
Introduction
Carcinosarcomas (CS) of the female genital tract are rare but very aggressive biphasic neoplasms composed of a mixture of carcinomatous (malignant epithelial) and sarcomatous (malignant mesenchymal) components [1]. CS can arise in different organs of the female reproductive tract but are mostly seen in the uterus, where they account for less than 3% of all uterine malignancies [2, 3], and in the ovaries, where they account for 5% of ovarian cancers [4].
Uterine carcinosarcomas (UCS) and ovarian carcinosarcomas (OCS) are usually diagnosed in postmenopausal women at a median age of 65 years, frequently are at advanced stage when detected, and carry a poor prognosis [3]. 5-year survival rates have been reported at 50% at the early stages but only 10% for stage IV CS [5, 6].
Data on molecular genetic alterations, gene expression status, and epigenetic profiles of UCS and OCS are scarce and the few studies reported are based on small numbers of tumours [7–9]. Mutations of the tumour protein gene (TP53) are assumed to be the most frequent alteration, observed in 50% of analysed tumours [7, 10, 11]. Other mutations, reported at lower frequencies, affect the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha gene (PI3K3CA), the ki-ras2 kirsten rat sarcoma viral oncogene homolog (KRAS), the catenin beta 1 gene (CTNNB1), and the neuroblastoma RAS viral (V-Ras) oncogene homolog (NRAS) gene [7, 8, 12].
Dysregulation of chromatin remodelling genes has been shown in CS indicating their importance in CS tumourigenesis [7]. In UCS, the genes involved in chromatin modification include those encoding AT-rich interactive domain-containing proteins (ARID1A and ARID1B), histone methyltransferase mixed-lineage leukaemia protein 3 (MLL3), histone deacetylase modifier speckle-type POZ (SPOP), and chromatin assembly factor bromodomain adjacent to zinc finger domain 1A (BAZ1A), all of which are mutated at frequencies varying from 18% to 36% [7]. Moreover, genes involved in chromosome dynamics were also found mutated, including those encoding DNA binding proteins, BCL6 corepressor (BCOR) and CCCTC-binding factor (CTCF), histone acetyl transferase E1A binding protein P300 (EP300), epigenetic activator zinc finger homeobox 3 (ZFHX3), and the nucleosome remodeling chromo domain helicase DNA binding protein 4 (CHD4) [12]. Some of these genes, including BCOR and CHD4, have been identified as mutated also in OCS [8].
Because both UCS and OCS may carry mutations in the histone genes H2 and H3, mutations that may facilitate epithelial-mesenchymal transition (EMT), this has been proposed to lie at the heart of their role in sarcomatous transformation [8, 9]. However, since the genetic basis of these tumours still remains largely unexplored, we performed molecular genetic investigations hoping to gain more knowledge about the pathogenesis of this type of cancer.
To this aim we checked the mutation status of the isocitrate dehydrogenase 1 and 2 genes (IDH1 and IDH2), telomerase reverse transcriptase (TERT) gene, the proto oncogenes BRAF, HRAS, KRAS, and NRAS, the histone H3F3A, CTNNB1, and PIK3CA, and TP53 in a series of CS arising in the uterus and ovaries. We also investigated the methylation status of the promoter of O6-methylguanine-DNA methyltransferase gene (MGMT).
To obtain more insight into the role of chromatin regulation genes and their pathways, we analysed the expression status of the high mobility group AT-Hook genes (HMGA1 and HMGA2), the pseudogenes HMGA1P6 and HMGA1P7, and the fragile histidine triad (FHIT), lin-28 homolog A (LIN28A) and metastasis associated 1 (MTA1) genes, as well as these genes’ possible regulation by miRNAs such as let-7a, let-7d, miR26a, miR16, miR214, and miR30c.
Results
Mutation and methylation analyses
All tumours analysed for IDH1, IDH2, TERT, CTNNB1, BRAF, H3F3A, KRAS, HRAS, NRAS, PIK3CA, and TP53 mutation status gave informative results. Whereas no tumour showed a mutated sequence for IDH1, IDH2, TERT, BRAF, H3F3A, HRAS,NRAS or CTNNB, a few were found to be mutated in KRAS, PIK3CA, and/or TP53. An overview of the findings is shown in Table 1. We identified a c.175G>A KRAS mutation in one of 16 UCS (case 8; Table 1). PIK3CA mutations were found in five of 16 UCS but in none of the OCS. More specifically, a c.3073A>G mutation was detected in case 8, a c.1637A>G in case 9, a c.3140A>G in cases 11 and 16, and a c.1634A>G in case 12 (Table 1). TP53 was found mutated in 12 of 16 UCS (cases 1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 16, and 17; 75% of the uterine CS) and in three of ten OCS (cases 18, 19, and 22; 30%). Details about the TP53 mutations are listed in Table 1. The expression of aberrant TP53 was confirmed by immunohistochemistry (Figure 1).
Table 1: Mutation status of KRAS, CTNNB1, PIK3CA, and TP53 and TP53 protein expression
| Case/lab no | Diagnosis | KRAS | CTNNB1 | PIK3CA | TP53 | TP53 carcinoma | TP53 sarcoma |
|---|---|---|---|---|---|---|---|
| 1/03–113 | UCS | - | - | - | c.722C>T | aberrant + | aberrant + |
| 2/03–221 | UCS | - | - | - | c.383_388delCTGCCC | WT | aberrant + |
| 3/08–1637 | UCS | - | - | - | rs28934578 (ARG175HIS) | aberrant + | aberrant + |
| 4/03–684 | UCS | - | - | - | rs28934578 (ARG175HIS) | aberrant + | aberrant + |
| 5/03–1023 | UCS | - | - | - | c.722C>A | aberrant + | aberrant + |
| 6/08–521 | UCS | - | - | - | c.818G>A | aberrant + | missing |
| 7/05–1309 | UCS | - | - | - | - | ||
| 8/0992–160 | UCS | c.175G>A | - | c.3073A>G | c.817C>T | ||
| 9/1002–102 | UCS | - | - | c.1637A>G | c.844C>G | aberrant + | aberrant + |
| 10/1002–186 | UCS | - | - | - | c.794T>C | aberrant + | aberrant + |
| 11/00–701 | UCS* | - | - | c.3140A>G | c.817C>T | aberrant + | aberrant + |
| 12/02–819 | UCS* | - | - | c.1634A>G | - | WT | WT |
| 13/06–539 | OCS | - | - | - | - | aberrant – | aberrant – |
| 14/1002–356 | UCS | - | - | - | - | ||
| 15/02–873 | UCS | - | - | - | - | aberrant + | aberrant + |
| 16/01–73 | UCS | - | - | c.3140A>G | c.215C>G | ||
| 17/06–1577 | UCS | - | - | - | c.558T>A | aberrant - | aberrant + |
| 18/08–974 | OCS | - | - | - | c.503A>C | aberrant + | aberrant + |
| 19/009–90 | OCS | - | - | - | c.815T>G | aberrant + | aberrant – |
| 20/01–139 | OCS | - | - | - | - | aberrant + | aberrant + |
| 21/008–35 | OCS | - | - | - | - | WT | WT |
| 22/0992–0288 | OCS | - | - | - | c.393_395delCAA | aberrant + | aberrant + |
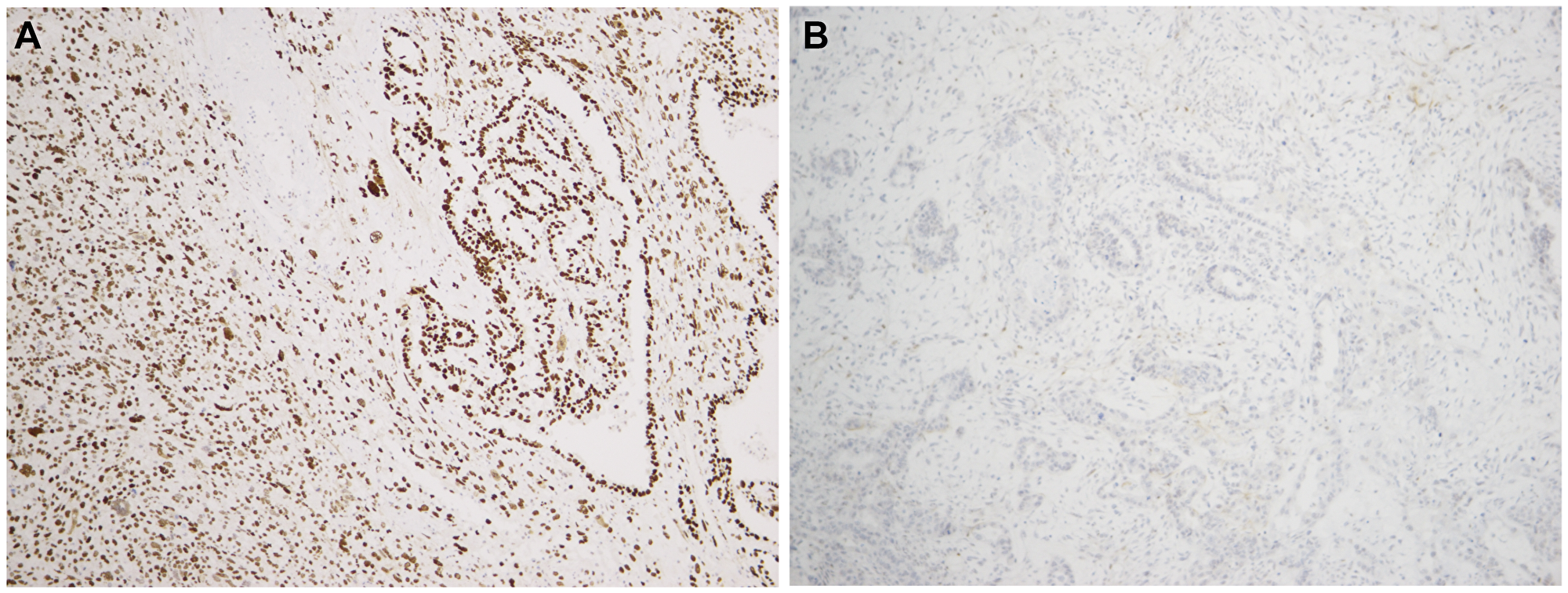
Figure 1: p53 immunostaining in two uterine carcinosarcomas showing the two aberrant patterns, i.e. diffuse strong expression and entirely negative expression in panels (A and B), respectively.
No MGMT promoter methylation was detected in the present series, suggesting that the gene is not involved in CS tumourigenesis.
Expression analyses
An overview of the expression status for the genes and miRNAs investigated is given in Tables 2 and 3. HMGA1 was found expressed in UCS and OCS (Figure 2A). HMGA1P6 was expressed in seven of 15 UCS and, at high levels, in all OCS (Figure 2B). HMGA1P7 was not expressed in UCS but was expressed in six of ten OCS (Figure 2B). HMGA2 was expressed at high levels in both uterine and ovarian CS (Figure 2C). FHIT was found normally expressed in both UCS and OCS (Figure 2D). LIN28A was found upregulated in six of 15 UCS and in most OCS (nine of ten) (Figure 2E). MTA1 was found overexpressed in UCS, whereas no substantial overexpression was identified in OCS (Figure 2F).
Table 2: Overview of the expression status of genes and miRNAs investigated in the CS
| Case/lab no | Histology | HMGA1 | HMGA2 | FHIT | LIN28A | HMGA1P6 | HMGA1P7 | MTA1 | Let-7a | Let-7d | miR26a | miR16 | miR214 | miR30c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/03–113 | UCS | ↑ | ↑ | ↑ | ↑ | ↑ | N/A | ↑ | ↓ | ↓ | - | - | ↓ | ↓ |
| 2/03–221 | UCS | ↑ | ↑ | ↑ | - | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | - | ↓ |
| 3/08–1637 | UCS | ↑ | ↑ | ↑ | ↑ | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 4/03–684 | UCS | ↑ | ↑ | ↑ | - | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 5/03–1023 | UCS | ↑ | ↑ | ↑ | ↑ | ↑ | N/A | ↑ | ↓ | ↓ | - | ↓ | ↓ | ↓ |
| 6/08–521 | UCS | ↑ | ↑ | ↑ | - | ↑ | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 7/05–1309 | UCS | ↑ | ↑ | ↑ | ↑ | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 8/0992–0160 | UCS | ↑ | ↑ | ↑ | - | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 9/1002–0102 | UCS | ↑ | ↑ | ↑ | - | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 10/1002–186 | UCS | ↑ | ↑ | ↑ | - | ↑ | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 11/00–701 | UCS* | ↑ | ↑ | ↑ | - | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 12/02–819 | UCS* | ↑ | ↑ | ↑ | ↑ | - | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 13/06–539 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | N/A | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | - |
| 17/06–1577 | UCS | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ | - |
| 18/08–974 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ | - |
| 19/09–90 | OCS | ↑ | ↑ | ↓ | - | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | - |
| 20/01–139 | OCS | ↑ | ↑ | ↓ | - | ↑ | N/A | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ |
| 21/08–35 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | - |
| 22/0992–0288 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ |
| 23/03–568 | UCS | ↑ | ↑ | ↑ | ↑ | ↑ | N/A | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ |
| 24/01–104 | UCS* | ↑ | ↑ | ↑ | - | ↑ | N/A | ↑ | ↓ | ↓ | - | - | ↓ | ↓ |
| 25/01–1056 | UCS* | ↑ | ↑ | ↑ | - | ↑ | N/A | ↑ | ↓ | ↓ | ↓ | - | ↓ | ↓ |
| 26/05–268 | OCS | ↑ | ↑ | ↓ | ↑ | - | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ |
| 27/05–1076 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | N/A | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ |
| 28/02–1150 | OCS | ↑ | ↑ | ↓ | ↑ | ↑ | N/A | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ |
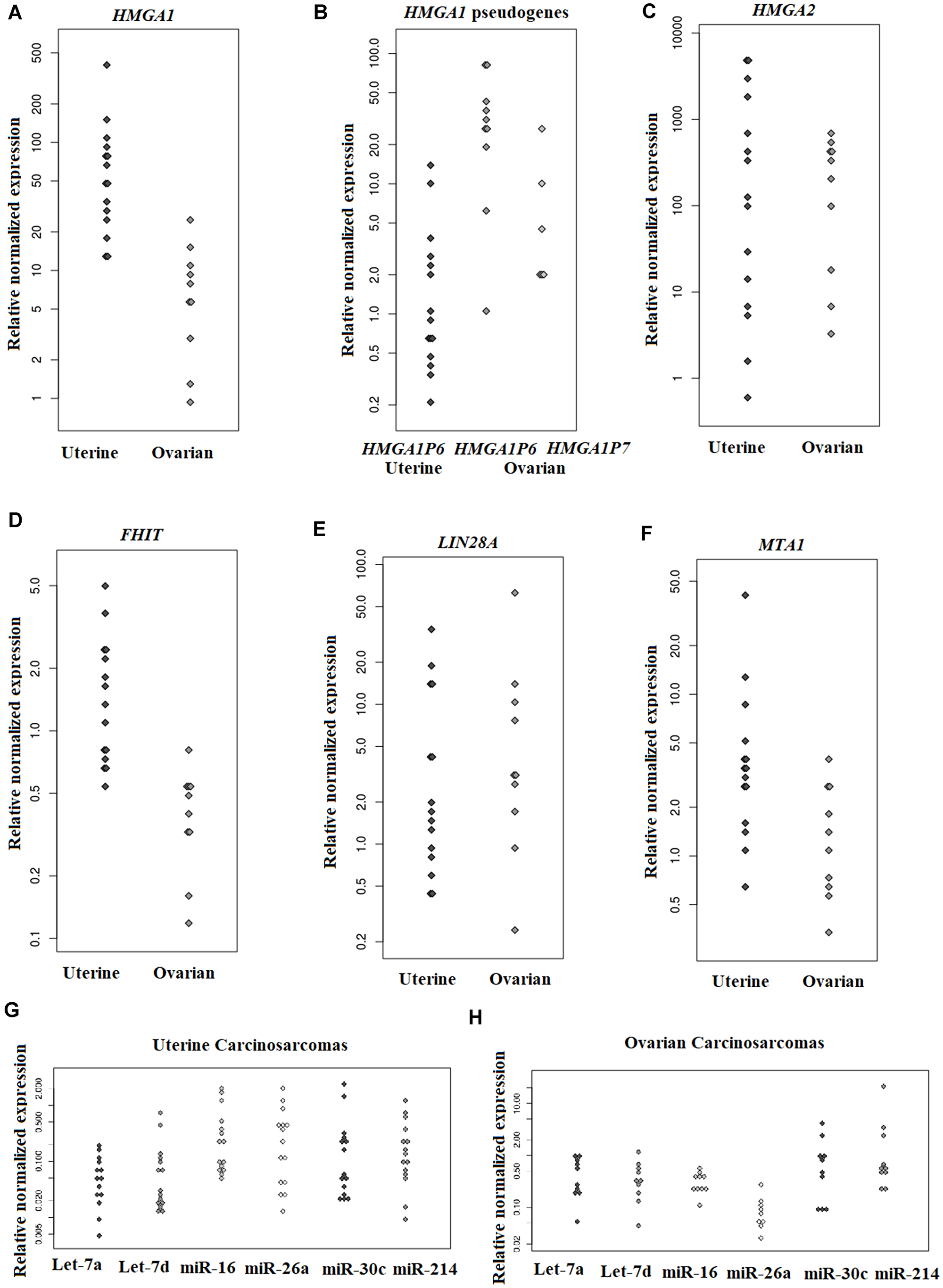
Figure 2: Genes and miRNA expression levels in uterine and ovarian carcinosarcomas assessed by Real-Time PCR. The relative expression of HMGA1 (A), HMGA1 pseudogenes (B), HMGA2 (C), FHIT (D), LIN28A (E), MTA1 (F) in uterine and ovarian CS; let-7a, let-d, miR16, miR-26a, miR-30c, and miR-214 in UCS (G) and in OCS (H).
The miRNAs let-7a, let-7d, miR-16, miR26a, and miR-30c were found downregulated in both UCS and OCS. miR-214 was downregulated in all UCS, whereas it was upregulated in three out of ten ovarian tumours but downregulated in the remaining seven (Table 3; Figures 2G and 2H). The Mann-Whitney U Test for statistical analysis was used to compare uterine and ovarian carcinosarcomas for gene and miRNA expression. No significantly different expression between the two tumour types (p > 0.05) was seen for any of the genes or miRNAs examined.
Table 3: Mean and median of genes and miRNA expression
| Gene | UCS | OCS | ||
|---|---|---|---|---|
| Mean | Median | Mean | Median | |
| HMGA1 | 81.3 | 47.1 | 8.5 | 7.8 |
| HMGA1P6 | 2.6 | 0.9 | 39.7 | 32.4 |
| HMGA1P7 | 5.1 | 2.0 | ||
| HMGA2 | 1146.2 | 117.7 | 279.2 | 310.2 |
| FHIT | 1.7 | 1.2 | 0.4 | 0.4 |
| LIN28A | 1.7 | 1.2 | 12.3 | 3.3 |
| MTA1 | 6.4 | 3.3 | 1.7 | 1.3 |
| miRNA | ||||
| let-7a | 0.06 | 0.04 | 0.5 | 0.6 |
| let-7d | 0.12 | 0.03 | 0.4 | 0.3 |
| miR-16 | 0.4 | 0.2 | 0.3 | 0.3 |
| miR26a | 0.4 | 0.21 | 0.09 | 0.07 |
| miR-30c | 0.3 | 0.05 | 1.16 | 0.1 |
| miR-214 | 0.2 | 0.1 | 3.5 | 0.6 |
We performed 3′ RACE-PCR on three tumours (cases 4, 18, and 25) that lacked 3′ sequences. In case 4, an UCS, exon 3 of HMGA2 was fused with part of the third intron, 78 kb downstream from the exon 3/intron 3 splicing site (Figure 3A). Case 25, a UCS, showed an in-frame fusion between HMGA2 (exon 3) and the Homo sapiens helicase (DNA) B (HELB; NM_033647; exon 3) located in the same chromosomal region (12q14.3) but 467 Kb distally (Figure 3B). Case 18, an OCS, did not give informative sequencing results.
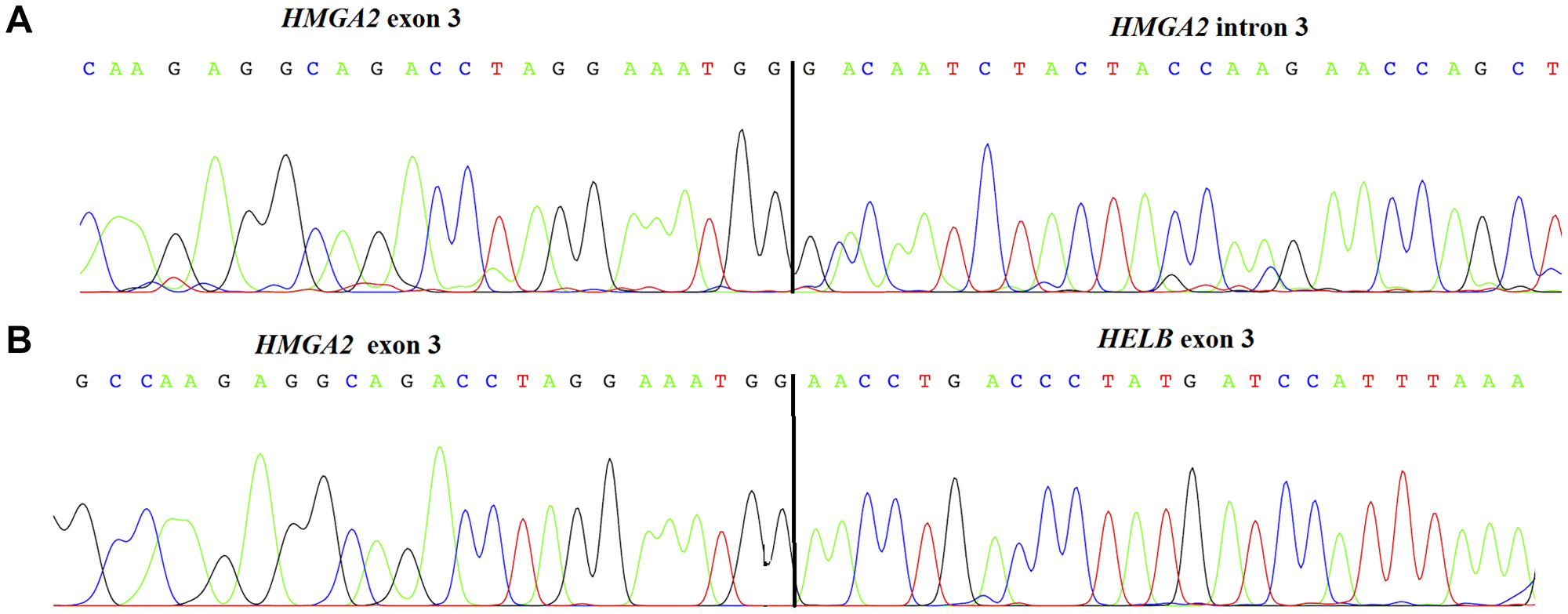
Figure 3: Chromatogram and sequence of HMGA2 truncated transcript found in an uterine carcinosarcoma (case 4) showing the junction between exon 3 and the intronic region (A). Chromatogram of HMGA2 truncated transcript found in a uterine carcinosarcomas (case 25) showing a fusion between HMGA2 and HELB (B).
DISCUSSION
In the present study, mutations in KRAS, PIK3CA, and TP53 were found in 6%, 31%, and 75% of UCS, respectively, in line with previous findings [7, 8, 10]; (COSMIC database https://cancer.sanger.ac.uk/cosmic). In OCS, KRAS and PIK3CA were not mutated, whereas 30% of OCS carried TP53 mutations.
Genetic alterations of TP53 have been thoroughly investigated in human cancer [13]. It is known that TP53 mutations occur during CS tumourigenesis, causing the gene to lose its tumour suppressive function, indicating its role as an early pathogenetic driver [8, 14]. The distribution pattern of TP53 mutations found by us was in line with that found in previous studies [7]. Alterations in TP53 were previously observed in most UCS and OCS analysed [8, 9]. The TP53 mutations targeted the core of the DNA-binding domain, resulting in loss of its regulatory function on gene expression and accumulation of non-functional p53 protein. We validated p53 expression by immunohistochemistry, finding a correlation between TP53 mutational status and p53 expression pattern. The latter analysis showed equal expression of the protein in both components (carcinomatous and sarcomatous) suggesting that there is no leading components for p53 expression as a “driving force” of tumourigenesis.
HMGA1 and HMGA2 are members of the high-mobility group AT-hook family and are involved in a variety of biological processes from chromosome dynamics to gene regulation [15]. They are usually expressed during embryonic development [15, 16] but not in adult normal tissues [17]. The genes were found overexpressed and/or targeted as part of the pathogenesis of many different tumours, both benign [18] and malignant [19], including mesenchymal [20] and epithelial [21] ones. HMGA proteins are involved in different pathogenic processes, but exert their main tumourigenic effect activating and sustaining epithelial-mesenchymal transition (EMT) [22]. We found HMGA1 overexpressed in both UCS and OCS. Interestingly, HMGA2 was expressed at higher levels than its homologue in UCS as well as in OCS. The mechanisms of regulation of these two genes are not fully understood, but non-coding RNA dysregulation and chromosomal alterations are the two main causes leading to upregulation of HMGA1 and HMGA2 in cancer [18, 20, 23, 24]. The HMGA1-targeting miRNAs let-7a [24], miR-26a [21], miR-16 [25], and miR-214 [26] were downregulated in CS of both sites in the present study, giving the impression that these cancers do not differ from other malignancies in this regard. The HMGA1 pseudogenes HMGA1P6 and HMGA1P7 were found to be implicated in the downregulation of the aforementioned miRNAs [27] and the overexpression of HMGA1. The HMGA1P6 and HMGA1P7 pseudogenes conserve seed matches for the HMGA1-targeting miRNAs and operate as decoys for these miRNAs, contributing to HMGA1 overexpression [28]. In UCS, only HMGA1P6 was expressed, while both HMGA1P6 and HMGA1P7 were expressed at high levels in OCS. The findings suggest that these pseudogenes may contribute to HMGA1 deregulation in gynaecological CS.
The mechanisms leading to expression of HMGA2 are still partly obscure, but interaction between miRNAs and the HMGA2 3′untranslated region (3′UTR) seems to be crucial [29]. It has been shown that the HMGA2 3′UTR has many regulatory sequences which are targeted by different families of miRNAs [29], and it is thought that miRNA-dependent repression is the main mechanism controlling HMGA2 expression [30–32]. We observed upregulation of HMGA2 with miRNA downregulation in both UCS and OCS, providing another piece of evidence that the interaction between the two is important also in gynaecological CS. Another indication pointing in the same direction has been the identification of disrupted forms of HMGA2, due to rearrangements of chromosomal band 12q15 (the band where the gene is located), that are consistently seen in different benign mesenchymal tumours but also in some malignant neoplasms such as ovarian carcinomas and leukemia [20, 33–36]. These alterations involve exon 3 and cause deletion of downstream regions leading to a truncated transcript that can evade miRNA-dependent gene silencing. As we have seen a 3′ rearranged form of HMGA2 in only two of 15 UCS and one of ten OCS, we hypothesize that mechanism(s) other than HMGA2-rearrangements may be active in these tumours.
The HMGA2-targeting miRNAs let-7a, let-7d, miR-30c, and miR-26a were found highly downregulated in all UCS examined. Only let-7a, let-7d, and miR-26a were downregulated in OCS, whereas miR-30c was normally expressed.
Allegedly, LIN28A causes downregulation of the let-7 family of miRNAs, inhibiting the maturation of both pri- and pre-let-7 [37]. The gene was found expressed in both UCS and OCS, suggesting possible involvement in the downregulation of let-7 miRNAs in CS generally.
Expression of FHIT and miR-30c has been shown to be inversely correlated with HMGA2 expression in lung cancer [31] and squamous cell carcinoma of the vulva [38]. FHIT and miR-30c downregulation causes HMGA2 upregulation promoting EMT [31, 38]. We did not find any similar correlation between FHIT and miR-30c in the CS analysed, as FHIT was normally expressed while miR30c was highly downregulated in UCS, whereas FHIT was downregulated while miR30c was normally expressed in OCS. We therefore suggest that other/additional mechanisms and/or genes are involved in the pathway leading to overexpression of HMGA2 in this tumour type. More specifically, there could be other molecules than FHIT involved in miR30c downregulation.
MTA1 has emerged as one of several highly deregulated oncogenes in human cancer, possibly because of its dual nature as corepressor and coactivator [39]. The MTA1 protein forms the NuRD chromatin remodeling complex and regulates expression of a wide range of genes involved in carcinogenesis such as HIFα [40] and ERα [41]. MTA1 is regulated by miR-30c and miRNA downregulation is associated with MTA1 upregulation in endometrial [42] and ovarian [43] cancer. In UCS, we found the same inverse correlation reported by others [42, 43] where MTA1 is overexpressed and miR-30c downregulated, whereas the expression levels of miR-30c and MTA1 in our series of OCS were generally normal.
In conclusion, our analyses showed that miRNAs responsible for HMGA expression are downregulated in CS of the female genital tract. The downregulation was more pronounced in UCS compared to OCS (the mean was 10-fold lower). This may explain the consistently higher levels of HMGA1 and HMGA2 in UCS compared to OCS. Future studies should be focused on seeing if mutations in the above-mentioned genes are present in both tumour components, i.e., the sarcomatous and carcinomatous areas, or only in one of them. Unfortunately, in our tumours these parts were so intermingled that it was not possible to separate them and run parallel tests.
Materials and Methods
Tumour material
The material consisted of fresh samples from 16 UCS and ten OCS surgically removed at The Norwegian Radium Hospital between 2000 and 2010. Four of the uterine carcinosarcomas were previously karyotyped and tested by comparative genomic hybridization (CGH) for chromosomal aberrations and genomic imbalances [44]. For historical reasons and to facilitate relevant electronic searches, we refer to all tumours arising in the uterine adnexa as ovarian throughout the manuscript; this should not be interpreted as reflecting certainty that they arise from cells of the ovary and not from the fallopian tube. All samples had a minimum of 50% of tumor cell content, the majority >80%; no difference was noted between uterine and ovarian tumors. The study was approved by the Regional Committee for Medical and Health Research Ethics, South-East Norway (REK Sør-Øst; http://helseforskning.etikkom.no).
DNA and RNA extraction and cDNA synthesis
DNA extraction was performed using the Maxwell 16 extractor (Promega, Madison, WI, USA) and Maxwell 16 Tissue DNA Purification kit (Promega) according to the manufacturer’s recommendations. RNA extraction was performed using the miRNeasy kit (Qiagen, Hilden, Germany) and QIAcube (Qiagen). The concentration was measured with QIAxel (Qiagen). One microgram of extracted RNA was reverse-transcribed in a 20 μL reaction volume using the iScript Advanced cDNA Synthesis kit according to the manufacturer’s instructions (Bio-Rad Laboratories, Oslo, Norway).
Mutational and methylation analyses
Mutational analyses of IDH1, IDH2, TERT, CTNNB1, BRAF, H3F3A, and TP53 were performed according to previously described protocols [45, 46]. Primers for HRAS, KRAS, NRAS, and PIK3CA are listed in Table 4. The mutational analyses were performed using M13-linked PCR primers designed to flank and amplify targeted sequences. The thermal cycling for HRAS and NRAS included an initial step at 95° C for 10 min followed by 35 cycles at 96° C for 3 sec, 58° C for 15 sec, 30 sec at 68° C, and a final step at 72° C for 2 min. The thermal cycling for KRAS was set to 94° C for 30 sec followed by 35 cycles of 7 sec at 98° C, 30 sec at 54° C, 1 min at 77° C, and a final step at 68° C for 5 min. The thermal cycling for PIK3CA was set to 95° C for 10 min followed by 35 cycles of 3 sec at 96° C, 15 sec at 62° C, 30 sec at 68° C, and a final step at 72° C for 2 min. Direct sequencing was performed using a 3500 Genetic Analyzer (Applied Biosystems). The BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and BLAT (https://genome-euro.ucsc.edu/cgi-bin/hgBlat) programs were used for computer analysis of sequence data.
Table 4: Primers used for molecular investigations
| Primer name | Sequence | Position | Gene | Accession number |
|---|---|---|---|---|
| Mutational analyses | ||||
| HRAS-EXON2FW | 5′-CATTAAGAGCAAGTGGGGGCG-3′ | 5973–5993 | HRAS | NG_007666.1 |
| HRAS-EXON2REV | 5′-CGAGGGACTCCCCTCCTCTA-3′ | 6466–6485 | HRAS | NG_007666.1 |
| HRAS-EXON3FW | 5′-AGGGGCATGAGAGGTACCAG-3′ | 6516–6535 | HRAS | NG_007666.1 |
| HRAS-EXON3REV | 5′-CATCCAGGACATGCGCAGA-3′ | 6871–6889 | HRAS | NG_007666.1 |
| KRAS-EXON2FW | 5′-AAGGTACTGGTGGAGTATTTG-3′ | 10439–10459 | KRAS | NG_007524.1 |
| KRAS-EXON2REV | 5′-ATGAAAATGGTCAGAGAAACC-3′ | 10707–10727 | KRAS | NG_007524.1 |
| KRAS-EXON3FW | 5′-TTGAAGTAAAAGGTGCACTG-3′ | 28457–28475 | KRAS | NG_007524.1 |
| KRAS-EXON3REV | 5′-AATTACTCCTTAATGTCAGCTT-3′ | 28710–28731 | KRAS | NG_007524.1 |
| NRAS EX 2 FW | 5′-GGCTCGCCAATTAACCCTGA-3′ | 5681–5700 | NRAS | NG_007572.1 |
| NRAS EX 2 REV | 5′-TCCGACAAGTGAGAGACAGGA-3′ | 5876–5886 | NRAS | NG_007572.1 |
| NRAS EX 3 FW | 5′-GCATTGCATTCCCTGTGGTTT-3′ | 7841–7871 | NRAS | NG_007572.1 |
| NRAS EX 3 REV | 5′-GTGTGGTAACCTCATTTCCCCA-3′ | 8150–8171 | NRAS | NG_007572.1 |
| PIK3CA- Ex10F1 | 5′-ATCATCTGTGAATCCAGAGGGGAA-3′ | 74619–74642 | PIK3CA | NG_027450.2 |
| PIK3CA- Ex10R1 | 5′- CATGCTGAGATCAGCCAAATTCAG-3′ | 74868–74891 | PIK3CA | NG_012113.2 |
| PIK3CA- Ex21F1 | 5′-CATCATTTGCTCCAAACTGACCAA-3′ | 90528–90551 | PIK3CA | NG_012113.2 |
| PIK3CA- Ex21R1 | 5′-TCATGGATTGTGCAATTCCTATGC-3′ | 90922–90945 | PIK3CA | NG_012113.2 |
| Expression analyses | ||||
| HMGA2-846F1 | 5′ –CCACTTCAGCCCAGGGACAACCT- 3′ | 846–868 | HMGA2 | NM_003483.4 |
| HMGA2-1021R1 | 5′ -CCTCTTGGCCGTTTTTCTCCAGTG- 3′ | 1021–1044 | HMGA2 | NM_003483.4 |
| HMGA2-1112R1 | 5′ –CCTCTTCGGCAGACTCTTGTGAGGA3′ | 1112–1136 | HMGA2 | NM_003483.4 |
| HMGA2F1 | 5′-TCAGAAGAGAGGACGCGG-3′ | 883–900 | HMGA2 | NM_003483.4 |
| HELBR1 | 5′-CTTCAAATCAGTCATTCTTTGGGT- 3′ | 66306281–66306304* | HELB | NM_033647.4 |
| HMGA2F4 | 5′ -AAAAACAAGAGTCCCTCTAAAGCA- 3′ | 977–1000 | HMGA2 | NM_003483.4 |
| HELBR4 | 5′-TTGCAGTTTCCGAAGATAATGGA- 3′ | 693–715 | HELB | NM_033647.4 |
Methylation-specific quantitative polymerase chain reaction (MSP-qPCR) analysis of the MGMT promoter was performed as reported earlier [45].
Real-Time polymerase chain reaction (Real-Time PCR)
Expression level of the selected genes and miRNAs was assessed by Real-Time PCR using the CFX96 Touch Real-Time detection system (Bio-Rad Laboratories, Oslo, Norway). The reactions were carried out in triplicate using the TaqMan Universal Master Mix II with UNG (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s protocol. Human Universe Reference Total RNA (Clontech, Mountain View, CA, USA) was used as internal reaction control. The Human Ovary Total RNA (MVP Total RNA Human Ovary, Agilent Technologies, Santa Clara, CA, USA) and one sample of normal uterus tissue were used as reference for relative expression normalization. Two stably expressed known genes (housekeeping genes) were used as references as these were previously evaluated as stable in gynaecological tumours [47]. The Real-Time data were analysed with Bio-Rad CFX manager 3.1 (Bio-Rad). The normalized expression was calculated using the 2-ΔΔCt (Livak) method [48].
One μg of extracted total RNA was reverse-transcribed in a 20 μL reaction volume using iScript Advanced cDNA Synthesis Kit according to the manufacturer’s instructions (Bio-Rad Laboratories, Oslo, Norway). Gene expression was assessed with Real-Time PCR using the TaqMan Gene Expression Assays (Applied Biosystems) for the following genes: HMGA1 (Hs_00852949_g1), HMGA2 (Hs_04397751_m1), FHIT (Hs_00179987_m1), LIN28A (Hs_00702808_Gh), HMGA1P6 (ARYMJHZ), and HMGA1P7 (Hs04232395_m1). The UBC (Hs01871556_m1) and TBP (Hs00427620_m1) genes were used as references.
Ten ng of total RNA were reverse transcribed using the TaqMan microRNA Reverse Transcription Kit (Applied Biosystems) following the manufacturer’s protocol. miRNA expression was assessed with Real-Time PCR using the TaqMan microRNA assays (Applied Biosystems) for let-7a (RT: 000377), let-7d (RT: 002283), miR-26a (RT: 000405), miR-16 (RT: 000391), miR-214 (TM: 002306), and miR30c (TM:000419). The RNU6B gene (TM:001093) was used as a reference as it was previously validated as stable in different gynaecological tumours [38, 49].
Reverse transcriptase-polymerase chain reaction (RT-PCR)
cDNA equivalent to 10 ng RNA was amplified using the Takara Premix Ex Taq (Takara-Bio, Europe/SAS, Saint-Germain-en-Laye, France). The primers used for PCR reactions are listed in Table 4. The primer combination HMGA2-846F1 and HMGA2-1021R1 was used to amplify the region between exons 1 and 3, whereas the primer combination HMGA2-846F1 and HMGA2-1112R1 was used for exons 1 to 5 (Table 4). The PCR cycling program was previously reported [35].
3′ Rapid amplification of cDNA ends – PCR (3′ RACE–PCR)
For 3′-RACE-PCR, 100 ng of total RNA were reverse-transcribed in a 20 μL reaction volume using a previously described protocol [45]. To validate the fusion between HMGA2 (exon 3) and HELB (exon 3), RT-PCR was performed with specific primer combinations for the two genes. The PCR cycling program was: 30 sec at 94° C followed by 35 cycles of 7 sec at 98° C and 1 min at 55° C, 1 min at 72° C, and a final step at 72° C for 2 min.
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections were analysed for p53 protein expression in 19 tumours from which material was available using the Dako EnVision™ Flex+ System (K8012; Dako, Glostrup, Denmark). Epitope unmasking was carried out in a high pH solution. Sections were incubated with a 0.3% hydrogen peroxide (H2O2) solution for 5 min to block endogenous tissue peroxidase activity. Sections were then incubated with a mouse monoclonal p53 primary antibody (clone DO-1, catalogue #sc-126, Santa Cruz Biotechnology, Santa Cruz CA, USA) and treated with EnVision™ Flex+ mouse linker (15 min) and EnVision™ Flex/HRP enzyme (30 min), stained for 10 min with 3`3 diaminobenzidine tetrahydrochloride (DAB), counterstained with haematoxylin, dehydrated, and mounted in Richard-Allan Scientific Cyto seal XYL (Thermo Fisher Scientific, Waltham, MA, USA). Positive control consisted of colon carcinoma.
CONFLICTS OF INTEREST
None.
GRANT SUPPORT
This work was supported by grants from the John and Inger Fredriksen Foundation, the Anders Jahre foundation through UNIFOR (University of Oslo), and the South-East Norway Regional Health Authority (Helse Sør-Øst).
References
1. D’Angelo E, Prat J. Pathology of mixed Müllerian tumours. Best Pract Res Clin Obstet Gynaecol. 2011; 25:705–18. https://doi.org/10.1016/j.bpobgyn.2011.05.010. [PubMed].
2. Singh R. Review literature on uterine carcinosarcoma. J Cancer Res Ther. 2014; 10:461–68. [PubMed].
3. Berton-Rigaud D, Devouassoux-Shisheboran M, Ledermann JA, Leitao MM, Powell MA, Poveda A, Beale P, Glasspool RM, Creutzberg CL, Harter P, Kim JW, Reed NS, Ray-Coquard I. Gynecologic Cancer InterGroup (GCIG) consensus review for uterine and ovarian carcinosarcoma. Int J Gynecol Cancer. 2014; 24:S55–60. https://doi.org/10.1097/IGC.0000000000000228. [PubMed].
4. Rauh-Hain JA, Gonzalez R, Bregar AJ, Clemmer J, Hernández-Blanquisett A, Clark RM, Schorge JO, Del Carmen MG. Patterns of care, predictors and outcomes of chemotherapy for ovarian carcinosarcoma: A National Cancer Database analysis. Gynecol Oncol. 2016; 142:38–43. https://doi.org/10.1016/j.ygyno.2016.04.025. [PubMed].
5. George EM, Herzog TJ, Neugut AI, Lu YS, Burke WM, Lewin SN, Hershman DL, Wright JD. Carcinosarcoma of the ovary: natural history, patterns of treatment, and outcome. Gynecol Oncol. 2013; 131:42–45. https://doi.org/10.1016/j.ygyno.2013.06.034. [PubMed].
6. Vitale SG, Laganà AS, Capriglione S, Angioli R, La Rosa VL, Lopez S, Valenti G, Sapia F, Sarpietro G, Butticè S, Tuscano C, Fanale D, Tropea A, Rossetti D. Target Therapies for Uterine Carcinosarcomas: Current Evidence and Future Perspectives. Int J Mol Sci. 2017; 18:E1100. https://doi.org/10.3390/ijms18051100. [PubMed].
7. Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, Angiuoli SV, Sausen M, Kann L, Shukla M, Makar R, Wood LD, Diaz LA
8. Zhao S, Bellone S, Lopez S, Thakral D, Schwab C, English DP, Black J, Cocco E, Choi J, Zammataro L, Predolini F, Bonazzoli E, Bi M, et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2016; 113:12238–43. https://doi.org/10.1073/pnas.1614120113. [PubMed].
9. Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, Ling S, Soslow RA, Broaddus RR, Zuna RE, Robertson G, Laird PW, et al, and Cancer Genome Atlas Research Network. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell. 2017; 31:411–23. https://doi.org/10.1016/j.ccell.2017.02.010. [PubMed].
10. Growdon WB, Roussel BN, Scialabba VL, Foster R, Dias-Santagata D, Iafrate AJ, Ellisen LW, Tambouret RH, Rueda BR, Borger DR. Tissue-specific signatures of activating PIK3CA and RAS mutations in carcinosarcomas of gynecologic origin. Gynecol Oncol. 2011; 121:212–17. https://doi.org/10.1016/j.ygyno.2010.11.039. [PubMed].
11. de Jong RA, Nijman HW, Wijbrandi TF, Reyners AK, Boezen HM, Hollema H. Molecular markers and clinical behavior of uterine carcinosarcomas: focus on the epithelial tumor component. Mod Pathol. 2011; 24:1368–79. https://doi.org/10.1038/modpathol.2011.88. [PubMed].
12. McConechy MK, Hoang LN, Chui MH, Senz J, Yang W, Rozenberg N, Mackenzie R, McAlpine JN, Huntsman DG, Clarke BA, Gilks CB, Lee CH. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J Pathol Clin Res. 2015; 1:173–85. https://doi.org/10.1002/cjp2.18. [PubMed].
13. Zhang W, Edwards A, Flemington EK, Zhang K. Significant Prognostic Features and Patterns of Somatic TP53 Mutations in Human Cancers. Cancer Inform. 2017; 16:1176935117691267. https://doi.org/10.1177/1176935117691267. [PubMed].
14. Semczuk A, Ignatov A, Obrzut B, Reventos J, Rechberger T. Role of p53 pathway alterations in uterine carcinosarcomas (malignant mixed Müllerian tumors). Oncology. 2014; 87:193–204. https://doi.org/10.1159/000363574. [PubMed].
15. Cleynen I, Van de Ven WJ. The HMGA proteins: a myriad of functions (Review). Int J Oncol. 2008; 32:289–305. https://doi.org/10.3892/ijo.32.2.289. [PubMed].
16. Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M, Simeone A, Fusco A. High level expression of the HMGI (Y) gene during embryonic development. Oncogene. 1996; 13:2439–46. [PubMed].
17. Rogalla P, Drechsler K, Frey G, Hennig Y, Helmke B, Bonk U, Bullerdiek J. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am J Pathol. 1996; 149:775–79. [PubMed].
18. Panagopoulos I, Gorunova L, Bjerkehagen B, Lobmaier I, Heim S. Fusion of the TBL1XR1 and HMGA1 genes in splenic hemangioma with t(3;6)(q26;p21). Int J Oncol. 2016; 48:1242–50. https://doi.org/10.3892/ijo.2015.3310. [PubMed].
19. Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, D’Armiento J, Chada K. HMGA2 is a driver of tumor metastasis. Cancer Res. 2013; 73:4289–99. https://doi.org/10.1158/0008-5472.CAN-12-3848. [PubMed].
20. Agostini A, Gorunova L, Bjerkehagen B, Lobmaier I, Heim S, Panagopoulos I. Molecular characterization of the t(4;12)(q27~28;q14~15) chromosomal rearrangement in lipoma. Oncol Lett. 2016; 12:1701–04. https://doi.org/10.3892/ol.2016.4834. [PubMed].
21. Sekimoto N, Suzuki A, Suzuki Y, Sugano S. Expression of miR26a exhibits a negative correlation with HMGA1 and regulates cancer progression by targeting HMGA1 in lung adenocarcinoma cells. Mol Med Rep. 2017; 15:534–42. https://doi.org/10.3892/mmr.2016.6053. [PubMed].
22. Fedele M, Fusco A. HMGA and cancer. Biochim Biophys Acta. 2010; 1799:48–54. https://doi.org/10.1016/j.bbagrm.2009.11.007. [PubMed].
23. Wu A, Wu K, Li J, Mo Y, Lin Y, Wang Y, Shen X, Li S, Li L, Yang Z. Let-7a inhibits migration, invasion and epithelial-mesenchymal transition by targeting HMGA2 in nasopharyngeal carcinoma. J Transl Med. 2015; 13:105. https://doi.org/10.1186/s12967-015-0462-8. [PubMed].
24. Liu K, Zhang C, Li T, Ding Y, Tu T, Zhou F, Qi W, Chen H, Sun X. Let-7a inhibits growth and migration of breast cancer cells by targeting HMGA1. Int J Oncol. 2015; 46:2526–34. https://doi.org/10.3892/ijo.2015.2949. [PubMed].
25. Kaddar T, Rouault JP, Chien WW, Chebel A, Gadoux M, Salles G, Ffrench M, Magaud JP. Two new miR-16 targets: caprin-1 and HMGA1, proteins implicated in cell proliferation. Biol Cell. 2009; 101:511–24. https://doi.org/10.1042/BC20080213. [PubMed].
26. Chandrasekaran KS, Sathyanarayanan A, Karunagaran D. MicroRNA-214 suppresses growth, migration and invasion through a novel target, high mobility group AT-hook 1, in human cervical and colorectal cancer cells. Br J Cancer. 2016; 115:741–51. https://doi.org/10.1038/bjc.2016.234. [PubMed].
27. Esposito F, De Martino M, Forzati F, Fusco A. HMGA1-pseudogene overexpression contributes to cancer progression. Cell Cycle. 2014; 13:3636–39. https://doi.org/10.4161/15384101.2014.974440. [PubMed].
28. Esposito F, De Martino M, Petti MG, Forzati F, Tornincasa M, Federico A, Arra C, Pierantoni GM, Fusco A. HMGA1 pseudogenes as candidate proto-oncogenic competitive endogenous RNAs. Oncotarget. 2014; 5:8341–54. https://doi.org/10.18632/oncotarget.2202. [PubMed].
29. Kristjánsdóttir K, Fogarty EA, Grimson A. Systematic analysis of the Hmga2 3′ UTR identifies many independent regulatory sequences and a novel interaction between distal sites. RNA. 2015; 21:1346–60. https://doi.org/10.1261/rna.051177.115. [PubMed].
30. Liu Y, Liang H, Jiang X. MiR-1297 promotes apoptosis and inhibits the proliferation and invasion of hepatocellular carcinoma cells by targeting HMGA2. Int J Mol Med. 2015; 36:1345–52. https://doi.org/10.3892/ijmm.2015.2341. [PubMed].
31. Suh SS, Yoo JY, Cui R, Kaur B, Huebner K, Lee TK, Aqeilan RI, Croce CM. FHIT suppresses epithelial-mesenchymal transition (EMT) and metastasis in lung cancer through modulation of microRNAs. PLoS Genet. 2014; 10:e1004652. https://doi.org/10.1371/journal.pgen.1004652. [PubMed].
32. Lin Y, Liu AY, Fan C, Zheng H, Li Y, Zhang C, Wu S, Yu D, Huang Z, Liu F, Luo Q, Yang CJ, Ouyang G. MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1. Sci Rep. 2015; 5:9995. https://doi.org/10.1038/srep09995. [PubMed].
33. Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat Genet. 1995; 10:436–44. https://doi.org/10.1038/ng0895-436. [PubMed].
34. Bartuma H, Hallor KH, Panagopoulos I, Collin A, Rydholm A, Gustafson P, Bauer HC, Brosjö O, Domanski HA, Mandahl N, Mertens F. Assessment of the clinical and molecular impact of different cytogenetic subgroups in a series of 272 lipomas with abnormal karyotype. Genes Chromosomes Cancer. 2007; 46:594–606. https://doi.org/10.1002/gcc.20445. [PubMed].
35. Agostini A, Panagopoulos I, Davidson B, Trope CG, Heim S, Micci F. A novel truncated form of HMGA2 in tumors of the ovaries. Oncol Lett. 2016; 12:1559–1563. https://doi.org/10.3892/ol.2016.4805. [PubMed].
36. Nyquist KB, Panagopoulos I, Thorsen J, Roberto R, Wik HS, Tierens A, Heim S, Micci F. t(12;13)(q14;q31) leading to HMGA2 upregulation in acute myeloid leukaemia. Br J Haematol. 2012; 157:769–71. https://doi.org/10.1111/j.1365-2141.2012.09081.x. [PubMed].
37. Wang T, Wang G, Hao D, Liu X, Wang D, Ning N, Li X. Aberrant regulation of the LIN28A/LIN28B and let-7 loop in human malignant tumors and its effects on the hallmarks of cancer. Mol Cancer. 2015; 14:125. https://doi.org/10.1186/s12943-015-0402-5. [PubMed].
38. Agostini A, Brunetti M, Davidson B, Trope CG, Heim S, Panagopoulos I, Micci F. Expressions of miR-30c and let-7a are inversely correlated with HMGA2 expression in squamous cell carcinoma of the vulva. Oncotarget. 2016; 7:85058–62. https://doi.org/10.18632/oncotarget.13187. [PubMed].
39. Sen N, Gui B, Kumar R. Role of MTA1 in cancer progression and metastasis. Cancer Metastasis Rev. 2014; 33:879–89. https://doi.org/10.1007/s10555-014-9515-3. [PubMed].
40. Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J. 2006; 25:1231–41. https://doi.org/10.1038/sj.emboj.7601025. [PubMed].
41. Kang HJ, Lee MH, Kang HL, Kim SH, Ahn JR, Na H, Na TY, Kim YN, Seong JK, Lee MO. Differential regulation of estrogen receptor α expression in breast cancer cells by metastasis-associated protein 1. Cancer Res. 2014; 74:1484–94. https://doi.org/10.1158/0008-5472.CAN-13-2020. [PubMed].
42. Kong X, Xu X, Yan Y, Guo F, Li J, Hu Y, Zhou H, Xun Q. Estrogen regulates the tumour suppressor MiRNA-30c and its target gene, MTA-1, in endometrial cancer. PLoS One. 2014; 9:e90810. https://doi.org/10.1371/journal.pone.0090810. [PubMed].
43. Wang X, Qiu LW, Peng C, Zhong SP, Ye L, Wang D. MicroRNA-30c inhibits metastasis of ovarian cancer by targeting metastasis-associated gene 1. J Cancer Res Ther. 2017; 13:676–82. https://doi.org/10.4103/jcrt.JCRT_132_17. [PubMed].
44. Micci F, Teixeira MR, Haugom L, Kristensen G, Abeler VM, Heim S. Genomic aberrations in carcinomas of the uterine corpus. Genes Chromosomes Cancer. 2004; 40:229–46. https://doi.org/10.1002/gcc.20038. [PubMed].
45. Agostini A, Panagopoulos I, Andersen HK, Johannesen LE, Davidson B, Tropé CG, Heim S, Micci F. HMGA2 expression pattern and TERT mutations in tumors of the vulva. Oncol Rep. 2015; 33:2675–80. https://doi.org/10.3892/or.2015.3882. [PubMed].
46. Malcikova J, Tausch E, Rossi D, Sutton LA, Soussi T, Zenz T, Kater AP, Niemann CU, Gonzalez D, Davi F, Gonzalez Diaz M, Moreno C, Gaidano G, et al, and European Research Initiative on Chronic Lymphocytic Leukemia (ERIC) — TP53 network. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia. 2018; 32:1070–80. https://doi.org/10.1038/s41375-017-0007-7. [PubMed].
47. Kowalewska M, Danska-Bidzinska A, Bakula-Zalewska E, Bidzinski M. Identification of suitable reference genes for gene expression measurement in uterine sarcoma and carcinosarcoma tumors. Clin Biochem. 2012; 45:368–71. https://doi.org/10.1016/j.clinbiochem.2012.01.001. [PubMed].
48. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–08. https://doi.org/10.1006/meth.2001.1262. [PubMed].
49. Agostini A, Brunetti M, Davidson B, Tropé CG, Heim S, Panagopoulos I, Micci F. Genomic imbalances are involved in miR-30c and let-7a deregulation in ovarian tumors: implications for HMGA2 expression. Oncotarget. 2017; 8:21554–60. https://doi.org/10.18632/oncotarget.15795. [PubMed].