cancer cell number as much as possible. The range of combinational doses were chosen in a way that using the highest concentrations no detectable amount of living cells remained and then used the same curve fitting method as in the case of IC50 value determination, only had the cutoff at 5% viability instead of 50 when determining the IC95 value. CI95 values are derived from IC95 values in the same way as in the case of CI95 values. In this study, we considered a given drug combination effective based on two major factors: the IC95 value (which represents efficacy) and the CI95 value (which demonstrates the synergism or antagonism of the compounds when applied together).
IC and CI values for the myeloma cell lines can be found in Supplementary Table 1A–1C.
Effect of driver-targeting compounds on the RPMI8226 cell line
We designed 75 combinations of 16 inhibitors. Mutated KRAS activity, which may be crucial in this system, was blocked by the farnesyl transferase inhibitor tipifarnib [33]. We used the pan-CDK inhibitor dinaciclib, being under investigation in clinical trials [34–36]. CUDC101 and CUDC907 are HDAC inhibitors. CUDC101 also inhibits EGFR, while CUDC907 has an effect on PI3Kα [23, 37, 38]. PF-03084014 is a gamma secretase inhibitor that blocks NOTCH signaling [39]. To block the androgen receptor (AR) we used an analogue of flutamide (Vichem Flutamide analogue) [40]. To target BCL2 we used GX15-070 (obatoclax) and ABT-263 [41–43]. We targeted the tankyrase system with the TRKA inhibitor AG879 and used Dp44mT as a topoisomerase II inhibitor [44, 45]. We used AGI-6780 to inhibit IDH1/2 and MG-132 to block the proteasome [46, 47]. MG-132 is one of the analogues of bortezomib, a promising therapeutic agent for multiple myeloma [48, 49]. To inhibit JAKs we used SB1317 which has also an effect against FLT3 [50]. Trametinib is an FDA-approved MEK inhibitor [51]. Finally, for FGFR inhibition, we used XL999, which inhibits FLT3, PDGFRs and VEGFR1-2-3 as well as FGFR1-2-3 [52] and an FGFR2–FGFR3-selective inhibitor developed by Vichem Ltd. (Vichem FGFR Inhibitor). For all 75 combinations, 50 were bicomponent, while 25 consisted of three different compounds. When CI95 was used as a basis to define synergism among two or three compounds we found that 48 out of 75 designed combinations (64%) were synergistic. Furthermore, 38 out of the 48 synergistic combinations (79%) had a CI value < 0.1 which means strong synergism. Regarding the ratio of the synergistic–non-synergistic combinations with respect to the number of drugs used, 26 out of 50 (52%) double combinations were synergistic and 22 out of 25 triple combinations were synergistic (88%). In Figure 2 we present a combination with high synergy as well as a low IC95 value.
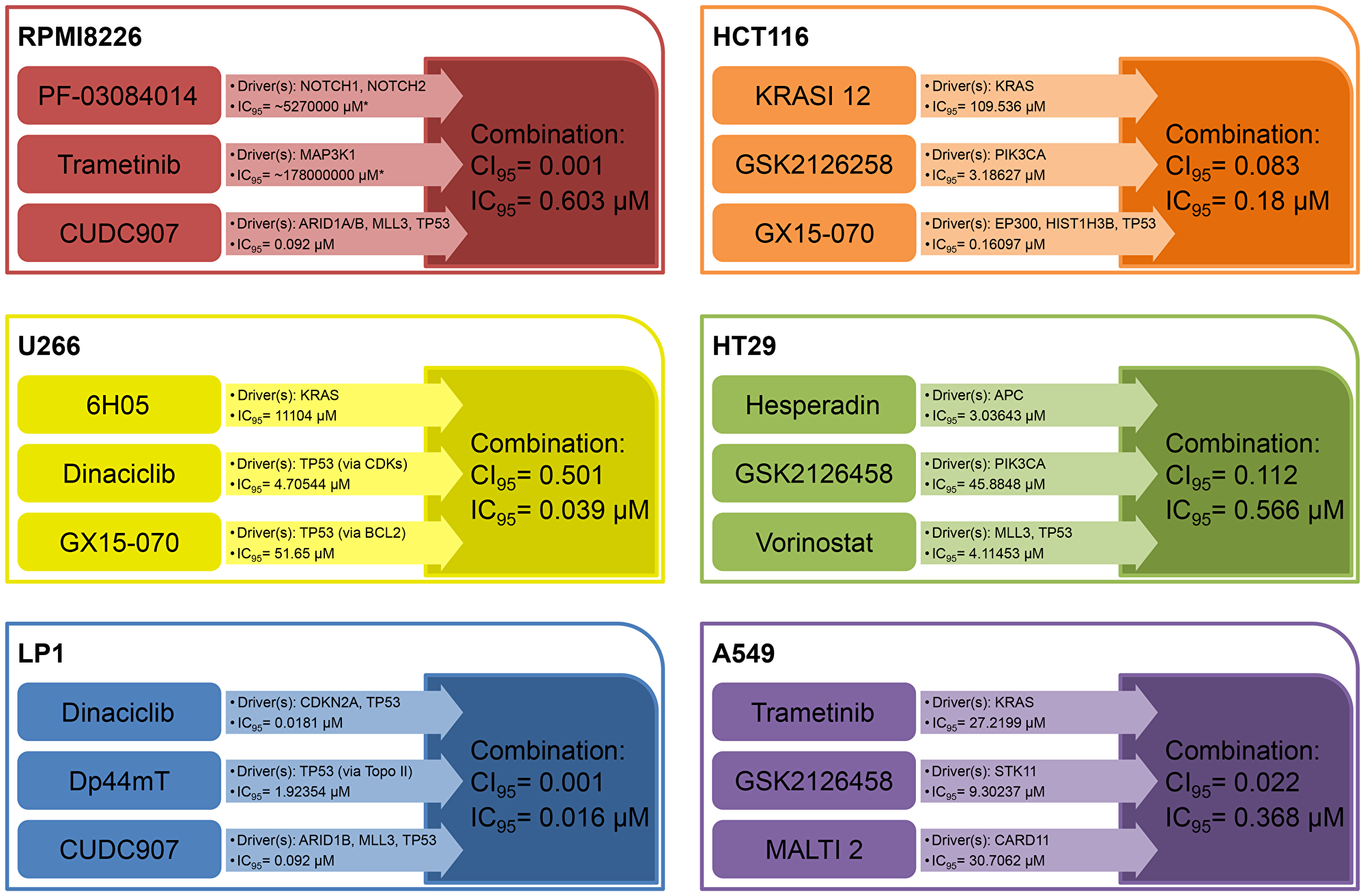
Figure 1: Selected synergistic combination examples for each cell line. Here we show one example of synergistic combination therapy for each cell line used. We applied IC95 values as an indicator of near-total effectiveness in killing tumor cells. CI95 values < 1 indicate synergism between drugs. Combinations are more effective than monotherapies with regard to the magnitude of the IC95 values and/or the number of drivers knocked out. Abbreviations: IC95: 95% cell killing; CI95: Combination Index associated with IC95; KRASI 12: KRAS Inhibitor 12; MALTI: MALT1 Inhibitor 2. *Indicates that this value is a computational estimation which indicates that there’s no appreciable effect biologically.
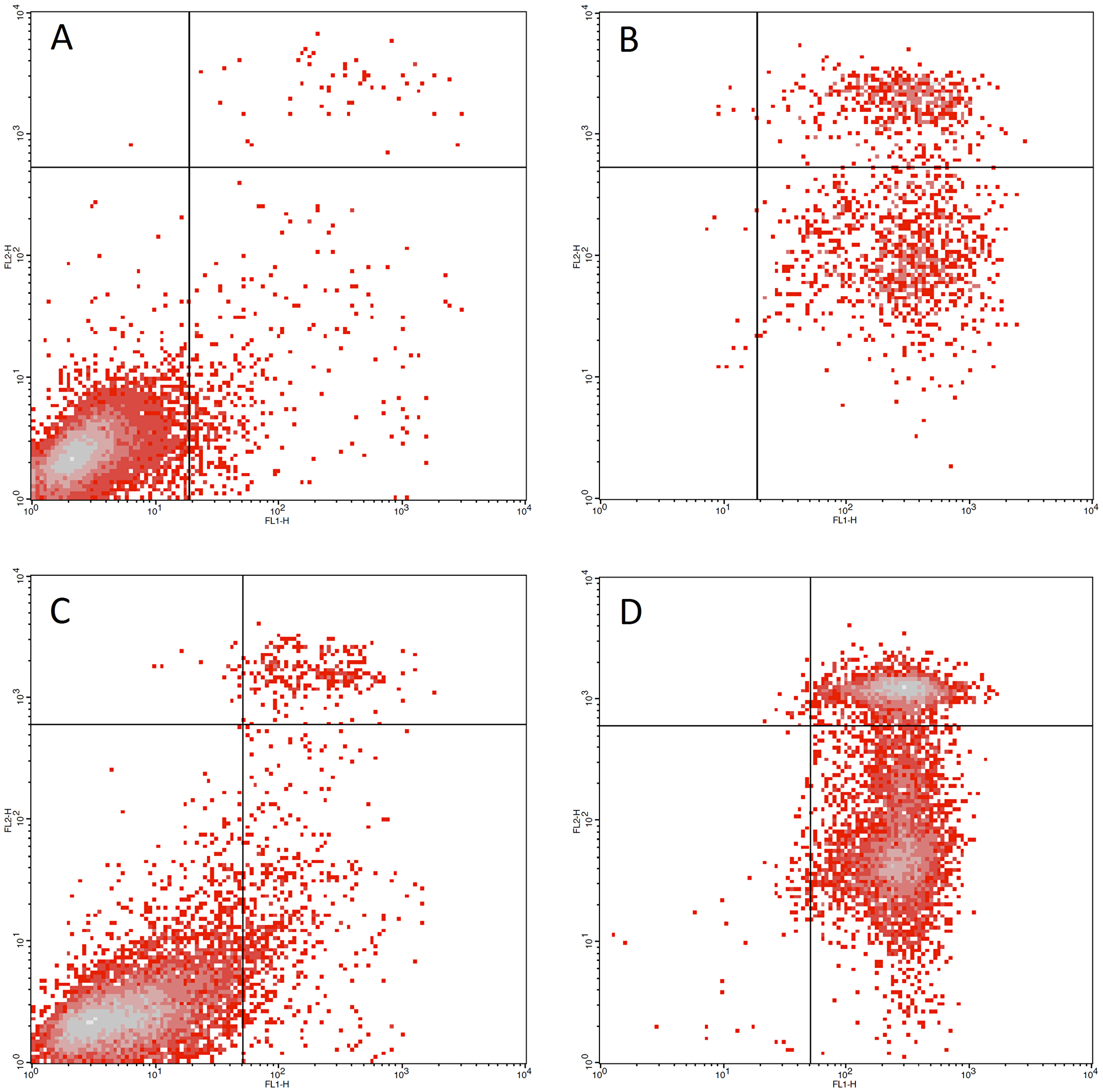
Figure 2: Low dose combination therapy effects on multiple myeloma cell lines. (A) LP1 cell line untreated control (B) LP1 cell line treated with 0.122 μM dinaciclib + MK2206 + CUDC907. 0.61% viable cells were detected (C) RPMI8226 cell line untreated control (D) RPMI8226 cell line treated with 0.122 μM dinaciclib + trametinib + PF-3084014. 1.26% viable cells were detected.
Effect of driver-targeting compounds on the U266 cell line
Here we defined a highly similar set of driver genes and targets as on RPMI8226, however, U266 proved to be a much more resilient model system. To improve the effects on KRAS, proteasome and JAK targets, we incorporated compounds with different mechanisms of action or a slightly altered inhibitory profile and used the novel experimental KRAS inhibitor 6H05 [53], carfilzomib, a covalently binding bortezomib analogue [49], and XL019 (a more JAK-specific inhibitor) [54]. We also focused on the PI3K-AKT-mTOR pathway. We used three PI3K-mTOR inhibitors (GSK2126458 [55], XL765 [56] and PP242 [18]) and a highly selective allosteric AKT inhibitor, MK2206 [57–60]. We also used a pan-Aurora kinase inhibitor, Danusertib [61], and a PLK inhibitor, BI2536 [62, 63]. We targeted the Wnt pathway by inhibiting Porcupine with IWP-2 [64] and used a SET-SETD system-specific experimental inhibitor, NF279 [65]. We also used oltipraz, which interferes with the NF2 system by inhibiting NFE2L2 [66] with kinase inhibitors. Out of the designed 102 combinations, 74 combinations were double, and 28 were triple. According to CI95 value, 25 out of 102 combinations were synergistic (25%), 15 double (15%) and 10 triple (10%) combinations. Figure 1 depicts one of the successful combinations.
Rates of synergistic combinations on the LP1 cell line
The LP1 cell line has some mutational qualities in common with the RPMI8226 and U266 lines. The ARID1B, CDKN1A and caspase 8 genes are mutant but the KRAS, NOTCH1, JAK and CDH1 genes are not. Out of the 86 designed combinations, 65 contained two and 21 had three drugs. We found that 48 combinations out of 86 were synergistic (56%) with close to 100% cell killing. 23 out of the 48 synergistic combinations (48%) had a CI value < 0.1. The ratio of synergistic to non-synergistic combinations was as follows: 31 out of 65 (48%) double combinations and 17 out of 21 (81%) triple combinations were synergistic. For a successful combination see Figure 1.
Apoptotic effect of combinations at low doses in multiple myeloma models
Using FACS analysis, we investigated the beneficial (apoptotic) cell killing potential of the synergistic combinations at low concentrations looking for the ratio of apoptotic cells. We present concentration values predicted to produce maximal detectable cell killing based on the data extracted from viability assays on multiple myeloma cell lines and performed measurements for dose series that covered these values in order to identify the lowest value capable of achieving maximal cell death. In Figure 1, we show the apoptosis-inducing effect of selected successful combinations used on the LP1 (Figure 1A and 1B) and RPMI8226 cell lines (Figure 1C and 1D). The first combination (Figure 1B) targeted the CDK family, AKT1 and HDAC family driver targets of the LP1 myeloma cell line, while the second combination affected the CDK family, the MEK1/2 complex and NOTCH via gamma secretase. At a concentration of 0.122 nM, both combinations pushed the myeloma cells into early or late apoptosis in comparison to untreated controls.
Solid tumor models
Parallel to the studies on myeloma we investigated the effectiveness of our method on solid tumor models using three adherent cell line models with well-established driver status: HCT116 and HT29 colon carcinoma cell lines and the A549 lung adenocarcinoma cell line.
Targeting actually present versus absent driver genes
Prior to the combination studies we conducted an experiment on the HCT116 cell line to explore the difference between the effectiveness of the compounds of the DriverHit Library on the actually present driver-related proteins and molecules having no target in the given model. According to this, the first group of targeted proteins were products of driver genes associated with HCT116, whereas the second cohort of targets were not involved in the pathologic proliferation of HCT116 cells. We performed an unpaired t-test using the pIC50 values of the compounds. The two-tailed P-value turned out to be 0.0015. This indicates that prior to drug selection a careful analysis of the actually present and targetable driver genes has to be performed.
Combinations used on colon cancer cell lines
To determine and characterize driver genes in HCT116 and HT29 colon cell lines we used the COSMIC database. Subsequently, we designed 77 for HCT116 and 82 drug combinations for HT29. Of the 77 HCT116 combinations 37 contained two and 40 had three drugs. Forty three combinations out of 77 proved to be synergistic (56%). Three out of 43 synergistic combinations (7%) had a CI value < 0.1. Sixteen of 37 double combinations (43%) and 27 of 40 triple combinations (68%) were synergistic. Out of the 82 combinations for HT29 55 had two and 27 had three drugs. Using CI95 as the basis for evaluation of synergism between compounds we found that 57 combinations out of 82 were synergistic (70%). Ten out of 57 synergistic combinations (18%) had a CI value < 0.1 representing an extremely strong synergism. The number of synergistic combinations was 31 (56%) in double and 26 (96%) in the case of triple combinations. Figure 1 shows synergistic combinations for each cell line. For the full data set for HCT116 and HT29 see Supplementary Table 1D and 1E.
Driver-targeted combinations in the NSCLC model
In order to increase the diversity of models we also performed experiments on the A549 lung adenocarcinoma cell line. Out of 55 combinations for A549 28 contained two and 27 had three drug compounds. Forty six out of 55 combinations proved to be synergistic (84%). Twenty out of 46 synergistic combinations (43%) had CI values < 0.1. The number of synergistic combinations depending on the number of drugs included was 23 (82%) in the case of double and 23 (85%) in the case of triple combinations. For all combinations see Supplementary Table 1F. A representative combination is shown in Figure 1.
Synergistic combinations on multiple myeloma patient-derived surviving cultures
For our experimental purposes we produced balanced patient-derived myeloma-stroma cell co-cultures and performed in vitro combination treatments in 9 cases (the balanced co-culture models were developed by F. Uher who passed the know-how to Vichem Ltd., unpublished results). Samples that were transformed to stable co-cultures originated from patients before or under treatment. We focused on already approved drugs or compounds being still in clinical trials. We used bortezomib and MG-132 as proteasome inhibitor monotherapy controls therefore the combinations used were comparable with the clinically applied therapies. Combinations were used at 0.5 and 0.5 μM concentrations and proteasome inhibitors were applied at 1 μM. We managed to effectively shift the cell killing profile from stromal cells to myeloma cells in most cases. A selection of our results is shown in Table 2. Corresponding monotherapy results are shown in Supplementary Table 2.
Table 2: Patient-derived surviving culture combination therapies
| Patient No. | Drug combinations [0.5 μM + 0.5 μM] | Targets | Drivers | Myeloma cells | Stromal cells | ||||
|---|---|---|---|---|---|---|---|---|---|
| Expected Inh % | Meas. Inh % | Ratio | Expected Inh % | Meas. Inh % | Ratio | ||||
| P 1 | GSK2126458 + Danusertib | PI3K-mTOR; AURKA; AURKB | PTEN, TP53 | 20.8 | 40.5 | 1.95 | 5.3 | 14.9 | 2.81 |
| P 1 | MG-132 | proteasome | 10.1 | 6.8 | |||||
| P 2 | GSK2126458 + Danusertib | PI3K-mTOR; AURKA; | PTEN, TP53 | 36 | 51.4 | 1.43 | 10.3 | 10.3 | 1 |
| P 2 | Danusertib + CUDC907 | AURKA; HDACs | MLL3, ASXL1, TP53 | 32.8 | 43.4 | 1.33 | 18.4 | 18 | 0.98 |
| P 2 | Danusertib + Dinaciclib | AURKA; CDKs | CDKN2A, CEBPA, TP53 | 27 | 31.1 | 1.15 | 15.2 | 39.3 | 2.59 |
| P 2 | MG-132 | proteasome | 16.3 | 16.7 | |||||
| P 3 | GSK2126458 + Danusertib | PI3K-mTOR; AURKA; AURKB | PTEN, TP53 | 24.6 | 40.5 | 1.65 | 5.7 | 35.2 | 6.23 |
| P 3 | CUDC907 + Danusertib | HDACs; AURKA; AURKB | MSH2, MLL3, TP53 | 22.15 | 29.1 | 1.31 | 6.2 | 23.2 | 3.74 |
| P 3 | MG-132 | proteasome | 10.9 | 16.8 | |||||
| P 5 | Nintedanib + MG-132 | FGFR2; FGFR3; PDGFR; VEGFR2; FGFR; VEGFR; PDGFR; proteasome | FGFR3, PDGFRA, TP53 | 32.8 | 47.4 | 1.45 | 37.8 | 32.5 | 0.86 |
| P 5 | Dinaciclib + Nintedanib | CDKs; FGFR2; FGFR3; PDGFR; VEGFR2; FGFR; VEGFR; PDGFR | FGFR3, PDGFRA, TP53 | 30.95 | 40.5 | 1.31 | 32.7 | 61.6 | 1.88 |
| P 5 | MG-132 | proteasome | 45.9 | 71.4 | |||||
| P 6 | Nintedanib + Bortezomib | FGFR2; FGFR3; FLT3; PDGFR; VEGFR2; proteasome | FGFR3, PDGFRA, TP53 | 20.1 | 40.4 | 2.01 | 41.2 | 14 | 0.34 |
| P 6 | Bortezomib | proteasome | 33 | 79.1 | |||||
DISCUSSION
Myeloma cell lines and patient-derived surviving cultures are multiple myeloma models, while HCT116 and HT29 are colon and A549 is lung adenocarcinoma cell lines. As a result, we intend to discuss the two groups separately due to their distinctive driver gene characteristics.
Synergistic driver targeting combinations in multiple myeloma models
Recent developments in myeloma treatment have led to new and more effective therapeutic protocols. Approved drugs constantly increase, the growing number of newly discovered actionable targets and the enhanced efficacy of novel compounds pave the way to prolonged survival and better quality of life. Similarly, to other tumors, it is becoming evident that targeting only one driver-related pathogenic protein probably never lead to cure but will merely generate therapy resistance and failure. Therefore, a careful assembly of potentially effective drug combinations based on a precise detection of a vulnerable targets will be necessary to improve our treatment results. In our in vitro experiments we found that blocking 2–5 cancer pathways using only 2–3 drugs was sufficient to reach maximal levels of cell killing at extraordinarily low doses due to synergisms among drugs (Figure 3).
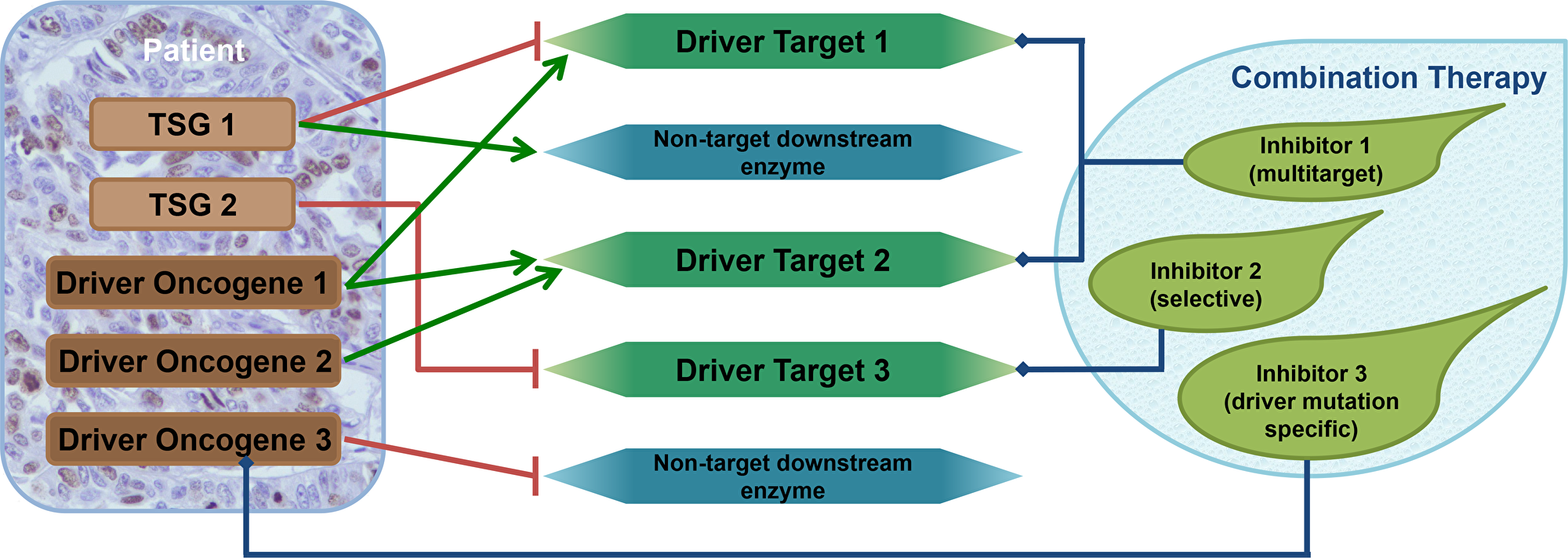
Figure 3: Combination therapy design using the driver gene concept. Tumors usually contain 2–8 driver genes. Tumor suppressor genes (TSG) harbor loss-of-function mutations, while oncogenes possess gain-of-function mutations. Oncogenes can be inhibited directly or via the downstream partners that they stimulate. TSG functional loss may be corrected by inhibiting the downstream partners they are supposed to inhibit naturally. These downstream elements are the driver targets. Although exact protein-selective small molecule inhibitors exist the majority of them have multiple protein targets. The diverse selectivity profiles of the inhibitors and the fact that different driver genes can share their driver targets indicate that more than one driver gene can be blocked by each compound. This confirms our finding that tumors with more than three driver genes can be handled using a mixture of merely three compounds.
We tested drug combinations in order to identify synergistic drug combinations for myeloma cell lines and surviving cultures. We found that drug combinations targeting driver gene-related targets tend to be synergistic and we determined novel targets for use together with the canonical ones. These new potent targets, not yet investigated in the context of multiple myeloma, were revealed as targets through identification of the specific driver genes and their related proteins.
Modern treatment of multiple myeloma is based on five pillars: a) immunomodulatory drugs (IMiDs) (i.e. lenalidomide, pomalidomide and thalidomide); b) proteasome inhibitors (i.e. bortezomib, MG-132 and carfilzomib); c) traditional cytotoxic drugs (i.e. doxorubicine); d) HDAC inhibitors (i.e., vorinostat or panobiostat) and e) corticosteroids (e.g., dexamethasone and prednisone). CDK inhibitors have also been extensively studied in clinical trials. Because IMiDs, cytotoxics and steroids do not target the signal transduction pathways of cancer cells only HDAC, proteasome and CDK inhibitors were considered as targeted compounds in recent myeloma studies [67].
Combinations of HDAC inhibitors
Combination of the HDAC inhibitor CUDC907 with the BCL2 inhibitor GX15-070 at extremely low doses resulted in synergism and total cell killing in all three cell lines. The combinations of CUDC907 with tipifarnib also proved to be successful, although this did not allow for dosages as low as those used with the HDAC plus BCL2 inhibitor combinations. When used as a combination partner, the androgen receptor (AR) inhibitor flutamide analogue also improved the therapeutic efficacy of CUDC907. HDAC and CDK inhibitors were also synergistic in all cell lines. CUDC101, another HDAC inhibitor was synergistic with Dp44mT, a topoisomerase inhibitor. This finding could be observed in all cell lines with the exception of U266. With a reference to the triple combinations, HDAC inhibitors tended to synergize with combinations containing a CDK inhibitor and a cell line-specific inhibitor, such as PF-03084014 (a NOTCH inhibitor), trametinib (a MEK1/2 inhibitor), 6H05 (an allosteric KRAS inhibitor), BI2536 (a PLK inhibitor), GSK2126458 (a PI3K-mTOR dual inhibitor) and MKK2206 (an allosteric AKT inhibitor) (See Supplementary Table 1A–1C). The HDAC inhibitor CUDC907 showed synergism when used with the AURKA inhibitor danusertib on several surviving cultures.
Combinations of proteasome inhibitors
In the case of U266 and patient derived co-cultures we were able to successfully enhance the effect of proteasome inhibitors in combinations but we were not successful to find synergistic partners when experimenting with the LP1 cell line. We found only one synergistic partner (CUDC101, an HDAC inhibitor) in studies with the RPMI8226 cell line. However, we showed, that targeting HDACs, PLK1, KRAS, CDKs, AURKA and FGFRs may provide significant benefits in the improvement of IC95 values for proteasome inhibitors. The results of our experiments on surviving cultures demonstrate that these new targets improve myeloma cell killing rather than stromal cell killing.
Combinations of CDK inhibitors
We found the inhibition of CDKs to be exceptionally beneficial in all investigated multiple myeloma models. Dinaciclib, a pan-CDK inhibitor, synergizes with the vast majority of driver target inhibitors. The most potent partners for CDK inhibitors in multiple myeloma are the HDAC inhibitors CUDC101 and CUDC907, the MEK1/2 inhibitor trametinib, the NOTCH inhibitor PF-03084014, the BCL2 inhibitors GX15-070 and ABT-263, the IDH1/2 inhibitor AGI-5180, the FLT3 and JAK2 inhibitor SB1317 and also the FGFR-targeting inhibitors XL999 and nintedanib. Dinaciclib was also part of synergistic duos and trios with inhibitors affecting the PI3K-AKT-mTOR pathway. Dinaciclib was synergistic with the PI3K-mTOR dual inhibitor GSK2126458 in experiments on the LP1 cell line and in patient-derived surviving cultures, as well as with AKT1 inhibitors MK2206 and PP242. Dinaciclib also synergized with the proteasome inhibitor MG-132 and the AURKA inhibitor danusertib. FACS results of experiments on the RPMI8226 and LP1 cell lines also confirmed that CDK inhibitors can play a key role in combination therapy.
Novel driver targets identified in multiple myeloma
Our results raised new potential targets in multiple myeloma therapy. These targets may offer effective therapeutic surfaces for use in combination with previously known targets. These new targets to be considered in multiple myeloma treatment are the NOTCHs, BCL2, the IDHs, AR, NF2, Porcupine, the JAKs, MEK1/2 and the Aurora kinases.
Synergistic combinations in solid tumor models
The HCT116 and A549 cell lines both have activating KRAS mutations, while KRAS in HT29 is not mutated. We used farnesyl transferase inhibitors (lonafarnib and tipifarnib), allosteric KRAS inhibitors and downstream MEK1/2 inhibitors to eliminate the effect of mutated KRAS. The most successful targets and combinations covered the HDACs, the CHEKs, the PI3K-mTOR axis and BCL2 in all three cell lines. However, A549 has a wild-type TP53 and was therefore less sensitive to the blockage of TP53 TSG targets and more sensitive to the MALT1 inhibitor and JNK inhibitor 1. Results of combination therapies for these models are shown in Supplementary Table 1D (HCT116), 1E (HT29) and 1F (A549). With regard to the PI3K-AKT-mTOR pathway, PI3K and the PI3K-mTOR dual inhibitors were more effective than the AKT1 inhibitors. These measurements suggest that AKT1 inhibition could successfully complements PI3K-mTOR inhibition. The pan-HDAC and PI3K inhibitor CUDC907 and the BCL2 inhibitor GX15-070 were highly potent when applied as monotherapies because each of them targets 2–5 drivers at the same time.
Materials and Methods
For information on Materials and Methods see Supplementary Text 1.
Conclusions
Concentrating on the individual driver gene pattern of a tumor may contribute to an effective selection method to design synergistic combination therapies. In our in vitro models we demonstrated that combination therapies based on the individual driver gene patterns are more efficient than monotherapies in the majority of cases. We concluded that strong synergisms among compounds in driver-targeted drug combinations may result in shorter treatment periods and/or may help to reduce unwanted toxic side effects, because it may allow us to use remarkably lower doses. Because of the common targets in drivers matched with the different inhibitory profiles of the particular drugs used, even double and triple combinations may be highly effective in extremely low concentrations. In each case we proposed crucial driver combinations which affect 3–5 important cancer pathways in a given system. By using the method of targeting tumor specific driver gene sets we would like to form a common rationale for combination therapy design in different types of cell lines or patient derived primary cultures.
ACKNOWLEDGMENTS
We would like to thank the scientific guidance of Manfred Auer and his group, Ferenc Uher and the technical support of collegues at the Oncompass Medicine Hungary, Ltd. We would like to thank for the employees of Central Hospital of Southern Pest National Institute of Hematology and Infectious Diseases, especially to Eva Karaszi for the technical support of the FACS measuerements in surviving cultures.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research project has been supported by the European Commission under the 7th Framework Programme HEALTH.2011.2.4.1-2: Translational research on cancers with poor prognosis. Contract n°: 278570 (OPTATIO).
This project has also received funding from the European Union’s Seventh Framework Programme HEALTH.2011.2.1.1-2 Proteins and their interactions in health and disease. Contract n°: 278568 (PRIMES).
The project was also supported by the Hungarian National Research, Development and Innovation Office within the framework of the National Oncogenomic and Precision Oncotherapy Program (ID n° NVKP_16-1-2016-0005). Project no. NVKP_16-1-2016-0005 has been implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the NVKP_16 funding scheme.
References
1. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013; 339:1546–58. https://doi.org/10.1126/science.1235122. [PubMed].
2. Chen Y, Hao J, Jiang W, He T, Zhang X, Jiang T, Jiang R. Identifying potential cancer driver genes by genomic data integration. Sci Rep. 2013; 3:3538. https://doi.org/10.1038/srep03538. [PubMed].
3. Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, Pineda M, Gindin Y, Jiang Y, Reinhold WC, Holbeck SL, Simon RM, Doroshow JH, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013; 73:4372–82. https://doi.org/10.1158/0008-5472.CAN-12-3342. [PubMed].
4. Peták I, Schwab R, Orfi L, Kopper L, Kéri G. Integrating molecular diagnostics into anticancer drug discovery. Nat Rev Drug Discov. 2010; 9:523–35. https://doi.org/10.1038/nrd3135. [PubMed].
5. Ross JS, Wang K, Elkadi OR, Tarasen A, Foulke L, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Chmielecki J, Ali SM, Elvin J, et al. Next-generation sequencing reveals frequent consistent genomic alterations in small cell undifferentiated lung cancer. J Clin Pathol. 2014; 67:772–76. https://doi.org/10.1136/jclinpath-2014-202447. [PubMed].
6. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al, and Australian Pancreatic Cancer Genome Initiative, and ICGC Breast Cancer Consortium, and ICGC MMML-Seq Consortium, and ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21. https://doi.org/10.1038/nature12477. [PubMed].
7. Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci USA. 2015; 112:118–23. https://doi.org/10.1073/pnas.1421839112. [PubMed].
8. Webster RM. Combination therapies in oncology. Nat Rev Drug Discov. 2016; 15:81–2. https://doi.org/10.1038/nrd.2016.3. [PubMed].
9. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013; 108:479–85. https://doi.org/10.1038/bjc.2012.581. [PubMed].
10. Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, Lipson EJ, Chapman PB, Diaz LA Jr, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013; 2:e00747. https://doi.org/10.7554/eLife.00747. [PubMed].
11. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347:78–81. https://doi.org/10.1126/science.1260825. [PubMed].
12. Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012; 486:537–40. https://doi.org/10.1038/nature11219. [PubMed].
13. Nathanson DA, Gini B, Mottahedeh J, Visnyei K, Koga T, Gomez G, Eskin A, Hwang K, Wang J, Masui K, Paucar A, Yang H, Ohashi M, et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014; 343:72–76. https://doi.org/10.1126/science.1241328. [PubMed].
14. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012; 487:505–09. https://doi.org/10.1038/nature11249. [PubMed].
15. Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015; 21:560–62. https://doi.org/10.1038/nm.3854. [PubMed].
16. Troiani T, Napolitano S, Vitagliano D, Morgillo F, Capasso A, Sforza V, Nappi A, Ciardiello D, Ciardiello F, Martinelli E. Primary and acquired resistance of colorectal cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway activation and can be overcome by combined MEK/EGFR inhibition. Clin Cancer Res. 2014; 20:3775–86. https://doi.org/10.1158/1078-0432.CCR-13-2181. [PubMed].
17. Crunkhorn S. Cancer: combination therapy for lung cancer. Nat Rev Drug Discov. 2016; 15:532. [PubMed].
18. Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008; 4:691–99. https://doi.org/10.1038/nchembio.117. [PubMed].
19. Pénzes K, Baumann C, Szabadkai I, Orfi L, Kéri G, Ullrich A, Torka R. Combined inhibition of AXL, Lyn and p130Cas kinases block migration of triple negative breast cancer cells. Cancer Biol Ther. 2014; 15:1571–82. https://doi.org/10.4161/15384047.2014.956634. [PubMed].
20. Torka R, Pénzes K, Gusenbauer S, Baumann C, Szabadkai I, Őrfi L, Kéri G, Ullrich A. Activation of HER3 interferes with antitumor effects of Axl receptor tyrosine kinase inhibitors: suggestion of combination therapy. Neoplasia. 2014; 16:301–18. https://doi.org/10.1016/j.neo.2014.03.009. [PubMed].
21. Szokol B, Gyulavári P, Kurkó I, Baska F, Szántai-Kis C, Greff Z, Orfi Z, Peták I, Pénzes K, Torka R, Ullrich A, Orfi L, Vántus T, Kéri G. Discovery and biological evaluation of novel dual EGFR/c-Met inhibitors. ACS Med Chem Lett. 2014; 5:298–303. https://doi.org/10.1021/ml4003309. [PubMed].
22. Perez-Lopez ÁR, Szalay KZ, Türei D, Módos D, Lenti K, Korcsmáros T, Csermely P. Targets of drugs are generally, and targets of drugs having side effects are specifically good spreaders of human interactome perturbations. Sci Rep. 2015; 5:10182. https://doi.org/10.1038/srep10182. [PubMed].
23. Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, Atoyan R, Qu H, Yin L, Samson M, Zifcak B, Ma AW, DellaRocca S, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012; 18:4104–13. https://doi.org/10.1158/1078-0432.CCR-12-0055. [PubMed].
24. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011; 471:467–72. https://doi.org/10.1038/nature09837. [PubMed].
25. Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008; Chapter 10:11. [PubMed].
26. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004; 10:789–99. https://doi.org/10.1038/nm1087. [PubMed].
27. Kéri G, Székelyhidi Z, Bánhegyi P, Varga Z, Hegymegi-Barakonyi B, Szántai-Kis C, Hafenbradl D, Klebl B, Muller G, Ullrich A, Erös D, Horváth Z, Greff Z, et al. Drug discovery in the kinase inhibitory field using the Nested Chemical Library technology. Assay Drug Dev Technol. 2005; 3:543–51. https://doi.org/10.1089/adt.2005.3.543. [PubMed].
28. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27–55. https://doi.org/10.1016/0065-2571(84)90007-4. [PubMed].
29. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010; 70:440–46. https://doi.org/10.1158/0008-5472.CAN-09-1947. [PubMed].
30. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The Tao of myeloma. Blood. 2014; 124:1873–79. https://doi.org/10.1182/blood-2014-05-578732. [PubMed].
31. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335–48. https://doi.org/10.1038/nrc3257. [PubMed].
32. Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, Middha S, Asmann Y, Schmidt J, Braggio E, Keats JJ, Fonseca R, Bergsagel PL, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012; 120:1060–66. https://doi.org/10.1182/blood-2012-01-405977. [PubMed].
33. Margolin KA, Moon J, Flaherty LE, Lao CD, Akerley WL 3rd, Othus M, Sosman JA, Kirkwood JM, Sondak VK. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438). Clin Cancer Res. 2012; 18:1129–37. https://doi.org/10.1158/1078-0432.CCR-11-2488. [PubMed].
34. Manohar SM, Rathos MJ, Sonawane V, Rao SV, Joshi KS. Cyclin-dependent kinase inhibitor, P276-00 induces apoptosis in multiple myeloma cells by inhibition of Cdk9-T1 and RNA polymerase II-dependent transcription. Leuk Res. 2011; 35:821–30. https://doi.org/10.1016/j.leukres.2010.12.010. [PubMed].
35. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010; 9:2344–53. https://doi.org/10.1158/1535-7163.MCT-10-0324. [PubMed].
36. Lloyd AC, Obermüller F, Staddon S, Barth CF, McMahon M, Land H. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev. 1997; 11:663–77. https://doi.org/10.1101/gad.11.5.663. [PubMed].
37. Cai X, Zhai HX, Wang J, Forrester J, Qu H, Yin L, Lai CJ, Bao R, Qian C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010; 53:2000–9. https://doi.org/10.1021/jm901453q. [PubMed].
38. Wagner T, Brand P, Heinzel T, Krämer OH. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim Biophys Acta. 2014; 1846:524–38. https://doi.org/10.1016/j.bbcan.2014.07.010. [PubMed].
39. Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, Randolph S, Smeal T. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010; 9:1618–28. https://doi.org/10.1158/1535-7163.MCT-10-0034. [PubMed].
40. Koch DC, Jang HS, O’Donnell EF, Punj S, Kopparapu PR, Bisson WH, Kerkvliet NI, Kolluri SK. Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-β1. Oncogene. 2015; 34:6092–104. https://doi.org/10.1038/onc.2015.55. [PubMed].
41. Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, Bornmann W, Kantarjian H, Viallet J, Samudio I, Andreeff M. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res. 2008; 68:3413–20. https://doi.org/10.1158/0008-5472.CAN-07-1919. [PubMed].
42. Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008; 68:3421–28. https://doi.org/10.1158/0008-5472.CAN-07-5836. [PubMed].
43. Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer. 2014; 13:98. https://doi.org/10.1186/1476-4598-13-98. [PubMed].
44. Rende M, Pistilli A, Stabile AM, Terenzi A, Cattaneo A, Ugolini G, Sanna P. Role of nerve growth factor and its receptors in non-nervous cancer growth: efficacy of a tyrosine kinase inhibitor (AG879) and neutralizing antibodies antityrosine kinase receptor A and antinerve growth factor: an in-vitro and in-vivo study. Anticancer Drugs. 2006; 17:929–41. https://doi.org/10.1097/01.cad.0000224459.13651.fd. [PubMed].
45. Dixon KM, Lui GY, Kovacevic Z, Zhang D, Yao M, Chen Z, Dong Q, Assinder SJ, Richardson DR. Dp44mT targets the AKT, TGF-β and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br J Cancer. 2013; 108:409–19. https://doi.org/10.1038/bjc.2012.582. [PubMed].
46. Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013; 340:622–26. https://doi.org/10.1126/science.1234769. [PubMed].
47. Tsubuki S, Saito Y, Tomioka M, Ito H, Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996; 119:572–76. https://doi.org/10.1093/oxfordjournals.jbchem.a021280. [PubMed].
48. Halasi M, Pandit B, Gartel AL. Proteasome inhibitors suppress the protein expression of mutant p53. Cell Cycle. 2014; 13:3202–06. https://doi.org/10.4161/15384101.2014.950132. [PubMed].
49. Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, Orlowski RZ. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007; 110:3281–90. https://doi.org/10.1182/blood-2007-01-065888. [PubMed].
50. William AD, Lee AC, Goh KC, Blanchard S, Poulsen A, Teo EL, Nagaraj H, Lee CP, Wang H, Williams M, Sun ET, Hu C, Jayaraman R, et al. Discovery of kinase spectrum selective macrocycle (16E)-14-methyl-20-oxa-5,7,14,26-tetraazatetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8(27),9,11,16,21,23-decaene (SB1317/TG02), a potent inhibitor of cyclin dependent kinases (CDKs), Janus kinase 2 (JAK2), and fms-like tyrosine kinase-3 (FLT3) for the treatment of cancer. J Med Chem. 2012; 55:169–96. https://doi.org/10.1021/jm201112g. [PubMed].
51. Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011; 39:23–31. https://doi.org/10.3892/ijo.2011.1015. [PubMed].
52. You WK, Sennino B, Williamson CW, Falcón B, Hashizume H, Yao LC, Aftab DT, McDonald DM. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer Res. 2011; 71:4758–68. https://doi.org/10.1158/0008-5472.can-10-2527. [PubMed].
53. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013; 503:548–51. https://doi.org/10.1038/nature12796. [PubMed].
54. Forsyth T, Kearney PC, Kim BG, Johnson HW, Aay N, Arcalas A, Brown DS, Chan V, Chen J, Du H, Epshteyn S, Galan AA, Huynh TP, et al. SAR and in vivo evaluation of 4-aryl-2-aminoalkylpyrimidines as potent and selective Janus kinase 2 (JAK2) inhibitors. Bioorg Med Chem Lett. 2012; 22:7653–58. https://doi.org/10.1016/j.bmcl.2012.10.007. [PubMed].
55. Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, Luengo JI, Newlander KA, Parrish CA, Ridgers LH, Sarpong MA, Schmidt SJ, Van Aller GS, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010; 1:39–43. https://doi.org/10.1021/ml900028r. [PubMed].
56. Yu P, Laird AD, Du X, Wu J, Won KA, Yamaguchi K, Hsu PP, Qian F, Jaeger CT, Zhang W, Buhr CA, Shen P, Abulafia W, et al. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014; 13:1078–91. https://doi.org/10.1158/1535-7163.MCT-13-0709. [PubMed].
57. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010; 9:1956–67. https://doi.org/10.1158/1535-7163.MCT-09-1012. [PubMed].
58. Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, Robertson GP, Yan L, Rubin E, Yang JM. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011; 71:2654–63. https://doi.org/10.1158/0008-5472.CAN-10-2889. [PubMed].
59. Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008; 27:5511–26. https://doi.org/10.1038/onc.2008.246. [PubMed].
60. Wu X, Renuse S, Sahasrabuddhe NA, Zahari MS, Chaerkady R, Kim MS, Nirujogi RS, Mohseni M, Kumar P, Raju R, Zhong J, Yang J, Neiswinger J, et al. Activation of diverse signalling pathways by oncogenic PIK3CA mutations. Nat Commun. 2014; 5:4961. https://doi.org/10.1038/ncomms5961. [PubMed].
61. Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007; 6:3158–68. https://doi.org/10.1158/1535-7163.MCT-07-0444. [PubMed].
62. Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007; 17:316–22. https://doi.org/10.1016/j.cub.2006.12.037. [PubMed].
63. Yim H, Erikson RL. Plk1-targeted therapies in TP53- or RAS-mutated cancer. Mutat Res Rev Mutat Res. 2014; 761:31–39. https://doi.org/10.1016/j.mrrev.2014.02.005. [PubMed].
64. Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009; 5:100–07. https://doi.org/10.1038/nchembio.137. [PubMed].
65. Ibanez G, Shum D, Blum G, Bhinder B, Radu C, Antczak C, Luo M, Djaballah H. A high throughput scintillation proximity imaging assay for protein methyltransferases. Comb Chem High Throughput Screen. 2012; 15:359–71. https://doi.org/10.2174/138620712800194468. [PubMed].
66. Iida K, Itoh K, Kumagai Y, Oyasu R, Hattori K, Kawai K, Shimazui T, Akaza H, Yamamoto M. Nrf2 is essential for the chemopreventive efficacy of oltipraz against urinary bladder carcinogenesis. Cancer Res. 2004; 64:6424–31. https://doi.org/10.1158/0008-5472.CAN-04-1906. [PubMed].
67. Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012; 135: 9–43. https://doi.org/10.1038/nrclinonc.2012.15. [PubMed].