
Introduction
Few of the extensive number of mutations compiled for human tumors, since the advent of exome sequencing, have impacted our understanding of tumor biology like those in human diffuse low grade gliomas. These tumors represent a unique cancer subgroup largely because of a distinctive subset of variants, which includes isocitrate dehydrogenase 1/2 (IDH1/IDH2) [1–3]. These variants create a neomorphic enzyme that generates a metabolite, 2-hydroxyglutarate (2-HG), not normally detectable in the human brain [4]. The metabolite disrupts function of proteins involved in epigenetic modification and interrupts the differentiation process [5, 6]. Examination of other tumor types has revealed a role for IDH1/2 variants in the development of these cancers as well [7–9]. Yet, there is a clear need to further delineate the significance of these mutations with regard to the biology of tumors in general and particularly in the clinic.
Despite a less aggressive clinical course than other malignant gliomas, such as glioblastoma, the diffuse glioma subtype, oligodendroglioma, remains an incurable disease with a range of survival times after the primary diagnosis. Oligodendroglioma exhibits a high frequency of IDH1/2 variants, particularly in cases harboring the signature genetic event, 1p/19q codeletion [3]. The putative targets of 1p/19q codeletion, tumor suppressor genes FUBP1 and CIC, along with TERT promoter and IDH1/2 variants, generate a unique constellation of genetic aberrations which distinguish these tumors not only from other gliomas, but from most other human cancers [10–12]. However, our understanding of the biology of these tumors and the molecular mechanisms at play during therapy are extremely limited due until recently to the lack of appropriate models that reflect the human disease. Previously, we orthotopically engrafted a human oligodendroglioma in two successive implantations [13]. Other in vivo models of oligodendroglioma have been described, but only recently has a more detailed analysis of the underlying genetic alterations been reported [14–17].
In this work, a complete analysis of the exome sequence dataset from a recurrent primary oligodendroglioma (recurrent O2010) and the derived xenograft (xenograft1) was performed. The single nucleotide variants (SNVs) and insertions/deletions (InDels) detected were subsequently analyzed in DNA from the tumor at the primary diagnosis in 2005 (primary O2005) and a second-generation xenograft (xenograft2). A subset of potential driver mutations was identified, which provides insight into the initiating events in oligodendroglioma as well as a candidate tumor suppressor pathway on chromosome 4.
Results
Copy number changes and copy neutral loss of heterozygosity (cnLOH) in oligodendroglioma evolution
Recurrent O2010 and the derived xenograft (xenograft1) had been previously analyzed for gross chromosomal aberrations by array comparative genomic hybridization (CGH) [13]. Array CGH revealed the characteristic genomic profiles for oligodendroglioma, which included the 1p/19q codeletion as well as chromosome 4, 6q and 14q losses that are common in higher grade oligodendrogliomas [18]. The tumor genome at a gross level appeared to be highly stable upon passage through mice, and an increase in ploidy for chromosomes 1 and 19 was not evident based on fluorescence in situ hybridization analysis (Miletic, H., data not shown, [19]). One major difference was a gain of the chromosomal arm 4q in xenograft1, which could be the result of the outgrowth of a subclone that was either present but previously undetectable in recurrent O2010, or it represented the generation of a new clone altogether in the mouse.
A more cryptic form of genomic instability, copy neutral loss of heterozygosity (cnLOH), has been previously described in oligodendrogliomas. In cnLOH, the chromosomes appeared to be diploid but were homozygous, indicating loss with subsequent duplication of a remaining chromosome. Because the predominant clone in recurrent O2010 had lost 4q, a subclone would only be distinguishable if it retained heterozygosity at 4q. Sequencing datasets for recurrent O2010 and xenograft1 were therefore analyzed for copy number and regions of heterozygosity and homozygosity. Analysis of SNVs revealed regions of homozygosity correlating with chromosomal losses on 1p, 19q, 4, 6q, and 14q in recurrent O2010 as expected [13]. LOH was also observed on chromosomes with two copies, such as 9p and 12. The cnLOH at 9p was previously observed in oligodendroglioma and was associated with loss of CDKN2A expression in approximately 30% of the cases examined [20].
LOH mapped similarly in the xenograft across the genome, including the cnLOH at 9p and 12. Genomic sequencing also revealed cnLOH on 4q indicating that loss of one chromosome had occurred with subsequent duplication of the remaining arm (Figure 1; [21]). Thus whether the xenograft represented a subclone from the original tumor or a new variant that developed during the engraftment process cannot be differentiated. However, these regions of cnLOH were consistent with an underlying level of genomic instability that may promote tumor development through loss of wild type and subsequent duplication of chromosomal regions harboring mutated tumor suppressor genes or oncogenes.
![Deletion of chromosome 4 in recurrent O2010 (A) and copy neutral LOH 4q in xenograft1 (B). LOH and CNV analysis was performed using the CEQer software [21] on the whole exome sequencing data from both tumors and data from patient blood as the control. Shown is the Log2 ratio of LOH/AI data according to the formula: ratio = MaxCovAllele / (CovAllele1 + CovAllele2), whereas MaxCovAllele is the allele with the highest coverage. Small green dots indicate copy loss of CNV exons, and horizontal green bars highlight individual copy loss of CNV regions. Small yellow dots show copy neutral regions. Big blue circles indicate either a loss of heterozygosity (-1 copy loss), an allelic imbalance (copy gain) or a copy neutral imbalance. Big grey circles indicate a conserved heterozygosity.](https://oncotarget-cdn.atl1.digitaloceanspaces.com/article/26950/figure/1029792/26950-1029792-1-PB.png)
Figure 1: Deletion of chromosome 4 in recurrent O2010 (A) and copy neutral LOH 4q in xenograft1 (B). LOH and CNV analysis was performed using the CEQer software [21] on the whole exome sequencing data from both tumors and data from patient blood as the control. Shown is the Log2 ratio of LOH/AI data according to the formula: ratio = MaxCovAllele / (CovAllele1 + CovAllele2), whereas MaxCovAllele is the allele with the highest coverage. Small green dots indicate copy loss of CNV exons, and horizontal green bars highlight individual copy loss of CNV regions. Small yellow dots show copy neutral regions. Big blue circles indicate either a loss of heterozygosity (-1 copy loss), an allelic imbalance (copy gain) or a copy neutral imbalance. Big grey circles indicate a conserved heterozygosity.
Exome sequencing reveals a stable xenograft genome
We performed in-depth analysis of the exome sequencing datasets to compare the genomes of the primary tumor/xenograft pair (recurrent O2010/xenograft1) and to identify putative genes targeted by losses of chromosomes 4q, 6q, and 14q which typically occur in higher grade oligodendrogliomas [18].
Exome sequencing revealed 80 somatic variants, 73 single nucleotide variants (SNVs) and 7 small insertions and deletions (InDels) with potential effects on protein function in recurrent O2010. This number of variants ranks in a higher range for oligodendroglioma, but is in overall agreement with the number observed in tumors previously treated with radiation and chemotherapy, as in this case [10, 22, 23]. Of these, 71 SNVs and all 7 InDels were also present in the xenograft. The variants found in both included IDH1 (NM_005896: c.395G>A; R132H), CIC (NM_015125: c.4421T>G; p.Val1474Gly - missense), and FUBP1 (NM_003902:c.1307_1310delTAGA; p.Ile436fs), as previously reported. FUBP1 on chromosome 1p sustained a small deletion, which created a truncation/frameshift [13]. Variants in tumor associated genes not typically associated with oligodendroglioma were also found, such as missense variants in TET1 (NM_030625:c.4399G>A; p.Glu1467Lys) and TGFBR2 (NM_003242:c.426G>C; p.Glu142Asp as well as a frameshift variant in ARID1A (NM_006015:c. 2290_2291insC; p.Gln766fs), a gene that is also located at 1p36 and frequently mutated in endometriosis associated ovarian cancers [24]. Variants in several genes were observed on chromosomes exhibiting homozygosity and/or loss, including COL25A1, HS3ST1, NDST4, and ZCCHC4 on chromosome 4 and ATXN2, NUAK1, RASAL1 and ZC3H12 on chromosome 12. However, no variant or amplification was found in PDGFRα on 4q, a gene involved in the development of human oligodendroglioma, or in CDKN2A on 9p, one of the most frequently altered genes in diverse cancer types.
Only 7 novel variants were found in the xenografts. These variants could potentially be newly acquired, or they were either undetectable by our methods or in the tumor sections available. All together, these results indicate that the xenograft was genomically highly stable and reflects the genetic make-up of recurrent O2010.
Deep sequencing distinguishes potential initiating events from progression events
As recurrent O2010 had been treated, it was of interest to identify variants present before therapy, which could help to define new driver initiating versus progression or passenger mutations. In addition, whether variants in the xenograft were already present either in primary O2005 or recurrent O2010 was of interest in terms of potential selective advantages during engraftment.
Deep sequencing was performed for all variants with DNAs from the patient’s blood and all tumors. Deep sequencing confirmed the presence of 81/87 variants in recurrent O2010, while 6 variants identified originally in xenograft1 (out of the 7 by exome sequencing exclusively in xenograft1 detected ones) were not present in recurrent O2010 (Table 1), also not at low frequency (Figure 2).
Table 1: Variants exclusively found in xenografts using deep sequencing
| Position | Gene | Coding region change | Amino acid change | Allele frequency in xenograft1 | Allele frequency in xenograft2 | Effect |
| chr10: 69748523 | HERC4 | NM_015601:c. 1703T>C | NM_015601:p. Val568Ala | 47% | 48% | missense |
| chr1: 179497498 | AXDND1 | NM_144696.4:c. 2647C>T | NM_144696.4:p. Arg883* | 28% | 31% | stop gained |
| chr13: 52440005 | CCDC70 | NM_031290.2:c. 491G>A | NM_031290.2:p. Trp164* | 31% | 34% | stop gained |
| chr1: 52838968 | ORC1 | NM_001190819.1:c. 2456C>G | NM_001190819.1:p. Ser819Cys | 100% | 99% | missense |
| chr6: 161501997 | MAP3K4 | NM_005922.2:c. 2182G>A | NM_005922.2:p. Glu728Lys | 26% | 27% | missense |
| chr4: 26426323 | RBPJ | NM_015874:c. 706_707delAA | NM_015874:p. Lys236fs | 95% | 95% | frameshift |
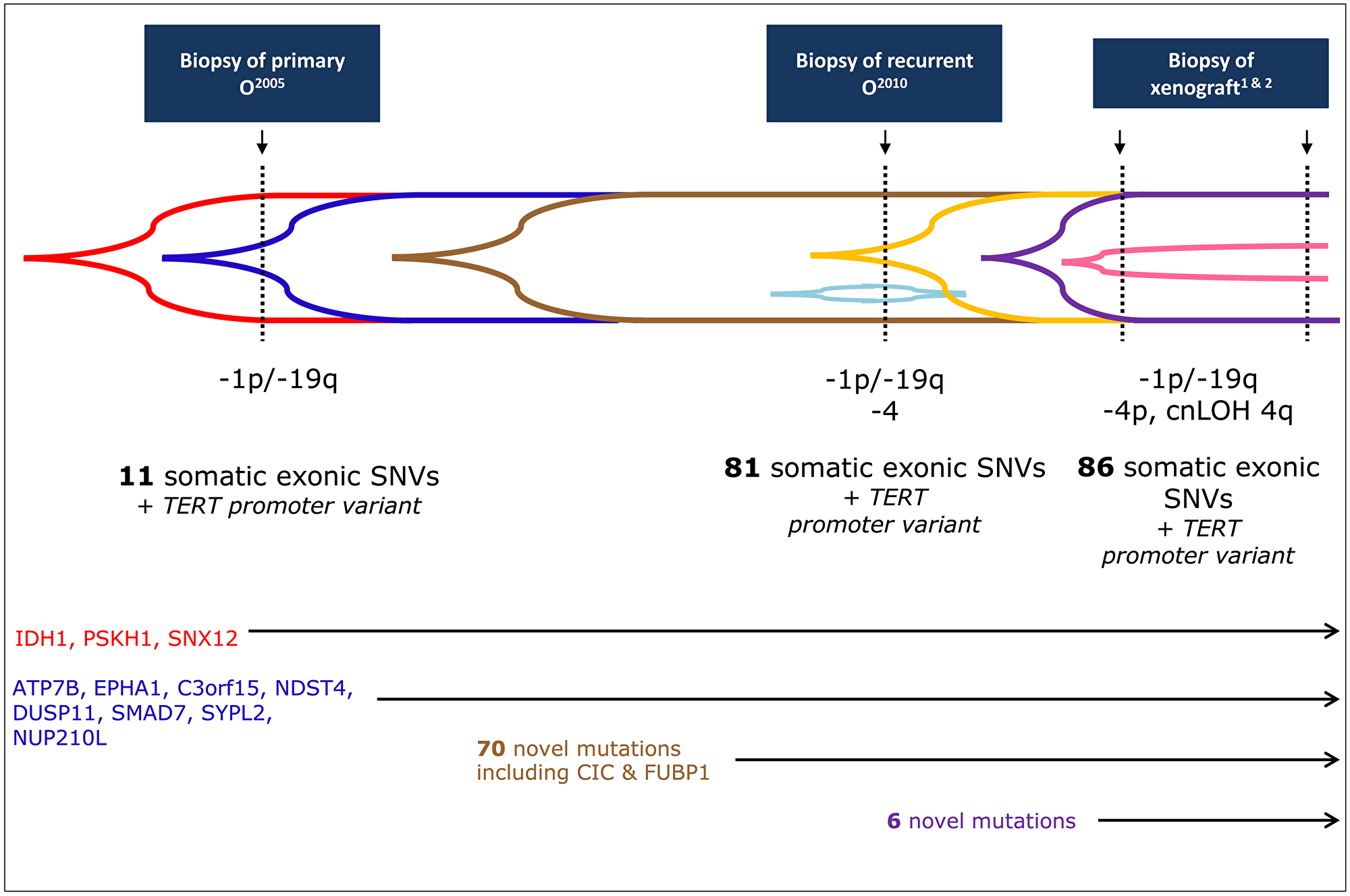
Figure 2: Codeletion of 1p/19q occurs before variants in FUBP1 and CIC. Timeline for the acquisition of all genetic events in primary O2005, recurrent O2010, and xenografts1&2, including array CGH. Exome sequencing was performed on recurrent O2010 and xenograft1, and these datasets were used to generate the set of variants (total = 87) to be further analyzed by deep sequencing. Deep sequencing detected a subset of these variants in primary O2005 (n = 11) and was used to generate an allele frequency for each variant in each tumor. Only 3/11 (red) variants, including IDH1 R132H (NM_005896:c.395G>A), were considered to be in the majority of the cells in primary O2005 (allele frequency ≥ 50%). The other 8 (blue) were detectable in primary O2005 but at lower frequencies (< 50%). In addition to these 11 variants, 70 novel variants occurred in the recurrent O2010 (total = 81, 70 new, distinguished by brown, gold, and light blue). Of these, 63 were present in the majority of the cells (brown). Included are variants in CIC (NM_015125:c.4421T>G) and FUBP1 (NM_003902:c.1307_1310delTAGA). A single variant, ARHGAP6 NM_013423:c.455G>C (light blue), was found only in recurrent O2010. In the xenografts, only 6 new variants (purple, ≥ 50% and pink, < 50%, perhaps latest changes) appeared in addition to those from recurrent O2010 (n = 86; 6 new, 1 lost). Additional targeted Sanger sequencing revealed the presence of a variant in TERT promoter in all tumors.
Only 11/87 somatic variants were also present in the primary O2005 (Table 2), so that the other mutations (n = 76) were considered to be due to progression and/or treatment. All variants found in primary O2005 were confirmed using Sanger sequencing. With the exception of one variant (one of two identified by exome sequencing), ARHGAP6 (NM_013423:c.455G>C; p.Arg152Pro) on the X chromosome, all variants observed in recurrent O2010 were confirmed in both xenograft1 and xenograft2. One small InDel, NUP210L (NM_207308:c.2072delT; p.Leu691fs), was also detected in the DNA from the patient’s blood, but with an allele frequency of only 1.1%. This result is possibly due to a sequencing error.
Table 2: Variants found in primary O2005 using deep sequencing
| Position | Gene | Coding region change | Amino acid change | Allele frequency | Effect |
| chr1: 110019535 | SYPL2 | NM_001040709:c. 392C>G | NM_001040709:p.Ala131Gly | 19.8% | missense |
| chr13: 52539048 | ATP7B | NM_000053:c.1829C>T | NM_000053:p.Pro610Leu | 3% | missense |
| chr16: 67943302 | PSKH1 | NM_006742:c.650G>A | NM_006742:p.Arg217Gln | 63% | missense |
| chr2: 209113112 | IDH1 | NM_005896:c.395G>A | NM_005896:p.Arg132His | 37% | missense |
| chr2: 73996452 | DUSP11 | NM_003584:c.575A>G | NM_003584:p.Asp192Gly | 20% | missense |
| chr3: 119462961 | C3orf15 | NM_033364:c.1820G>A | NM_033364:p.Arg607Gln | 11% | missense |
| chr4: 115754766 | NDST4 | NM_022569:c.2392C>G | NM_022569:p.Leu798Val | 17.5% | missense |
| chr7: 143095768 | EPHA1 | NM_005232:c.1262C>A | NM_005232:p.Ala421Asp | 10% | missense |
| chrX: 70280873 | SNX12 | NM_001256188:c.470G>A | NM_001256188:p.Arg157His | 56% | missense |
| chr1: 154067525 | NUP210L | NM_207308:c.2072delT | NM_207308:p.Leu691fs | 1.2% | frameshift |
| chr18: 46447974 | SMAD7 | NM_001190823:c.484_485insT | NM_001190823:p.Pro162fs | 8.3 | frameshift |
Of the genes already associated with the development of oligodendroglioma (IDH1/IDH2, CIC, and FUBP1), remarkably only the mutation in IDH1 (NM_005896:c.395G>A) was present in the primary O2005. FUBP1 and CIC variants were not present, even though 1p/19q codeletion had been detected by array CGH in primary O2005 (Supplementary Figure 1). Thus, FUBP1 and CIC variants appeared to occur secondary to the losses of 1p and 19q during tumor progression and were not necessary changes for tumor initiation in this case.
In addition to the FUBP1 frameshift variant, we also found a loss-of-function mutation of the known tumor suppressor gene ARID1A on 1p in the recurrent O2010 but not in primary O2005. In the TCGA dataset, alterations were detected in ARID1A in low grade glioma (SNV: 3.9%, 11/283; deletion: 0.4%, 1/283) and glioblastoma (SNV: 0.7%, 2/281) [25, 26]. As this variant emerged following treatment of the patient, the relationship to tumor development remains unclear. CIC, FUBP1, and ARID1A variants were all maintained through xenograft2 and in a homozygous state.
The TERT promoter is mutated
Most variants in the TERT promotor occurring in human gliomas are one of two changes: NM_198253.2:c.-124G>A (C228T) and NM_198253.2: c.-146G>A (C250T) located in a common SNV rs2853669 [12, 27, 28]. In oligodendrogliomas, a variant in the TERT promoter was found in 78% of cases (n = 45), and of these, 43% were the C250T variant [12]. To determine whether the TERT promoter also harbored a variant in our system, we performed targeted Sanger sequencing on DNA from each of the tumor samples. C250T (NM_198253.2: c.-146G>A; p.Cys250Thr) was found in all samples and was heterozygous.
Temporal acquisition of variants reconstructed based on allele frequency
Allele frequency based on the sequencing reads was also used to more precisely reconstruct the temporal acquisition of variants (Figures 2 and 3). We identified 10 variants that displayed a significantly lower frequency where the variant first appeared, than in the tumor that followed. Eight were variants that were already detectable in primary O2005: the missense variants in ATP7B (NM_000053:c.1829C>T; allele frequency, AF 3%); C3orf15 (NM_033364:c.1820G>A; AF, 10%); DUSP11 (NM_003584:c.575A>G; AF, 10%); EPHA1 (NM_005232:c.1262C>A; AF 10%); NDST4 (NM_022569:c.2392C>G; AF 20%); SYPL2 (NM_001040709:c.392C>G; AF 20%) and the frame shift mutations in NUP210L (NM_207308:c. 2072delT; AF 1%) and SMAD7 (NM_001190823:c.484_485insT; AF 10%). The remaining two variants initially appeared in recurrent O2010 and became predominant in xenograft1: ZAK (NM_016653:c.1618C>T; p.Gln540*; AF 20%; Figure 3) and the InDel in KPNA1 (NM_002264:c.179_181delAAG; p.lu60del; AF 20%). Only two variants, PSKH1 (NM_006742.2:c.650G>A; p.Arg217Gln), and SNX12 (NM_001256188:c.470G>A; p.Arg157His) were present in ≥ 50% of reads as early as primary O2005. Variants in these genes and IDH1 exhibited an allele frequency that remained stable across all tumor samples. They were considered to be heterozygous and uniformly distributed amongst tumor cells, indicating that they had occurred early in tumor development.
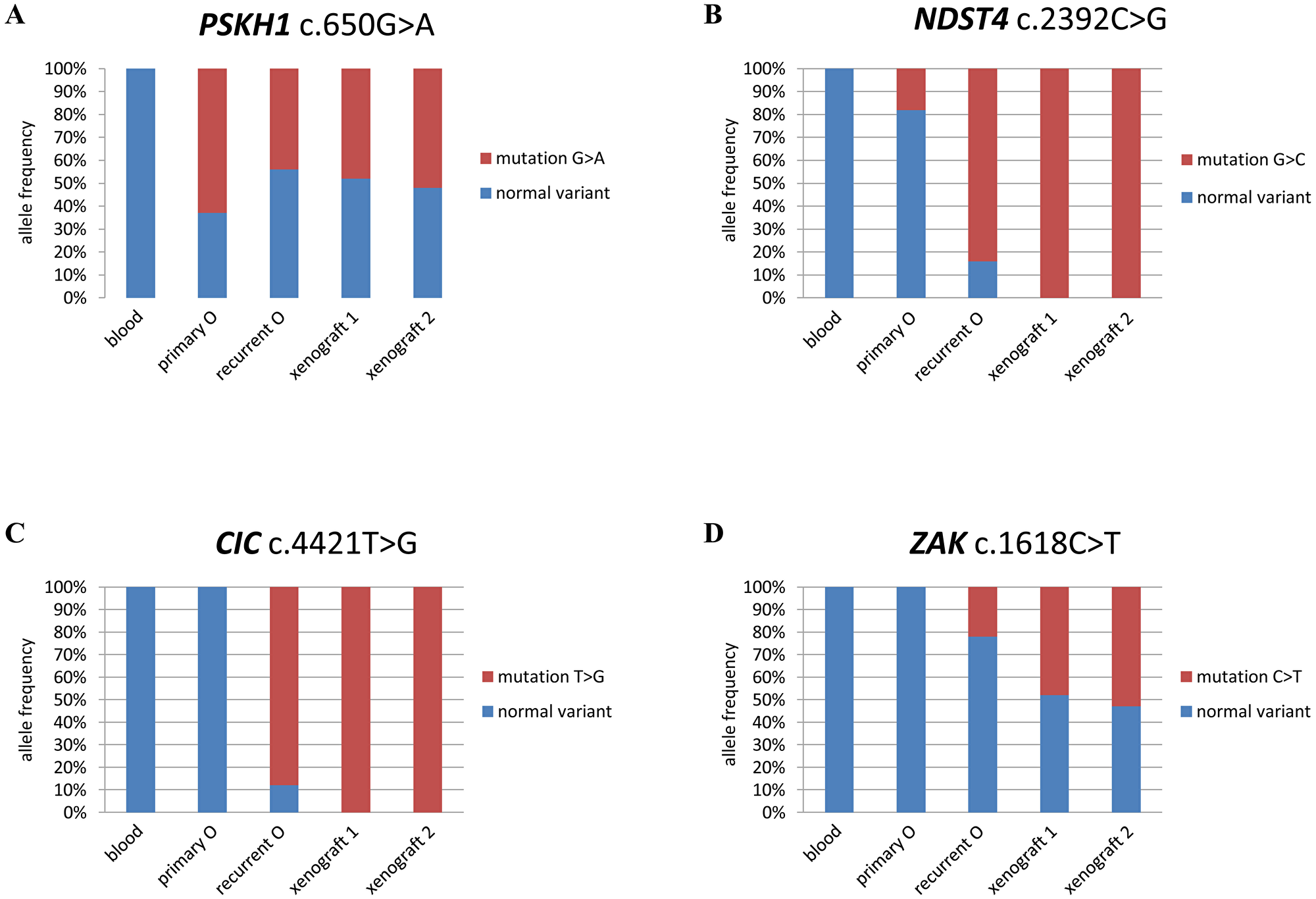
Figure 3: Allele frequency distinguishes between early and late variants. Graphic representation of allele frequency (% of reads for variant indicated) for variants in 4 genes in the tissue sample indicated. (A) PSKH1; (B) NDST4; (C) CIC; and (D) ZAK.
Variants that exhibited an allele frequency of < 50% were considered to have occurred later and in a subset of tumor cells. Any increase in allele frequency could be an indication that a subclone bearing the variant had been selected for. Examples of such variants were NDST4 (NM_022569:c.2392C>G; p.Leu798Val) and ZAK (NM_016653:c.1618C>T; p.Gln540*), which were first detected in primary O2005 and recurrent O2010, respectively (Figure 3). NDST4 represented an interesting case because of the changing status of chromosome arm 4q. In primary O2005, where 4q was diploid, the mutation was detected at a frequency of ~20%. In recurrent O2010, the allele frequency of the NDST4 variant was 80%. This result was consistent with selection of tumor cells harboring reduction to homozygosity at NDST4/chromosome arm 4q. The allele frequency of the variant increased to 100% in xenograft1 in the absence of contaminating non-neoplastic human cells. In xenograft1 with cnLOH on the 4q arm, a normal variant of NDST4 was no longer present, indicating that duplication of the remaining mutated allele had occurred.
NDST4 on 4q is not frequently mutated in human oligodendrogliomas
Chromosomal losses 4q and 14q have been associated with higher grade oligodendroglioma. NDST4 was a candidate tumor suppressor gene on chromosome 4 as the NDST4 previous variant was already detectable in the primary tumor and the wild-type allele was lost in recurrent O2010, indicating a loss of function mechanism. To investigate if alterations in NDST4 might play a role in the development of oligodendroglioma, we performed Sanger sequencing to analyze all coding exons of the gene in our own cohort of primary oligodendrogliomas (n = 15). We found no variants in coding regions of NDST4 in this tumor set; only known polymorphisms and variants in intronic sequences were observed. However, only 3 cases in our cohort had sustained loss of chromosome 4. In TCGA datasets, 8 missense mutations and deletions in NDST4 were found in lower grade gliomas, while 17 missense and 1 truncating variant were found in glioblastoma [25, 26]. All together the somatic mutation frequency was 0.6% [25, 26]. Thus, although NDST4 remains a gene of interest in tumor development and indeed might play a role in the progression from primary O2005 to recurrent O2010, NDST4 mutation seems to be a rare event in oligodendroglioma.
DISCUSSION
Exome sequencing coupled with array CGH performed on a rare oligodendroglioma xenograft revealed a highly stable genome relative to the recurrent human tumor from which it was derived. Using deep sequencing (and targeted Sanger sequencing for the TERT promoter), we identified a set of early and potential driver variants (n = 12) already present in genes in primary O2005 which included IDH1 and TERT, but surprisingly not FUBP1 and CIC. Allele frequencies for 10 of these 12 variants was ≤ 50%, but increased for some variants indicating possible selection of tumor cell subpopulations in response to normal progression, treatment, or engraftment. Finally, the increase in allele frequency of the NDST4 variant in the tumor series was consistent with the selection of a tumor suppressor gene.
The overall number of variants (n = 82 - including TERT) in recurrent O2010 was significantly higher compared to primary O2005 (n = 12 - including TERT) and the average for primary oligodendrogliomas, but was consistent with cases from patients who had undergone treatment [10, 11]. All variants were also detected in xenograft1, along with 6 new changes including 5 SNVs and 1 small InDel. These 6 changes in xenograft1 might have spontaneously occurred during progression in xenograft1 or were previously undetectable but selected for during engraftment into the mouse. Only one variant in ARHGAP6 was exclusively found in recurrent O2010 but at a low allele frequency (12%).
Through the deep sequencing analysis emerged a sequence of mutational events that may be more generally relevant to the development of a subset of human oligodendrogliomas. FUBP1 and CIC variants first appeared in recurrent O2010. Thus, the scenario for inactivation of these tumor suppressor genes in some cases may be loss of heterozygosity before selection of cells with variants. It has been previously suggested that haploinsufficiency at 1p is tumor promoting [29]. In oligodendrogliomas, haploinsufficiency occurs with a high frequency in the context of an IDH1/IDH2 variant [3]. In such cases, loss in combination with epigenetic modification may sufficiently knock down expression of key target genes to promote uncontrolled proliferation and/or inhibit differentiation. Variants in FUBP1 and CIC may further stabilize the phenotype established by haploinsufficiency along with epigenetic changes.
In support of this scenario, results from several studies indicate that concurrent variants in FUBP1 and CIC appear infrequently. In two early studies, only ~10% of human oligodendrogliomas with codeletion of 1p and 19q were found to harbor simultaneous variants [10, 22]. Our own study from 2013 corroborated these results. FUBP1 and CIC were both mutated in only 12% (2/17) of oligodendrogliomas examined, and all had sustained a variant in IDH1 [30]. In the dataset from the TCGA, simultaneous variants occurred in a higher proportion of tumors, 24% (13/53) [25, 26]. However, while we used deep sequencing in our analysis, there are other possibilities that might account for the low frequency of simultaneous mutations in primary tumors, such as technique and sample bias due to tumor heterogeneity or small undetectable subclones.
Although ultimately it is the accumulation of genetic events that results in cancer, the putative timeline of our oligodendroglioma might provide insight for the contribution of IDH, TERT and 1p/19 codeletion to the development of oligodendroglioma. First, the results might support studies indicating that IDH mutation occurs early in a stem/progenitor cell type with subsequent p53 mutations or 1p/19 codeletion defining the path to astrocytoma or oligodendroglioma. IDH mutation appears to be a gatekeeper, triggering uncontrolled proliferation which renders cells vulnerable to additional tumor promoting genetic events [31]. Second, 1p/19q codeletion might drive the development of oligodendroglioma through mechanisms other than loss of FUBP1 and CIC [32]. Examination of expression datasets for oligodendroglioma have led to the identification of a network of genes in the codeleted region of 1p/19q that may promote the development of oligodendroglioma. Finally, we can wonder whether TERT or IDH mutations have a more direct impact on genomic instability. TERT promoter mutations have been shown to extend cellular lifespan but do not necessarily prevent shortening of telomeres [33]. These shorter telomeres however later in tumor development contribute to genomic instability. A second possibility is mutation of the IDH gene itself. IDH mutation promotes epigenetic modification of DNA, and methylation has been implicated in chromosomal instability, although more generally through global hypomethylation [34]. The fact that the incidence of IDH mutation is so high in 1p/19q codeleted oligodendrogliomas and that cnLOH is also commonly observed further supports this possible role for IDH mutation in cancer [3, 20].
Finally, deep sequencing performed on primary O2005 importantly also enabled us to distinguish potential driver from passenger variants in recurrent O2010. NDST4 on chromosome 4q emerged as a potential tumor suppressor gene as it was one of the few genes with a variant already detectable in primary O2005. Unlike IDH1 or PSKH1, this variant was probably present only in a subpopulation of cells in primary O2005 which later became predominant. The protein is involved in heparan sulfate synthesis, and the distribution of heparan sulfate is altered in tumorigenesis. Heparan sulfate synthesis has also been shown to be necessary for growth factor activity. Furthermore, heparan sulfates contribute to cell-cell and cell-extracellular matrix interactions forming barriers for tumor invasion [35]. Interestingly, two additional genes mutated in recurrent O2010, EXTL3 (chromosome 8) and HS3ST1 (chromosome 4), are also involved in heparan sulfate synthesis. These genes are altered in many cancers and like NDST4, frequent alterations are present in skin cutaneous melanoma and lung cancer [25, 26]. Therefore, alteration of heparan sulfate synthesis could be important in tumor biology.
In conclusion, we have used exome sequencing datasets from a unique series of a recurrent human tumor and the derived xenografts to construct an evolutionary genetic profile. Although the primary oligodendroglioma exhibited 1p/19q codeletion, the variants detected did not include changes in FUBP1 and CIC. Only a few variants, in addition to IDH1 R132H and TERT C250T, were observed in the primary tumor that gained predominance and might represent interesting candidate genes for oligodendroglioma development. Because mutations in several of these genes are rare in oligodendrogliomas, and human cancer in general, some of these variants may simply represent hits in the so-called “hills” of the oligodendroglioma genome landscape [31]. Functional experiments are therefore required to determine whether the genes identified in this study are in fact true driver genes in human cancer.
Materials and Methods
Ethics statement
Patient material was obtained from surgeries performed at the Haukeland University Hospital (Bergen, Norway). Written consent was obtained from all patients to use tissues and clinical data for research purposes. Procedures were approved for the project (project number 013.09) by the Regional Ethics Committee (Bergen, Norway). All animal protocols were approved by authorities in an AAALAC accredited facility at the Haukeland University Hospital and in accordance with the national regulations of Norway (project numbers 2010 2658 and 2011 3079).
Patient and xenograft tumors
Tissue was obtained from a recurrent primary tumor from 2010 (oligodendroglioma WHO grade III; recurrent O2010), and implanted as previously described [13]. Xenograft tissue was harvested at the first implantation to obtain the first-generation xenograft (xenograft1). Cells from this tumor were reimplanted to obtain the second-generation xenograft (xenograft2). Portions of all three tumors were harvested and stored at –80°C for molecular analysis. Tissue from the original tumor (oligodendroglioma WHO grade III; primary O2005) was obtained from archived paraffin embedded surgical material in 2005.
DNA isolation
DNAs were isolated from patient material that had been immediately frozen in liquid nitrogen subsequent to surgery. Frozen sections were prepared, and every fifteenth section was stained with H&E to ensure > 60% tumor cell content. As a control, DNA was obtained from frozen patient blood (–20°C) by isolating nuclei with Buffer C1 from the Genomic DNA Buffers Kit (Qiagen; Germantown, MD, USA). Samples were treated with proteinase K overnight at 50°C in ATL buffer (Qiagen) and then DNAse free RNAseA (Fermentas/Thermo Fisher Scientific; Waltham, MA, USA) for 5 min at room temperature. Reactions were extracted with phenol:chloroform:isoamyl alcohol 25:24:1 saturated with 10 mM tris, pH 8.0, 1 mM EDTA (Sigma; St. Louis, MO, USA) and precipitated in 2.5M NH4OAc and 2.5 volumes of 100% ethanol. DNAs were resuspended in Nuclease-Free water (Qiagen). DNA was extracted from FFPE tissue samples using the QIAamp DNA FFPE Tissue Kit (Qiagen) according to the manufacturer’s protocol.
Exome sequencing
Agilent SureSelect target enrichment kit (SureSelect Human All Exon 50 Mb kit, Agilent; Santa Clara, CA, USA) was used for exome library construction according to the manufacturer’s protocol. Captured DNA libraries were sequenced on the Illumina HiSeq2000 platform at a coverage of 100× (Illumina; San Diego, CA, USA).
Data analysis
Briefly, the genomes derived from human tissues were aligned against the human genome hg19. Analysis of the sequencing reads from the xenografts underwent a cleaning process to eliminate contaminating mouse reads. Two alignment steps were performed. In the first step, the sequencing reads were aligned against the human (hg19) and the mouse (mm10) genomes separately. Reads with better mapping results (higher mapping quality) in the mouse genome than in the human genome were deleted. In the second step, surviving reads were aligned against a chimeric genome, which was obtained through the concatenation of the human and mouse genomes. The resulting BAM file was then separated into those reads that mapped to the human or the mouse genome using SAMtools (version 0.1.18) [36, 37]. The cleaning process was therefore accomplished by cross-referencing reads with a high mapping quality in the other set and eliminating them.
The pipeline for all the samples was the following: alignment using BWA (version 0.7.2) [38], and post-alignment processing using GATK [39] for local realignment, duplicate removal and score recalibration, and finally variant calling between each pair of samples using VarScan (version 2.3.2) [40]. SAMtools (version 0.1.18) was used to create the needed pileup file [36, 37]. False positives were eliminated by a false positive filtering (strandedness (0.1); maximum mismatch quality sum (100); minimum average distance to effective 3’end (0.1) [40].
After a statistical filtering for somatic variants (for details see Supplementary Material), we selected for mutation type (non-synonymous substitution, start and stop codon gain or loss, and frame-shifts), to focus on variants with potential effects at the protein level. All called variants were hand-curated, which included an evaluation using the Integrative Genomics Viewer provided by the Broad Institute [41, 42], applying the BLAT - BLAST- like Alignment Tool [43] of the UCSC Genome Browser [44], and including additional information from the UCSC Genome Browser, such as pseudogenes and self-chain or repetitive regions. Variants were determined to be damaging/deleterious and disease causing by several prediction software programs, including PolyPhen-2, Mutation Taster, Mutation Assessor, PROVEAN and SIFT [45–51]. Details of the variant selection process are described in Supplementary Materials.
PCR and targeted sequencing
All variants were additionally verified using Sanger and/or deep sequencing. Primers were designed against regions of interest using Primer3 software (version 4.1.0) [52, 53]. Primers and conditions used are shown in Supplementary Data. A first/preliminary analysis of the xenograft sequencing data revealed a large number of novel variants that were suspected to be due to contamination with mouse tissue rather than novel variants occurring during engraftment. Therefore, for validation of variants, we designed all primers to bind to human sequences only. Validation of primers was performed on normal human control DNA obtained from blood and DNA from mouse tissue to verify that primers would only yield a PCR-product in human but not mouse DNA.
For ultra-deep sequencing, PCR was carried out using a proofreading polymerase (Phusion® Hot Start Flex DNA Polymerase; New England Biolabs; Ipswich, MA, USA) according to the manufacturer’s instructions under the following cycling conditions: initial denaturation step: 98°C, 30 sec; amplification step (34×): 98°C, 10 sec, 61.5 – 66.5°C, 30 sec, 72°C, 30 sec; final extension: 72°C, 10 min. The correct size of PCR products was confirmed by agarose gel electrophoresis (2.0% agarose impregnated with GelRed; 1 h, 110 V) and purified from contaminating nucleotides and primers by PEG precipitation. Libraries were sequenced to an average coverage of 36,000× on an Illumina HiSeq2000 Next Generation Sequencer (Illumina; San Diego, CA, USA). Results were analyzed using CLC Genomics Workbench Version 7.5. Reads were aligned to the human reference genome (hg19) and variants were called using the Basic Variant Detection algorithm (minimum coverage 1000×; minimum count 2, minimum frequency 1%; the base quality filter was used with default values, no direction or position filters were applied).
Variants found in primary O2005 were validated using Sanger’s dye terminator method on an ABI 3130x1 DNA Analyzer (Applied Biosystems/Thermo Fisher Scientific). Products were cleaned with rAPid alkaline phosphatase (Roche Diagnostics GmbH; Mannheim, Germany) and exonuclease I treatment (New England Biolabs) at 37°C for 1 h followed by inactivation at 80°C for 20 min.
Array comparative genomic hybridization
Array CGH was performed on Agilent’s SurePrint G3 Human CGH Microarray Kit 2x400K (Design ID021850, Agilent, Santa Clara, CA, USA) according to the manufacturer’s protocol. Scanning of the hybridized arrays was carried out using the Agilent High-Resolution Microarray scanner. Raw data were processed by the Feature Extraction 9.5 (Agilent) software and normalization was performed using the default settings. Feature Extraction files were imported into Genomic Workbench 7.0 (Agilent Technologies) for visualization and analysis. After diploid centralization and GC correction, aberrations were called using the ADM2 algorithm with a threshold setting of 20, centralization on with threshold of 25 and an aberration filter min Probes = 5 and minAvgAbsLogRatio = 0.35 for amplifications and deletions [13].
Abbreviations
AF: allele frequency; CGH: comparative genomic hybridization; cnLOH: copy neutral loss of heterozygosity; InDel: insertions/deletions; LOH: loss of heterozygosity; O: oligodendroglioma; SNV: single nucleotide variants..
ACKNOWLEDGMENTS
The authors are deeply indebted to the patients who generously donated the tissues necessary to perform this study.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
The study was supported by the Norwegian Cancer Society (PK01-2009-0349 to JMN), Deutsche Krebshilfe (70112014 to LK, BK), Helse-Sørøst (2017082 to PCH), Helse-Vest, the Haukeland University Hospital, Fonds National Recherche, Luxembourg, and the Centre de Recherche Public de la Santé, Luxembourg.
References
1. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321:1807–12. https://doi.org/10.1126/science.1164382. [PubMed].
2. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008; 116:597–602. https://doi.org/10.1007/s00401-008-0455-2. [PubMed].
3. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009; 360:765–73. https://doi.org/10.1056/NEJMoa0808710. [PubMed].
4. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009; 462:739–44. https://doi.org/10.1038/nature08617. [PubMed].
5. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010; 18:553–67. https://doi.org/10.1016/j.ccr.2010.11.015. [PubMed].
6. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012; 483:474–8. https://doi.org/10.1038/nature10860. [PubMed].
7. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, Eskandarpour M, Presneau N, Hogendoorn PC, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011; 224:334–43. https://doi.org/10.1002/path.2913. [PubMed].
8. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S, Aston W, Green CL, Gale RE, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet. 2011; 43:1262–5. https://doi.org/10.1038/ng.994. [PubMed].
9. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009; 361:1058–66. https://doi.org/10.1056/NEJMoa0903840. [PubMed].
10. Bettegowda C, Agrawal N, Jiao Y, Sausen M, Wood LD, Hruban RH, Rodriguez FJ, Cahill DP, McLendon R, Riggins G, Velculescu VE, Oba-Shinjo SM, Marie SK, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011; 333:1453–5. https://doi.org/10.1126/science.1210557. [PubMed].
11. Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J, Birol I, Chesnelong C, Chiu R, Chuah E, Corbett R, Docking R, Firme M, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012; 226:7–16. https://doi.org/10.1002/path.2995. [PubMed].
12. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA
13. Klink B, Miletic H, Stieber D, Huszthy PC, Campos Valenzuela JA, Balss J, Wang J, Schubert M, Sakariassen PO, Sundstrom T, Torsvik A, Aarhus M, Mahesparan R, et al. A novel, diffusely infiltrative xenograft model of human anaplastic oligodendroglioma with mutations in FUBP1, CIC, and IDH1. PLoS One. 2013; 8:e59773. https://doi.org/10.1371/journal.pone.0059773. [PubMed].
14. Kelly JJ, Blough MD, Stechishin OD, Chan JA, Beauchamp D, Perizzolo M, Demetrick DJ, Steele L, Auer RN, Hader WJ, Westgate M, Parney IF, Jenkins R, et al. Oligodendroglioma cell lines containing t(1;19)(q10;p10). Neuro Oncol. 2010; 12:745–55. https://doi.org/10.1093/neuonc/noq031. [PubMed].
15. Wakimoto H, Tanaka S, Curry WT, Loebel F, Zhao D, Tateishi K, Chen J, Klofas LK, Lelic N, Kim JC, Dias-Santagata D, Ellisen LW, Borger DR, et al. Targetable signaling pathway mutations are associated with malignant phenotype in IDH-mutant gliomas. Clin Cancer Res. 2014; 20:2898–909. https://doi.org/10.1158/1078-0432.CCR-13-3052. [PubMed].
16. Srikanth M, Kim J, Das S, Kessler JA. BMP signaling induces astrocytic differentiation of clinically derived oligodendroglioma propagating cells. Mol Cancer Res. 2014; 12:283–94. https://doi.org/10.1158/1541-7786.MCR-13-0349. [PubMed].
17. Navis AC, Niclou SP, Fack F, Stieber D, van Lith S, Verrijp K, Wright A, Stauber J, Tops B, Otte-Holler I, Wevers RA, van Rooij A, Pusch S, et al. Increased mitochondrial activity in a novel IDH1-R132H mutant human oligodendroglioma xenograft model: in situ detection of 2-HG and α-KG. Acta Neuropathol Commun. 2013; 1:18. https://doi.org/10.1186/2051-5960-1-18. [PubMed].
18. Kitange G, Misra A, Law M, Passe S, Kollmeyer TM, Maurer M, Ballman K, Feuerstein BG, Jenkins RB. Chromosomal imbalances detected by array comparative genomic hybridization in human oligodendrogliomas and mixed oligoastrocytomas. Genes Chromosomes Cancer. 2005; 42:68–77. https://doi.org/10.1002/gcc.20108. [PubMed].
19. Nigro JM, Takahashi MA, Ginzinger DG, Law M, Passe S, Jenkins RB, Aldape K. Detection of 1p and 19q loss in oligodendroglioma by quantitative microsatellite analysis, a real-time quantitative polymerase chain reaction assay. Am J Pathol. 2001; 158:1253–62. https://doi.org/10.1016/S0002-9440(10)64076-X. [PubMed].
20. Idbaih A, Ducray F, Dehais C, Courdy C, Carpentier C, de Bernard S, Uro-Coste E, Mokhtari K, Jouvet A, Honnorat J, Chinot O, Ramirez C, Beauchesne P, et al, and POLA Network. SNP array analysis reveals novel genomic abnormalities including copy neutral loss of heterozygosity in anaplastic oligodendrogliomas. PLoS One. 2012; 7:e45950. https://doi.org/10.1371/journal.pone.0045950. [PubMed].
21. Piazza R, Magistroni V, Pirola A, Redaelli S, Spinelli R, Redaelli S, Galbiati M, Valletta S, Giudici G, Cazzaniga G, Gambacorti-Passerini C. CEQer: a graphical tool for copy number and allelic imbalance detection from whole-exome sequencing data. PLoS One. 2013; 8:e74825. https://doi.org/10.1371/journal.pone.0074825. [PubMed].
22. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012; 3:709–22. https://doi.org/10.18632/oncotarget.588. [PubMed].
23. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014; 343:189–93. https://doi.org/10.1126/science.1239947. [PubMed].
24. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363:1532–43. https://doi.org/10.1056/NEJMoa1008433. [PubMed].
25. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088. [PubMed].
26. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095. [PubMed].
27. Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, Costello JC, Theodorescu D, Cech TR. Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015; 347:1006–10. https://doi.org/10.1126/science.1260200. [PubMed].
28. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013; 4:2185. https://doi.org/10.1038/ncomms3185. [PubMed].
29. Henrich KO, Schwab M, Westermann F. 1p36 tumor suppression--a matter of dosage? Cancer Res. 2012; 72:6079–88. https://doi.org/10.1158/0008-5472.CAN-12-2230. [PubMed].
30. Eisenreich S, Abou-El-Ardat K, Szafranski K, Campos Valenzuela JA, Rump A, Nigro JM, Bjerkvig R, Gerlach EM, Hackmann K, Schrock E, Krex D, Kaderali L, Schackert G, et al. Novel CIC point mutations and an exon-spanning, homozygous deletion identified in oligodendroglial tumors by a comprehensive genomic approach including transcriptome sequencing. PLoS One. 2013; 8:e76623. https://doi.org/10.1371/journal.pone.0076623. [PubMed].
31. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA
32. Gladitz J, Klink B, Seifert M. Network-based analysis of oligodendrogliomas predicts novel cancer gene candidates within the region of the 1p/19q co-deletion. Acta Neuropathol Commun. 2018; 6:49. https://doi.org/10.1186/s40478-018-0544-y. [PubMed].
33. Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, Ryu J, Darzacq X, Bastian BC, Hockemeyer D. Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science. 2017; 357:1416–20. https://doi.org/10.1126/science.aao0535. [PubMed].
34. Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003; 300:455. https://doi.org/10.1126/science.1083557. [PubMed].
35. Tzeng ST, Tsai MH, Chen CL, Lee JX, Jao TM, Yu SL, Yen SJ, Yang YC. NDST4 is a novel candidate tumor suppressor gene at chromosome 4q26 and its genetic loss predicts adverse prognosis in colorectal cancer. PLoS One. 2013; 8:e67040. https://doi.org/10.1371/journal.pone.0067040. [PubMed].
36. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011; 27:2987–93. https://doi.org/10.1093/bioinformatics/btr509. [PubMed].
37. Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–95. https://doi.org/10.1093/bioinformatics/btp698. [PubMed].
38. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–9. https://doi.org/10.1093/bioinformatics/btp352. [PubMed].
39. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303. https://doi.org/10.1101/gr.107524.110. [PubMed].
40. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:568–76. https://doi.org/10.1101/gr.129684.111. [PubMed].
41. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013; 14:178–92. https://doi.org/10.1093/bib/bbs017. [PubMed].
42. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29:24–6. https://doi.org/10.1038/nbt.1754. [PubMed].
43. Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002; 12:656–64. https://doi.org/10.1101/gr.229202. [PubMed].
44. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. https://doi.org/10.1101/gr.229102. [PubMed].
45. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–9. https://doi.org/10.1038/nmeth0410-248. [PubMed].
46. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11:361–2. https://doi.org/10.1038/nmeth.2890. [PubMed].
47. Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007; 8:R232. https://doi.org/10.1186/gb-2007-8-11-r232. [PubMed].
48. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118. https://doi.org/10.1093/nar/gkr407. [PubMed].
49. Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012; 7:e46688. https://doi.org/10.1371/journal.pone.0046688. [PubMed].
50. Choi Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB ’12). 2012; 414–7. https://doi.org/10.1145/2382936.2382989. Available from: http://provean.jcvi.org/downloads/provean.acmbcb.pdf.
51. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073–81. https://doi.org/10.1038/nprot.2009.86. [PubMed].
52. Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007; 23:1289–91. https://doi.org/10.1093/bioinformatics/btm091. [PubMed].
53. Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3 - new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. https://doi.org/10.1093/nar/gks596. [PubMed].